
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In search of molecules displaying ferromagnetic exchange: multiple-decker Ni12 and Ni16 complexes from the use of pyridine-2-amidoxime†
Constantinos G.
Efthymiou
ab,
Luís
Cunha-Silva
c,
Spyros P.
Perlepes
b,
Euan K.
Brechin
d,
Ross
Inglis
*d,
Marco
Evangelisti
 *e and
Constantina
Papatriantafyllopoulou
*a
*e and
Constantina
Papatriantafyllopoulou
*a
aSchool of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland. E-mail: constantina.papatriantafyllopo@nuigalway.ie; Tel: +353 91 493462
bDepartment of Chemistry, University of Patras, 26504 Patras, Greece
cREQUIMTE-LAQV, Department of Chemistry and Biochemistry, Faculty of Sciences, University of Porto, 4169-007 Porto, Portugal
dEastCHEM School of Chemistry, The University of Edinburgh, David Brewster Road, EH9 3FJ Edinburgh, UK. E-mail: r.inglis@ed.ac.uk; Tel: +44 (0)11 650470
eInstituto de Ciencia de Materiales de Aragón (ICMA), CSIC-Universidad de Zaragoza, Departamento de Fisica de la Materia Condensada, 50009 Zaragoza, Spain. E-mail: evange@unizar.es; Tel: +34-876-55-3342
First published on 27th September 2016
Abstract
The use of pyridine-2-amidoxime (pyaoxH2) in Ni chemistry has provided access to a dodecanuclear complex and a hexadecanuclear Ni cluster, namely [Ni12(pyaox)6(pyaoxH)6(MeOH)2Cl2]Cl4·5MeOH (1·5MeOH) and [Ni16(pyaox)8(pyaoxH)8(MeOH)4](SO4)4·10H2O·26MeOH (2·10H2O·26MeOH). Complex 1·5MeOH was isolated by the reaction of NiCl2·6H2O, pyaoxH2 and NaOMe in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:2 molar ratio in MeOH in 60% yield. Treatment of NiSO4·6H2O with pyaoxH2 and NEt3 in a 1:1:2 molar ratio in MeOH afforded 2·10H2O·26MeOH in good yield (65%). The two compounds display a multi-decker configuration based on stacked Ni4 layers, {Ni4(pyaox)2(pyaoxH)2}2+x (x = 3, 1·5MeOH; x = 4, 2·10H2O·26MeOH); each deck consists of two square planar and two octahedral NiII centres. The number of decks observed in 1·5MeOH and 2·10H2O·26MeOH depends on the nature of the inorganic anion that is present in the reaction system, which provides elements of synthetic control towards new high nuclearity NiII species. 2·10H2O·26MeOH is the first structurally characterized complex of any metal displaying a quadruple-decker configuration, being also the highest nuclearity metal cluster bearing pyaoxH2 and the highest nuclearity NiII cluster with any type of 2-pyridyl oxime. Each cluster cation displays ferromagnetic exchange between the octahedral NiII ions resulting in a spin ground state of S = 6 for 1 and S = 8 for 2. Magnetothermal studies have been performed and discussed for both clusters.
:1:2 molar ratio in MeOH in 60% yield. Treatment of NiSO4·6H2O with pyaoxH2 and NEt3 in a 1:1:2 molar ratio in MeOH afforded 2·10H2O·26MeOH in good yield (65%). The two compounds display a multi-decker configuration based on stacked Ni4 layers, {Ni4(pyaox)2(pyaoxH)2}2+x (x = 3, 1·5MeOH; x = 4, 2·10H2O·26MeOH); each deck consists of two square planar and two octahedral NiII centres. The number of decks observed in 1·5MeOH and 2·10H2O·26MeOH depends on the nature of the inorganic anion that is present in the reaction system, which provides elements of synthetic control towards new high nuclearity NiII species. 2·10H2O·26MeOH is the first structurally characterized complex of any metal displaying a quadruple-decker configuration, being also the highest nuclearity metal cluster bearing pyaoxH2 and the highest nuclearity NiII cluster with any type of 2-pyridyl oxime. Each cluster cation displays ferromagnetic exchange between the octahedral NiII ions resulting in a spin ground state of S = 6 for 1 and S = 8 for 2. Magnetothermal studies have been performed and discussed for both clusters.
Introduction
It has been over 20 years since the discovery that the discrete [MnIII8MnIV4O12(O2CMe)16(H2O)4] molecule could possess an energy barrier to its spin vector reversal that was large enough to observe magnetic hysteresis, the classical property of a magnet.1 This discovery constituted the commencement of a new era in transition metal cluster chemistry, sparking off intense interest in the synthesis and characterization of new coordination compounds that could behave like magnets at very low temperatures in the absence of an applied magnetic field. Such species are now known as single-molecule magnets (SMMs)2 with their properties originating from intrinsic structural and electronic characteristics of their metal cores. The potential applications of SMMs range from high-density information storage, molecular spintronics, to qubits for quantum computation.3–5 Furthermore, several other quantum mechanical phenomena have been identified in SMMs, such as spin-phonon coupling,6 spin state entanglement,5 spin parity effects,7 both thermally assisted and pure quantum tunneling of the magnetization (QTM),8 and quantum phase interference.7b,9Clusters containing MnIII ions have proven to be the most fruitful source of SMMs to date. This is not surprising considering that: (1) the first and most well-known SMM comes from Mn cluster chemistry, a fact that activated intense research in this area, and (2) the d4 MnIII ion displays significant anisotropy, an inherent requirement for the generation of new SMMs.2 However, SMMs are also known for 3d metal ions other than Mn, e.g. VIII, FeII, FeIII, NiII, lanthanide ions, actinide ions, and combinations of 3d with 4d, 5d or 4f paramagnetic metal ions.2,10 Among them, NiII has shown much promise in the synthesis of both SMMs11 and spin phonon traps,12 due to its significant single-ion anisotropy and its paramagnetic nature when confined within a highly symmetric cage.
The isolation of new SMMs remains a hot research field, not only for a better understanding of the phenomenon but also for bringing these species closer to technological applications. In fact, the chances of identifying novel types of coordination clusters with improved or new magnetic properties will be increased by the development of new reaction systems with suitable organic ligands or combinations of organic and inorganic ligands. The organic ligand often dictates the cluster symmetry, metal topology and the number of metal ions that are present, which in turn affects the nature of the magnetic exchange interactions and the intermolecular interactions between the polynuclear molecules in the crystal. One fertile approach towards the synthesis of new polynuclear metal complexes with interesting magnetic properties is the use of oxime ligands, since oximates are good bridging groups fostering formation of coordination clusters.13 We and others have been investigating a number of oxime-based ligands, and one broad family of the latter has been the 2-pyridyl oximes (Scheme 1).14 The anions of 2-pyridyl oximes are versatile ligands for a variety of research objectives; the activation of 2-pyridyl oximes by 3d-metal centres towards further reaction is also becoming an exciting area of research.15 Such ligands have been key “players” in several areas of single-molecule and single-chain magnetism.16
 | ||
| Scheme 1 General structural formula and abbreviations of the 2-pyridyl oximes discussed in the text. | ||
Restricting further discussion to nickel, our groups have been engaged in the use of 2-pyridyl oximate ligands for NiII cluster synthesis for many years. The first step of this approach included the synthesis and characterization of complexes of pao−,17 mpko−18 and ppko−,17e,18c,d,19i.e., complexes bearing anionic ligands in which R contains no additional donor site. In a second step, we have been studying NiII complexes with anionic ligands in which R contains a donor site, i.e., clusters of (py)pko−17a,20 and pyaoxH−/pyaox2−.21 In particular, the ligand pyaoxH2 (pyridine-2-amidoxime; IUPAC name: N-hydroxy-pyridine-2-carboxamidine) belongs to the class of amidoximes. Although the versatile chemistry of conventional oximes has been extensively studied over the years in both the organic chemistry and coordination chemistry literature, amidoximes, RC(NH2) = NOH, have so far received substantially less attention.22 The presence of the amino functionality is expected – due to its coordination capability, potential for deprotonation and hydrogen bonding effects – to alter the coordination behaviour of this ligand (and hence the identity of the resultant NiII complexes) in comparison with that of the pao−, mpko−, ppko−, (py)pko−, etc., ligands.
