
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCO-releasing molecule (CORM) conjugate systems
Anna Christin
Kautz
,
Peter C.
Kunz
and
Christoph
Janiak
 *
*
Institut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität, Universitätsstr. 1, D-40225 Düsseldorf, Germany. E-mail: janiak@uni-duesseldorf.de
First published on 18th October 2016
Abstract
The development of CORMs (CO-releasing molecules) as a prodrug for CO administration in living organisms has attracted significant attention. CORMs offer the promising possibility of a safe and controllable release of CO in low amounts triggered by light, ligands, enzymes, etc. For the targeting of specific tissues or diseases and to prevent possible side effects from metals and other residues after CO release, these CORMs are attached to biocompatible systems, like peptides, polymers, nanoparticles, dendrimers, protein cages, non-wovens, tablets, and metal–organic frameworks. We discuss in this review the known CORM carrier conjugates, in short CORM conjugates, with covalently-bound or incorporated CORMs for medicinal and therapeutic applications. Most conjugates are nontoxic, show increasing half-lives of CO release, and make use of the EPR-effect, but still show problems because of a continuous background of CO release and the absence of an on/off-switch for the CO release.
 Anna Christin Kautz | Anna Christin Kautz studied business chemistry at Heinrich-Heine-University in Düsseldorf and received her Diploma in 2012. Her current research (dissertation) focuses on metal–organic frameworks and encapsulation of drugs, for example, CO releasing molecules (CORMs). |
 Peter C. Kunz | Peter C. Kunz studied Chemistry at Heinrich-Heine-University in Düsseldorf where he received his Diploma in 1999 on studying the nickel-catalysed copolymerisation of ethene and CO. Under the guidance of Wolfgang Kläui he completed his Dissertation in 2003 on biomimetic tripodal ligands. In 2004 he stayed with Roger Alberto as Postdoc in Zürich, where he investigated organometallic Re and Tc complexes and focused on medicinal bioinorganic topics. From 2004 to 2010 he worked on his Habilitation at Heinrich-Heine-University in Düsseldorf and as an extraordinary lecturer in the Pharmaceutical Chemistry Department. Since 2012, he has been a chemistry and physics teacher at Annette-von-Droste-Hülshoff-Gymnasium in Düsseldorf. |
 Christoph Janiak | Christoph Janiak studied Chemistry at TU Berlin and the Univ. of Oklahoma, followed by a postdoc at Cornell Univ. and at BASF AG and an associate professorship at the Univ. of Freiburg. Since 2010, he has been a full professor of Bioinorganic Chemistry and catalysis at the University of Düsseldorf with further research interests in metal and porous organic frameworks (MOFs, COFs) and metal nanoparticles and catalysis. |
Introduction
In recent years, carbon monoxide releasing molecules (CORMs) have attracted much attention due to their possibility to deliver low amounts of carbon monoxide in a controllable therapeutic way.1For decades, carbon monoxide (CO) has been known as “the silent killer”, as first described by Haldane, because of its strong affinity to bind to hemoglobin and therefore it was only seen as an obstacle to oxygen transport in the blood system.2,3 However, it has been proven that CO is not only toxic but also occurs in small amounts within the human body (produced by the oxidation of heme to biliverdin catalyzed by heme oxygenase) and plays important physiological roles as a small molecule messenger (such as stimulating guanylyl cyclase to form cyclic guanosine monophosphate (cGMP), activating cGMP, inhibiting the DNA binding activity of holo-NPAS2 (neuronal Per-Arnt-Sim-domain protein 2)) like nitric oxide or hydrogen sulfide.4–7 In addition, in low concentrations, carbon monoxide has a beneficial effect on cardiovascular diseases, inflammatory disorders, tumor growth, bacterial infections, and organ transplantations.8,9
In order to achieve the release of CO at specific sites in tissue or organs with a controllable and tunable quantity, Motterlini et al. proposed the pharmaceutical use of carbon monoxide releasing molecules (CORMs), a chemically bound form of CO as a prodrug.10 This initial work described the biochemical properties of transition metal carbonyl complexes, like the commercially available [Mn2(CO)10] (CORM-1) and [RuCl2(CO)3]2 (CORM-2) (Fig. 1).10 Over a period of time, more CORMs, like the water-soluble [Ru(CO)3Cl(glycinate)] (CORM-3) and [Mn(CO)3(tpm)]PF6 (CORM-L1, tpm: tris(pyrazolyl) methane), were synthesized and analyzed in terms of their CO-releasing properties (Fig. 1).11–13
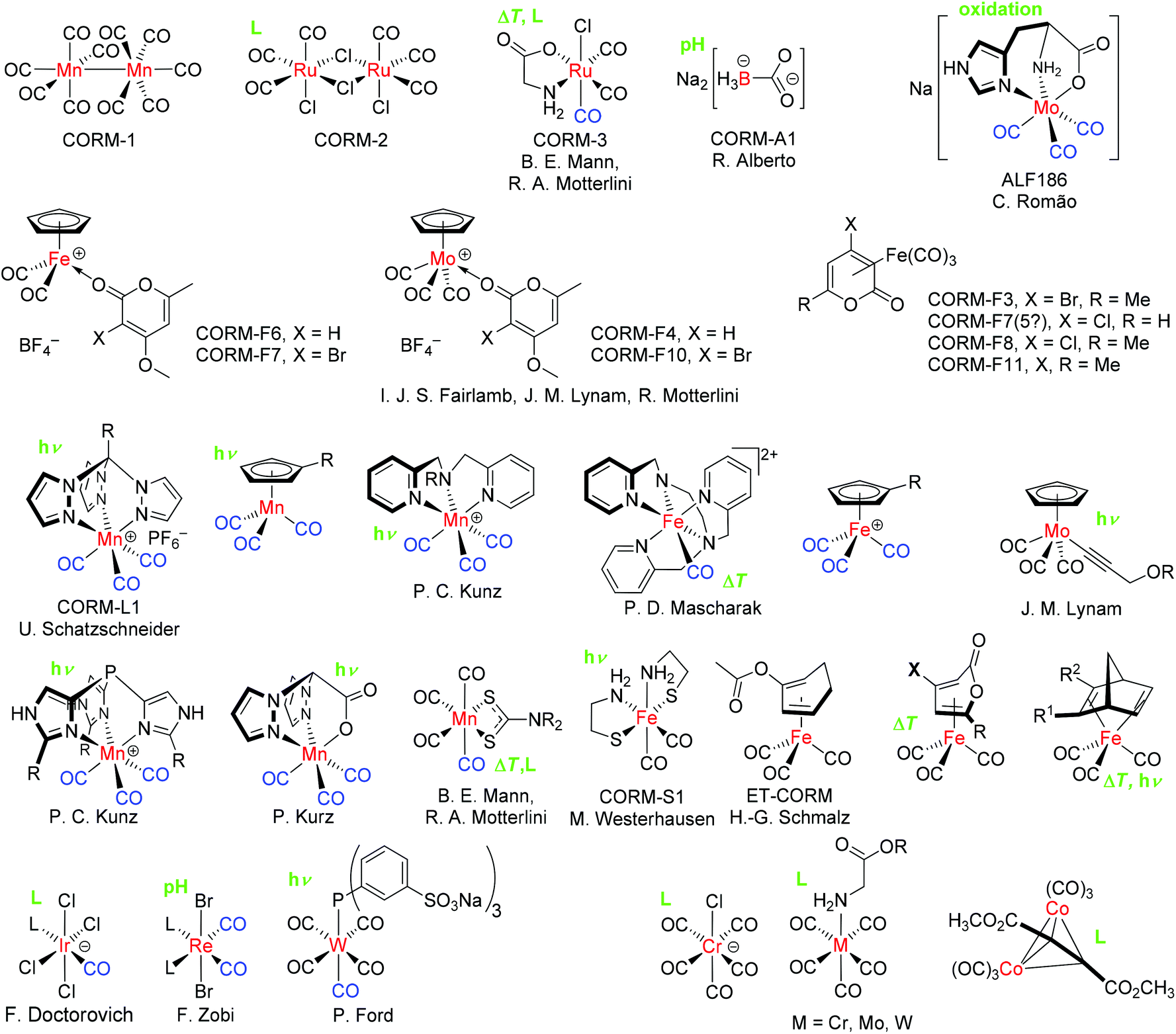 | ||
| Fig. 1 Examples of some CO-releasing molecules (CORMs) with their accepted acronym (CORM-A1, CORM-2 etc.), principal investigator, and specific release trigger, where known (in green: hν = photolysis, ΔT = temperature increase, pH change, oxidation or ligand, L substitution). For CORMs with a spontaneous or unclear release trigger, no indication is given. The released or replaceable CO ligands are depicted in blue.17 | ||
The CO release of CORMs can be initiated through diverse specific triggers.1 One possible mechanism leading to the CO release of CORMs is photochemical external activation by different wavelengths of light. These CORMs are often called PhotoCORMs and examples include CORM-1 and CORM-L1.13 CORM-2 releases CO spontaneously through ligand substitution/exchange by forming a new bond with, for example, sulfur.1 In particular for application under physiological conditions, the trigger temperature may be important. Some CORMs, like CORM-3, release CO also by a trigger combination, e.g., by thermal degradation and ligand substitution. Other triggers are degradation by enzymes (ET-CORMs, ET: enzyme triggered), pH change, and oxidation.1,14 The prototypical oxidatively triggered CORM is ALF186.15,16 Unfortunately, it is also possible that this oxidation trigger is responsible for the background or steady CO release of other CORMs in solution, which are thought to be triggered by ligand exchange. Only one electron is necessary to start the release and this oxidation trigger could hide behind several triggers. Fig. 1 provides a summarizing overview of CORMs and their specific release triggers.
Vibrational spectroscopy (infrared, IR, and Raman) is the simplest and quickest method to analyze CORMs and to prove if the CO groups are bound to the transition metal complex. The aforementioned CORM-L1, for example, shows the typical two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands at 1941 and 2047 cm−1 for a facial coordination of the tpm ligand to the manganese tricarbonyl unit.12 In contrast to the inert CORM-L1, the IR spectra of CORM-3 show pH-dependency. After the addition of aqueous HCl, the IR spectrum shows three bands at 2134, 2070, and 2052 cm−1, because of the reversible conversion of [Ru(CO)3Cl(NH2CH2CO2)] to [Ru(CO)3Cl2(NH2CH2CO2H)]. According to the added amount of NaOH, the two bands of a cis-dicarbonyl ruthenium fragment in a basic solution are 2058 and 1985 cm−1 (1 equivalent of NaOH) or 2044 and 1968 cm−1 (2 equivalents of NaOH), respectively. In basic solution, the reversible reactions of all the CORM-3 derivates, like [Ru(CO)3Cl2L], with OH− leads to [Ru(CO)2(COOH)Cl2L] compounds showing the two mentioned CO stretching bands.18,19
O bands at 1941 and 2047 cm−1 for a facial coordination of the tpm ligand to the manganese tricarbonyl unit.12 In contrast to the inert CORM-L1, the IR spectra of CORM-3 show pH-dependency. After the addition of aqueous HCl, the IR spectrum shows three bands at 2134, 2070, and 2052 cm−1, because of the reversible conversion of [Ru(CO)3Cl(NH2CH2CO2)] to [Ru(CO)3Cl2(NH2CH2CO2H)]. According to the added amount of NaOH, the two bands of a cis-dicarbonyl ruthenium fragment in a basic solution are 2058 and 1985 cm−1 (1 equivalent of NaOH) or 2044 and 1968 cm−1 (2 equivalents of NaOH), respectively. In basic solution, the reversible reactions of all the CORM-3 derivates, like [Ru(CO)3Cl2L], with OH− leads to [Ru(CO)2(COOH)Cl2L] compounds showing the two mentioned CO stretching bands.18,19
There are a plethora of methods known in order to detect the CO upon release from the compounds, with the so-called myoglobin assay being the most widely used technique and therefore something like a “gold standard”. In this protocol, first reported by Motterlini et al., the CORM analyte is added to a buffered aqueous solution containing sodium dithionite (Na2S2O4) and deoxy-myoglobin (Mb) under an atmosphere of nitrogen gas.10 The buffers used (pH = 7.4) can be 3-(N-morpholino)-propanesulfonic acid (MOPS), phosphate-buffered saline (PBS), potassium phosphate (KPI), Krebs Henseleit buffer (KHB), or others.10,20,21 Sodium dithionite is used to reduce any Fe(III) in oxymyoglobin to desoxymyoglobin. During CO release from the CORM, the formation of carboxy-myoglobin (Mb-CO) is observed by a decreasing intensity of the absorption band of myoglobin (556 nm) and two increasing absorption bands of the carboxy-myoglobin (541 nm, 578 nm) in the visible spectra (eqn (1)). By this method, CO release over time can be followed easily and the resultant half-life for the decay of the CO complex as well as the amount of CO released can be calculated. In general, the myoglobin assay is performed using conditions found in living organisms (pH 7.4, 37 °C).10,22 It should be noted, however, that the reducing agent, sodium dithionite, also induces CO release from several CORMs, namely CORM-2 and CORM-3.20 Ligand-labile CORMs, such as CORM-3 and its analogs, are also susceptible to CO abstraction by heme proteins.23
| Mb + CO → Mb − CO | (1) |
| λmax = 556 nm 541, 578 nm |
Fluorescent probes, electrochemical sensing, gas chromatography, and gas-phase IR spectroscopy are other used methods to measure the CO release of CORMs.24–28 Fluorescent probes for the measurement of the CO release of CORMs can detect CO in lower concentrations compared to the myoglobin assay, but they are not suitable for short-time kinetic measurements as they have relatively long response times (about one hour).24,25 Electrochemical sensors can determine the CO concentration directly in solution and do not require anaerobic conditions. A disadvantage of this method is the restricted view on the release kinetics of CO due to the indirect measurement of the electrochemical oxidation of CO. Furthermore, additional studies on the influence of electrochemical reactions on the CO release are not available.26 Using gas chromatography as a direct CO sensing method, various detectors, like GC-RGD (reduction gas detector),27 GC-MS,28 and GC-TCD (thermal conductivity detector), are available. However, for GC-TDC, the samples have to be taken out of the reaction mixture or from the headspace, thereby making this method not suitable for continuous, long-term measurements.26 An outstanding, high-resolution method for the measurement of the CO release is gas-phase IR spectroscopy. It is suited for the determination of CO released in the gas phase as well as in solution (ATR technique). A further advantage of this method is that there is no need for additives, like a buffer or sodium dithionite reductant.26
In addition to the cytotoxicity and stability issues of CORMs in solution, their delivery to specific targets and the suitable half-life of CO release are also essential for therapeutic applications. Indeed, half-lives constitute a fundamental problem of CORMs. Compared to the perhaps less practicable and controllable CO gas, which is administered by inhalation (t1/2 = 3–7 h under normoxic conditions in humans),8 the half-lives of some of the above-mentioned unprotected CORMs (t1/2 = 1 min in PBS buffer at 37 °C and pH 7.4 for CORM-1, -2, and -3; t1/2 = 3.6 min in human plasma for CORM-3)29,30 are often low. Such half-lives are relatively short considering the CORMs have to circulate in body fluids to reach the target tissue or organs before the CO is fully released.31
Furthermore, there is a wide range of different specific therapeutic applications and so their transport, interactions with tissue compounds, and a suitable degradation of the CORMs needs to be addressed for the controlled and effective use of CORMs within the human body.22 Furthermore, the degradation products of the CORM after CO release could have side effects on the organism, which one needs to be aware of and which has to be controlled.31
These problems might be addressed using a conjugated CORM carrier or guest–host systems, which could open doors for the future applications of CORMs and offer new aspects in medicinal chemistry.32 Covalent binding or encapsulating CORMs in a protective host structure should increase their half-lives, especially for CORMs where the CO release is induced by ligand exchange reactions. Also, the carrier or host structure should be easier and better adapted to the relevant application and through the corresponding trigger of the specific CORM, CO release should still be controllable. A pre-condition for the carrier system is still its non-toxicity as well as the non-toxicity of the remaining CORM-fragments after CO release. Furthermore, due to covalent binding or incorporation of the CORM, specific delivery to a biological target could become possible through the carrier and the CORM stability under physiological conditions would also be increased.31
Target control could be exerted through the tissue specific accumulation of conjugated CORM carrier systems, such as CORM-functionalized nanoparticles or polymers. This can in general be achieved by two different strategies: (a) by “active” decoration of the material surface, e.g., with site-directing proteins, or (b) by “passive” accumulation based on the enhanced permeability and retention (EPR) effect.33–35 The growth of tumors (>2 mm) and especially their supporting blood vessels is very fast and leads to destabilization and higher permeability of the newly formed blood vessel tissue compared to normal tissue.36 Thereby, holes in the order of about 50 nm and junctions of about 500 nm arise (healthy tissue has pores sized 5–8 nm). These pores allow polymers, polymer conjugates, functionalized polymers, and nanoparticles to pass through. The lymphatic system is not able to remove these foreign molecules, which leads to their accumulation.37 Consequently, polymer–CORM conjugates can accumulate within the tumor tissue and effectively release the CO load at the target site.
