
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctional polyoxometalates from solvothermal reactions of VOSO4 with tripodal alkoxides – a study on the reactivity of different “tris” derivatives†
Olaf
Nachtigall
,
Adelheid
Hagenbach
,
Jelena
Wiecko
,
Dieter
Lentz
,
Ulrich
Abram
and
Johann
Spandl
*
Freie Universität Berlin, Institut für Chemie und Biochemie, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: jspandl@chemie.fu-berlin.de
First published on 8th December 2016
Abstract
We report a study on the structure directing effects of functional groups and counterions. The aim was to find a facile and high yielding synthetic procedure to obtain polyoxometalate (POM) building blocks for post-functionalisation. Therefore, solvothermal reactions of VOSO4 with various tris(hydroxymethyl)methane derivatives in alkaline methanolic solutions were investigated. In doing so, new POM fragments were isolated and characterised. The binding modes of the functionalised tripodal alkoxides turned out to be surprisingly different.
Introduction
Early transition metals like vanadium tend to form anionic metal-oxygen clusters with well-defined structures. A common trend is to introduce heterometals by so-called “capping” to these polyoxometalates (POMs), meaning that some metal ions are replaced by ions of another metal within the metalate structure.1–7 Thereby, the overall charge of the cluster is often reduced and physical characteristics are altered depending on the added metal. Recent examples are vanadium capped Dawson-type tungstates or molybdates.1–3 Especially the [P2V3W15O62]6− aggregate drew a lot of attention. Therein, three W(VI) ions of the [P2W18O62]9− are replaced by three V(V) ions. These heterometals form a [V3O13] fragment within the Dawson-type tungstate and introduce a new reactive side to the polyoxometalate.Another way to tune the properties of POMs is the functionalisation with organic ligands. An established method is to use 2-(hydroxymethyl)propane-1,3-diol derivatives, which are also referred to as tris(hydroxymethyl)methane (tris) derivatives.8–13 As depicted in Fig. 1, those tripodal alkoxide ligands are typically found capping tetrahedral voids at the surface of vanadium-based POMs. Thereby, the whole structure is stabilised and the charge of the aggregate is reduced, as oxo groups are replaced by alkoxo groups. By employing such organic ligands, even neutral aggregates may be obtained.
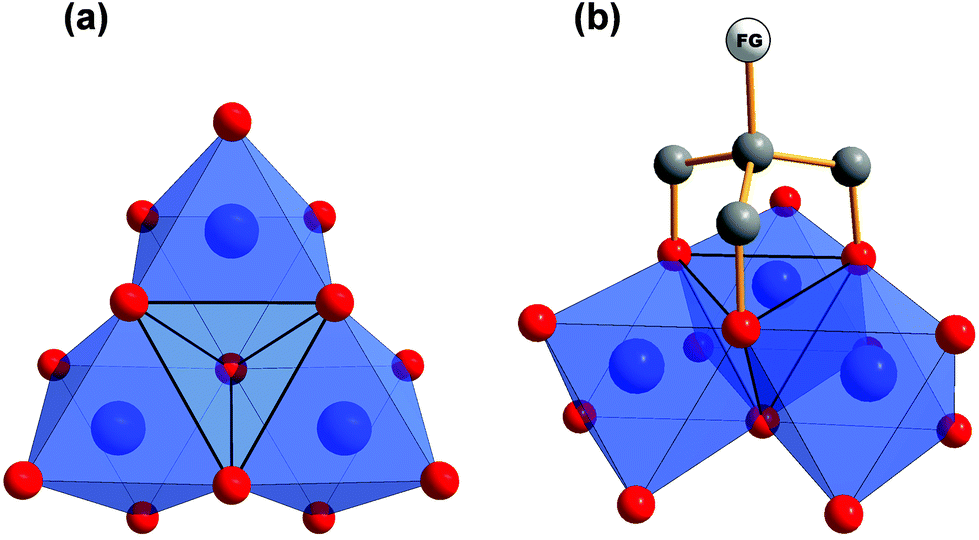 | ||
| Fig. 1 (a) Polyhedra plot of a [V3O13] using Diamond software.38 The tetrahedral void in the centre is highlighted. (b) Tripodal “tris” ligand introducing a functional group (FG) by capping the tetrahedral void. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red; carbon atoms, grey. | ||
Our aim was to synthesise versatile building blocks which can be modified by post-functionalisation. Earlier studies showed that interesting coordination complexes, metal–organic frameworks (MOFs), or other hybrid nanoscale systems may be constructed that way.14–20 To achieve this goal, we have tried to introduce different functionalities at the surface of our POMs by using the functional “tris” derivatives shown in Fig. 2.
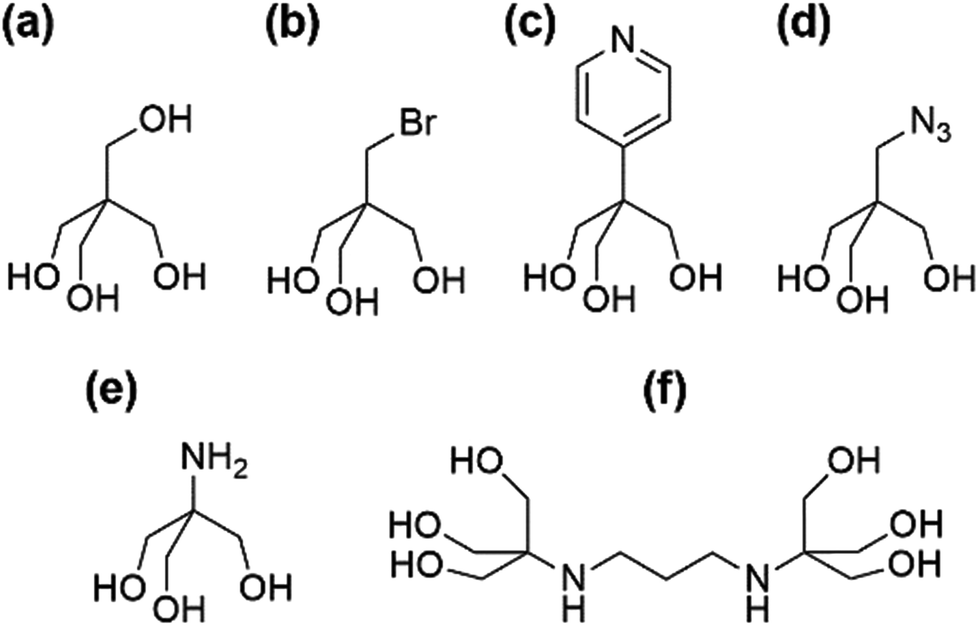 | ||
| Fig. 2 Functional “tris” derivatives. (a) Tris-CH2OH (pentaerythritol). (b) Tris-CH2Br (pentaerythritol monobromide.) (c) Tris-pyridine (2-(hydroxymethyl)-2-(pyridin-4-yl)-propane-1,3-diol). (d) Tris-CH2N3 (2-(azidomethyl)-2-(hydroxymethyl)-propane-1,3-diol). (e) Tris-NH2 (“TRIS”, tris(hydroxymethyl)aminomethane). (f) Tris-NH-(CH2)3-NH-tris (bis–tris propane). | ||
The well-known pentaerythrithol (Fig. 2a) is bearing a fourth hydroxymethyl group, which could be useful for silyl or sulfonyl ether formations. The additional bromomethyl group of pentaerythritol monobromide (Fig. 2b) could react with alcoholates in Williamson ether syntheses. More promising are the pyridyl (Fig. 2c) and azido (Fig. 2d) functionalities. The pyridyl group is readily applicable in coordination chemistry while the azido group has a potential synthetic application as 1,3-dipole in cycloadditions like the commonly copper catalysed Huisgen reaction. The most established functional “tris” derivative in POM chemistry is tris(hydroxymethyl)-aminomethane (Fig. 2e) which is often directly referred to as “TRIS”. Usually, it is further functionalised by amide couplings before being attached to POMs. Its commercially available dimer “bis–tris propane” (Fig. 2f) is not very valuable for post-functionalisation, but was used for reasons of comparison.