In 2008, our groups employed pyaoxH2 in Ni cluster chemistry and proved that it can be a fruitful method for the synthesis of high nuclearity species.21 Nevertheless, the use of pyaoxH2 for the synthesis of polynuclear Ni compounds remains limited.21,23–25 In particular, so far there have been reports of only salts of the cations [Ni4(pyaox)2(pyaoxH)2L4]2+ (L = imidazole, 1-methylimidazole, pyridine, 3-methylpyridine, 4-methylpyridine),23,24 [Ni8(pyaox)4(pyaoxH)4L′4] (L′ = H2O, imidazole),23,24 [Ni12(pyaox)6(pyaoxH)6(H2O)2(N3)2]4+ and [Ni12(pyaox)6(pyaoxH)6(H2O)2]6+.21,23,24 The cations have single-(Ni4), double-(Ni8) and triple-decker (Ni12) structures, where each deck is composed of the same tetranuclear cationic unit {Ni4(pyaox)2(pyaoxH)2}2+ (proved to be present in solution by ESI-MS spectrometry23,24) and features two square planar and two octahedral NiII centres.
In this work, we expand the above mentioned family of multiple-decker Ni clusters by reporting the syntheses, crystal structures and magnetic and heat capacity properties of two such species, namely [Ni12(pyaox)6(pyaoxH)6(MeOH)2Cl2]Cl4·5MeOH (1·5MeOH) and [Ni16(pyaox)8(pyaoxH)8(MeOH)4](SO4)4·10H2O·26MeOH (2·10H2O·26MeOH). The two clusters are high-spin featuring both the singly (pyaoxH−) and doubly deprotonated (pyaox2−) forms of pyaoxH2. 2·10H2O·26MeOH is the first example of a metal cluster with a quadruple-decker topology, being also the highest nuclearity metal cluster, in general, containing any form of the pyaoxH2 ligand and the highest nuclearity NiII cluster with any type of 2-pyridyl oxime. In addition, its nuclearity is the highest that has been observed so far in metal clusters bearing 2-pyridyl oximes.17c,26 The number of the decks in 1·5MeOH and 2·10H2O·26MeOH, and hence the nuclearity of the products, depend on the nature of the inorganic anion that is present in the reaction system, providing elements of synthetic control towards new ferromagnetic species. Portions of this work have been previously communicated.21
Experimental section
Materials and physical measurements
All manipulations were performed under aerobic conditions using materials (reagent grade) and solvents as received. The organic ligand pyaoxH2 was synthesized following the reported method.27 Elemental analyses (C, H, N) were performed by the University of Patras microanalysis service. IR spectra (4000–400 cm−1) were recorded using a Perkin Elmer 16PC FT-IR spectrometer with samples prepared as KBr pellets. The magnetic susceptibility measurements were carried out on a Quantum Design SQUID magnetometer MPMS-XL 7 operating between 1.8 and 300 K for dc-applied fields ranging from 0 to 5 T. Microcrystalline samples were dispersed in vaseline in order to avoid torquing of the crystallites. The sample mulls were contained in a calibrated gelatin capsule held at the center of a drinking straw that was fixed at the end of the sample rod. Alternating current (ac) susceptibility measurements were carried out under an oscillating ac field of 3.5 Oe and frequencies ranging from 0.1 to 1500 Hz. The heat capacity measurements were carried out for temperatures down to 0.3 K by using a Quantum Design 14T-PPMS, equipped with a 3He cryostat. The experiments were performed on thin pressed pellets (ca. 1 mg) of a polycrystalline sample, thermalized by ca. 0.2 mg of Apiezon N grease, whose contribution was subtracted by using a phenomenological expression.Synthetic details
Single-crystal X-ray crystallography
Suitable single-crystals of 1·5MeOH and 2·10H2O·26MeOH were mounted on a Hampton Research CryoLoop using FOMBLIN Y perfluoropolyether vacuum oil (LVAC 25/6) purchased from Aldrich,28 with the help of a Stemi 2000 stereomicroscope equipped with Carl Zeiss lenses. Data were collected at 150(2) K on a Bruker X8 Kappa APEX II charge-coupled device (CCD) area-detector diffractometer (Mo Kα graphite-monochromated radiation, λ = 0.71073 Å) controlled by the APEX2 software package,29 and equipped with an Oxford Cryosystems Series 700 cryostream monitored remotely using the software interface, Cryopad.30 Images were processed using the software package SAINT+,31 and data were corrected for absorption by the multi-scan semi-empirical method implemented in SADABS.32 The structures were solved by the direct methods of SHELXS-97,33 and refined by full-matrix least squares on F2 using SHELXL-97.34All non-hydrogen atoms were directly located from difference Fourier maps and successfully refined with anisotropic displacement parameters. For both compounds, hydrogen atoms attached to carbon and to the hydroxido groups were located at their idealised positions and included in the structural model in subsequent refinement cycles in riding-motion approximation with Uiso = 1.2 (aromatic carbon) or 1.5 (–CH3 and –OH groups) of Ueq of the parent atom. Hydrogen atoms associated with the nitrogen atoms were markedly visible in difference Fourier maps and were included in the structures with the N–H and H⋯H distances restrained to 0.90(1) and 1.50(1) Å, respectively, and with Uiso = 1.5 × Ueq(N).
For 2·10H2O·26MeOH the hydrogen atoms belonging to the uncoordinated water molecules could not be directly located from difference Fourier maps and no attempt was made to place these in approximate calculated positions; they have been included in the empirical formula of the compound.
Beside the solvent molecules located in both the structures (5 MeOH in compound 1, as well as 10 H2O and 26 MeOH in 2), considerable smeared-out electron densities were found in the empty spaces available in the two structures, which prevented a sensible location and refinement of other solvent molecules. A search for the total potential solvent area using the software package PLATON35 revealed the presence of large voids housing the disordered molecules. The two data sets were then analysed using the Squeeze,36 and the reflection lists arising from this procedure were used for the final structural refinements.