In this review, we summarize all, at least to the best of our knowledge, CORM carrier conjugates described so far. “Small molecule” CORMs have already been the subject of various reviews.13,18,22,31 In terms of the CORM conjugates, we mainly present the systems that are based on already existing molecular carbonyl compounds, whereby either the covalently-bound CORM in the conjugate has a direct molecular parent CORM or the molecular CORM is non-covalently encapsulated into a carrier. Accordingly, we have organized the CORMs in this review into conjugates with a covalently-bound CORM-fragment, conjugates with non-covalently-encapsulated CORMs, and conjugate systems that combine both approaches. Additionally, we discuss their potential in therapeutic applications with respect to their biocompatibility and their CO release characteristics (trigger, half-life, measuring method, etc.).
Covalently-bound CORM conjugates
CORMs@peptides
Schatzschneider et al. were the first to covalently connect CORMs to a substrate when they attached the photoactive CORM-L1 fragment [Mn(CO)3(tris(pyrazolyl)methane)]+ using Pd-catalyzed Sonogashira cross-coupling and copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC, “click reaction”) to amino acids and peptides. These CORM conjugates showed target specific uptake in cancer cells. For example, the synthesized CO-releasing peptide compound utilized the five amino-acid sequence Thr-Phe-Ser-Asp-Leu (Fig. 2), which is part of the tumor suppressor protein p53. The Mn(CO)3(tpm)@peptide conjugate released 1.7 mol CO per mol Mn (as assessed by myoglobin assay in phosphate-buffered solution, with the addition of sodium dithionite) and presented CO IR vibration bands at 2048 and 1941 cm−1, which are typical bands for the CORM-L1 moiety.38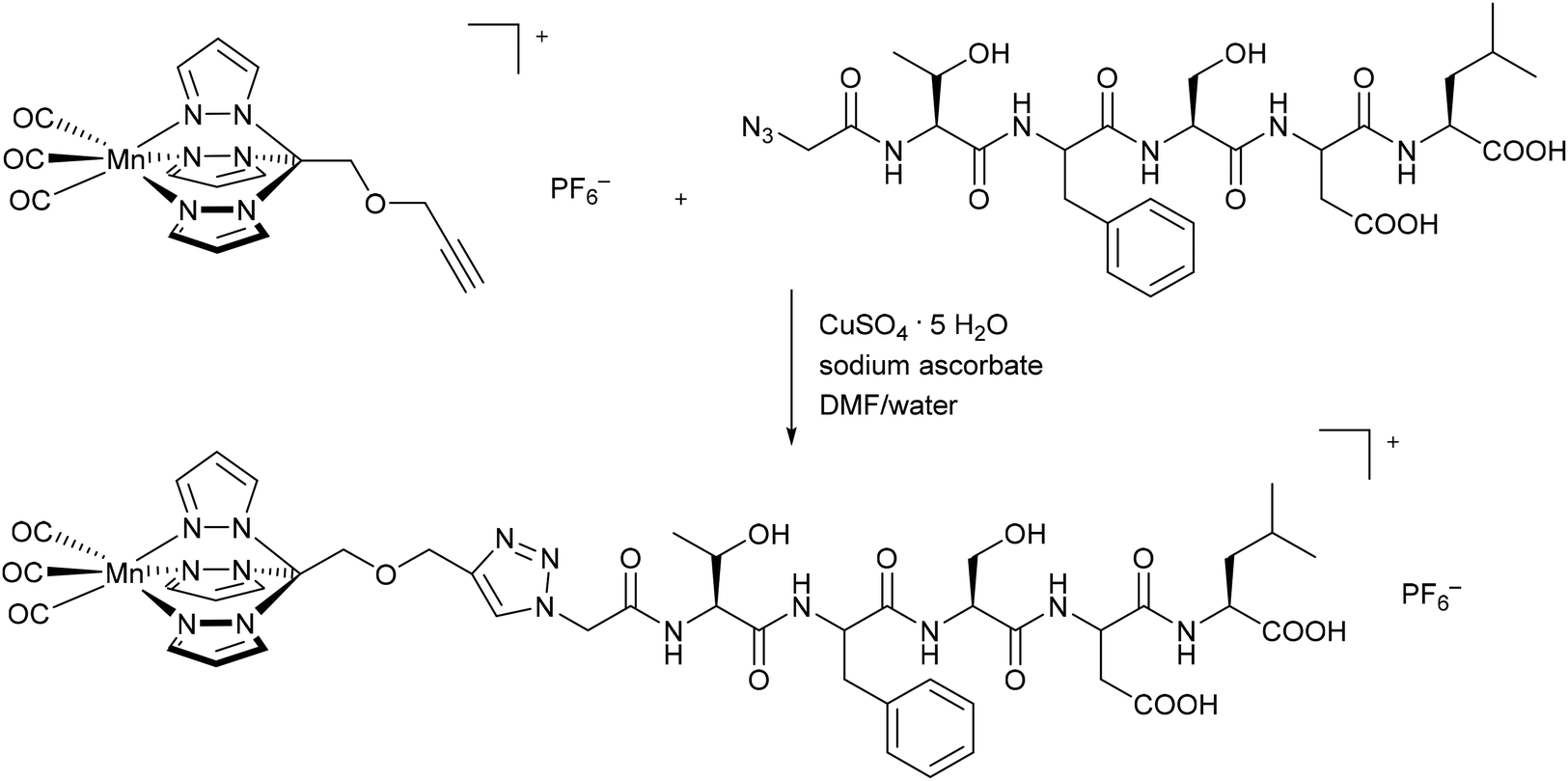 | ||
| Fig. 2 Synthesis of a covalent CORM@peptide conjugate via copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition of a [Mn(CO)3(tris(pyrazolyl)methane)]+ fragment to the peptide sequence in the tumor suppressor protein p53 by Schatzschneider et al.38 | ||
In addition to CORM-L1, Schatzschneider et al. also attached a Mo(CO)4-bipyridine fragment through a functional aldehyde group to peptides (Fig. 3). Here, a milder and catalyst-free conjugation method was chosen to covalently bind the CORM to the peptide. In this approach, oxime ligation of the aldehyde-functionalized metal carbonyl complex to the N-terminus of the peptide was used. The peptide of the so-synthesized CO-releasing compound was a sequence of TGF-β (transforming growth factor). This signal molecule inhibits the growth of tumors and was chosen for the potential tumor targeting.39
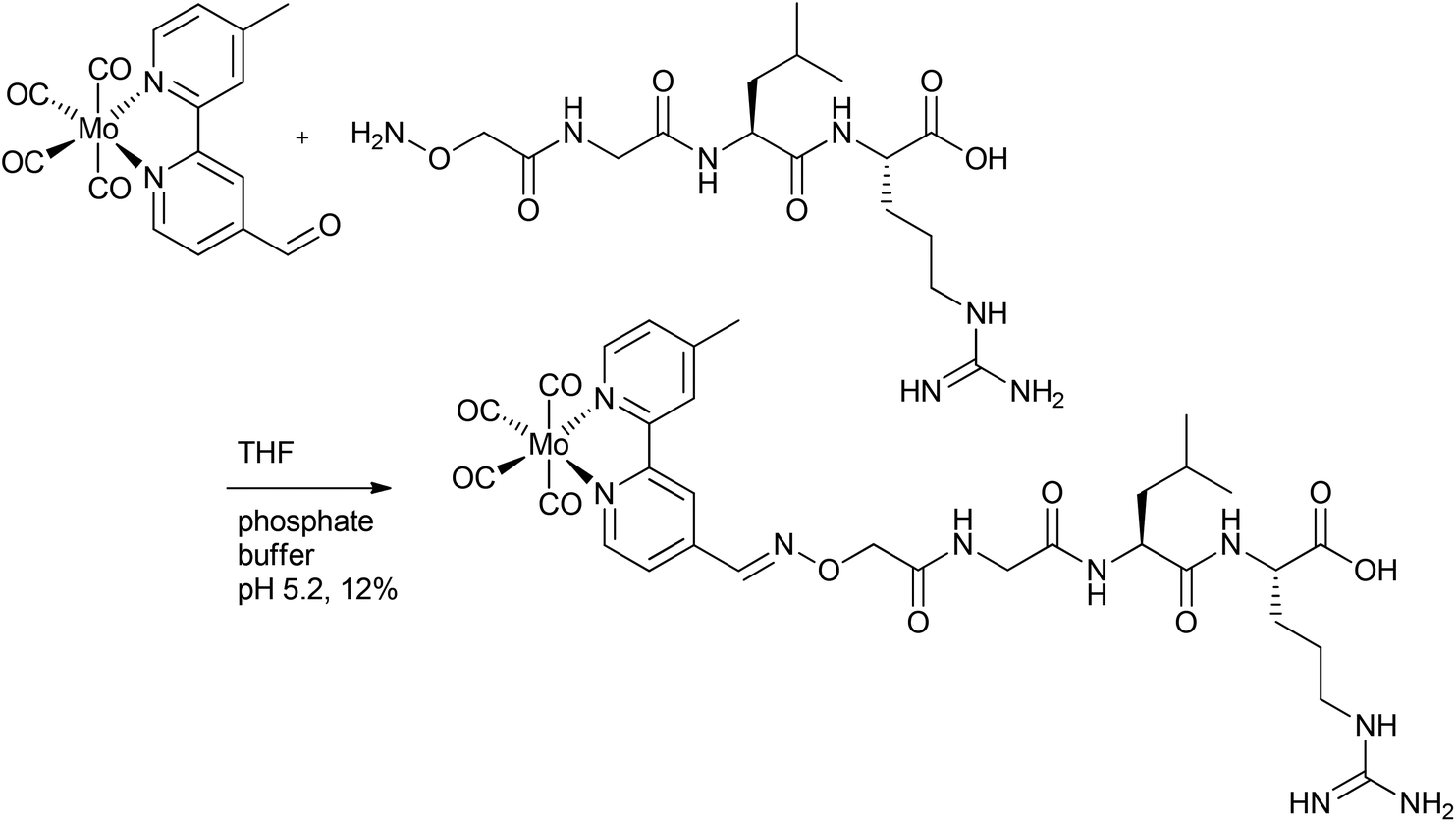 | ||
| Fig. 3 Synthesis of a covalent CORM@peptide conjugate via catalyst-free oxime ligation of a Mo(CO)4-bipyridine fragment to a peptide sequence of TGF-β (transforming growth factor) by Schatzschneider et al.39 | ||
Here again, IR spectroscopy was used to indicate successful ligation of the metal carbonyl fragment (through the presence of three CO vibration bands at 2015, 1861 and 1812 cm−1). Although the CO release was mainly triggered by light, the Mo-compound also released CO spontaneously due to a ligand exchange in the dark (t1/2 = 123 ± 38 min for the compound in Fig. 3). Upon stimulation by light excitation at 468 nm, the half-life of the conjugate was approximately 40 min (in phosphate-buffered myoglobin/dimethyl sulfoxide solution, with the addition of sodium dithionite), which was thereby a bit faster compared to excitation at 387 nm. Due to the CO release of the CORM peptide conjugate upon excitation above 400 nm, a deeper tissue penetration at longer wavelengths is possible, which is beneficial compared to the other conjugate systems. In addition, the capability to target tumors is favorable.39
Stupp et al. covalently attached CORMs to peptides, deriving a CORM-3@peptide conjugate (Fig. 4). The synthesized one-dimensional (1D) nanofibers (diameter 8.2 nm) consisted of an alkyl tail attached to a peptide sequence with a CORM-3 fragment. The peptide sequence had a β-sheet-configuration to self-assemble into nanofibers and a β-aspartic acid component for binding the Ru-carbonyl fragment (instead of glycine in CORM-3). This CORM-3-type conjugate released CO after ligand exchange. Stupp et al. proposed an easier use of CORM-3 conjugates in medical applications in tissue compared to photoactive CORM-L1 conjugates because of the possible harm that UV light causes to healthy tissue and cells.40
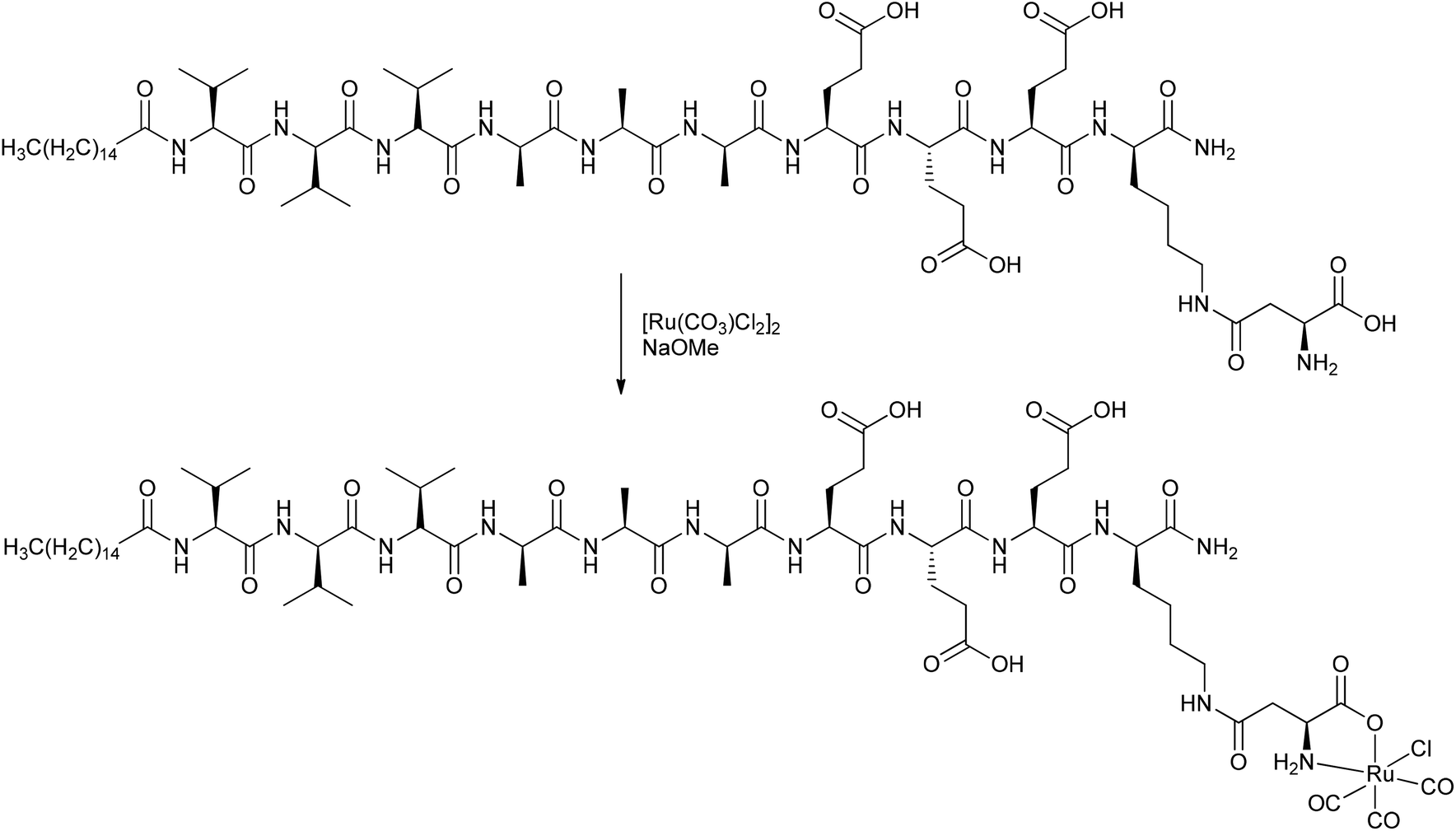 | ||
| Fig. 4 Synthesis of a covalent CORM@peptide conjugate of an amphiphilic peptide with a CORM-3 fragment by Stupp et al.40 | ||
Similar to the parent compound CORM-3, the CO vibration bands of the conjugate were pH-dependent. Under acidic conditions, the IR spectrum showed slightly shifted bands at 2140, 2060, and 1986 cm−1, while in the base there were only two bands at 2023 and 1951 cm−1 present, but the bands were also shifted due to the linkage. The amphiphilic CORM-3@peptide had, within a margin of error, the same half-life of CO release (as assessed by assay in phosphate-buffered myoglobin, with the addition of sodium dithionite; t1/2 = 2.16 ± 0.05 min for CORM-3; t1/2 = 2.14 ± 0.17 min for the conjugate) and the same cardio-protective activity as CORM-3. In order to increase the half-life, Stupp et al. formed self-assembling peptide gels of this compound. They argued that, due to the decreased water accessibility upon nanofiber bundling, the half-life of CO release would be effectively increased to 17.8 ± 1.4 min. Hence, the advantage of this amphiphilic CORM-3@peptide is its possible administration in the aqueous solution state and, as a gel, the prospects of the gel material remaining at the injection site in diseased tissue, thereby preventing any side effects occurring in other tissues.40
The described CORM@peptides conjugates are advantageous due to their great biocompatibility and their wide range of design options. In further developments, the peptides could maybe also be used for the conjugation of receptors for the specific targeting of the CORM conjugate, so that localized applications, such as in tumors, would become possible.