Results and discussion
Functionalised tetranuclear polyoxoalkoxovanadates
Functional groups on tripodal ligands can have a major impact on the POM structure. We found that solvothermal reactions of VOSO4 with tripodal ligands and tetramethylammonium hydroxide in methanol at 125 °C usually yield blue tetranuclear polyoxoalkoxovanadates. Within these planar structures, V(IV) ions are octahedrally coordinated by oxo ligands (Fig. 3a). Four [VO6] units build a [V4O16] fragment by being connected via common edges (Fig. 3b). This fragment has already been reported for vanadium in oxidation states between +II to +IV.21,22 Also, such [V4] aggregates were functionalised with tripodal ligands before.23,24 This POM structure can be considered as an extract of the well-known decavanadate structure25,26 or other polyoxovanadates.27–29 From another perspective, it is an extension of the [V3O13] fragment by an additional [VO6] building block.30–32 Other known [V4] aggregates are the non-planar butterfly complexes.33–37 | ||
| Fig. 3 (a) Vanadium–oxygen framework of [V4O16] aggregates. (b) Polyhedra plot of [V4O16] fragment. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red. | ||
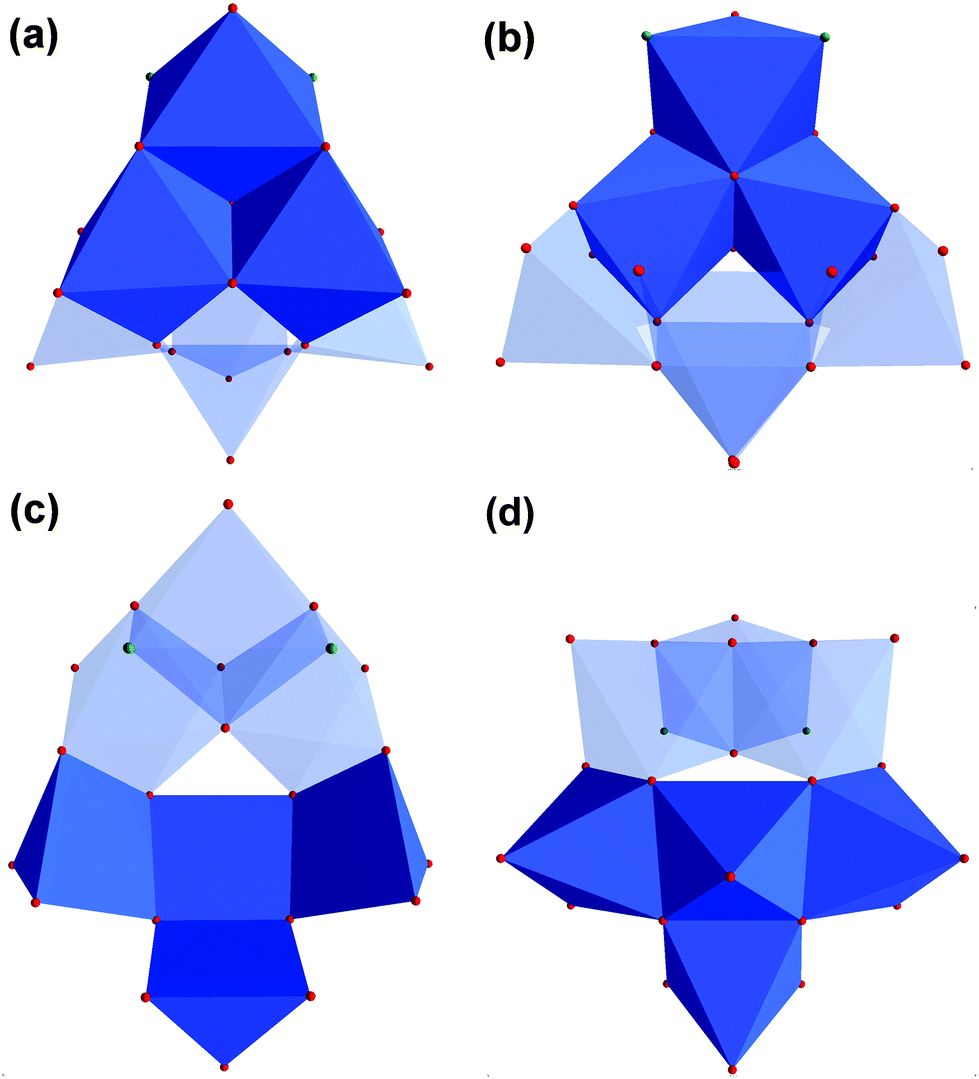
For our examples, two tripodal ligands are capping the tetrahedral voids on both sides of the aggregates. Bidentate sulfato and methanol ligands are bound to the corners of the POM structure. The different functional groups (R) are pointing away from the vanadium–oxygen framework (Fig. 4a–d). The composition of these ionic compounds was determined to be [N(CH3)4]2[V4O4(CH3OH)2(SO4)2((OCH2)3C-R)2] (R: –CH2OH (I), –CH2Br (II), –C5H4N (III), –CH2N3 (IV)).