Full details can be found in Table 1 and the cif files.
| 1·5MeOH | 2·10H2O·26MeOH | |
|---|---|---|
| a Including solvent molecules. b Graphite monochromator. c R 1 = ∑(|Fo| − |Fc|)/∑|Fo|. d wR2 = [∑[w(Fo2 − Fc2)2]/∑[wFo2)2]]1/2. | ||
| Formulaa | C79H94Cl6N36Ni12O19 | C126H228N48Ni16O72S4 |
| M w | 2769.12 | 4635.16 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| a/Å | 14.8718(8) | 16.6742(9) |
| b/Å | 15.1738(7) | 17.3665(17) |
| c/Å | 15.3809(8) | 19.7073(10) |
| a/° | 60.525(2) | 106.175(3) |
| β/° | 73.084(3) | 113.293(2) |
| γ/° | 69.854(2) | 99.202(4) |
| V/Å 3 | 2805.3(3) | 4792.3(6) |
| Z | 1 | 1 |
| T/K | 150(2) | 150(2) |
| λ /Å | 0.71073 | 0.71073 |
| D c/g cm−3 | 1.639 | 1.606 |
| μ(Mo Kα)/mm−1 | 2.182 | 1.672 |
| Reflections collected | 147901 |
321514 |
| Independent reflections | 14885 |
21715 |
| R int | 0.0422 | 0.0375 |
|
R
1c |
0.0596 | 0.0461 |
| wR2d |
0.2006 | 0.1148 |
| Goodness of fit on F2 | 1.047 | 1.138 |
| Δρmax/min/e Å−3 | 4.818/−0.908 | 1.194/−0.700 |
Results and discussion
Synthetic parameters and IR spectra
Our groups have strong interest in the employment of 2-pyridyl oxime ligands as a route to high nuclearity 3d transition metal clusters and SMMs.14,17–21 As a part of this program, we recently decided to explore pyaoxH2 in NiII cluster chemistry. Thus, a variety of experiments were performed to study how the different synthetic parameters affect the identity and/or crystallinity of the isolated product. In particular, treatment of NiCl2·6H2O with one equivalent of pyaoxH2 and two equivalents of NaOMe in MeOH gave a dark brown solution from which the dodecanuclear cluster [Ni12(pyaox)6(pyaoxH)6(MeOH)2Cl2]Cl4·5MeOH (1·5MeOH) was obtained in 60% yield. Its formation can be summarized by eqn (1):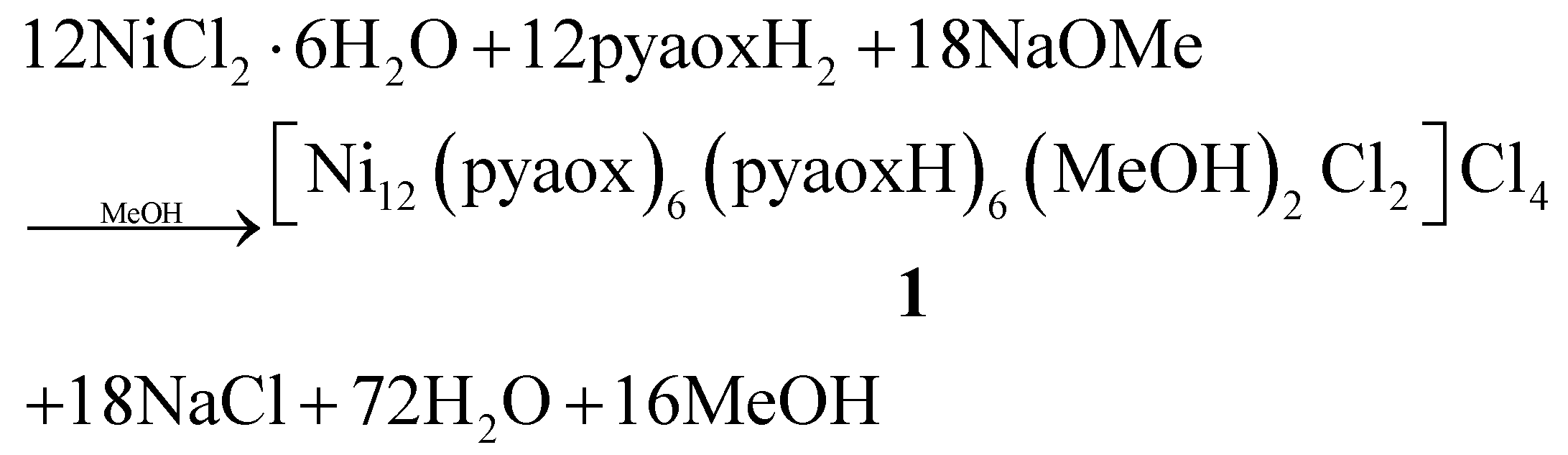 | (1) |
The methoxide ions act as proton acceptors to facilitate formation of pyaox2− and pyaoxH− ligands. The reaction procedure involved a slight excess of NaOMe over that required by eqn (1), and this might have proven beneficial in shifting a possible equilibrium to the right. The reaction mixtures which lead to 1 should be filtered before crystallization to remove NaCl which is insoluble in MeOH. The product also needs to be washed with a small amount of H2O to ensure complete removal of NaCl.
The determination of the crystal structure of 1 with single crystal X-ray crystallography revealed that the twelve metal ions have an unprecedented triple-decker topology with each “deck” containing two square planar and two octahedral NiII atoms. The intriguing structural and magnetic features of 1 (vide infra) prompted us to further study this reaction system exploring the ability of pyaoxH2 to deliver other novel polynuclear NiII compounds. In the context of this investigation, we decided to employ different counterions possessing higher charge than Cl− hoping that they could stabilize higher nuclearity clusters. Indeed, this turned out to be the case and the treatment of NiSO4·6H2O with one equivalent of pyaoxH2 and two equivalents of NEt3 in MeOH gave a dark brown solution from which the higher nuclearity cluster [Ni16(pyaox)8(pyaoxH)8(MeOH)4](SO4)4 (2) was obtained in 65% yield. Its formation can be summarized by eqn (2):
 | (2) |
:pyaoxH2 ratio gave the same Ni16 compound. The nature of the base and the crystallization method are not crucial for the identity of the product and affect only its crystallinity; we were able to isolate 2 (IR evidence) by using a plethora of bases such as NaOMe, NaOH, NH3etc.