CORMs@polymers
Following the aforementioned covalently CORM@peptide bound bio-conjugates, Hubbell et al. developed the first nanosized technical polymer conjugate for CORMs. To achieve a slow diffusion of CO to the targeted tissue, polymeric micelles with a conjugated CORM-3 component were synthesized. The CO-releasing micelles were built from a triblock copolymer (PEG-bl-OrnRu-bl-nBu) of poly(ethylene glycol), poly[Ru(CO)3Cl(ornithinate acrylamide)] and poly(n-butylacrylamide) (Fig. 5).41 As the PEG-parts were hydrophilic and the nBu-blocks hydrophobic spherical, micelles with a hydrodynamic diameter in the range of 30–40 nm were formed (Fig. 6).41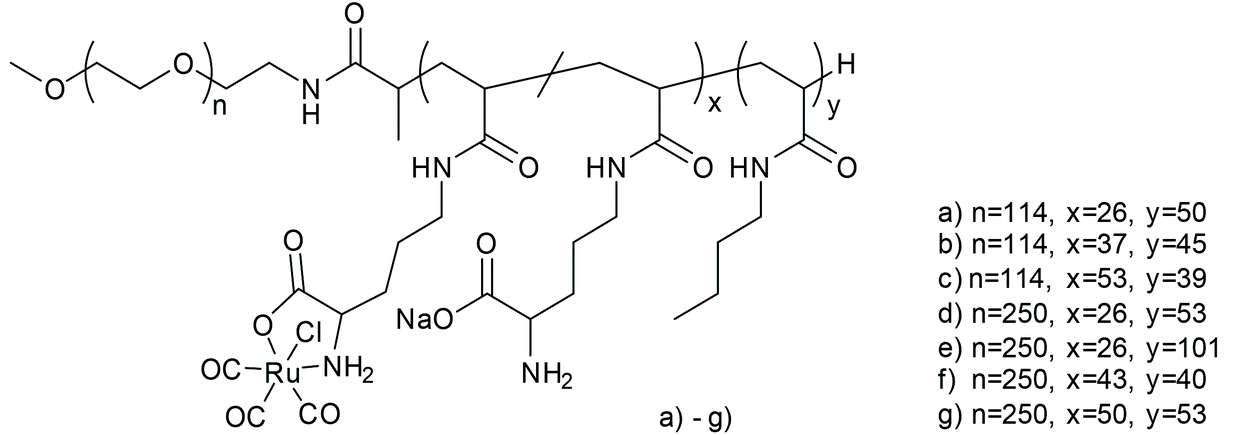 | ||
| Fig. 5 Structure of the triblock copolymer PEG-bl-OrnRu-bl-nBu by Hubbell et al. [PEG: poly(ethylene glycol), OrnRu: poly[Ru(CO)3Cl(ornithinate acrylamide)], nBu: poly(n-butylacrylamide)].41 | ||
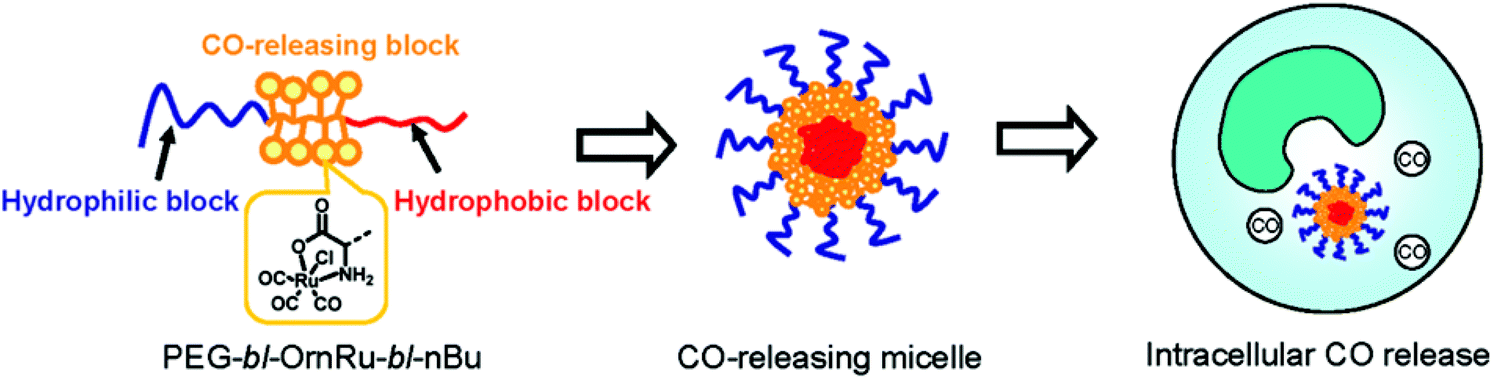 | ||
| Fig. 6 Scheme of the CORM-3@micelle system by Hubbell et al. Reprinted with permission from ref. 41. Copyright 2010 American Chemical Society. | ||
The micelles were stable in water as well as serum and released CO slower than CORM-3 itself (conditions: myoglobin assay in aqueous PBS buffered myoglobin, sodium dithionite, and 10 mmol L−1 cysteine). CORM-3 released 0.12 mol CO per mol CORM over 120 min. From the micelles, only 0.05 mol CO per mol CORM was released over the same time. The steric barrier of the PEG shell hampered the diffusion of an exogenous release trigger (such as thiol-containing compounds, like cysteine) in the micelles. In the IR spectra, again the typical three CO bands were slightly shifted toward 2135, 2049, and 1968 cm−1. The micelles showed a reduced toxicity compared to the parent CORM, owing to the PEG (as assessed by in vitro MTT-assay against THP-1 Blue™ cells) and a high loading capacity of 2500 releasable CO molecules per micelle, as determined by myoglobin assay. Furthermore, their size was controllable and they were easy to formulate.41 It cannot be excluded, however, that the micelles slowed the CO diffusion to the outside myoglobin assay or that part of the CO had already been lost from the CORM during the micelle preparation. Further, the myoglobin assay commonly leads to low CO content values due to a number of diverse reasons, especially for slow releasers. Therefore, sometimes the lower CO release value is an artifact of a slow CO liberation and not an incomplete CO release, as was shown by Berends and Kurz in a detailed spectroscopic study on the light-triggered CO release from two manganese PhotoCORMs (including CORM-L1).42
In nearly the same manner, Boyer et al. synthesized the water-soluble diblock copolymer P(OEGA)-b-P(VB-R-CORM-2) with poly(oligoethylene glycol methyl ether acrylate) as well as poly(vinylbenzyl chloride) with different thiol moieties and a CORM-2 component (Fig. 7).43 The CO release from the polymer–Ru-CORM compounds occurred by a ligand exchange. The half-lives were dependent on the bound thiol component, but in general they were slower compared to the parent CORM-2 (as assessed by assay in phosphate-buffered myoglobin, with the addition of sodium dithionite). The half-lives were t1/2 = 180 min for P(OEGA)-b-P(VB-R1-CORM-2), t1/2 = 40 min for P(OEGA)-b-P(VB-R2-CORM-2), t1/2 = 90 min for P(OEGA)-b-P(VB-R3-CORM-2), and approximately 0.8 equivalents of CO were released.43
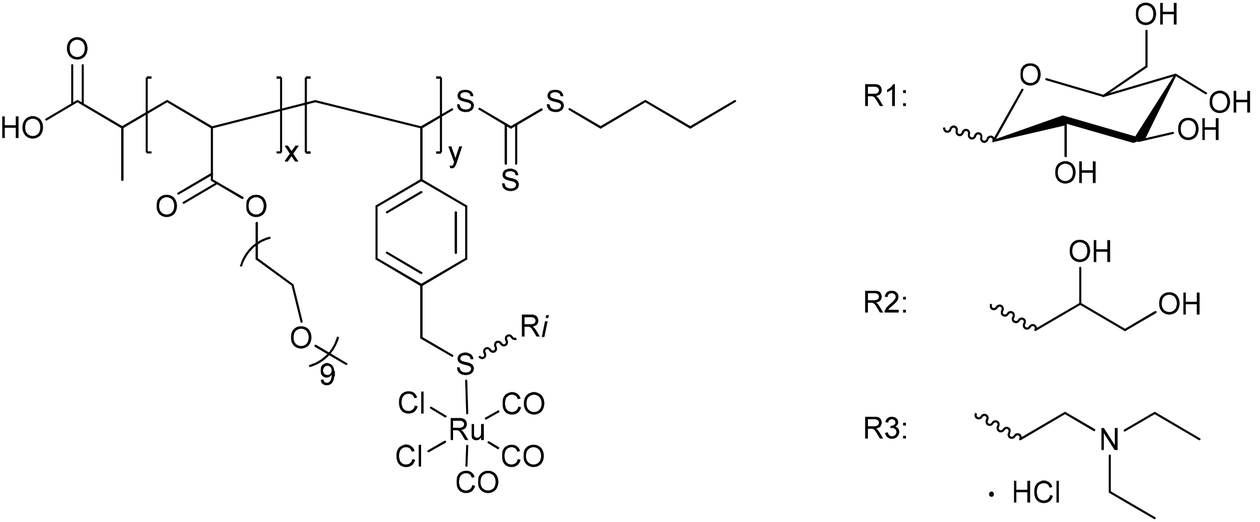 | ||
| Fig. 7 Structure of the diblock copolymer P(OEGA)-b-P(VB-R-CORM-2) synthesized by Boyer et al. [P(OEGA): poly(oligoethylene glycol methyl ether acrylate), P(VB): poly(vinylbenzyl chloride), R1: 1-thio-β-D-glucose sodium salt, R2: 1-thiolglycerol, R3: 2-diethylaminoethanethiol hydrochloride].43 | ||
The CO-releasing polymer-Ru-CORMs were developed due to the promising effects of the released CO against the bacterium Pseudomonas aeruginosa and the associated possible use of the polymer as an antibiotic. For this possible application, the polymers were tested as an inhibitor of the growth, biofilm formation, and cell viability of the bacterium. The growth of Pseudomonas aeruginosa was in fact reduced by about 90% compared to untreated cultures, with the best results obtained with P(OEGA)-b-P(VB-R2-CORM-2). In addition, this positive result for P(OEGA)-b-P(VB-R2-CORM-2) was supported by the inhibition of biofilm formation, which was also decreased by about 95%, and the cell viability tests, whereby the number of cells were reduced by up to 94% through the polymer–Ru-CORMs. All the results were compared to the parent CORM-2, where the better results for the polymer–Ru-CORMs showed the benefits of CORMs being bound to polymers.43
Following up these good performances, Boyer et al. also synthesized micelles of a slightly varied diblock copolymer P(OEGA)-b-P(4VP), again with the hydrophilic polymer poly(oligoethylene glycol methyl ether acrylate) and the hydrophobic polymer poly(4-vinylpyridine) with a CORM-2 component (Fig. 8). The aim of using these micelles was to improve the water solubility, enhance the half-lives, and to utilize the EPR-effect as in the work of Hubbell et al.41,44
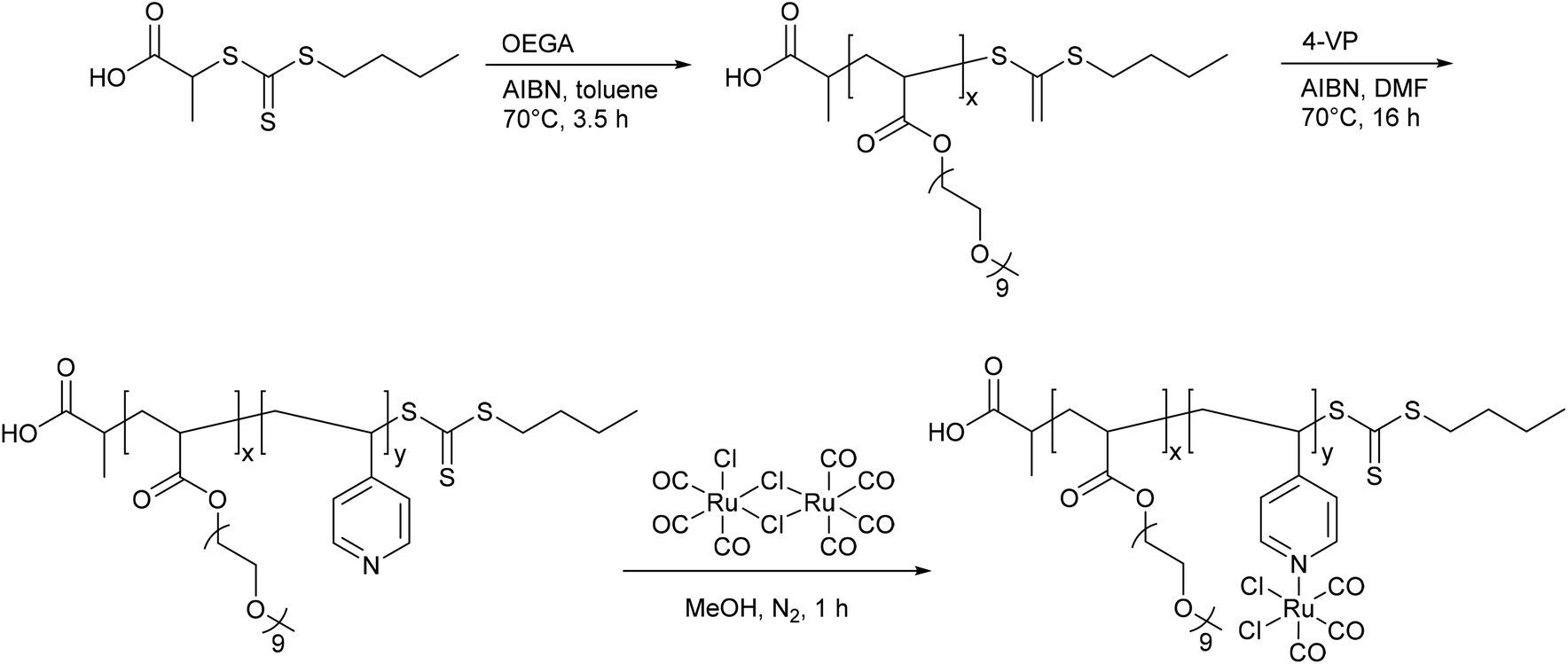 | ||
| Fig. 8 Structure of the diblock copolymer P(OEGA)-b-P(4VP-CORM-2) synthesized by Boyer et al. [P(OEGA): poly(oligoethylene glycol methyl ether acrylate), P(4VP): poly(4-vinylpyridine)].44 | ||
The synthesized CORM@polymer was, among other things, analyzed with IR spectroscopy to control the presence of the CO vibrational bands (1800–2100 cm−1), which were found to be comparable to those of free CORM-2 (2136, 2065, 2017 cm−1) and the above-mentioned diblock copolymer P(OEGA)-b-P(VB-R-CORM-2).43–45 The CORM conjugates by Boyer et al. released CO through a pH change, which was different from the cysteine trigger for the CORM@micelles synthesized by Hubbell et al. By increasing the pH value from 4 to 7 and then up to 9, the half-lives of CO release (as assessed by assay in phosphate-buffered myoglobin, with the addition of sodium dithionite) also increased from 30 to over 50 and up to 60 min, with a release of approximately 0.6 equivalents of CO per 4-vinylpyridine unit.44
A PhotoCORM@polymer conjugate system, including the light-triggered Mn(CO)3-fragment, was synthesized by Kunz et al. to selectively accumulate in tumor tissue due to the EPR-effect. The macromolecular carrier systems used were copolymers formed by the copolymerization of methacrylate or methacrylamide together with a linker unit that carried a bis(pyridylmethyl)amine ligand to bind the CORM-fragment (Fig. 9). A second PhotoCORM@polymer conjugate was extended with a polylactide (PLA) side chain.46
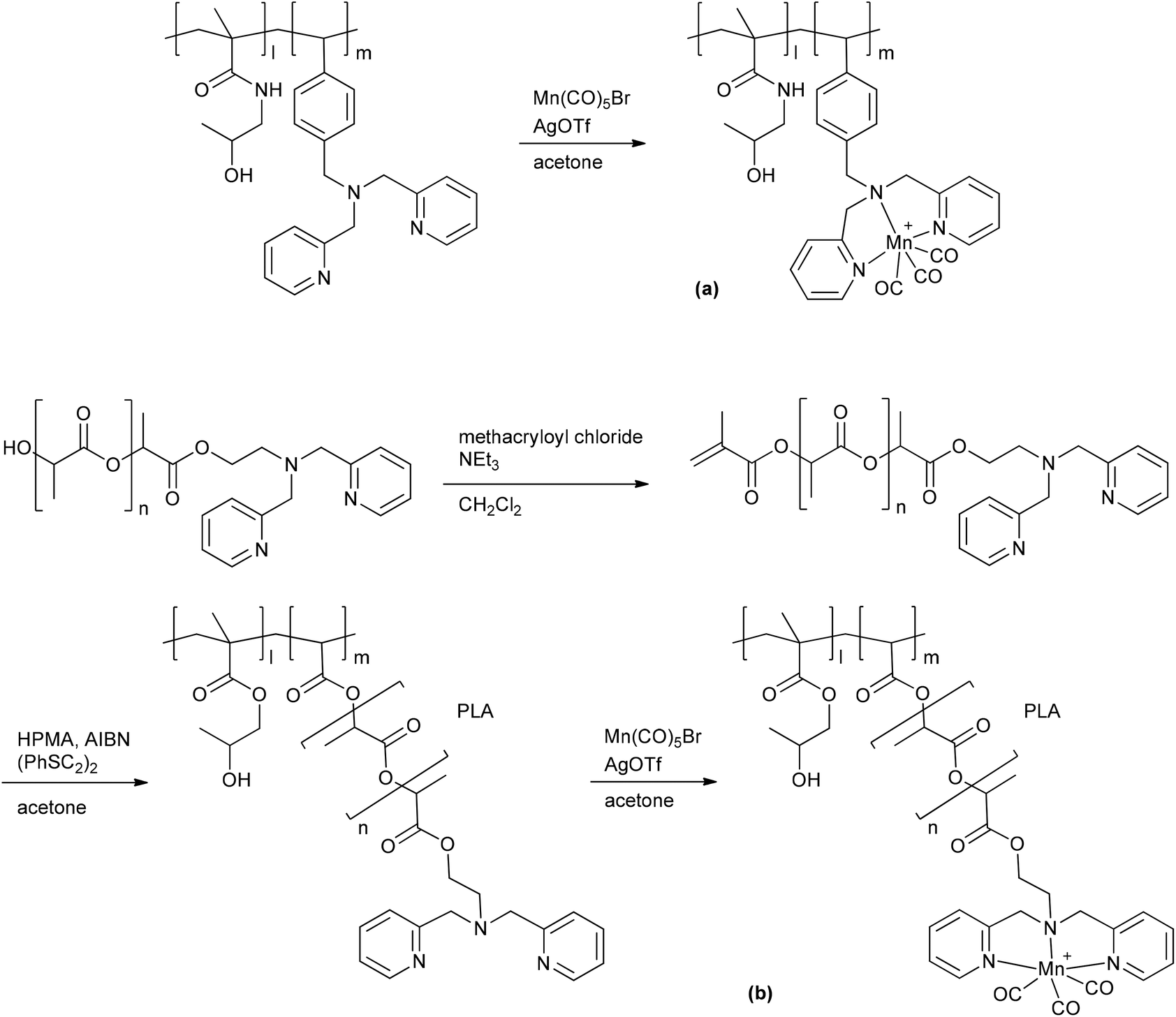 | ||
| Fig. 9 Synthesis of the PhotoCORM Mn(CO)3@polymer1 (a) and Mn(CO)3@polymer2 (b) conjugates by Kunz et al. (HPMA: N-(2-hydroxypropyl) methacrylamide, AIBN: azobisisobutyronitrile, PLA: polylactide).46 | ||