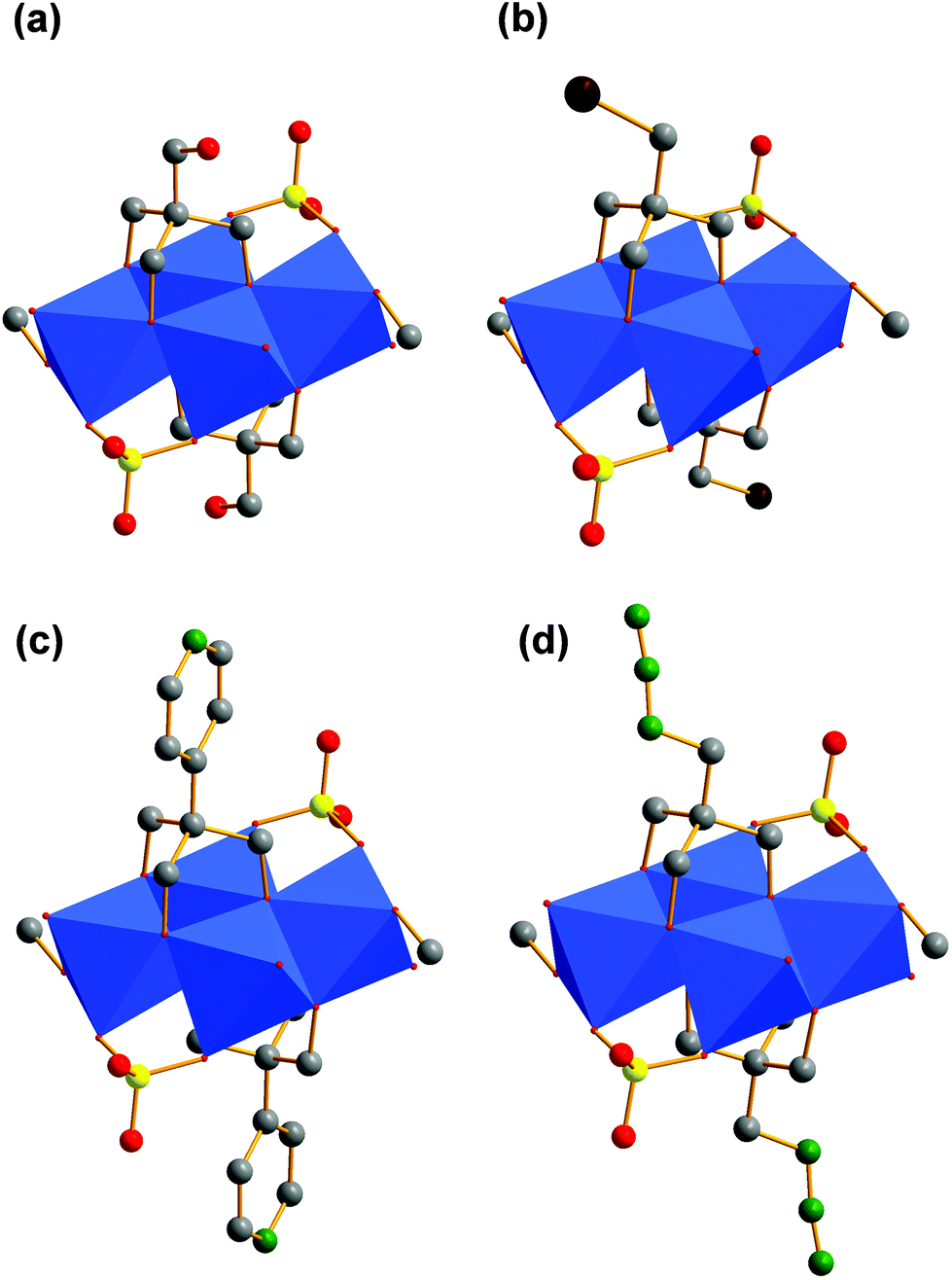 | ||
| Fig. 4 (a) Polyhedra plot of I. (b) Polyhedra plot of II. (c) Polyhedra plot of III. (d) Polyhedra plot of IV. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red; carbon atoms, grey; sulphur atoms, yellow; bromine atoms, brown; nitrogen atoms, green. | ||
Hexanuclear polyoxoalkoxovanadate
If the tripodal ligand with a functional amino group (tris-NH2) is heated with VOSO4 and tetramethylammonium hydroxide in methanol under solvothermal conditions at 125 °C, the grey compound [N(CH3)4]3[V6O9(SO4)3((OCH2)2(HOCH2)C-NH3)3] (V) is produced (Fig. 5). The colour change from blue to grey can be assigned to the different coordination sphere of vanadium. Inside the ionic compound V, the V(IV) ions are coordinated by oxo ligands in a square pyramidal manner (Fig. 6). The six [VO5] units are connected via common edges in a circular fashion. The overall structure of the [V6O18] fragment looks like a triangle with a triangular hole in the middle.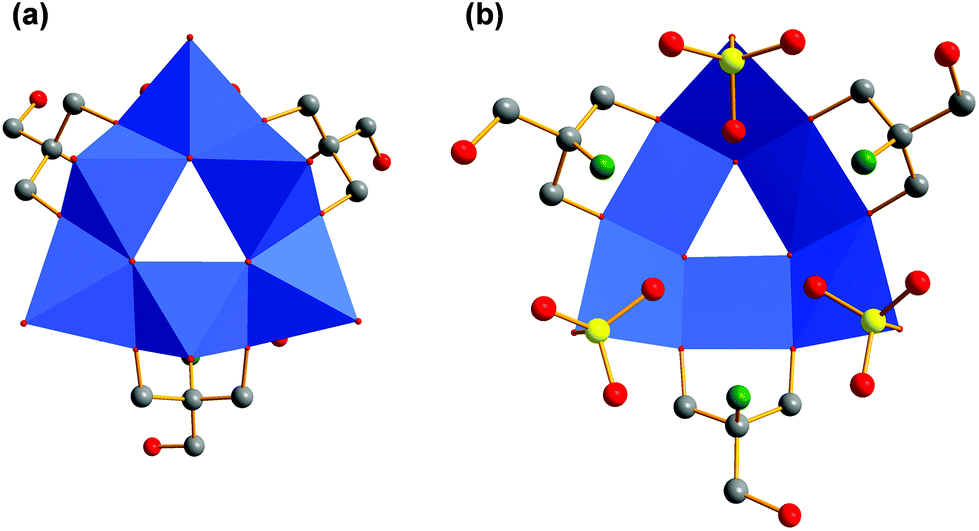 | ||
| Fig. 5 (a) Polyhedra plot of V from the outside of the bowl. (b) Polyhedra plot of V from the inside of the bowl. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red; carbon atoms, grey; sulphur atoms, yellow; nitrogen atoms, green. | ||
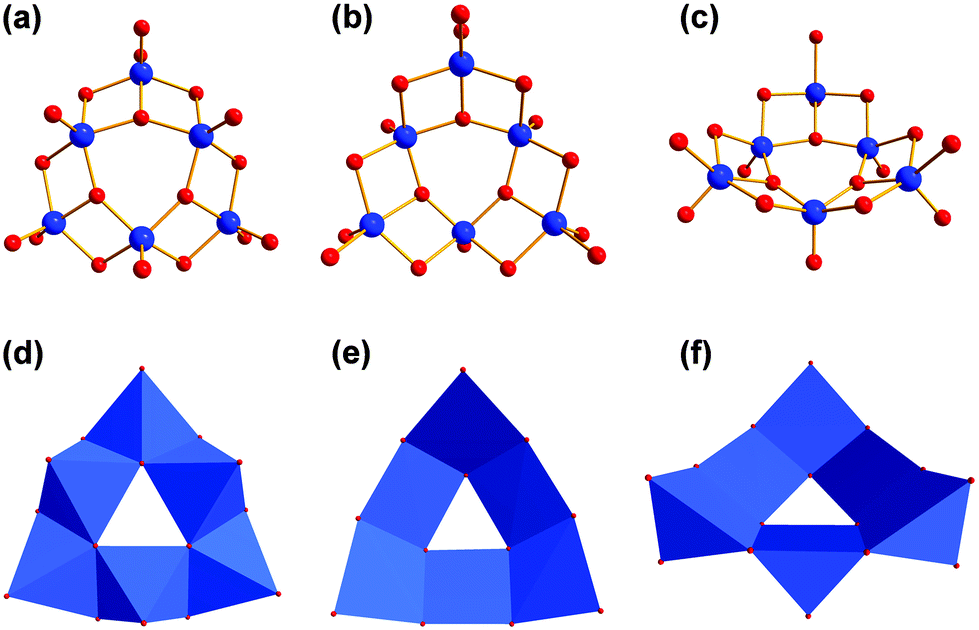 | ||