The IR spectra of dried 1·5MeOH and 2·10H2O·26MeOH exhibit a strong broad band at ∼3330 cm−1 assignable to the ν(OH) vibration of coordinated MeOH molecules, as well as solvent MeOH and H2O molecules, and to the ν(NH2)/ν(NH) vibrations of the pyaoxH/pyaox2− ligands. The broadness and low frequency of these bands are both indicative of strong hydrogen bonding. There are medium intensity bands at 1566 and 1116 cm−1 in the spectrum of the free pyaoxH2 ligand, which are assigned to ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)oxime and ν(N–O)oxime, respectively.27 The 1116 cm−1 band is shifted to a lower wavenumber (1103 cm−1 in 1·5MeOH; 1100 cm−1 in 2·10H2O·26MeOH). This shift is attributed to the coordination of the oximate nitrogen.37 The band is strong in the spectrum of the Ni16 cluster, because it also has a ν3(F2) [νd(SO)] character due to the ionic sulfates (Td symmetry). Somewhat to our surprise, the 1566 cm−1 band is shifted to a higher wavenumber in the complexes (1577 cm−1 in 1·5MeOH, 1580 cm−1 in 2·10H2O·26MeOH), overlapping with an aromatic stretch. This shift may be indicative of the oximate nitrogen coordination.37 Extensive studies on Schiff-base complexes (which also contain a CN bond) have shown38 that a change in the s character of the nitrogen lone pair occurs upon coordination such that the s character of the nitrogen orbital, which is involved in the CN bond, increases; this change in hybridization produces a greater CN stretching force constant relative to the free neutral ligand. The in-plane deformation band of the 2-pyridyl ring of free pyaoxH2 at 632 cm−1 shifts upwards in 1·5MeOH (646 cm−1) and 2·10H2O·26MeOH (645 cm−1), confirming the involvement of the ring-N atom in coordination.39
N)oxime and ν(N–O)oxime, respectively.27 The 1116 cm−1 band is shifted to a lower wavenumber (1103 cm−1 in 1·5MeOH; 1100 cm−1 in 2·10H2O·26MeOH). This shift is attributed to the coordination of the oximate nitrogen.37 The band is strong in the spectrum of the Ni16 cluster, because it also has a ν3(F2) [νd(SO)] character due to the ionic sulfates (Td symmetry). Somewhat to our surprise, the 1566 cm−1 band is shifted to a higher wavenumber in the complexes (1577 cm−1 in 1·5MeOH, 1580 cm−1 in 2·10H2O·26MeOH), overlapping with an aromatic stretch. This shift may be indicative of the oximate nitrogen coordination.37 Extensive studies on Schiff-base complexes (which also contain a CN bond) have shown38 that a change in the s character of the nitrogen lone pair occurs upon coordination such that the s character of the nitrogen orbital, which is involved in the CN bond, increases; this change in hybridization produces a greater CN stretching force constant relative to the free neutral ligand. The in-plane deformation band of the 2-pyridyl ring of free pyaoxH2 at 632 cm−1 shifts upwards in 1·5MeOH (646 cm−1) and 2·10H2O·26MeOH (645 cm−1), confirming the involvement of the ring-N atom in coordination.39
Description of structures
Representations of the molecular structure and the structural core of compound 1·5MeOH are shown in Fig. 1. Selected interatomic distances and angles are listed in Table 2. Compounds 1·5MeOH and 2·10H2O·26MeOH display related structures based on parallel Ni4 layers, {Ni4(pyaox)2(pyaoxH)2}2+x (x = 3, 1·5MeOH; x = 4, 2·10H2O·26MeOH). Thus, only the structure of compound 1·5MeOH will be discussed in detail and compared to that of 2·10H2O·26MeOH. | ||
| Fig. 1 Representations of the molecular structure of the cation of 1 (top) and its triple-decker structural core (bottom). | ||
| a Symmetry transformations used to generate equivalent atoms: ′: −x, −y, −z + 1. | |||
|---|---|---|---|
| Ni1–O4 | 2.0279(18) | Ni3–N11 | 2.250(2) |
| Ni1–O2 | 2.0293(19) | Ni4–O4 | 1.8306(18) |
| Ni1–N10′ | 2.048(2) | Ni4–N5 | 1.853(2) |
| Ni1–N11′ | 2.070(2) | Ni4–N12 | 1.861(2) |
| Ni1–O1 | 2.1705(19) | Ni4–N4 | 1.903(2) |
| Ni1–N17 | 2.278(2) | Ni5–O5 | 1.8263(19) |
| Ni2–O3 | 1.998(2) | Ni5–N15 | 1.846(2) |
| Ni2–O5 | 2.0033(19) | Ni5–N8 | 1.852(2) |
| Ni2–N16 | 2.031(2) | Ni5–N7 | 1.887(2) |
| Ni2–N17 | 2.089(2) | Ni6–O6 | 1.8227(18) |
| Ni2–O2 | 2.1729(19) | Ni6–N18 | 1.846(2) |
| Ni2–Cl1 | 2.5713(10) | Ni6–N2′ | 1.846(2) |
| Ni3–N14 | 2.013(2) | Ni6–N1′ | 1.888(2) |
| Ni3–O6 | 2.0320(18) | Ni2⋯Ni3 | 3.891(2) |
| Ni3–O1′ | 2.0589(19) | Ni2⋯Ni5 | 3.323(2) |
| Ni3–N13 | 2.076(2) | Ni2⋯Ni6 | 4.670(2) |
| Ni3–O1 M | 2.113(2) | Ni1⋯Ni3 | 3.141(2) |
| O4–Ni1–N10′ | 178.74(8) | N5–Ni4–N12 | 174.14(10) |
| O2–Ni1–N11′ | 172.77(8) | O4–Ni4–N4 | 172.22(9) |
| O1–Ni1–N17 | 175.06(7) | N12–Ni4–N4 | 102.89(10) |
| O5–Ni2–N16 | 175.01(9) | N15–Ni5–N8 | 173.30(11) |
| O2–Ni2–Cl1 | 172.98(5) | O5–Ni5–N7 | 170.12(10) |
| O3–Ni2–N17 | 169.24(8) | N15–Ni5–N7 | 102.09(11) |
| O6–Ni3–N13 | 173.75(9) | N18–Ni6–N2′ | 174.26(10) |
| O1M–Ni3–N11 | 172.46(9) | O6–Ni6–N1′ | 172.83(10) |
| N14–Ni3–N11 | 89.73(9) | N18–Ni6–N1′ | 102.40(10) |
Complex 1·5MeOH crystallizes in the triclinic space group P. Its structure consists of [Ni12(pyaox)6(pyaoxH)6Cl2(MeOH)2]4+ cations, Cl− counterions and MeOH lattice molecules. The centrosymmetric cation of 1·5MeOH is composed of six distorted octahedral (Ni1, Ni2, Ni3 and symmetry equivalents) and six square planar (Ni4, Ni5, Ni6 and symmetry equivalents) NiII atoms, which are held together through two η1:η1:η1:μ and four η1:η1:η2:μ3 pyaoxH− ligands, as well as two η1:η1:η1:η2:μ3 and four η1:η2:η1:η2:μ4 pyaox2− groups (Scheme 2). The coordination sphere of the NiII atoms is completed by two terminal chlorido ligands on Ni2 and Ni2′, and two terminal MeOH molecules on Ni3 and Ni3′. The twelve metal ions in 1·5MeOH are arranged into three layers, which are stacked one above the other forming an unusual, for 3d-metal cluster chemistry, triple-decker topology. Each “deck” contains two square planar and two octahedral NiII atoms, whereas the linkage of the central deck with each of the outer decks is achieved through two bridging oxygen atoms from two η1:η1:η2:μ3 pyaoxH− ligands and two bridging oximate nitrogen atoms from two η1:η2:η1:η2:μ4 pyaox2− groups. The four NiII atoms of the two outer layers are arranged in an almost planar topology with the largest deviation from the plane being 0.14 Å for Ni3 and Ni6, whereas the metal ions in the central layer are perfectly coplanar. The distance between two adjacent decks is ∼2.70 Å. The chromophores in 1·5MeOH are Ni(1,1′)(Npy)(Nox)2(Oox)3, Ni(2,2′)(Npy)(Nox)(Oox)3(Cl), Ni(3,3′)(Npy)(Nox)2(Oox)2(OMeOH) and Ni(4,4′/5,5′/6,6′)(Npy)(Nox)(Nam)(Oox), where the abbreviations “py”, “ox” and “am” are for the 2-pyridyl, oximate and deprotonated amino donor atoms, respectively. The strong ligand field, deprotonated amino donor atoms are coordinated only to the square planar NiII atoms and may be considered responsible for their geometry. The average Ni(4,5,6)-Npy, Nox, Oox bond distances are 1.893, 1.850 and 1.827 Å, respectively, while the average Ni(1,2,3)–Npy, Nox, Oox bond lengths are 2.052, 2.140 and 2.062 Å, respectively. The longer bonds involving Ni(1,2,3) compared to those involving Ni(4,5,6) are a consequence of the higher coordination number of Ni1, Ni2 and Ni3.
 | ||
| Scheme 2 The crystallographically established coordination modes of pyaoxH− and pyaox2− in complexes 1 and 2. | ||
The η1:η2:η1:η2:μ4 coordination mode of pyaox2− found in 1·5MeOH is extremely rare in the coordination chemistry of pyridine-2-amidoxime.21 In fact, this can be rationalised by considering the C–Nox bond lengths; the C–Nox and C–Nam distances for the η1:η2:η1:η2:μ4 pyaox2− ligands (average values: 1.345 and 1.322 Å) are longer and shorter, respectively, than those for the η1:η1:η1:η2:μ3 pyaox2− ligands (1.318 and 1.336 Å), indicating a decreased double bond character for the C–Nox bond and an increased double bond character for the C–Nam bond in the former.