These PhotoCORM@polymer conjugates had a hydrodynamic diameter of 10 nm and a manganese content of 8.9% without PLA and 2.8% with PLA. Due to their size, their distribution within the body is limited to the vascular system. Both conjugates showed nearly the same three CO vibration bands, i.e., at 2037, 1948, and 1934 cm−1versus 2036, 1945, and 1929 cm−1, which demonstrated that the CORM was still intact after the reaction. The metal carbonyl in the polymer conjugate still acted as PhotoCORM and released CO, with a half-life of 20 min (as assessed by assay in phosphate-buffered myoglobin/dimethyl sulfoxide solution, with the addition of sodium dithionite). Compound Mn(CO)3@polymer1 had no significant cytotoxicity, whereas Mn(CO)3@polymer2 was toxic, even though the individual polymer component parts and the CORM were not (as tested against Hct116 human colon carcinoma and HepG2 human hepatoma cells). These studies showed that the design of such polymeric carriers is easily adaptable to the needs of further applications, but that the cytotoxicity of such conjugates is more complex.46
A benefit shown by the above-mentioned CORM@polymer conjugates are their tremendous design options and thereby the possibility to match the application and the polymer. In addition polymers offer the possibility to adapt their size for use of the EPR-effect to accumulate the conjugates in diseased tissue. However, one has to pay attention to the toxicity of the polymer conjugate and the residues after CO release, as shown by Kunz et al.46
CORMs@nanoparticles
As not only polymers are applicable to the EPR-effect, other research groups have focused on nanoparticles as CORM conjugates. Schatzschneider et al., for example, studied the attachment of PhotoCORM-L1 via a copper-catalyzed azide–alkyne 1,3-dipolar cycloaddition (CuAAC, “click reaction”) to obtain functionalized silicium dioxide nanoparticles (Fig. 10). The major benefits of this nano-scale carrier system were the simple synthesis, controllable size distribution, and biocompatibility of the silicium dioxide nanoparticles.47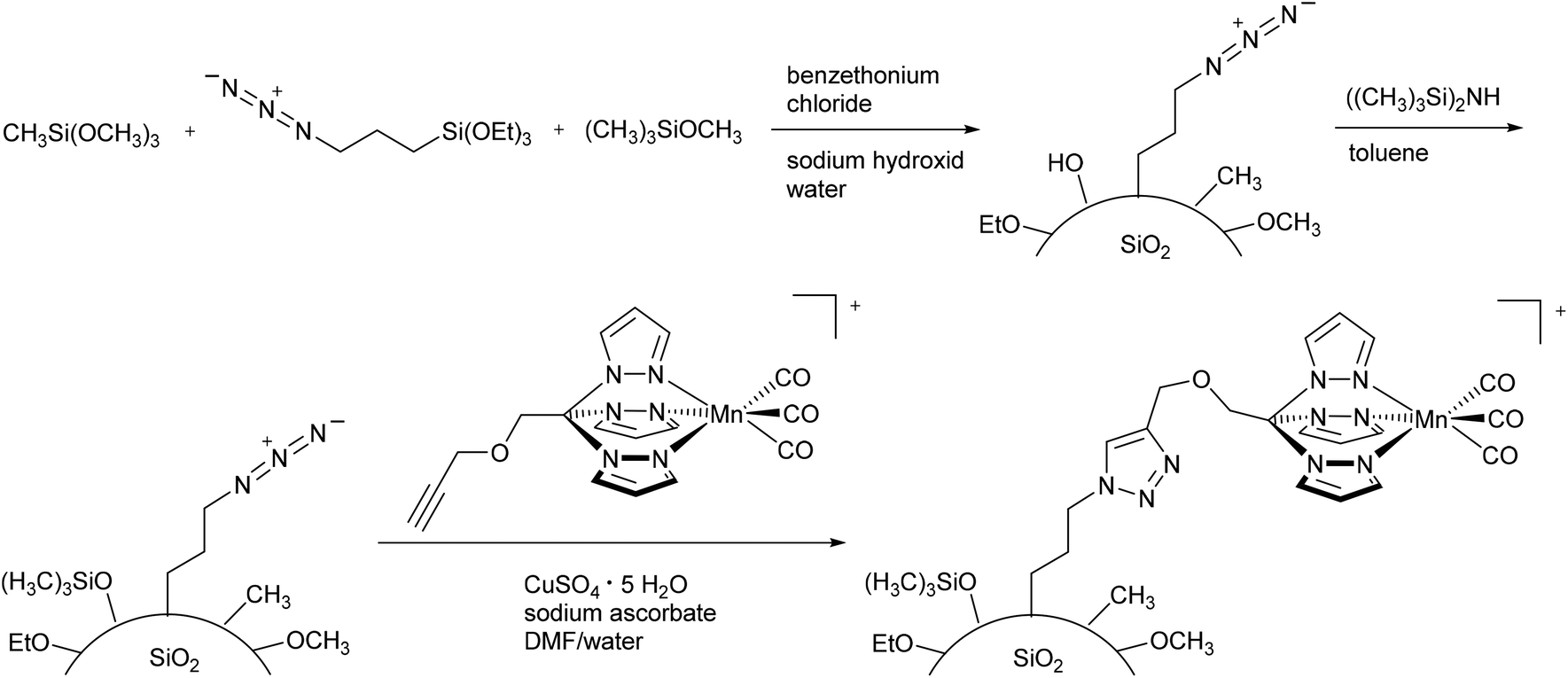 | ||
| Fig. 10 Synthesis of PhotoCORM-L1-functionalized silicium dioxide nanoparticles by Schatzschneider et al.47 | ||
Due to the mild synthesis conditions, the PhotoCORM-L1 was stable during the “click reaction”, as verified by IR (CO bands at 2050 and 1958 cm−1) and still tested as photoactive in the SiO2 composite material (cf.Fig. 10). The so-decorated PhotoCORM nanoparticles released 2 to 3 equivalents of CO per mole of complex upon irradiation (as assessed by myoglobin assay in phosphate-buffered aqueous dimethyl sulfoxide solution, with the addition of sodium dithionite). However, decoration of the nanoparticle surface gave only 22% surface functionalization due to the limited accessibility of most of the surface sites.47
More recently, Schatzschneider et al. attached the [Mn(CO)3(tris(pyrazolyl)methane)]+-fragment to azido-functionalized nanodiamonds using the aforementioned CuAAC click reaction (Fig. 11).48 The advantages of nanodiamonds are a good cellular uptake in order to achieve accumulation in diseased tissue and their non-toxicity.49,50 The resulting nanoscale carrier compounds had a content of 0.47 wt% manganese and a size of about 10 nm. Although the CORM-fragment was still photoactive, quantitative measurement of the CO release using the myoglobin assay (in phosphate-buffered solution, with the addition of dimethyl sulfoxide and sodium dithionite) was not possible due to the formation of cloudy dispersions. IR spectroscopy showed that the CO vibration bands at 2051 and 1961 cm−1 were identical to those of the parent compound CORM-L1 and were not shifted by attachment to the nanocarrier.48
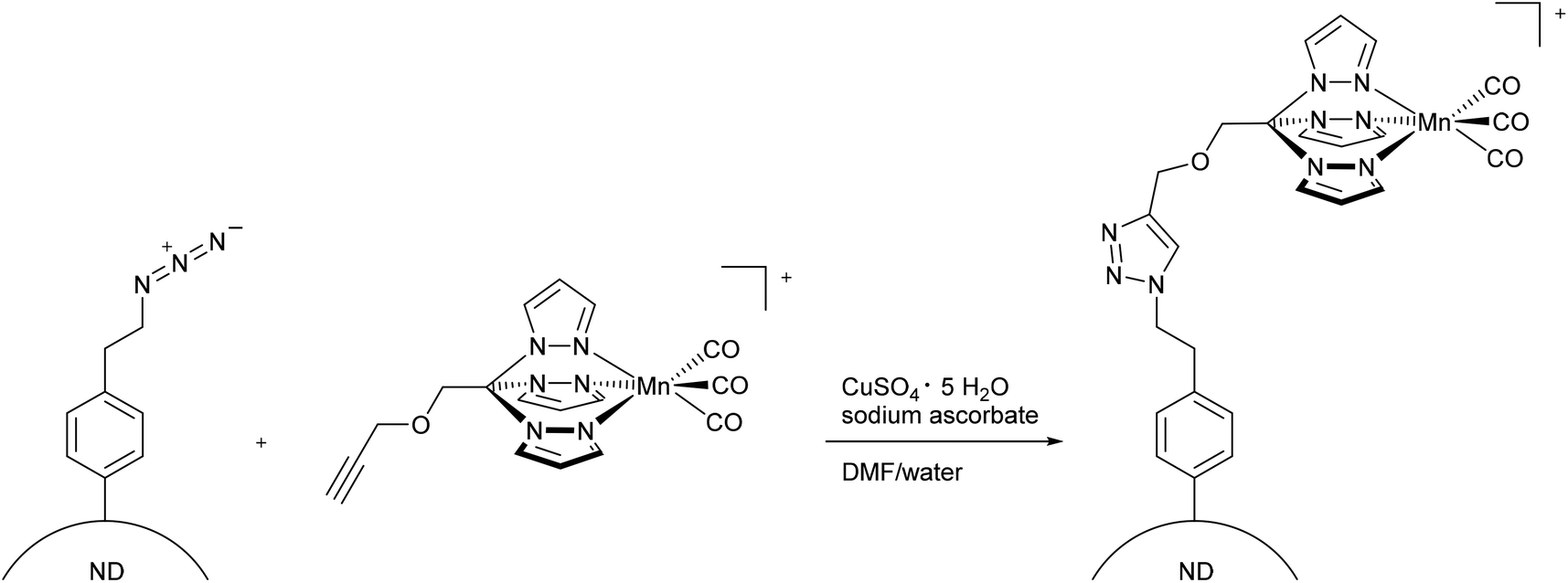 | ||
| Fig. 11 Synthesis of PhotoCORM-L1-functionalized nanodiamonds (ND) by Schatzschneider et al.48 | ||
In addition Kunz et al. immobilized CORM-3 analogs on the surface of maghemite (Fe2O3) iron oxide nanoparticles (IONPs) using D/L-dihydroxyphenylalaninato (DOPA) anchoring ligands (Fig. 12).51
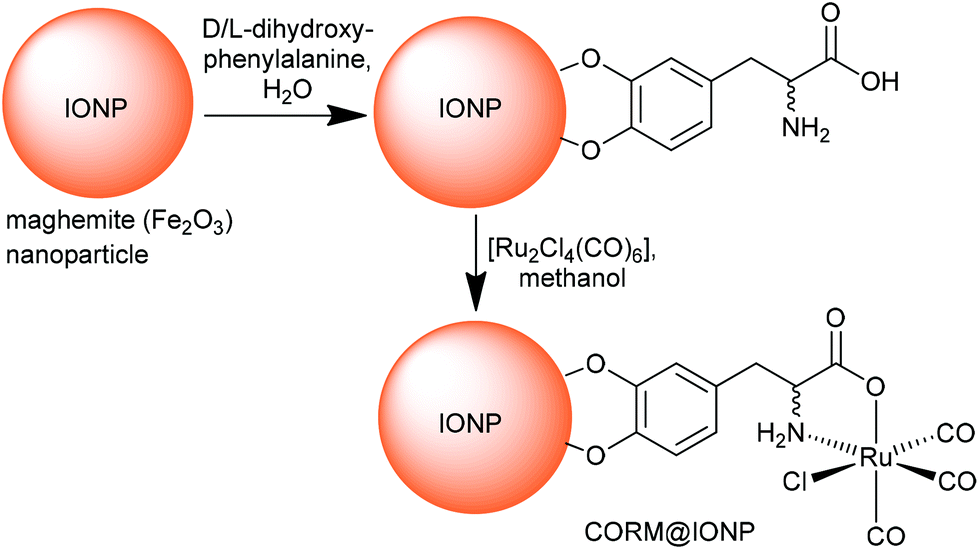 | ||
| Fig. 12 Synthesis of CORM-functionalized iron oxide nanoparticles (IONPs) using an Fe3+-affine catecholate linker with a CORM-3 analog. The maghemite IONPs had a hydrodynamic diameter of (8 ± 2) nm, which increased to (11 ± 3) nm after functionalization with D/L-dihydroxyphenylalanine. Redrawn from ref. 51 with permission from The Royal Society of Chemistry. | ||
Maghemite nanoparticles are readily accessible.52 The dihydroxyphenyl (catecholate) group strongly coordinates to Fe3+ on the IONP surface. Magnetic IONPs can be heated in an alternating magnetic field. This technique is, inter alia, applied in the hyperthermia/thermal ablation treatment of cancer tissue.53 The idea of combining CORMs with IONPs was that magnetic iron oxide nanoparticles decorated with temperature-sensitive CORMs on their surface could be triggered to release CO by heating of the IONP when stimulated by an external alternating magnetic field (Fig. 13).51
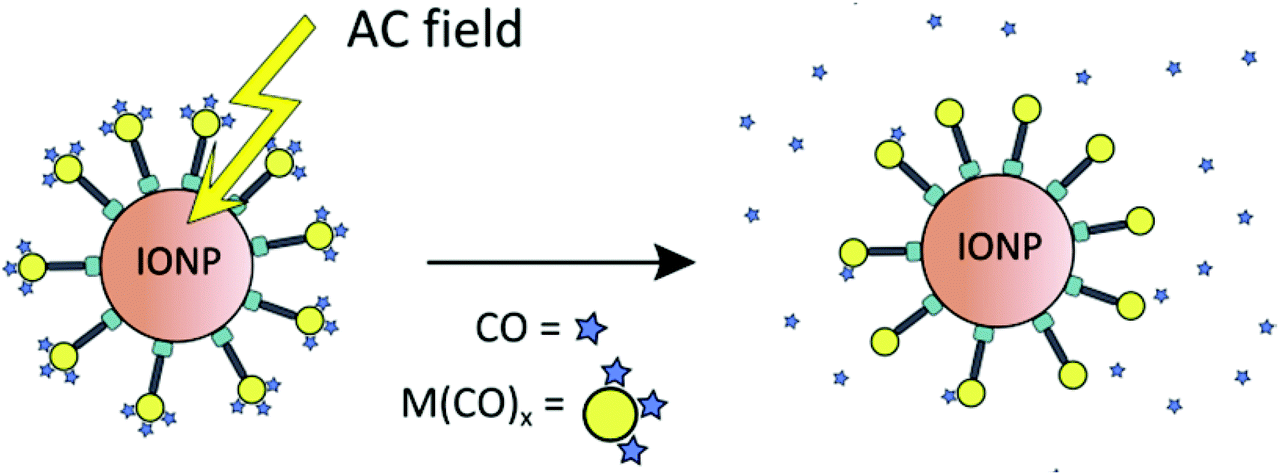 | ||
| Fig. 13 Schematic presentation of triggered CO-release from a temperature-sensitive CORM on the surface of magnetic iron oxide nanoparticles (IONPs) through heating by an external alternating magnetic field. Reprinted from ref. 51 with permission from The Royal Society of Chemistry. | ||
The proof-of-concept study showed that the half-life of CO release from [RuCl(CO3) (μ-DOPA)]@maghemite nanoparticles was halved from 13 ± 2 min to 7 ± 2 min by applying an external alternating magnetic field (31.7 kAm−1, 247 kHz, 25 °C, 39.9 mTesla; both at 25 °C solution temperature in MOPS-buffered myoglobin assay, with the addition of sodium dithionite). However, the ligand-labile CORM-3 analog (as CORM-3 itself)20,23 was already susceptible to CO substitution by the Na2SO3/protein environment of the myoglobin assay. In addition, the IONPs featured a low water solubility and were intensely dark colored, which made the CO-release measurement difficult using optical absorption, which is part of the standard protocol for the myoglobin assay.51 A subsequent study addressed this problem using a modified protocol for the myoglobin assay in which the CORM@IONP system was incorporated into polymer beads (see below).
CORMs@nanoparticles are also controllable in size, easy to synthesize and are applicable to the EPR-effect, same as the CORMs@polymers. However, different to the more often described PhotoCORM conjugate systems, the CORM@magnetic nanoparticle conjugate prepared by Kunz et al. offered a new trigger mechanism, whereby it was the first known conjugate to utilize highly localized heating for CO release, akin to hyperthermia treatment, through a distant alternating magnetic field. This is a benefit for controllable therapeutic applications as the CO release may be started and stopped at the specific target through an on/off alternating magnetic field.
CORM@dendrimers
A metallodendritic PhotoCORM was synthesized by Smith et al. that exhibited potential as a drug carrier in cellular systems. They made use of their inherent ability to accumulate in tumor tissue due to the EPR-effect. Therefore, the MnBr(CO)3-fragment was attached to a diaminobutane-polypyridyl dendrimeric scaffold. In addition, this strategy does not set small molecular degradation products free; after CO release, the remaining metal fragment is still bound to the macromolecular conjugate, which prevents possible side effects occurring (Fig. 14).54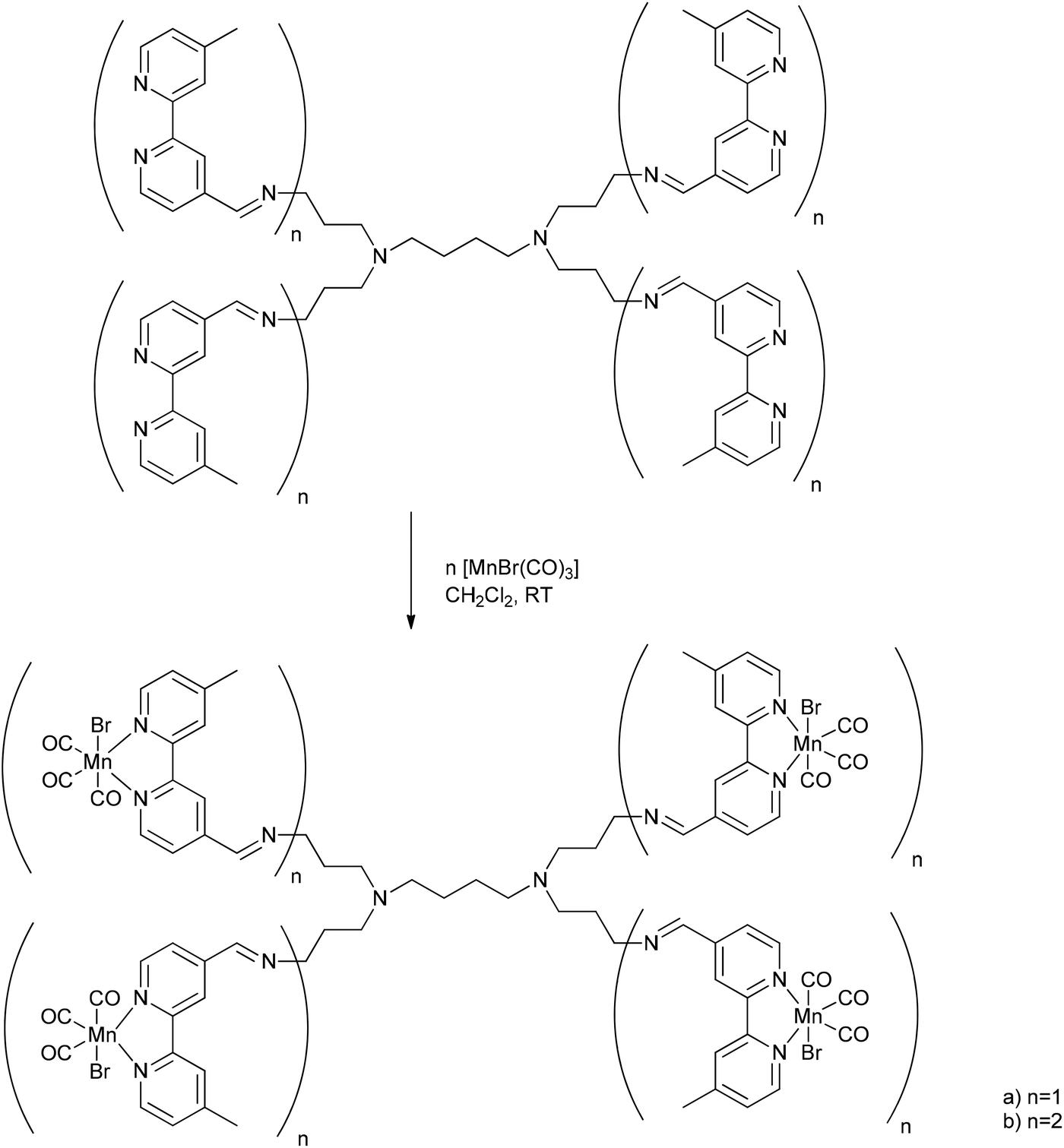 | ||