| Fig. 6 (a–c) Vanadium–oxygen framework of [V6O18] aggregate from different perspectives. (d–f) Polyhedra plot of [V6O18] aggregate from different perspectives. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red. | ||
Additionally, the structure is not planar but bowl-shaped (Fig. 6c). Three of these fragments associated would give the renown [V18O42] cage (Fig. 7).39,40 Two of the [V6O18] triangulars linked by boron were reported as well.41 The same [M6O18] motif is also known for other metals as part of larger architectures.42–45 Nevertheless, the single bowl-shaped [V6O18] fragment has not been published so far. There are other examples of similar [V5] or [V9] bowls.46,47
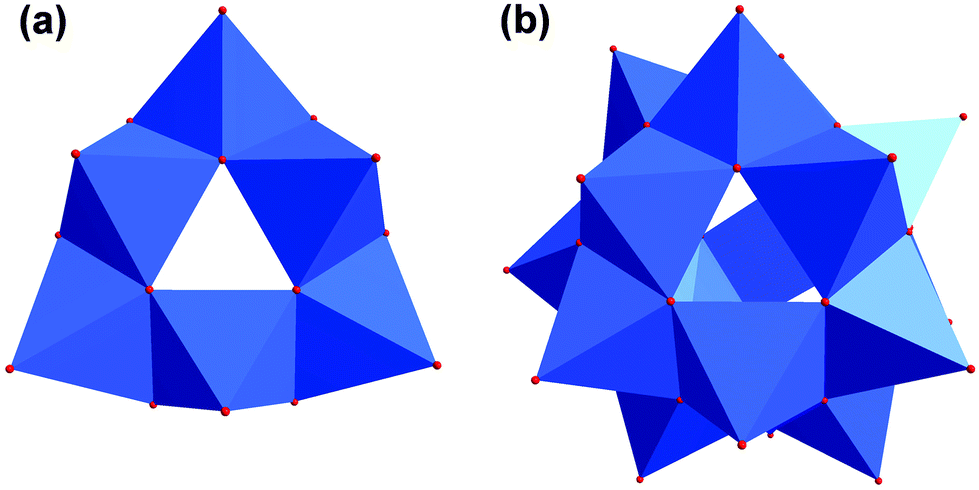 | ||
| Fig. 7 (a) Polyhedra plot of [V6O18] aggregate. (b) Polyhedra plot of [V18O42] aggregate. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red. | ||
Another difference to compounds I–IV is the fact that the organic ligands are only binding in a bipodal fashion (Fig. 5b). They bridge three vanadium atoms by sitting on the edges of the triangle. One hydroxymethyl group of each tris-NH2 protrudes unbound. The protonated amino groups are pointing to the middle of the bowl. As shown in Fig. 5b, they form hydrogen bonds to the sulfato groups of about 2.9 Å (N–O distance). The sulfato ligands bind only in a monodentate way to the vanadium ions at the corners of the triangular fragment. Methanol is not incorporated into the structure. The total charge of the fragment is −3.