The crystal structure of 1·5MeOH is stabilized by strong intramolecular hydrogen bonds between the O atoms of the terminally ligated MeOH molecules (O1M), which act as donors, and the terminal chlorido ligands, which act as acceptors [O1 M⋯Cl1 = 3.199(2) Å, H1 M⋯Cl1 = 2.256(2) Å, O1 M–H1 M–Cl1 = 167.19(2)°]. In addition, there is a network of intermolecular H bonds that involve: (1) the Cl− counterions (acceptors) and the NH2 groups (donors) of the organic ligands [N3⋯Cl3 = 3.245(2), H3B⋯Cl3 = 2.360(2) Å, N3–H3B–Cl3 = 166.93°; N9⋯Cl3 = 3.282(2) Å, H9B⋯Cl3 = 2.421(2) Å, N9–H9B–Cl3 = 159.83°] resulting in the formation of 1D chains, and 2) the coordinated chlorido ligands (acceptor) and the MeOH lattice molecules (donors) [O3 M⋯Cl1 = 3.237(2) Å, H9 M⋯Cl1 = 2.422(2) Å, O3 M–H9 M–Cl1 = 163.49°]. The cations of 1 are in close proximity with the shortest metal⋯metal separation between the adjacent Ni12 units being 4.797(2) Å (Ni5⋯Ni5).
Representations of the molecular structure and the structural core of compound 2·10H2O·26MeOH are shown in Fig. 2. Selected interatomic distances and angles are listed in Table S1 in the ESI.†
 | ||
| Fig. 2 Representations of the molecular structure of the cation of 2 (top) and its quadruple-decker structural core (bottom). | ||
Complex 2·10H2O·26MeOH crystallizes in the triclinic space group P. Its structure consists of [Ni16(pyaox)8(pyaoxH)8(MeOH)4]8+ cations, SO42− counterions, and MeOH and H2O lattice molecules. Similarly to 1·5MeOH, the NiII atoms in 2·10H2O·26MeOH are stacked in four layers, which form an unprecedented quadruple-decker topology. In particular, the inner decks in 2·10H2O·26MeOH are identical with the central deck of 1·5MeOH; thus, figuratively speaking, an internal deck has been added to the Ni12 cation of the latter to form a Ni16 cluster (2·10H2O·26MeOH). In addition, the external decks in 2·10H2O·26MeOH are similar to the corresponding decks in 1·5MeOH, with their only difference being the replacement of the chlorido ligands in the latter by terminal MeOH molecules in the former. The octahedral metal centres are Ni1, Ni2, Ni3 and Ni4 (and their symmetry equivalents) and the square planar ones are Ni5, Ni6, Ni7 and Ni8 (and their symmetry equivalents). The presence of many lattice molecules and counterions in the crystal structure of 2·10H2O·26MeOH results in the formation of a network of strong intermolecular hydrogen bonding interactions. Details of these interactions are listed in Table S2 in the ESI.† As it was the case for 1·5MeOH, the cations of 2·10H2O·26MeOH, are in close proximity with the shortest metal⋯metal separation between the adjacent Ni16 units being 4.671(2) Å (Ni1⋯Ni7).
1·5MeOH and 2·10H2O·26MeOH are new additions in the small family of high nuclearity, high spin Ni compounds with multiple-decker metal topology.23,24 In particular, 2·10H2O·26MeOH is the first structurally characterized complex of any metal displaying a quadruple-decker configuration. Compound 1·5MeOH can be described as a “trimer-of-tetramers”, while 2·10H2O·26MeOH as a “tetramer-of tetramers”, where the tetramer in both cases is the structural unit {Ni4(pyaox)2(pyaoxH)2}2+, which has been previously observed in a discrete tetranuclear cluster.23,24 The number of the decks, and hence the nuclearity and spin, of 1·5MeOH and 2·10H2O·26MeOH is affected by the nature (charge, shape) of the inorganic anion that is present in the reaction system providing the elements of synthetic control towards new high nuclearity NiII clusters. Both compounds contain the singly and doubly deprotonated pyridine-2-amidoxime ligand, with 2·10H2O·26MeOH being (1) the highest nuclearity metal cluster bearing this ligand, (2) one of the few highest nuclearity hexadecanuclear metal clusters with any 2-pyridyl oxime, in general,17c,26 and (3) the highest nuclearity NiII cluster with 2-pyridyl oximes. Furthermore, complex 2·10H2O·26MeOH joins only a very small family of Ni16 clusters with O- or/and N-ligation.40
Magnetic susceptibility studies
In order to probe the magnetic properties of the clusters, direct current (dc) magnetic susceptibility measurements were performed on polycrystalline samples of compounds 1 and 2 in the temperature range 2–300 K, with an applied magnetic field of 0.1 T.The collected data are plotted as χMT versus T in Fig. 3, which reveal room-temperature χMT values of 7.16 and 9.03 cm3 K mol−1 for 1 and 2, respectively. These are in good agreement with the χMT value expected (g = 2.10) for six non-interacting octahedral NiII ions (6.615 cm3 mol−1 K) in 1, and eight non-interacting octahedral NiII ions (8.82 cm3 mol−1 K) in 2. For both compounds, the value of χMT steadily increases to a maximum value (14.5 cm3 mol−1 K at 14 K, 1; 21.7 cm3 mol−1 K at 10 K, 2), and then sharply decreases to 10.8 and 14.0 cm3 mol−1 K at 2.0 K for 1 and 2, respectively. The profile of the χMT versus T plots is indicative of dominant ferromagnetic interactions between the paramagnetic ions within the cations of 1 and 2. The low temperature decrease of the χMT value is attributed to intermolecular antiferromagnetic interactions and/or zero-field splitting. The former is consistent with the crystal structure of the two compounds (vide supra).
 | ||
| Fig. 3 Plot of χMT as a function of T for complexes 1 (□) and 2 (○); the solid lines represent the fit of the data to the theoretical models given in the text. | ||
In order to fit the χMT data for 1·5MeOH, we modeled the Ni6 magnetic core with the 2J-model described in Fig. 4, left, (the octahedral NiII atoms define two triangles related by an inversion centre) employing the isotropic spin-Hamiltonian (1):
 | (1) |
 | (2) |
 | ||
| Fig. 4 Schematic presentation of the coupling scheme used to model susceptibility data for the cations of 1 (left) and 2 (right). | ||
Fig. S3 in the ESI† shows the dc-field magnetization data M(H) collected in the range 0–7 T at three temperatures between 2.3 and 20 K for 1 and 2. In both cases, M(H) initially rises quickly at the lowest temperatures, as expected for ferromagnetic exchange, and then keeps increasing slowly even at the highest applied fields, without reaching saturation. In accordance with the susceptibility data, this behaviour is attributed to intermolecular antiferromagnetic interactions, likely combined with some degree of magnetic anisotropy. For comparison, the calculated Brillouin curves were also plotted assuming uncorrelated molecules with S = 6 (1) and S = 8 (2) for the representative temperatures. For both compounds, it is observed that the experimental data tend to lie below the calculated curve, suggesting the influence either from antiferromagnetic intermolecular correlations or from low-lying excited spin states. The latter is more evident in 2 with respect to 1, as it is revealed from the larger deviation of the calculated curve and experimental data, which indicates that 2 does not display a well-defined ground state spin value. Keeping the values of S and g stable, detectable improvements are obtained only by adding an abnormally high uniaxial magnetic anisotropy term D in the case of 1. Since this applies also at the lowest investigated temperatures, in which the influence of excited states should be negligible, we conclude, in agreement with the susceptibility data, that antiferromagnetic interactions between cations are important for the two complexes.