| Fig. 14 Schematic structure of the PhotoCORM@dendrimer synthesized by Smith et al.54 | ||
Depending on the number of attached photoactive CORM-fragments, the CORM-dendrimers released between 8 and 15 CO ligands per dendrimer molecule (that is two CO ligands per CORM unit). The half-life in CO release was 14.5 to 16.8 min (as assessed by myoglobin assay in PBS buffered dimethyl sulfoxide solution, with the addition of sodium dithionite). The metallo-dendrimer compounds were stable in aqueous solution and air in the absence of light, and the CO bands in the conjugate were found at 2020, 1920, and 1900 cm−1.54
CO@MOFs
Another solid-storage material for CO was synthesized by Metzler-Nolte et al. The group used the biocompatible metal–organic frameworks (MOFs) MIL-88B(Fe) and NH2-MIL-88B(Fe) to adsorb CO at their accessible coordinative unsaturated metal sites (CUSs) (Fig. 15). A benefit of the MOFs could be their so-called breathing effect,55,56 which refers to the reversible swelling of the flexible MOF pores due to adsorption of a solvent. This characteristic could potentially be used for controlling the opening and closing of the MOF pores and thereby to control the CO release.57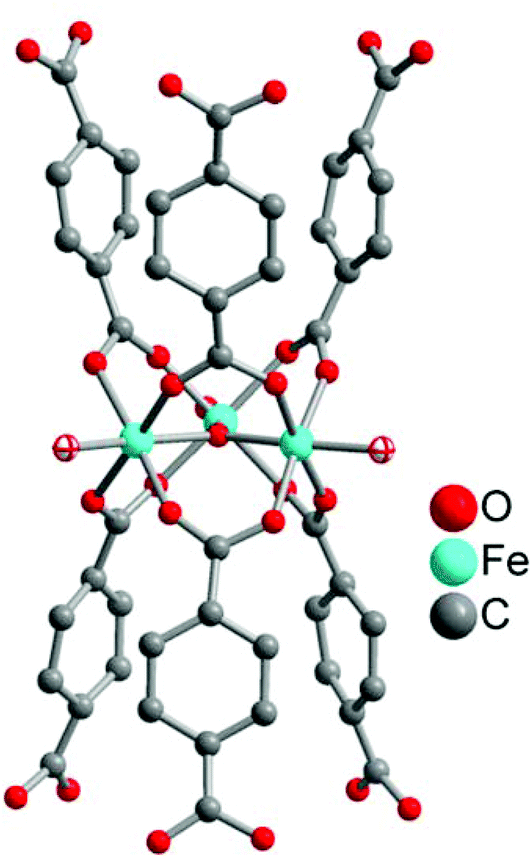 | ||
| Fig. 15 Structure of a μ3-oxo-bridged trinuclear secondary building unit of MIL-88B(Fe) and NH2-MIL-88B(Fe). The two terminal aqua ligands given with their ellipses are potentially coordinatively unsaturated metal sites (CUSs). Generated from the deposited cif-file (CCDC 285810, Refcode YEDKOI) for the isostructural Cr compound. | ||
After their microwave-assisted synthesis, the highly porous MOFs were activated at 550 K. Mössbauer spectroscopy showed that the terminal water molecules at the iron atoms (cf.Fig. 15) were first removed at around 450 K, giving coordinatively unsaturated FeIII species. Afterwards, the terminal chloride anions were reductively eliminated, generating FeII CUSs (cf.Fig. 15). Following this activation, the CO adsorption process for both MOFs was performed at 98 K. The IR spectrum of MIL-88B(Fe) showed the typical CO stretching vibrations at 2181 and 2167 cm−1 due to the different oxidation states of iron in the MOF. In comparison, NH2-MIL-88B(Fe) only showed one CO band at 2169 cm−1. The absence of the second band was explained by the less-stable FeIII binding strength to CO. The amino ligand, as an electron donor, lowers the Lewis acidity of the CUSs and thereby the binding strength between the iron species and the adsorbed CO. Probing the CO release with the myoglobin assay (at 37 °C in phosphate-buffered myoglobin, with the addition of sodium dithionite) showed also a difference for both MOFs. CO-loaded NH2-MIL-88B(Fe) released more CO with a higher half-life than MIL-88B(Fe) owing to more activated, accessible CUSs in the former MOF (0.69 μmol CO per mg NH2-MIL-88B(Fe), t1/2 = 76 min versus 0.36 μmol CO per mg MIL-88B(Fe), t1/2 = 38 min). This could be explained by the lower binding strength in NH2-MIL-88B(Fe), which facilitates the CO removal. Also, in both cases, CO release is accompanied by degradation of the MIL materials.57
CORM@protein cages
Other biocompatible and not cytotoxic (as assessed by in vitro MTT-assay against HEK293/KB-Fluc cells) conjugates, like the above-mentioned peptides, are self-assembled protein cages. Ueno et al. coordinated photoactive manganese-carbonyl moieties into a mutant of the iron storage protein complex ferritin in order to control the light-induced release of CO.58 Similar to the above-mentioned metallodendritic PhotoCORMs, the IR spectrum of the protein cage showed three CO vibration bands at 2028, 2011, and 1917 cm−1. The half-life of CO release after irradiation with visible light at 456 nm was found to be 2.5 ± 0.2 min (as assessed by myoglobin assay, PBS buffer, with sodium dithionite added), which is four times faster than that of CORM-1 under the same conditions (11.4 ± 0.8 min). In addition, the cellular uptake of this CORM conjugate was high (0.35% at 1.76 × 10−10 μmol per cell, 0.33% at 1.64 × 10−10 μmol per cell, as measured by ICP/MS with HEK293/KB-Fluc cells).58Encapsulated CORMs
CORMs@poly(L-lactide-co-D/L-lactide) (non-wovens)
A different approach related to the conjugation was used by Schiller et al. and the following CORM host systems, whereby the CORMs were not covalently linked to but were encapsulated into the scaffold and thereby the metal fragments remain trapped in the scaffold after CO release, which avoids toxic side effects. At first Schiller et al. incorporated the photoactive CORM-1 in biocompatible polymeric non-wovens (Fig. 16).59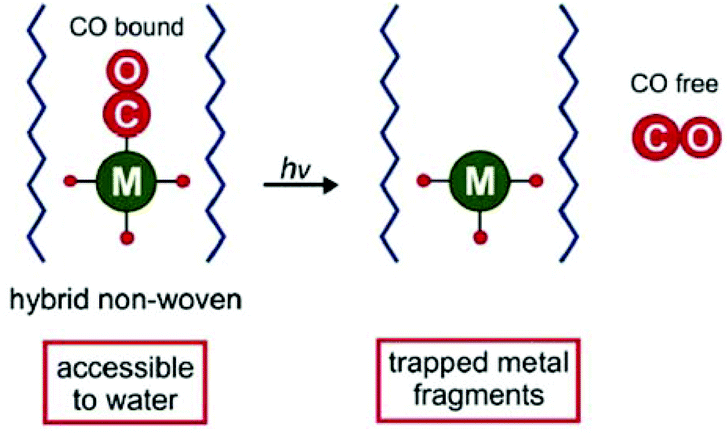 | ||
| Fig. 16 Scheme of the CORM@non-wovens by Schiller et al. Adapted from ref. 59 with permission from The Royal Society of Chemistry. | ||
By an electrospinning technique, CORM-1 was incorporated into the cyto-compatible polymer poly(L-lactide-co-D/L-lactide). Nanofibrous non-wovens with a diameter of about 1 μm and at most 14.8 wt% embedded, homogenous distributed CORM-1 were formed. This CO-releasing material was porous due to a marginal CO release during the synthesis procedure. The CO release measured by a portable CO detector depended on the wavelength of the light used for irradiation (365 nm: t1/2 = 309 s; 480 nm: t1/2 = 1289 s). At least 3.4 μmol of CO per mg sample was released. In experiments against 3T3 mouse fibroblast cells, the non-wovens were characterized as nontoxic. Furthermore, the leaching of CORM-1 out of the fibers in water as well as during the irradiation was low (0.1–2.4%) owing to the insolubility of CORM-1 in water. However, water insoluble CORM-1 is turned water soluble by encapsulation in the water-soluble polymer frame. The non-wovens are applicable in biological systems, such as in skin patches.59
The polymer poly(L-lactide-co-D/L-lactide) and electrospinning were also utilized by Schiller et al. to encapsulate the PhotoCORM dodecacarbonyl-tetrakis(μ3-propanethiolato) tetramanganese(I). Again, nanoporous fibers were formed, which were slightly larger (1.3–2.1 μm) than formed for the CORM-1@poly(L-lactide-co-D/L-lactide) conjugate. Cytotoxicity tests against 3T3 mouse fibroblast cells showed no cytotoxic effects of the CORM conjugate. Light-induced measurements with a LED lamp (14 mW cm−2) and a portable CO detector showed a dependency of the CO release on the wavelength of the light used for irradiation (365 nm: t30 min = 10.7 μmol of released CO per mg sample; 405 nm: t30 min = 8.1–8.3 μmol of released CO per mg sample). In addition, a laser-coupled glass fiber optical device was used with the CORM conjugate wrapped around the device head, such that different light intensities (2.1, 14, and 28 mW cm−2) could be applied at 405 nm. Variation of the intensity with the fiber optic showed that the CO release increased with the intensity (after 30 min, at 2.1 mW cm−2: 0.5 μmol of released CO per mg sample; 14 mW cm−2: 1.2 μmol; 28 mW cm−2: 2.5 μmol). Thereby, this method could allow the application of non-wovens not only for skin patches but also in living cells with high spatial resolution.60
CORM-2@cellulose acetate/PEG (tablets)