Mixed valence heptanuclear polyoxoalkoxovanadate
In avoidance of the structural impact of the free amino group of tris-NH2, we directly utilised its cross-linked dimer bis–tris propane (tris-NH-(CH2)3-NH-tris) for the reaction with VOSO4. Instead of getting chains of interlinked [V4] aggregates, the violet compound [N(CH3)4]4[V7O11(CH3O)(SO4)3((OCH2)2(HOCH2)C-NH-(CH2)3-NH-C(CH2OH)(CH2O)2)] (VI) of a discrete size was obtained (Fig. 8). Its [V7O21N2] framework, shown in Fig. 9a, comprises a square pyramidally all-oxo coordinated V(V) ion and six V(IV) ions (Fig. 9b). Three V(IV) ions have a square pyramidal coordination sphere and are bridged via two bidentate sulfato groups at their outer corners (Fig. 8a). They are connected to the V(V) via common edges (Fig. 10c and d). The remaining three V(IV) atoms are coordinated octahedrally and from a [V3] fragment (Fig. 10a and b).6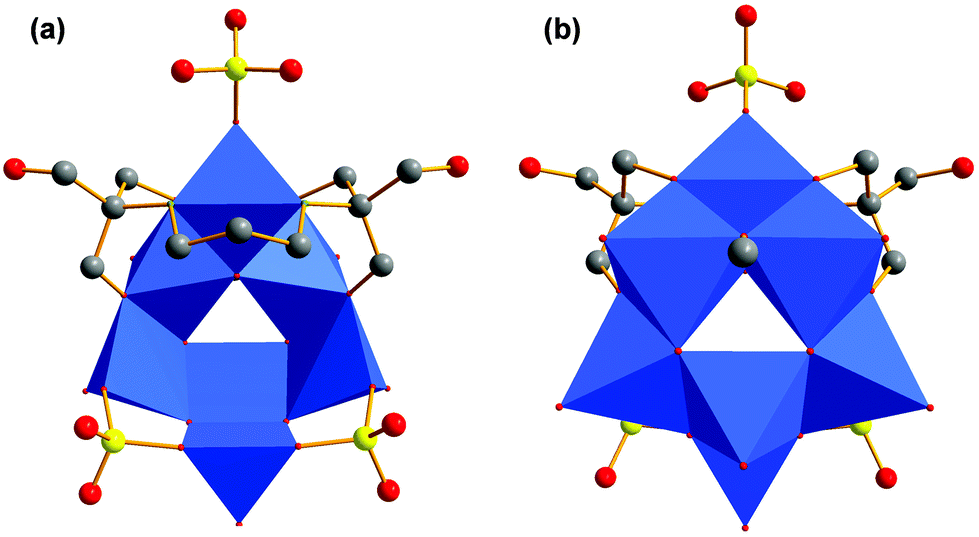 | ||
| Fig. 8 (a) Polyhedra plot of VI from the outside of the bowl. (b) Polyhedra plot of VI from the inside of the bowl. Colour code: vanadium atoms and [VO6] polyhedra, blue; oxygen atoms, red; carbon atoms, grey; sulphur atoms, yellow; nitrogen atoms, green. | ||
 | ||
| Fig. 9 (a) Vanadium–oxygen–nitrogen framework of [V7O21N2] aggregate. (b) Square pyramidally coordinated vanadium (V) ion. (c) Vanadium (IV) ion octahedrally coordinated by four oxygen and two nitrogen atoms. Colour code: vanadium atoms, blue; oxygen atoms, red; nitrogen atoms, green. | ||
 | ||
| Fig. 10 Polyhedra plots of [V7O21N2] fragment. (a) Polyhedra plot of octahedrally coordinated vanadium ions form the outside of the bowl. (b) Polyhedra plot of octahedrally coordinated vanadium ions form the inside of the bowl. (c) Polyhedra plot of square pyramidally coordinated vanadium ions form the inside of the bowl. (d) Polyhedra plot of square pyramidally coordinated vanadium ions form the outside of the bowl. Colour code: [VO6] polyhedra, blue; oxygen atoms, red; nitrogen atoms, green. | ||
Interestingly, the upper V(IV) ion is coordinated by four oxygen and two nitrogen atoms (Fig. 9c). There are few similar examples.48 All seven vanadium atoms form a new fragment. Like in substance V, the fragment is a bowl-shaped triangle with a triangular hole in the middle. The bis–tris propane ligand entangles the [V3] motif (Fig. 8a). It binds on each side via two hydroxymethyl and the secondary amino group. Again, hydroxymethyl groups protrude unbound. While two bidentate sulfato ligands are linked to the outer square pyramidally coordinated V(IV) ions, one monodentate sulfato ligand is bound to the outer octahedrally coordinated V(IV) ion (Fig. 8a). The inner V(IV) ions with an octahedral coordination sphere are bridged by a μ-methoxo ligand (Fig. 8b).
Amino functionalised tetranuclear polyoxoalkoxovanadate
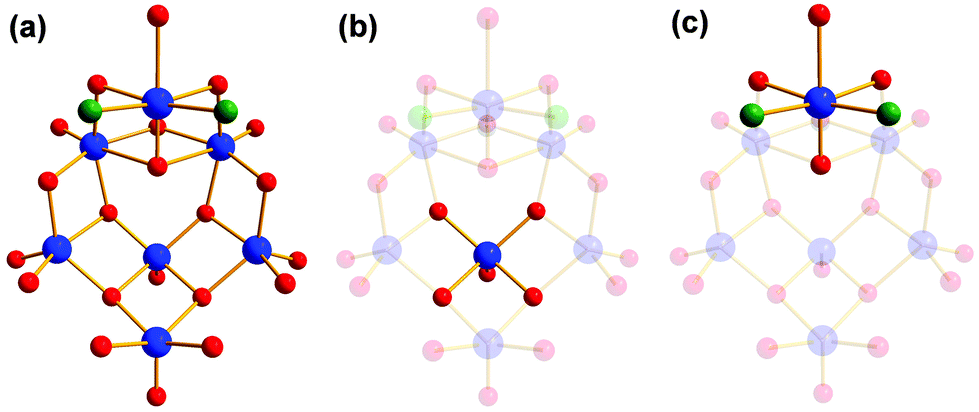
The reactions with other tris derivatives showed that a [V4] aggregate should be producible with tris-NH2. Hence, we tried to alter the reaction conditions slightly and varied the counterions. Besides other effects, the counterions influence the solubility of intermediates, byproducts, and products. This intervention in the highly complex equilibrium of possible side reactions can lead to different products. We set up series of reactions with equivalent concentrations of either LiOH, NaOH, KOH, RbOH, or CsOH. The reactions with LiOH, NaOH, RbOH, and CsOH gave amorphous insoluble products. Finally, we succeeded to synthesise an amino-functionalised [V4] building block by replacing N(CH3)4OH by an equivalent concentration of KOH. Instead of an ionic species like I–IV, the neutral aggregate [V4O4(CH3OH)2(SO4)2((OCH2)3C-NH3)2] (VII) was formed (Fig. 11). The two counterions are replaced by the protonation of the two functional amino groups.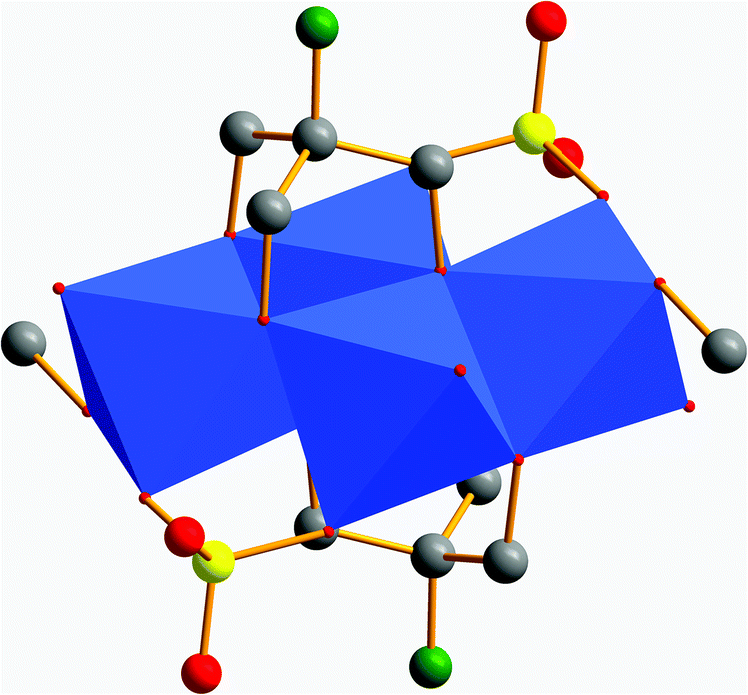 | ||
| Fig. 11 Polyhedra plot of VII. Colour code: [VO6] polyhedra, blue; oxygen atoms, red; carbon atoms, grey; sulphur atoms, yellow; nitrogen atoms, green. | ||
Comparison of different binding modes of tris-NH2
The results show that amino functionalised “tris” ligands can bind in various binding modes (Fig. 12). Tris-NH2 usually binds in a tripodal fashion capping tetrahedral voids on vanadium POMs with its oxygen atoms (Fig. 12a and b).17,49 In that way, either three or four vanadium ions are connected like in compound VII. This binding mode is found in most publications and is common for the majority of tris derivatives.8–13 | ||