It has recently been reported that the dominant factor which affects the magnetic properties of the oximato NiII compounds is the length of the bridging N–O bond.23,42 In particular, DFT calculations on an oximato bridged NiII compound revealed that N–O distances larger than 1.394 Å result in ferromagnetic interactions, while shorter ones propagate antiferromagnetic interactions. This observation has been explained by considering that with the decrease of the N–O bond, the spin population on the N and O atoms increases, thus enhancing the antiferromagnetic coupling between NiII ions. In compounds 1·5MeOH and 2·10H2O·26MeOH, the N–O distances are in the range of 1.401–1.425 Å, and thus the experimentally obtained ferromagnetic interactions are in excellent agreement with the previously reported theoretical calculations.
Specific heat measurements
Fig. 5 shows the collected specific heat C(T,H) data of 1 as a function of temperature for several applied fields. No particular feature is detected except for a very broad anomaly around 2 K at zero applied field, whose magnetic nature is proven by the fact that it quickly moves towards higher temperatures as the field H is increased. As typical for molecule-based materials,43 the contribution to the total C arising from lattice vibrations dominates as T increases above that of liquid helium. | ||
| Fig. 5 Specific heat C for 1 normalised to the gas constant R vs. T in the indicated temperature and field ranges. Inset: Magnetic contribution Cm as obtained by subtracting the lattice contribution (dashed line) to C. | ||
The lattice contribution can be described by the Debye model (dashed line in Fig. 5), which simplifies to a C/R = aT3 dependence (R is the gas constant) at the lowest temperatures, where a = 3.9 × 10−3 K−3 for 1. The magnetic contribution Cm (see inset in Fig. 5) is then obtained by subtracting the lattice contribution to the total C. This analysis reveals a bump centered at ≈9 K, that we associate with a Schottky-like anomaly, i.e., crystal-field splitting of the S = 6 multiplet. By decreasing the temperature, magnetic fluctuations within the 2D layers show up in the anomaly centered at ≈2 K. To properly associate these features with the magnetic contribution, we estimate the zero-field magnetic entropy  , shown in Fig. S4 in the ESI.† This tends at high-T to the value of 2.8 R, which is in fair agreement with the magnetic entropy expected, R ln(2S + 1) = 2.56 R (provided the spin value is S = 6) and consistent with the magnetization data. Since the lattice contribution is sensibly larger than Cm, the latter is affected by an important uncertainty. This inhibits us from providing anything other than a qualitative analysis of the experimental data.
, shown in Fig. S4 in the ESI.† This tends at high-T to the value of 2.8 R, which is in fair agreement with the magnetic entropy expected, R ln(2S + 1) = 2.56 R (provided the spin value is S = 6) and consistent with the magnetization data. Since the lattice contribution is sensibly larger than Cm, the latter is affected by an important uncertainty. This inhibits us from providing anything other than a qualitative analysis of the experimental data.
Specific heat data have been also collected for 2 and these are shown in Fig. S5 in the ESI† as a function of temperature for several applied magnetic fields. As it is the case for 1, the specific heat at high temperatures is dominated by a non-magnetic contribution, which is due to the thermal vibrations of the lattice. Furthermore, there is a clear magnetic contribution, which extends over a broad temperature range, i.e., from the lowest accessed T to well inside the range mainly determined by the lattice contribution to the specific heat. The fact that 2 does not display a well-defined ground state spin value (S ≈ 8, vide supra) prevents to determine reliably the magnetic contribution by subtracting the lattice specific heat from the total measured signal, as in the case of the Ni12 complex. As a result, a valuable analysis of the specific heat data is not feasible for 2.
Conclusions
The employment of pyaoxH2 in NiII chemistry has provided access to the new multiple-decker Ni complexes [Ni12(pyaox)6(pyaoxH)6(MeOH)2Cl2]Cl4·5MeOH (1·5MeOH) and [Ni16(pyaox)8(pyaoxH)8(MeOH)4](SO4)4·10H2O·26MeOH (2·10H2O·26MeOH) comprising stacked Ni4 layers, {Ni4(pyaox)2(pyaoxH)2}2+x (x = 3, 1·5MeOH; x = 4, 2·10H2O·26MeOH), with each layer containing two square planar and two octahedral NiII centres.Compound 1·5MeOH is a new addition to the small family of Ni12 clusters with a triple-decker topology.23,24 Compound 2·10H2O·26MeOH displays a quadruple-decker configuration, which has never been observed before in metal cluster chemistry. In addition, 2·10H2O·26MeOH is the highest nuclearity metal cluster bearing pyaoxH2 and joins only a very small family of Ni16 clusters with O- or/and N-ligation. It also belongs to the very small family of the hexadecanuclear homometallic or/and heterometallic clusters, which display the highest nuclearity that has been observed so far for compounds containing 2-pyridyl oximes, being the only NiII cluster in this category.17c,25
Dc and ac magnetic susceptibility and heat capacity measurements were performed on 1 and 2, which revealed that both display ferromagnetic exchange interactions resulting in a spin ground state of S = 6 for 1 and S = 8 for 2. Intercationic interactions, as revealed by single crystal X-ray crystallography, affect the magnetic properties preventing an in-depth analysis of the heat capacity measurements.
The number of decks in 1 and 2 is affected by the use of different inorganic anions with the bulkier and of a higher charge anion (SO42−vs. Cl−) being able to stabilize the higher nuclearity cluster. This could lead to the development of a rational method towards the increase of the nuclearity and the spin of ferromagnetic species. Based on this, we believe that we have seen just the tip of the iceberg in the NiII/pyaoxH2 compounds with a multiple decker topology. We have every reason to believe that higher nuclearity such species (e.g., Ni20, Ni24, etc.) are waiting to be discovered. Further studies towards this direction are currently in progress including the employment of higher charge counterions (e.g., PO43−). These studies will be reported in due course.
Acknowledgements
CP and CE thank the School of Chemistry, NUI Galway, for the financial support. RI thanks the Royal Society of Edinburgh and ME thanks Spanish MINECO (MAT2015-68204-R) for funding. LCS acknowledges the financial support by FEDER (Fundo Europeu de Desenvolvimento Regional) through PT2020, by FCT (Fundação para a Ciência e a Tecnologia) for the research centre REQUIMTE/LAQV (UID/QUI/50006/2013) and for the grant SFRH/BPD/111899/2015.Notes and references
- (a) R. Sessoli, H. L. Tsai, A. R. Schake, S. Wang, J. B. Vincent, K. Folting, D. Gatteschi, G. Christou and D. N. Hendrickson, J. Am. Chem. Soc., 1993, 115, 1804 CrossRef CAS; (b) R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141 CrossRef CAS.