Up to now, the therapy of gastrointestinal diseases using CORMs has been limited due to an insufficient controllable administration of either external (e.g., light) or internal stimuli, such as enzymes. Meinel et al. described the first CORM system suitable for oral administration using tablets as an easy and targeted way to deliver encapsulated CORMs (Fig. 17). The tablet core consisted of a citric acid buffer, coated (Eudragit E PO, sodium dodecyl sulfate, stearic acid, and talcum) sodium sulfite crystals, and CORM-2. The core was encased with a semi-permeable membrane of cellulose acetate/PEG.61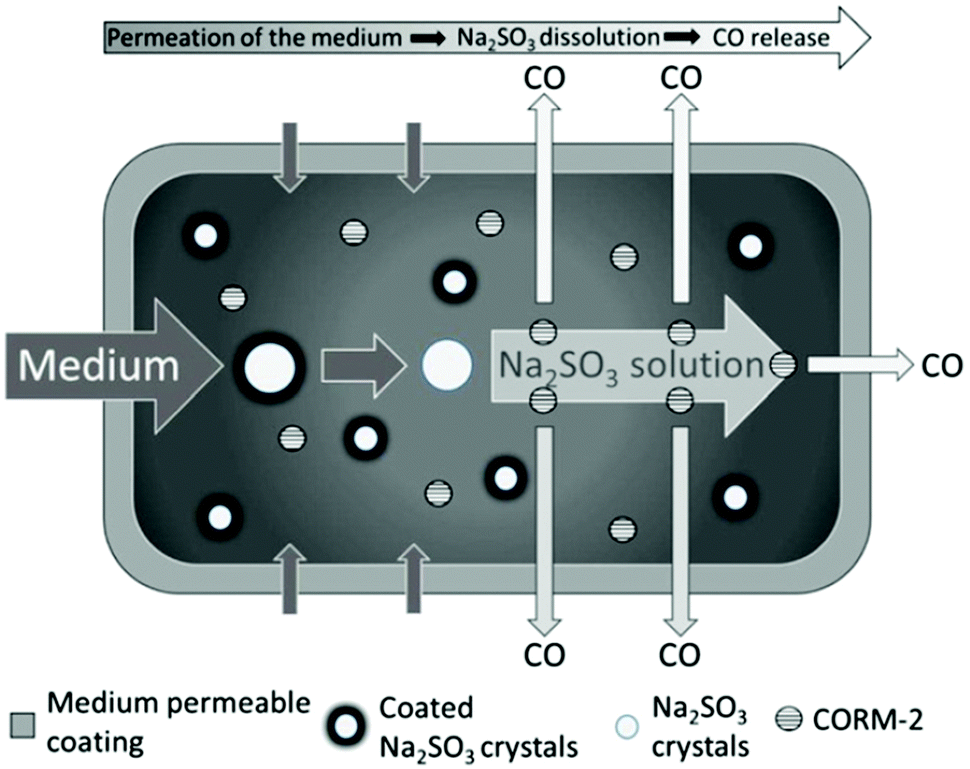 | ||
| Fig. 17 Scheme of the CORM@cellulose acetate/PEG system for oral administration of a tablet by Meinel et al. Reprinted from ref. 61 with permission from Elsevier. | ||
By the diffusion of water through the membrane, first the coating layer around the sodium sulfite crystals and subsequently the crystals themselves dissolved. The released sulfite then triggered CO release from CORM-2 through ligand substitution. The coating around the Na2SO3 crystals separates the sulfite trigger and CORM-2 during storage, so that no CO is released before water is added. Only in the presence of water does the sulfite enter into solution, where it interacts with the CORM in a CO-ligand displacement reaction. The system delivered CO for up to 10 h, with a nearly linear release kinetic between 30 and 240 min, as measured by an amperometric CO detection system (aqueous solutions, with the addition of sodium dithionite and simulated gastric or intestinal fluids). The release was slow due to the controlled flux of water into the tablet core, which could be controlled by varying the membrane thickness. This tablet system uses the advantage of a suitable CORM/trigger combination along with a preset microenvironment to ensure CORM stability during storage. Water then becomes a secondary trigger that activates the actual primary trigger for CO release. Thereby, the tablet is largely independent of the environment within the organism, e.g., the pH value or the ligands, such as in proteins. Further, the tablet also retains the resulting possibly toxic Ru-complex after CO delivery.61,62
In addition to the synthesis and in vitro analysis, the group of Meinel et al. also obtained for the first time in vivo data of these tablets for the prevention of colitis in mice. For this, the group changed the coating of the tablet core and the sodium sulfite crystals to cellulose acetate butyrate in order to increase the hydrophobicity of the coating. The tablets were then administered to mice by oral application, thereby only local and highly concentrated effects in the gastrointestinal tract were induced. For the analysis, the CO-hemoglobin (CO-Hb) formation was measured, which started 1–3 h after the administration of the tablets. The tablets showed protection from colitis by a reduced colon damage score and a maximal CO-Hb limit of 1.4% (safety threshold: 5–10% CO-Hb).8,63
MnCORM@Al-MCM-41 nanoparticles
Nanoparticles have high surface areas, may be biocompatible, are easy to synthesize, and can accumulate in tissue due to the EPR-effect. Because of this, Mascharak et al. chose nanoparticles of the mesoporous silica Al-MCM-41 as a host for the encapsulation of the PhotoCORM [Mn(pqa)(CO)3]ClO4 (pqa: (2-pyridylmethyl)(2-quinolylmethyl)amine) (Fig. 18).64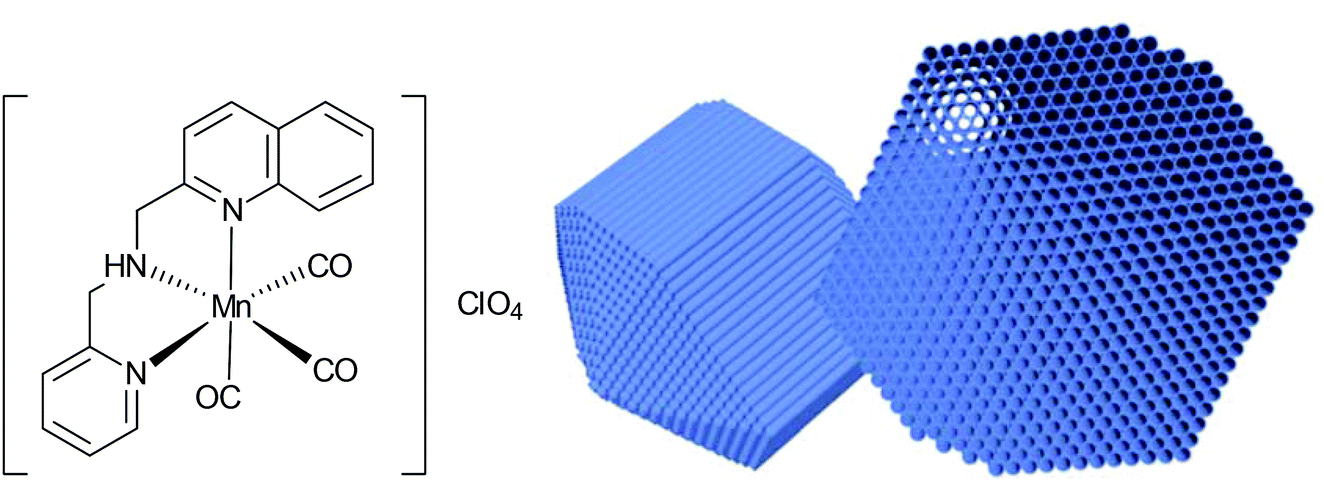 | ||
| Fig. 18 Structure of the incorporated CORM and schematic drawing of the host Al-MCM-41 used by Mascharak et al. Adapted in part from ref. 64 with permission of The Royal Society of Chemistry. | ||
By ion-exchange and diffusion over 48 h, the cationic CORM was incorporated into the negatively charged cylindrical pores of the Al-MCM-41 nanoparticles with a load of 2.06 wt% Mn. The strong interactions through the exchange were supported by leaching experiments in PBS (leaching of 2 wt% CORM after 24 h, 12 wt% after 60 h). The embedding of CORM again was confirmed by IR spectroscopy, whereby a marginal shift of the CO bands was observed compared to the parent CORM (CORM 2033 and 1928 cm−1; Mn-CORM@Al-MCM-41 2040 and 1947 cm−1). CO release in this Al-MCM-41 environment was said to be slower (albeit no times were given) compared to the parent CORM (myoglobin assay, PBS buffer, with the addition of sodium dithionite).64
ALF472@bioMOF-1/MCM-41-SO3H/SBA-15-SO3H
Barea et al. also worked on a diffusion approach into a micro- or mesoporous host like that performed by Mascharaks et al. with the MnCORM@Al-MCM-41 nanoparticles. Barea's group incorporated the photoactive CORM [Mn(tacn)(CO)3]Br (tacn: 1,4,7-triazacyclononane), also known as ALF472, into three different biocompatible porous inorganic or metal–organic framework (MOF) materials as a potential, controllable on/off-switch release system for the phototherapy of skin cancer. By a diffusion process over four days in darkness, the nontoxic, water-soluble, air-stable, cationic CORM was incorporated into the metal–organic framework bioMOF-1 {NH2(CH3)2)2[Zn8(adeninate)4(BPDC)6]·8DMF·11H2O (BPDC = 4,4′-biphenyldicarboxylate)} as well as the functionalized mesoporous silica structures MCM-41-SO3H and SBA-15-SO3H via cation exchange in DMF or water (Fig. 19).65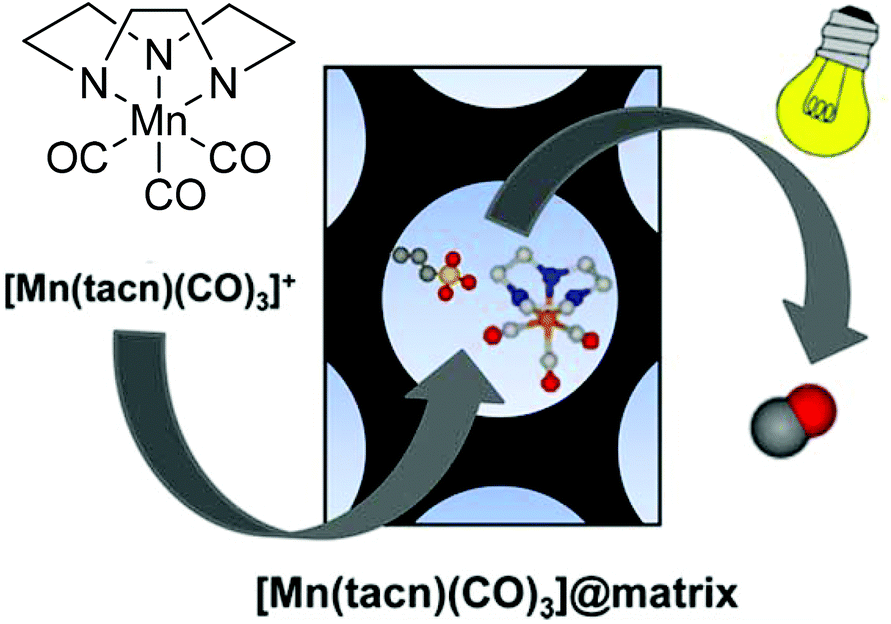 | ||
| Fig. 19 Scheme of the CORM@porous material system synthesized by Barea et al. Adapted with permission from ref. 65. Copyright 2016 American Chemical Society. | ||
The IR spectrum of the product showed only a marginal change of the CO bands after the diffusion process (2017 and 1895 cm−1 for CORM versus 2030 and 1925 cm−1 for the product). As the incorporation proceeded by a cation exchange process, no bromide was detected by EDX. In addition, the CORM load determined by EDX was nearly the same for ALF472@bioMOF-1 and ALF472@MCM-41-SO3H (0.25 mmol and 0.22 mmol ALF472+ per gram). ALF472@SBA-15-SO3H showed the highest CORM load, with 0.53 mmol ALF472+ per gram. This was also mirrored by the CO-release results (as assessed by myoglobin assay at 37 °C in PBS buffer, with the addition of sodium dithionite). In MCM-41-SO3H and SBA-15-SO3H, the release of CO was slower compared to that of the parent CORM (0.80 mmol CO per mmol of complex after 24 h for ALF472; 0.65 mmol CO per mmol of complex after 24 h for MCM-41-SO3H; 0.48 mmol CO per mmol of complex after 24 h for SBA-15-SO3H). In contrast, ALF472@bioMOF-1 released 0.82 mmol CO per mmol of complex but decomposed in water, as shown by the leaching experiments, where 85% of the zinc of the MOF and 75% of the manganese of the CORM were detected in solution after 6 h (ICP-OES). The leaching experiments also showed that ALF472@MCM-41-SO3H was the most potent material. In water, 80% of the CORM remained within the material over a period of three days and only a minor degradation of the mesoporous silica (2.9% silicon after 24 h in solution) was observed. Under physiological conditions, 85% of the CORM was leached after 1 h, which is promising for its applications.65
Covalently-bound CORM conjugates in combination with encapsulation
Polymer@CORM@iron oxide nanoparticles