| Fig. 12 Different binding modes of tris(hydroxymethyl)-aminomethane (“tris-NH2 or “TRIS”). (a) Tris-NH2 connecting three vanadium ions with its three hydroxymethyl groups. (b) Tris-NH2 connecting four vanadium ions with its three hydroxymethyl groups. (c) Tris-NH2 connecting four vanadium ions with two hydroxymethyl groups and its amino group. (d) Tris-NH2 connecting three vanadium ions with two of its hydroxymethyl groups. (e) Dimer of tris-NH2 (“bis–tris propane”) connecting five vanadium ions with four of its six hydroxymethyl groups and its two secondary amino groups. (f) Section of {e} focusing on the binding mode of a single tris-NH moiety within its dimer. The tris-NH moiety is connecting three vanadium ions with two hydroxymethyl groups and its amino group. Colour code: vanadium atoms, blue; oxygen atoms, red; nitrogen atoms, green; carbon atoms, grey. | ||
However, it turned out that tris-NH2 sometimes reacts differently than other ordinary tris derivatives like tris-CH3, tris-C2H5, or tris-CH2OH. That is the reason why tris-NH2 is often functionalised by amide couplings, alkylation, or protected with aldehydes before being reacted with vanadium sources.20,50,51
The complex reactivity of free tris-NH2 with vanadium is insufficiently investigated. Until now, there is only one more tripodal binding mode reported.52–54 As depicted in Fig. 12c, the amino group can be attached to the vanadium-based POM instead of one of the three alkoxo groups (Fig. 12c). The geometry of the bridged vanadium ions remains the same as in the common binding mode, but the tetrahedral void is not capped by the ligand. In both binding modes, vanadium is coordinated octahedrally.
We found two new binding modes of tris-NH2 which lead to unexpected vanadium structures. Fig. 12d shows that the ligand can also bind in a bipodal fashion with just two of the three alkoxo groups to vanadates. Thereby, three vanadium ions are connected, but in a more linear than triangular fashion. The coordination sphere of vanadium changes to a square pyramidal arrangement. Like in other known examples, the amino group is protonated.17
A second curious binding mode was found when we used “bis–tris propane” (tris-NH-(CH2)3-NH-tris), the commercially available dimer of tris-NH2 (Fig. 12e). We highlighted the binding mode of a single tris-NH moiety in Fig. 12f. It shows a third tripodal binding mode. Again, two of the three alkoxo groups are bridging three vanadium ions. In contrast to Fig. 12c, the amino group is attached to one of the outer vanadium ions. Interestingly, the outer vanadium ion that is not directly connected to the amino group is square pyramidally coordinated while the other two vanadium centres possess an octahedral coordination sphere.
Conclusions
In summary, we have shown the structure directing role of functional groups and counterions on our polyoxoalkoxovanadates. By using inexpensive VOSO4 as vanadium source, interesting POM structures were obtained under solvothermal conditions. The results indicate, that functional amino groups have a major impact on our POM structures while hydroxyl, bromo, pyridyl, and azide groups are tolerated. Both primary and even secondary amines can be incorporated within the metal–oxygen framework. This leads to a competition between amino and hydroxyl groups. Hence, the common tripodal binding mode of tris(hydroxymethyl)methane derivatives is challenged if amino groups are incorporated.Experimental
All compounds could be stored under argon atmosphere at room temperature, but hydrolysed slowly when being exposed to air. The structural analysis was performed on single crystals by X-ray crystallography. The unity of the batches was confirmed by the determination of the unit cells of various crystals. In addition, the crystalline and amorphous parts of the solid products were compared by IR spectroscopy and elemental analysis. The oxidation states of vanadium were calculated by valence sum analysis.55 All results were in good agreement. The results of the valence sum calulations, an overview of the crystallographic details, as well as the IR spectra with an assignment of the characteristic vibration modes of all compounds are supplied in the ESI.†Synthetic procedures
All commercially available compounds were used as received without further purification. Elemental compositions were determined on an ELEMENTAR vario EL III. Infrared measurements were performed on a Thermo-Nicolet Nexus 670 FTIR spectrometer with DuraSamplIR accessory in total reflection at room temperature. The IR spectra of all compounds and a table with selected characteristic vibrational frequencies for compounds I–VII are supplied in the ESI.†Tripodal ligands for compounds III (tris-pyridine)56,57 and IV (tris-CH2N3)58 were synthesised according to literature.