- For reviews and books, see: (a) R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011 RSC; (b) C. Papatriantafyllopoulou, E. Moushi, G. Christou and A. J. Tasiopoulos, Chem. Soc. Rev., 2016, 45, 1597 RSC; (c) M. Murrie and D. J. Price, Annu. Rep. Prog. Chem., 2007, 103, 20 RSC; (d) G. Aromi and E. K. Brechin, Struct. Bonding, 2006, 122, 1 CrossRef CAS; (e) D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, Oxford, 2006 Search PubMed; (f) C. J. Milios and R. E. P. Winpenny, Struct. Bonding, 2015, 164, 1 CrossRef CAS.
- (a) M. Affronte, J. Mater. Chem., 2009, 19, 1731 RSC; (b) L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179 CrossRef CAS PubMed.
- (a) G. A. Timco, T. B. Faust, F. Tuna and R. E. P. Winpenny, Chem. Soc. Rev., 2011, 40, 3067 RSC; (b) P. C. E. Stamp and A. Gaita-Ariño, J. Mater. Chem., 2009, 19, 1718 RSC.
- (a) S. Hill, R. S. Edwards, N. Aliaga-Alcalde and G. Christou, Science, 2003, 302, 1015 CrossRef CAS PubMed; (b) R. Tiron, W. Wernsdorfer, D. Foguet-Albiol, N. Aliaga-Alcalde and G. Christou, Phys. Rev. Lett., 2003, 91, 227203 CrossRef CAS PubMed (1–4).
- (a) A. Kovalev, L. Hayden, G. Bauer and Y. Tscherkovniak, Phys. Rev. Lett., 2011, 106, 147203 CrossRef PubMed; (b) D. A. Garanin and E. Chudnovsky, Phys. Rev., 2011, 1, 011005 Search PubMed; (c) M. Ganzhorn, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Nat. Nanotechnol., 2013, 8, 166 CrossRef PubMed.
- (a) W. Wernsdorfer, S. Bhaduri, C. Boskovic, G. Christou and D. N. Hendrickson, Phys. Rev. B: Condens. Matter, 2002, 65, 180403 CrossRef; (b) W. Wernsdorfer, N. E. Chakov and G. Christou, Phys. Rev. Lett., 2005, 95, 037203 CrossRef CAS PubMed.
- (a) G. Gatteschi and R. Sessoli, Angew. Chem., Int. Ed., 2003, 42, 268 CrossRef PubMed; (b) E. Del Barco, A. D. Kent, S. Hill, J. M. North, N. S. Dalal, E. M. Rumberger, D. N. Hendrickson, N. E. Chakov and G. Christou, J. Low Temp. Phys., 2005, 140, 119 CrossRef CAS; (c) P. C. E. Stamp, Nature, 1996, 383, 125 CrossRef CAS.
- (a) W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133 CrossRef CAS PubMed; (b) W. Wernsdorfer, M. Soler, G. Christou and D. N. Hendrickson, J. Appl. Phys., 2002, 91, 7164 CrossRef CAS.
- (a) J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078 RSC, and references therein; (b) D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110 CrossRef CAS PubMed; (c) K. R. Meihaus and J. R. Long, Dalton Trans., 2015, 44, 2517 RSC (Perspective).
- (a) A. Ferguson, J. Lawrence, A. Parkin, J. Sanchez-Benitez, K. V. Kamenev, E. K. Brechin, W. Wernsdorfer, S. Hill and M. Murrie, Dalton Trans., 2008, 6409 RSC; (b) E.-C. Yang, W. Wernsdorfer, L. N. Zakharov, Y. Karaki, A. Yamaguchi, R. M. Isidoro, G.-D. Lu, S. A. Wilson, A. L. Rheingold, H. Isimoto and D. N. Hendrickson, Inorg. Chem., 2006, 45, 529 CrossRef CAS PubMed; (c) A. Bell, G. Aromi, S. J. Teat, W. Wernsdorfer and R. E. P. Winpenny, Chem. Commun., 2005, 2808 RSC; (d) M. Moragues-Cánovas, M. Helliwell, L. Ricard, E. Riviére, W. Wernsdorfer, E. Brechin and T. Mallah, Eur. J. Inorg. Chem., 2004, 2219 CrossRef; (e) E.-C. Yang, W. Wernsdorfer, S. Hill, R. S. Edwards, M. Nakano, S. Maccagano, L. N. Zakharov, A. L. Rheingold, G. Christou and D. N. Hendrickson, Polyhedron, 2003, 22, 1727 CrossRef CAS; (f) S. T. Ochseinbein, M. Murrie, E. Rusanov, H. Stoeckli-Evans, C. Sekine and H.-U. Güdel, Inorg. Chem., 2002, 41, 5133 CrossRef; (g) H. Andres, R. Basler, A. J. Blake, C. Cadiou, G. Chabboussant, C. M. Grant, H.-U. Güdel, M. Murrie, S. Parsons, C. Paulsen, F. Semadini, V. Villar, W. Wernsdorfer and R. E. P. Winpenny, Chem. – Eur. J., 2002, 8, 4867 CrossRef CAS PubMed.
- S. Carretta, P. Santini, G. Amorretti, M. Affronte, A. Candini, A. Chirri, I. S. Tidmarsh, R. H. Laye, R. Shaw and E. J. L. McInnes, Phys. Rev. Lett., 2006, 97, 207201 CrossRef CAS PubMed.
- (a) P. Chaudhuri, Coord. Chem. Rev., 2003, 243, 143 CrossRef CAS; (b) A. J. L. Pombeiro and V. Yu Kukushkin, in Comprehensive Coordination Chemistry II, ed. J. A. McCleverty and T. J. Meyer, Elsevier, Amsterdam, 2004, vol. 7, pp. 631–637 Search PubMed; (c) C. J. Milios, S. Piligkos and E. K. Brechin, Dalton Trans., 2008, 1809 RSC (Perspective); (d) T. Pathmalingam, S. I. Gorelsky, T. J. Burchell, A.-C. Bédard, A. M. Beauchemin, R. Clérac and M. Murugesu, Chem. Commun., 2008, 2782 RSC; (e) C. J. Milios, A. Vinslava, W. Wernsdorfer, S. Moggach, S. Parsons, S. P. Perlepes, G. Christou and E. K. Brechin, J. Am. Chem. Soc., 2007, 129, 2754 CrossRef CAS PubMed; (f) L. F. Jones, A. Prescimone, M. Evangelisti and E. K. Brechin, Chem. Commun., 2009, 2023 RSC.
- For a comprehensive review, see: C. J. Milios, T. C. Stamatatos and S. P. Perlepes, Polyhedron, 2006, 25, 134 CrossRef CAS (Polyhedron Report).
- (a) H. Kumagai, M. Endo, M. Kondo, S. Kawata and S. Kitagawa, Coord. Chem. Rev., 2003, 237, 197 CrossRef CAS; (b) C. J. Milios, P. Kyritsis, C. P. Raptopoulou, A. Terzis, R. Vicente, A. Escuer and S. P. Perlepes, Dalton Trans., 2005, 501 RSC; (c) K. F. Konidaris, C. D. Polyzou, G. E. Kostakis, A. J. Tasiopoulos, O. Roubeau, S. J. Teat, E. Manessi-Zoupa, A. K. Powell and S. P. Perlepes, Dalton Trans., 2012, 41, 2862 RSC.