A ligand-labile CORM, such as CORM-3 and its analogs, has already been shown to be susceptible to CO substitution by the Na2SO3/protein environment of the myoglobin assay.20,23 Also, the IONPs used by Kunz et al. featured low water solubility and are intensely dark colored, which made CO-release measurement by optical absorption with the myoglobin assay difficult.51 These problems were addressed by Janiak et al. by encapsulation of the CORM@IONP system into polymer beads (Fig. 20).66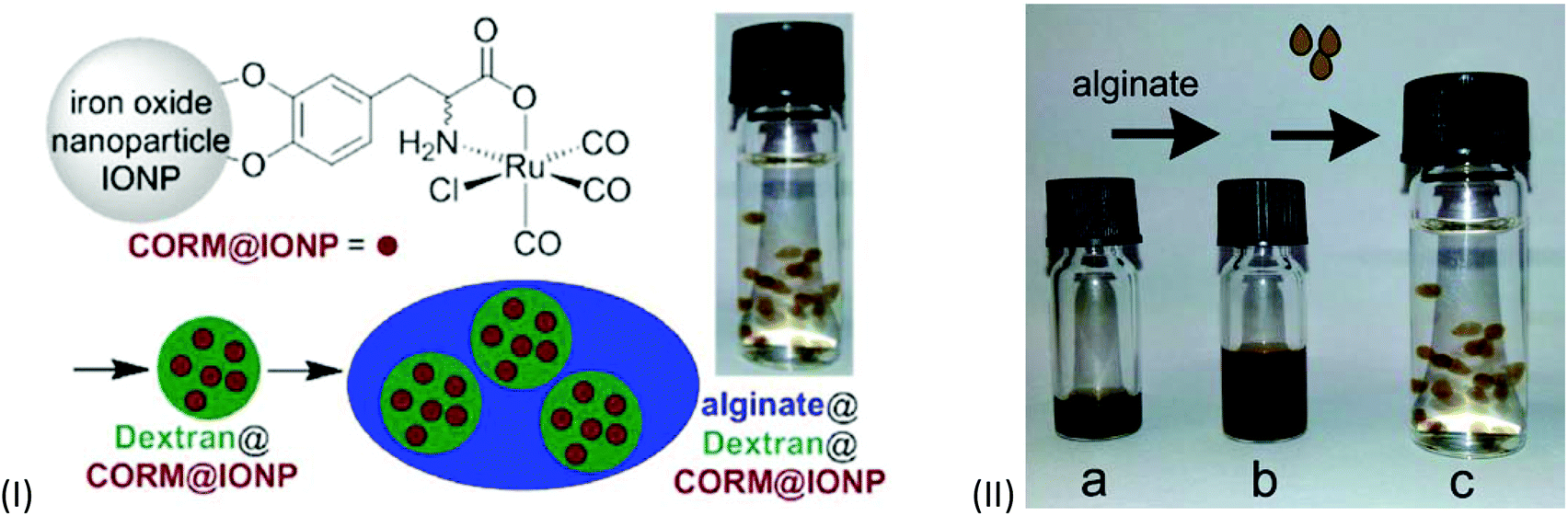 | ||
| Fig. 20 Scheme of the synthesis of CORM-functionalized iron oxide nanoparticles (IONPs) (IIa), encapsulated in a Dextran polymer coating (IIb) and collected into an alginate sphere (IIc). Adapted with permission from ref.66. Copyright 2015 American Chemical Society. | ||
In order to achieve water solubility, CORM@IONPs were coated with different polymers, e.g., dextrans. Using a 500 kDa dextran resulted in core–shell structures. Due to the polymer shell, the average diameter of the particles increased from 11 ± 3 nm to 85 ± 15 nm. TEM pictures showed that the several CORM-iron oxide nanoparticles are encapsulated into one polymer sphere. In addition, the half-life of CO release increased from 9 ± 1 min for CORM@IONP to 32 ± 2 min for dextran@CORM@IONP (both at 37 °C in MOPS-buffered myoglobin, with the addition of sodium dithionite). This is reasonable since the polymer shell is a barrier to the CO substituting Na2S2O4/myoglobin species used in the assay. Dextran@CORM@IONP showed no significant cytotoxic effects up to 100 μg mL−1 (with less than 4.77% growth inhibition) against the cell lines tested (i.e., A2780, Cal27, and HEK293).66
However, the now water-soluble polymer-coated iron oxide nanoparticles were still not yet suitable for the measurement of CO release using the myoglobin assay due to their still strong absorption in the UV/Vis region, but subsequent embedding of the dextran@CORM@IONP nanoparticles into alginate spheres allowed for magnetic separation as well as for applying the myoglobin assay.66
Sodium alginate is an anionic polysaccharide, found in the cell walls of brown algae. It is nontoxic and is already used in biomedical applications. The linear copolymer consists of β-D-mannuronate and α-L-guluronate blocks or sequences. A gel-like material is obtained by ionic cross-linkage of the carboxylate and Ca2+-ions via Coulomb interactions.67,68 Dripping a mixture of dextran@CORM@IONP and sodium alginate into a solution of CaCl2 led to stable hollow spheres of the Ca2+ cross-linked alginate gel containing dextran@CORM@IONP (Fig. 20).66
This alginate shell supported the dextran coating in protecting the CORM from rapid ligand-exchange-induced CO-displacement reactions, while still allowing for permeation of CO to the solution. The alginate shell also compartmented the highly absorbing iron oxide nanoparticles so that they could be easily separated magnetically from the myoglobin assay solution, such that the CO release studies became viable without interference in the optical path of the UV cell. For example, during the assay, the magnetic alginate particles could be kept at the bottom of the cuvette using a permanent magnet outside (Fig. 21).66
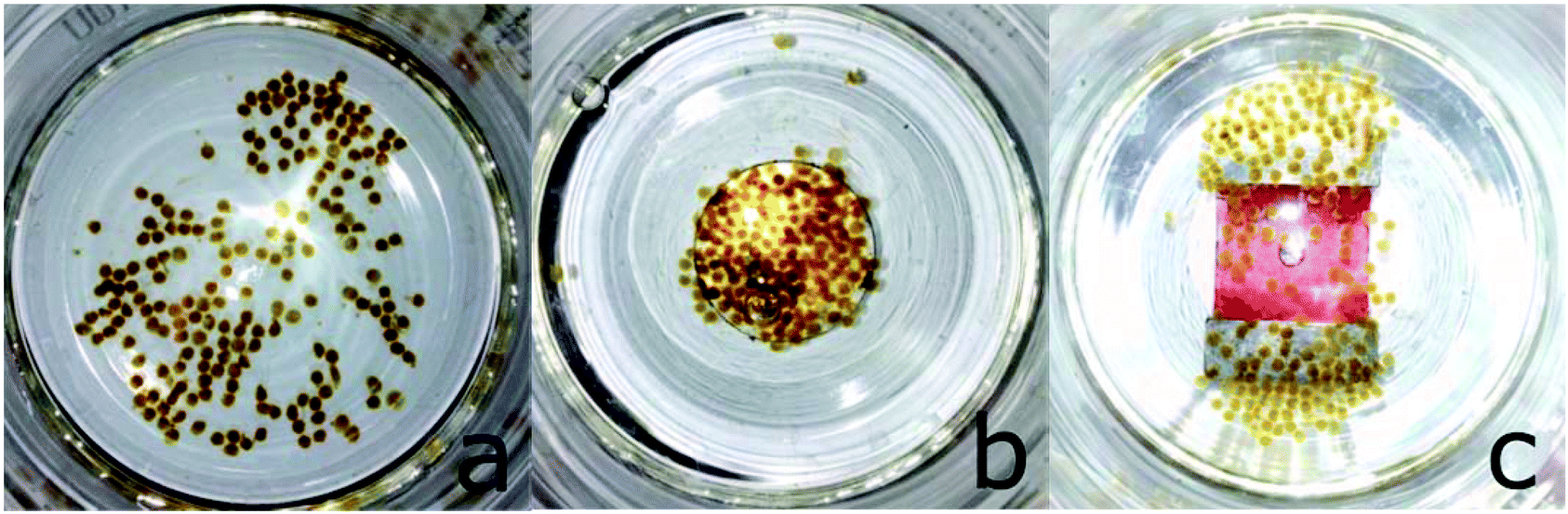 | ||
| Fig. 21 Alginate@dextran@CORM@IONP spheres (cf.Fig. 20) and their arrangement in magnetic fields. (a) No magnet, (b) round magnet, and (c) bar magnet under the glass vessel. Adapted with permission from ref. 66. Copyright 2015 American Chemical Society. | ||
The alginate spheres were magnetic, had diameters of 1–2 mm, and could be stored over a prolonged period of time in 3-(N-morpholino)propanesulfonic acid (MOPS) buffer. The half-life of CO release increased to 172 ± 27 min at 37 °C without and to 65 ± 5 min with the application of a magnetic AC-field (from 9 ± 1 min for CORM@IONP). From the temperature-variable kinetic measurements, an activation energy of 78 kJ mol−1 was determined for the CO release from the IONP-surface-bound CORM-3 analog in the alginate@dextran polymer shell.66
In a follow-up study, oxime-based CORMs69 were similarly covalently immobilized on IONPs, followed by the oximeCORM@IONP encapsulation in dextran and alginate (Fig. 22).70
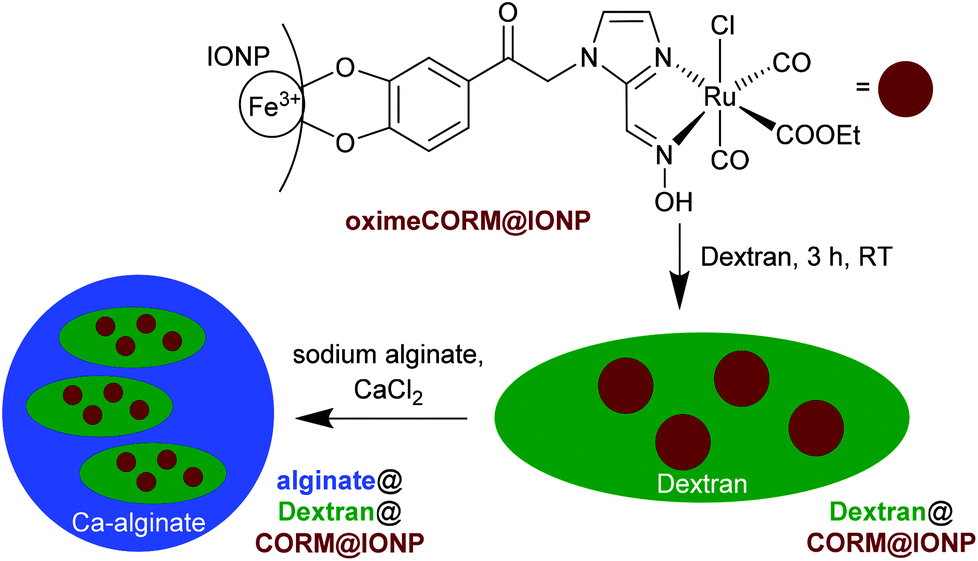 | ||
| Fig. 22 Schematic synthesis of the composite material alginate@dextran@oximeCORM@IONP, starting with immobilization of the oximeCORM on the maghemite nanoparticle surface, followed by encapsulating in dextran (molar mass 500 kDa) and alginate. Adapted from ref. 70 with permission from The Royal Society of Chemistry. | ||
The half-life of CO release from the composite material alginate@dextran@oximeCORM@IONP was 346 ± 83 min at 37 °C compared to the parent oximeCORM of 16 ± 1 min (as assessed by myoglobin assay, MOPS buffer, with the addition of sodium dithionite). Thus, the polymer coating again presented a remarkable stabilization with respect to a rapid CO-displacement reaction by Na2S2O4/protein during the myoglobin assay. Local magnetic heating of the IONP by application of an external alternating magnetic field (31.7 kAm−1, 247 kHz, 39.9 mTesla) decreased the half-life of CO release to 153 ± 27 min, but the solution temperature was not affected (37 °C). The activation energy for the CO release from alginate@dextran@oximeCORM@IONPs using an Arrhenius plot gave a value of 62(17) kJ mol−1.70
Conclusion/personal view
CORMs are typically small metal carbonyl molecules. In CORM conjugates, these CORMs have been covalently bound to peptides, polymers, nanoparticles, dendrimers, and protein cages or have been incorporated into non-wovens, tablets, or metal–organic frameworks to improve their stability, to target them to special tissues, to avoid possible side effects, to use the EPR-effect, or to allow for special triggers.In summary, all the described CORM conjugates, either with the CORM covalently bound or incorporated, show interesting options for biological applications. They are biocompatible and not cytotoxic or at least are less toxic than the parent CORM compounds. Although it has to be noted that for the presented conjugates, different divergent cytotoxicity tests and cell lines were used. Indeed these in vitro data of the CO release are only available for some conjugates and these results could differ from the in vivo profiles. For application of the conjugates, additional in vivo data should be analyzed and compared to the in vitro results as has appeared to have been done for the first time by the group of Meinel et al.63 The same as CORMs in solution, these conjugates play a key role in the pharmacokinetic parameters. They are prodrugs and their action is completely dependent on the in vivo constraints. In addition, it is necessary to analyze the remaining (metal) fragments or residues after CO release in order to detect if there are any side effects and to avoid them in future carrier designs.62,71,72
So far, the absence of side effects seems to be a benefit of the incorporated CORMs, where the CORM moieties after CO release are trapped in the system and cannot affect the tissue. This is not achieved by the exposed CORMs in the conjugates. There, it still has to be analyzed if the potentially cytotoxic ruthenium metal moiety reaches the tissue and acts toxic.