X-ray crystallography
Single crystals of I, III, V, VI, and VII were directly obtained after cooling the reaction mixtures to room temperature in the Teflon vessel. Since II and IV are very well soluble in methanol, the volume of the mother liquers had to be reduced under reduced pressure. Subsequent cooling in the freezer at −30 °C yielded crystals of II and IV. However, the crystal quality of II, V, and VI was rather poor. Recrystallisation of II from various solvents did not yield better crystals. Compounds V and VI are insoluble in allmost all organic solvents and decompose in water. Hence, recrystallisation of V and VI was not possible. The crystallisation of VI from the reaction mixture was improofed by an glassware inlay within the Teflon vessel.The intensities for the X-ray determinations for I, II, and III were collected on a STOE IPDS 2T instrument. The crystallographic experiments for compounds IV, V, VI, and VII were performed on a Bruker X8 Kappa APEX II diffractometer with a CCD-based detector. Mo Kα radiation (λ = 0.71073 Å) was used in all experiments, but for compound V. The crystal structure of V was determined using Cu radition (λ = 1.54178 Å). Standard procedures were applied for data reduction and absorption correction. All structures were solved by intrinsic or direct methods and refined by full-matrix least-squares on F2 using the SHELXTL and WinGX programme packages.59,60 Disordered water molecules in the crystal structures of II, IV, V, and VI were treated with PLATON SQUEEZE software.61 A twin matrix for compound II was determined with TwinRotMat.62
Crystallographic data for all structures in this publication have been deposited with the Cambridge Crystallographic Data Centre, CCDC 1432056–1432062 (I–VII), 12 Union Road, Cambridge CB21EZ, UK.
Acknowledgements
O. N. thanks the Stiftung der Deutschen Wirtschaft for a scholarship.References
- H. N. Miras, D. Stone, D.-L. Long, E. J. L. McInnes, P. Kögerler and L. Cronin, Inorg. Chem., 2011, 50, 8384–8391 CrossRef PubMed.
- H. N. Miras, M. Sorus, J. Hawkett, D. O. Sells, E. J. L. McInnes and L. Cronin, J. Am. Chem. Soc., 2012, 134, 6980–6983 CrossRef PubMed.
- P. Yin, T. Li, R. S. Forgan, C. Lydon, X. Zuo, Z. N. Zheng, B. Lee, D. Long, L. Cronin and T. Liu, J. Am. Chem. Soc., 2013, 135, 13425–13432 CrossRef PubMed.
- Y.-B. Liu, L.-W. Fu, W.-J. Duan, L.-N. Xiao, Y.-Y. Hu, D.-C. Zhao, H.-Y. Guo, X.-B. Cui and J.-Q. Xu, Inorg. Chem. Commun., 2014, 47, 5–8 CrossRef CAS.
- J. Tucher, S. Schlicht, F. Kollhoff and C. Streb, Dalton Trans., 2014, 43, 17029–17033 RSC.
- Z. Shi, Y. Zhou, L. Zhang, C. Mu, H. Ren, D. u. Hassan, D. Yang and H. M. Asif, RSC Adv., 2014, 4, 50277–50284 RSC.
- J.-H. Son and W. H. Casey, Chem. Commun., 2015, 51, 1436–1438 RSC.
- Q. Chen, D. P. Goshorn, C. P. Scholes, X.-L. Tan and J. Zubieta, J. Am. Chem. Soc., 1992, 114, 4667–4681 CrossRef CAS.
- M. I. Khan, Q. Chen, D. P. Goshorn and J. Zubieta, Inorg. Chem., 1993, 32, 672–680 CrossRef CAS.
- M. I. Khan, Q. Chen, H. Höpe, S. Parkin, C. J. O'Connor and J. Zubieta, Inorg. Chem., 1993, 32, 2929–2937 CrossRef CAS.
- M. I. Khan, Y.-S. Lee, C. J. O'Connor and J. Zubieta, J. Am. Chem. Soc., 1994, 116, 5001–5002 CrossRef CAS.
- L. J. Batchelor, R. Shaw, S. J. Markey, M. Helliwell and E. J. L. McInnes, Chem. – Eur. J., 2010, 16, 5554–5557 CrossRef CAS PubMed.
- L. J. Batchelor, E. Fitzgerald, J. Wolowska, J. J. W. McDouall and E. J. L. McInnes, Chem. – Eur. J., 2010, 16, 11082–11088 CrossRef CAS PubMed.
- P. R. Marcoux, B. Hasenknopf, J. Vaissermann and P. Gouzerh, Eur. J. Inorg. Chem., 2003, 2406–2412 CrossRef CAS.
- Y. Q. Hou and C. L. Hill, J. Am. Chem. Soc., 1993, 115, 11823–11830 CrossRef CAS.
- J. W. Han, K. I. Hardcastle and C. L. Hill, Eur. J. Inorg. Chem., 2006, 2598–2603 CrossRef CAS.
- D. Li, J. Song, P. C. Yin, S. Simotwo, A. J. Bassler, Y. Y. Aung, J. E. Roberts, K. I. Hardcastle, C. L. Hill and T. B. Liu, J. Am. Chem. Soc., 2011, 133, 14010–14016 CrossRef CAS PubMed.
- P. C. Yin, P. F. Wu, Z. C. Xiao, D. Li, E. Bitterlich, J. Zhang, P. Cheng, D. V. Vezenov, T. B. Liu and Y. G. Wei, Angew. Chem., Int. Ed., 2011, 50, 2521–2525 CrossRef CAS PubMed.
- K. Micoine, B. Hasenknopf, S. Thorimbert, E. Lacôte and M. Malacria, Org. Lett., 2007, 9, 3981–3984 CrossRef CAS PubMed.
- M. P. Santoni, A. K. Pal, G. S. Hanan, M. C. Tang, K. Venne, A. Furtos, P. Menard-Tremblay, C. Malveau and B. Hasenknopf, Chem. Commun., 2012, 48, 200–202 RSC.
- N. P. Luneva, S. A. Mironova, A. E. Shilov, M. Y. Antipin and Y. T. Struchkov, Angew. Chem., Int. Ed. Engl., 1993, 32, 1178–1179 CrossRef.
- I. S. Tidmarsh, E. Scales, P. R. Prearley, J. Wolowska, L. Sorace, A. Caneschi, R. H. Laye and E. J. L. McInnes, Inorg. Chem., 2007, 46, 9743–9753 CrossRef CAS PubMed.
- D. C. Crans, F. Jiang, J. Chen, O. P. Anderson and M. M. Miller, Inorg. Chem., 1997, 36, 1038–1047 CrossRef CAS PubMed.
- F. Jiang, O. P. Anderson, S. M. Miller, J. Chen, M. Mahroof-Tahir and D. C. Crans, Inorg. Chem., 1998, 37, 5439–5451 CrossRef CAS PubMed.
- F. J. C. Rossotti and H. Rossotti, Acta Chem. Scand., 1956, 10, 957–984 CrossRef CAS.
- H. T. Evans, Inorg. Chem., 1966, 5, 967–977 CrossRef CAS.
- Q. Chen and J. Zubieta, J. Chem. Soc., Chem. Commun., 1993, 1180–1182 RSC.
- S. Khanra, L. J. Batchelor, M. Helliwell, F. Tuna, E. J. L. McInnes and R. E. P. Winpenny, J. Mol. Struct., 2008, 890, 157–162 CrossRef CAS.
- E. Scales, L. Sorace, A. Dei, A. Caneschi, C. A. Muryn, D. Collison and E. J. L. McInnes, Chem. Sci., 2010, 1, 221–225 RSC.
- M. Hilbers, M. Meiwald and R. Mattes, Z. Naturforsch., B: Chem. Sci., 1996, 51, 57–67 CrossRef CAS.
- J. Spandl, I. Brüdgam and H. Hartl, Angew. Chem., Int. Ed., 2001, 40, 4018–4020 CrossRef CAS PubMed.
- J. Spandl, I. Brüdgam and H. Hartl, Z. Anorg. Allg. Chem., 2003, 629, 539–544 CrossRef CAS.
- D. D. Heinrich, K. Folting, W. E. Streib, J. C. Huffman and G. Christou, J. Chem. Soc., Chem. Commun., 1989, 1411–1413 RSC.
- J. R. Rambo, J. C. Huffman and G. Christou, J. Am. Chem. Soc., 1989, 111, 8027–8029 CrossRef CAS.
- S. L. Castro, Z. Sun, J. C. Bollinger, D. N. Hendrickson and G. Christou, J. Chem. Soc., Chem. Commun., 1995, 2517–2518 RSC.
- Z. Sun, C. M. Grant, S. L. Castro, D. N. Hendrickson and G. Christou, Chem. Commun., 1998, 721–722 RSC.
- S. L. Castro, Z. Sun, C. M. Grant, J. C. Bollinger, D. N. Hendrickson and G. Christou, J. Am. Chem. Soc., 1998, 120, 2365–2375 CrossRef CAS.
- H. Putz and K. Brandenburg, Diamond, Crystal Impact GbR, Bonn, Germany, 2014 Search PubMed.
- A. Müller, J. Döring, H. Bögge and E. Krickemeyer, Chimia, 1988, 42, 300 Search PubMed.
- A. Müller, R. Sessoli, E. Krickemeyer, H. Bögge, J. Meyer, D. Gatteschi, L. Pardi, J. Westphal, K. Hovemeier, R. Rohlfing, J. Döring, F. Hellweg, C. Beugholt and M. Schmidtmann, Inorg. Chem., 1997, 36, 5239–5250 CrossRef.
- P. Hermosilla-Ibáñez, W. Cañon-Mancisidor, J. Costamagna, A. Vega, V. Paredes-García, M. T. Garland, E. Le Fur, O. Cador, E. Spodine and D. Venegas-Yazigi, Dalton Trans., 2014, 43, 14132–14141 RSC.
- J. T. Rijssenbeek, D. J. Rose, R. C. Haushalter and J. Zubieta, Angew. Chem., Int. Ed. Engl., 1997, 36, 1008–1010 CrossRef CAS.
- T. C. Stamatatos, K. A. Abboud, W. Wernsdorfer and G. Christou, Angew. Chem., Int. Ed., 2007, 46, 884–888 CrossRef CAS PubMed.
- H. Chen, Y. Zhang, Z.-B. Yu and J. Sun, Dalton Trans., 2014, 43, 15283–15286 RSC.
- K. Kastner, J. T. Margraf, T. Clark and C. Streb, Chem. – Eur. J., 2014, 20, 12269–12273 CrossRef CAS PubMed.
- G. B. Karet, Z. Sun, D. D. Heinrich, J. K. McCusker, K. Folting, W. E. Streib, J. C. Huffman, D. N. Hendrickson and G. Christou, Inorg. Chem., 1996, 35, 6450–6460 CrossRef CAS PubMed.
- G. B. Karet, Z. Sun, W. E. Streib, J. C. Bollinger, D. N. Hendrickson and G. Christou, Chem. Commun., 1999, 2249–2250 RSC.
- K. Hegetschweiler, B. Morgenstern, J. Zubieta, P. J. Hagrman, N. Lima, R. Sessoli and F. Totti, Angew. Chem., Int. Ed., 2004, 43, 3436–3439 CrossRef CAS PubMed.
- C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388–4391 CrossRef CAS PubMed.
- P. Yin, A. Bayaguud, P. Cheng, F. Haso, L. Hu, J. Wang, D. Vezenov, R. E. Winans, J. Hao, T. Li, Y. Wei and T. Liu, Chem. – Eur. J., 2014, 20, 9589–9595 CrossRef CAS PubMed.
- A. Bayaguud, K. Chena and Y. Wie, CrystEngComm, 2016, 18, 4042–4045 RSC.
- P. Zabierowski, M. Radoń, J. Szklarzewicz and W. Nitek, Inorg. Chem. Commun., 2014, 41, 72–75 CrossRef CAS.
- G. Asgedom, A. Sreedhara, J. Kivikoski, J. Valkonen, E. Kolehmainen and C. P. Rao, Inorg. Chem., 1996, 35, 5674–5683 CrossRef CAS PubMed.
- D. F. Back, C. R. Kopp, G. M. de Oliveira and P. C. Piquini, Polyhedron, 2012, 3, 21–29 CrossRef.
- I. D. Brown and K. K. Wu, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 1957–1959 Search PubMed.
- W. Koenigs and G. Happe, Ber. Dtsch. Chem. Ges., 1903, 36, 2904–2910 CrossRef CAS.
- D. Menozzia, E. Biavardi, C. Massera, F.-P. Schmidtchen, A. Cornia and E. Dalcanal, Supramol. Chem., 2010, 22, 768–775 CrossRef.
- P. Pana, M. Fujitaa, W.-Y. Ooia, K. Sudeshb, T. Takaradaa, A. Gotoc and M. Maeda, Polymer, 2011, 52, 895–900 CrossRef.
- Bruker AXS Inc., Madison, Wisconsin, USA, 1998.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9–18 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1432056–1432062. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03638d |
| This journal is © The Royal Society of Chemistry 2017 |