For the CO release measurements, most research groups used a myoglobin assay to show the increased half-lives of CO release due to the covalent binding or incorporation of the CORMs into the conjugate. One problem that has emerged in the last few years with this method is the influence of dithionite on the CO release. With dithionite, CO release occurs without the stimulation of a trigger, which alters the results of the half-life as well as the amount of released CO calculated afterwards.20 To overcome this problem, commercial CO detectors, where the sensor part is brought in contact with the CORM system, could be used to follow the CO release. First in the work of Meinel et al.61 and later also in the work of Schiller et al.,59 the disassembly of such a commercial CO detector (Ei207D from Ei Electronics, Shannon, Ireland) was described, whereby the amperometric CO sensing part was achieved in a closed Erlenmeyer flask, with the rest of the CO detector remaining outside (connected by a cable to the sensor). Thereby, the CO release into the headspace (gas space) of the flask could be measured directly above the solution or solid CORM system.61 For focused light irradiation, the use of a laser-coupled fiber optical device could be advanced in the in vivo applications of PhotoCORMs.60
Many of the described conjugates are geared toward the enhanced permeability and retention (EPR) effect. The diameter of nanoparticles and the size of polymers can easily be controlled and adapted. Thereby, it may be possible to accumulate the CORM conjugates in the targeted tumor tissue due to the EPR-effect and then to release the CO there if a suitable trigger is available (see below).73
Although the design of all the conjugates seems to be promising, the CORMs at large still show problems for the therapeutic application of CO, irrespective of their assembly into a conjugate. For the light-triggered CORMs (and conjugates), such as CORM-L1, the trigger excitation with wavelengths under 500 nm (i.e., the blue spectral region) has only a low tissue penetration depth. For CORMs below the skin or inside tissue wavelengths of about 750 nm (i.e., the red spectral region), it would be highly desirable to trigger the release of CO without the harmful effects of UV radiation.74 However, longer wavelengths are not yet suitable for CORMs, and also cannot be adjusted through a conjugation of the CORMs. Exceptions to the case are the CORMs@non-wovens synthesized by Schiller et al., because they are designed for a surface application, e.g., to only act on the skin as patches, thereby a deep tissue penetration would not be necessary.
For most of the described conjugates, the release of CO at a specific target is still a problem, especially if they are used for local applications, such as in tumor tissues. Here, the tablets of Meinel et al. can be emphasized, because of their precise application for gastrointestinal diseases. Most of the CORM conjugates are more suitable for systemic applications, such as against inflammations, which need no specific targeting. An advantage of the solvent- or pH-triggered CORM conjugates (e.g., CORM-3) would then be the presumably longer lasting or delayed-release effect of CO, because the trigger needs to diffuse to or into the conjugate and will thereby trigger the release of CO over an extended time, thus avoiding burst releases of the parent CORM.
A common problem to all CORMs, irrespective if free or part of a conjugate, is the steady, spontaneous, low background release of CO, which cannot be completely stopped, even if no trigger seems to be present. It could be supposed that this background release is triggered by oxidation processes. Metal carbonyl-based CORMs are intrinsically thermodynamically unstable toward O2 and other oxidants, which could lead to this unavoidable, steady, low CO release.15 Indeed, for a tissue-targeted therapeutic application, it is highly desirable that the release only starts at this target tissue. For this, an on/off-switch of the CORM conjugate system would be essential. Yet, it may be difficult to control these triggers from outside an organism in a medicinal application. The trigger of a localized heating of CORM@iron oxide nanoparticles through an alternating magnetic AC-field was thus developed in this direction. However, even this system had the problem of a low background release of CO through the use of a temperature-sensitive CORM-3 derivative. The controlled release is also important for the treatment of diseases where a complete, immediate release of CO would be necessary, such as in tumor tissues.
Elaborate designs of CORM conjugates have been achieved, but more research is necessary to design CORM systems that have the possibility of a controllable on/off-switch for CO release. Only if this can be achieved may other measures, for example the addition of receptors to the conjugate for a specific targeting, seem plausible and sensible.
Further developments for the incorporation of CORMs could be MOFs with switchable linkers, which are able to “open/close” the MOF pores and channels by a trigger and thereby perhaps achieve a complete, controllable on/off-switch for the CO release.75 As described above, MOFs that exhibit the breathing effect55,56 have already been used for the conjugation of CO by the group of Metzler-Nolte et al.57
References
- C. C. Romão, W. A. Blättler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- C. G. Douglas, J. S. Haldane and J. B. Haldane, J. Physiol., 1912, 44, 274–304 Search PubMed.
- J. B. Haldane, Biochem. J., 1927, 21, 1068–1075 CrossRef CAS PubMed.
- T. Sjöstrand, Nature, 1949, 164, 580–581 CrossRef.
- L. Wu and R. Wang, Pharmacol. Rev., 2005, 57, 585–630 CrossRef CAS PubMed.
- A. K. Mustafa, M. M. Gadalla and S. H. Snyder, Sci. Signaling, 2009, 68, 1–8 Search PubMed.
- T. R. Johnson, B. E. Mann, J. E. Clark, R. Foresti, C. J. Green and R. Motterlini, Angew. Chem., Int. Ed., 2003, 42, 3722–3729 CrossRef CAS PubMed.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- S. W. Ryter, J. Alam and A. M. K. Choi, Physiol. Rev., 2006, 86, 583–650 CrossRef CAS PubMed.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–e24 CrossRef CAS PubMed.
- J. E. Clark, P. Naughton, S. Shurey, C. J. Green, T. R. Johnson, B. E. Mann, R. Foresti and R. Motterlini, Circ. Res., 2003, 93, e2–e8 CrossRef CAS PubMed.
- J. Niesel, A. Pinto, H. W. P. N'Dongo, K. Merz, I. Ott, R. Gust and U. Schatzschneider, Chem. Commun., 2008, 15, 1798–1800 RSC.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS.
- S. Romanski, B. Kraus, U. Schatzschneider, J.-M. Neudörfl, S. Amslinger and H.-G. Schmalz, Angew. Chem., Int. Ed., 2011, 50, 2392–2396 CrossRef CAS PubMed.
- J. D. Seixas, A. Mukhopadhyay, T. Santos-Silva, L. E. Otterbein, D. J. Gallo, S. S. Rodrigues, B. H. Guerreiro, A. M. L. Gonçalves, N. Penacho, A. R. Marques, A. C. Coelho, P. M. Reis, M. J. Romão and C. C. Romão, Dalton Trans., 2013, 42, 5985–5998 RSC.
- G. L. Bannenberg and H. L. Vieira, Epxert Opin. Ther. Pat., 2009, 19, 663–682 CrossRef CAS PubMed.
- P. C. Kunz, H. Meyer, A. Schmidt and C. Janiak, Nano-Sized Carriers for Carbon Monoxide Releasing Molecules, in EUROBIC 11, ed. J. M. Gonzaléz-Pérez, A. Matilla-Hernández and J. Niclós-Gutiérrez, Medimond, Bologna, Italy, 2013, p. 25–30 Search PubMed.
- T. R. Johnson, B. E. Mann, I. P. Teasdale, H. Adams, R. Foresti, C. J. Green and R. Motterlini, Dalton Trans., 2007, 1500–1508 RSC.
- J. D. Seixas, M. F. A. Santos, A. Mukhopadhyay, A. C. Coelho, P. M. Reis, L. F. Veiros, A. R. Marques, N. Penacho, A. M. L. Gonçalves, M. J. Romão, G. J. L. Bernardes, T. Santos-Silva and C. C. Romão, Dalton Trans., 2015, 44, 5058–5075 RSC.
- S. McLean, B. E. Mann and R. K. Poole, Anal. Biochem., 2012, 427, 36–40 CrossRef CAS PubMed.
- V. S. Stoll and J. S. Blanchard, Methods Enzymol., 2009, 463, 43–56 CAS.
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014, 50, 3644–3660 RSC.
- T. Santos-Silva, A. Mukhopadhyay, J. D. Seixas, G. J. L. Bernandes, C. C. Romão and M. J. Romao, J. Am. Chem. Soc., 2011, 133, 1192–1195 CrossRef CAS PubMed.
- L. Yuan, W. Y. Lin, L. Tan, K. B. Zheng and W. M. Huang, Angew. Chem., Int. Ed., 2013, 52, 1628–1630 CrossRef CAS PubMed.
- B. W. Michel, A. R. Lippert and C. J. Chang, J. Am. Chem. Soc., 2012, 134, 15668–15671 CrossRef CAS PubMed.
- M. Klein, U. Neugebauer, A. Gheisari, A. Malassa, T. M. Jazzazi, F. Froehlich, M. Westerhausen, M. Schmitt and J. Popp, J. Phys. Chem. A, 2014, 118, 5381–5390 CrossRef CAS PubMed.
- H. J. Vreman and D. K. Stevenson, Anal. Biochem., 1988, 168, 31–81 CrossRef CAS PubMed.
- M. Balazy and H. Jiang, Acta Haematol., 2000, 103, 78–83 CrossRef.
- R. Motterlini, B. E. Mann and R. Foresti, Expert Opin. Invest. Drugs, 2005, 14, 1305–1318 CrossRef CAS PubMed.
- R. Mottelini, B. E. Mann, T. R. Johnson, J. E. Clark, R. Foresti and C. J. Green, Curr. Pharm. Des., 2003, 9, 2525–2539 CrossRef.
- S. García-Gallego and G. J. L. Bernardes, Angew. Chem., Int. Ed., 2014, 53, 9712–9721 CrossRef PubMed.
- U. Schatzschneider, Br. J. Pharmacol., 2015, 172, 1638–1650 CrossRef CAS PubMed.
- J. N. Talbert and J. M. Goddard, Colloids Surf., B, 2012, 93, 8–19 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- L. D. D'Andrea, A. Romanelli, R. Di Stasi and C. Pedone, Dalton Trans., 2010, 39, 7625–7635 RSC.
- E. Neuse, Met.-Based Drugs, 2008, 2008, 1–19 CrossRef PubMed.
- H. Pfeiffer, A. Rojas, J. Niesel and U. Schatzschneider, Dalton Trans., 2009, 4292–4298 RSC.
- H. Pfeiffer, T. Sowik and U. Schatzschneider, J. Organomet. Chem., 2013, 734, 17–24 CrossRef CAS.
- J. B. Matson, M. J. Webber, V. K. Tamboli, B. Weber and S. I. Stupp, Soft Matter, 2012, 8, 6689–6692 RSC.
- U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed.
- H.-M. Berends and P. Kurz, Inorg. Chim. Acta, 2012, 380, 141–147 CrossRef CAS.
- D. Nguyen, T.-K. Nguyen, S. A. Rice and C. Boyer, Biomacromolecules, 2015, 16, 2776–2786 CrossRef CAS PubMed.
- D. Nguyen, N. N. M. Adnan, S. Oliver and C. Boyer, Macromol. Rapid Commun., 2016, 37, 739–744 CrossRef CAS PubMed.
- B. F. G. Johnson, R. D. Johnston and J. Lewis, J. Chem. Soc. A, 1969, 792–797 RSC.
- N. E. Brückmann, M. Wahl, G. J. Reiß, M. Kohns, W. Wätjen and P. C. Kunz, Eur. J. Inorg. Chem., 2011, 29, 4571–4577 CrossRef.
- G. Dördelmann, H. Pfeiffer, A. Birkner and U. Schatzschneider, Inorg. Chem., 2011, 50, 4362–4367 CrossRef PubMed.
- G. Dördelmann, T. Meinhardt, T. Sowik, A. Krueger and U. Schatzschneider, Chem. Commun., 2012, 48, 11528–11530 RSC.
- O. Faklaris, V. Joshi, T. Irinopoulou, P. Tauc, M. Sennour, H. Girard, C. Gesset, J.-C. Arnault, A. Thorel, J.-P. Boudou, P. A. Curmi and F. Treussart, ACS Nano, 2009, 3, 3955–3962 CrossRef CAS PubMed.
- V. Vaijayanthimala and H.-C. Chang, Nanomedicine, 2009, 4, 47–55 CrossRef CAS PubMed.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- G. A. van Ewijk, G. J. Vroege and A. P. J. Philip, J. Magn. Magn. Mater., 1999, 201, 31–33 CrossRef CAS.
- A. J. Cole, V. C. Yang and A. E. David, Trends Biotechnol., 2011, 29, 323–332 CrossRef CAS PubMed.
- P. Govender, S. Pai, U. Schatzschneider and G. S. Smith, Inorg. Chem., 2013, 52, 5470–5478 CrossRef CAS PubMed.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- G. Férey and C. Serre, Chem. Soc. Rev., 2009, 38, 1380–1399 RSC.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill, M. Muhler, R. A. Fischer, Y. Wang, U. Schatzschneider and N. Metzler-Nolte, Chem. – Eur. J., 2013, 19, 6785–6790 CrossRef CAS PubMed.
- K. Fujita, Y. Tanaka, S. Abe and T. Ueno, Angew. Chem., Int. Ed., 2016, 55, 1056–1060 CrossRef CAS PubMed.
- C. Bohlender, S. Gläser, M. Klein, J. Weisser, S. Thein, U. Neugebauer, J. Popp, R. Wyrwa and A. Schiller, J. Mater. Chem. B, 2014, 2, 1454–1463 RSC.
- S. Gläser, R. Mede, H. Görls, S. Seupel, C. Bohlender, R. Wyrwa, S. Schirmer, S. Dochow, G. U. Reddy, J. Popp, M. Westerhausen and A. Schiller, Dalton Trans., 2016, 45, 13222–13233 RSC.
- C. Steiger, T. Lühmann and L. Meinel, J. Controlled Release, 2014, 189, 46–53 CrossRef CAS PubMed.
- I. C. Winburn, K. Gunatunga, R. D. McKernan, R. J. Walker, I. A. Sammut and J. C. Harrison, Basic Clin. Pharmacol. Toxicol., 2012, 111, 31–41 CAS.
- C. Steiger, K. Uchiyama, T. Takagi, K. Mizushima, Y. Higashimura, M. Gutmann, C. Hermann, S. Botov, H.-G. Schmalz, Y. Naito and L. Meinel, J. Controlled Release, 2016, 239, 128–136 CrossRef CAS PubMed.
- M. A. Gonzales, H. Han, A. Moyes, A. Radinos, A. J. Hobbs, N. Coombs, S. R. J. Oliver and P. K. Mascharak, J. Mater. Chem. B, 2014, 2, 2107–2113 RSC.
- F. J. Carmona, S. Rojas, P. Sánchez, H. Jeremias, A. R. Marques, C. C. Romão, D. Choquesillo-Lazarte, J. A. R. Navarro, C. R. Maldonado and E. Barea, Inorg. Chem., 2016, 55, 6525–6531 CrossRef CAS PubMed.
- H. Meyer, F. Winkler, P. Kunz, A. M. Schmidt, A. Hamacher, M. U. Kassack and C. Janiak, Inorg. Chem., 2015, 54, 11236–11246 CrossRef CAS PubMed.
- K. I. Draget, Alginates, in Handbook of hydrocolloidsm, ed. G. O. Phillips and P. A. Williams, 2009, pp. 807–828 Search PubMed.
- S. N. Pawar and K. J. Edgar, Biomaterials, 2012, 33, 3279–3305 CrossRef CAS PubMed.
- L. Oresmaa, H. Tarvainen, K. Machal and M. Haukka, Dalton Trans., 2012, 41, 11170–11175 RSC.
- H. Meyer, M. Brenner, S.-P. Höfert, T.-O. Knedel, P. C. Kunz, A. M. Schmidt, A. Hamacher, M. U. Kassack and C. Janiak, Dalton Trans., 2016, 45, 7605–7615 RSC.
- H. Song, H. Zhao, Y. Qu, Q. Sun, F. Zhang, Z. Du, W. Liang, Y. Qi and P. Yang, J. Periodontal Res., 2011, 46, 48–57 CrossRef CAS PubMed.
- M. Desmard, K. S. Davidge, O. Bouvet, D. Morin, D. Roux, R. Foresti, J. D. Ricard, E. Denamur, R. K. Poole, P. Montravers, R. Motterlini and J. Boczkowski, FASEB J., 2009, 23, 1023–1031 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- F. Vasefi, N. MacKinnon and D. L. Farkas, Hyperspectral and Multispectral Imaging in Dermatology, in Imaging in Dermatology, ed. M. R. Hamblin, P. Avci and G. K. Gupta, Academic Press, Elsevier, 2016, p. 187 Search PubMed.
- S. Castellanos, F. Kapteijn and J. Gascon, CrystEngComm, 2016, 18, 4006–4012 RSC.
| This journal is © The Royal Society of Chemistry 2016 |