- (a) T. C. Stamatatos, D. Foguet-Albiol, C. C. Stoumpos, C. P. Raptopoulou, A. Terzis, W. Wernsdorfer, S. P. Perlepes and G. Christou, Polyhedron, 2007, 26, 2165 CrossRef CAS; (b) T. C. Stamatatos, D. Foguet-Albiol, C. C. Stoumpos, C. P. Raptopoulou, A. Terzis, W. Wernsdorfer, S. P. Perlepes and G. Christou, J. Am. Chem. Soc., 2005, 127, 15380 CrossRef CAS PubMed; (c) A. Escuer, G. Vlahopoulou and F. A. Mautner, Inorg. Chem., 2011, 50, 2717 CrossRef CAS PubMed; (d) A. M. Mowson, T. N. Nguyen, K. A. Abboud and G. Christou, Inorg. Chem., 2013, 52, 12320 CrossRef CAS PubMed; (e) T. Nguyen, W. Wernsdorfer, K. A. Abboud and G. Christou, J. Am. Chem. Soc., 2011, 133, 20688 CrossRef CAS PubMed; (f) T. N. Nguyen, M. Shiddiq, T. Ghosh, K. A. Abboud, S. Hill and G. Christou, J. Am. Chem. Soc., 2015, 137, 7160 CrossRef CAS PubMed; (g) T. N. Nguyen, W. Wernsdorfer, M. Shiddiq, K. A. Abboud, S. Hill and G. Christou, Chem. Sci., 2016, 7, 1156 RSC.
- (a) T. C. Stamatatos, A. Escuer, K. A. Abboud, C. P. Raptopoulou, S. P. Perlepes and G. Christou, Inorg. Chem., 2008, 47, 11825 CrossRef CAS PubMed; (b) T. C. Stamatatos, K. A. Abboud, S. P. Perlepes and G. Christou, Dalton Trans., 2007, 3861 RSC; (c) C. Papatriantafyllopoulou, T. C. Stamatatos, C. G. Efthymiou, L. Cunha-Silva, F. A. A. Paz, S. P. Perlepes and G. Christou, Inorg. Chem., 2010, 49, 9743 CrossRef CAS PubMed; (d) S. Zhang, L. Zhen, B. Xu, R. Inglis, K. Li, W. Chen, Y. Zhang, K. F. Konidaris, S. P. Perlepes, E. K. Brechin and Y. Li, Dalton Trans., 2010, 3563 RSC; (e) T. C. Stamatatos, C. Papatriantafyllopoulou, E. Katsoulakou, C. P. Raptopoulou and S. P. Perlepes, Polyhedron, 2007, 26, 1830 CrossRef CAS.
- (a) C. D. Polyzou, H. Nikolaou, C. Papatriantafyllopoulou, V. Psycharis, A. Terzis, C. P. Raptopoulou, A. Escuer and S. P. Perlepes, Dalton Trans., 2012, 13755 RSC; (b) C. Papatriantafyllopoulou, G. Aromi, A. J. Tasiopoulos, V. Nastopoulos, C. P. Raptopoulou, S. J. Teat, A. Escuer and S. P. Perlepes, Eur. J. Inorg. Chem., 2007, 2761 CrossRef CAS; (c) C. G. Efthymiou, C. P. Raptopoulou, A. Terzis, S. P. Perlepes, A. Escuer and C. Papatriantafyllopoulou, Polyhedron, 2010, 29, 627 CrossRef CAS; (d) C. Papatriantafyllopoulou, M. Estrader, C. G. Efthymiou, D. Dermitzaki, K. Gkotsis, A. Terzis, C. Diaz and S. P. Perlepes, Polyhedron, 2009, 28, 1652 CrossRef CAS; (e) D. Dermitzaki, C. P. Raptopoulou, V. Psycharis, A. Escuer, S. P. Perlepes and T. C. Stamatatos, Dalton Trans., 2014, 14520 RSC; (f) C. Papatriantafyllopoulou, C. P. Raptopoulou, A. Terzis, J. F. Janssens, S. P. Perlepes and E. Manessi-Zoupa, Z. Naturforsch., B: Chem. Sci., 2007, 62, 1123 CrossRef CAS.
- (a) C. G. Efthymiou, A. A. Kitos, C. P. Raptopoulou, A. Escuer, S. P. Perlepes and C. Papatriantafyllopoulou, Polyhedron, 2009, 28, 3177 CrossRef CAS; (b) C. Papatriantafyllopoulou, C. G. Efthymiou, C. Raptopoulou, A. Terzis, E. Manessi-Zoupa and S. P. Perlepes, Spectrochim. Acta, Part A, 2008, 70, 718 CrossRef PubMed; (c) C. Papatriantafyllopoulou, T. C. Stamatatos, W. Wernsdorfer, S. J. Teat, A. J. Tasiopoulos, A. Escuer and S. P. Perlepes, Inorg. Chem., 2010, 49, 10486 CrossRef CAS PubMed.
- (a) E. Moushi, C. G. Efthymiou, S. P. Perlepes and C. Papatriantafyllopoulou, Int. J. Inorg. Chem., 2011, 606271 Search PubMed; (b) T. C. Stamatatos, E. Diamantopoulou, C. P. Raptopoulou, V. Psycharis, A. Escuer and S. P. Perlepes, Inorg. Chem., 2007, 46, 2350 CrossRef CAS PubMed.
- C. Papatriantafyllopoulou, L. F. Jones, T. D. Nguyen, N. Matamoros-Salvador, L. Cunha-Silva, F. A. Almeida Paz, J. Rocha, M. Evangelisti, E. K. Brechin and S. P. Perlepes, Dalton Trans., 2008, 3153 RSC.
- D. S. Boltin, N. A. Bokach and V. Yu. Kukushkin, Coord. Chem. Rev., 2016, 313, 62 CrossRef.
- H.-Z. Kou, G.-Y. An, C.-M. Ji, B.-W. Wang and A.-L. Cui, Dalton Trans., 2010, 9604 RSC.
- C.-M. Ji, H.-J. Yang, C.-C. Zhao, V. Tangoulis, A.-L. Cui and H.-Z. Kou, Cryst. Growth Des., 2009, 9, 4607 CAS.
- G.-Y. An, H.-B. Wang, A.-L. Cui and H.-Z. Kou, New J. Chem., 2014, 38, 5037 RSC.
- (a) L. Alcazar, M. Font-Bardia and A. Escuer, Eur. J. Inorg. Chem., 2015, 1326 CrossRef CAS; (b) J. Jankolovits, C. M. Andolina, J. W. Kampf, K. N. Raymond and V. L. Pecoraro, Angew. Chem., Int. Ed., 2011, 50, 9660 CrossRef CAS PubMed.
- E. Bernasek, J. Org. Chem., 1957, 22, 1263 CrossRef CAS.
- T. Kottke and D. Stalke, J. Appl. Crystallogr., 1993, 26, 615 CrossRef.
- APEX2, Data Collection Software Version 2.1-RC13, Bruker AXS, Delft, The Netherlands, 2006 Search PubMed.
- Cryopad, Remote monitoring and control, Version 1.451, Oxford Cryosystems, Oxford, United Kingdom, 2006 Search PubMed.
- SAINT+, Data Integration Engine v. 7.23a ©, Bruker AXS, Madison, Wisconsin, USA, 1997–2005 Search PubMed.
- G. M. Sheldrick, SADABS v.2.01, Bruker/Siemens Area Detector Absorption Correction Program, Bruker AXS, Madison, Wisconsin, USA, 1998 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for Crystal Structure Refinement, University of Göttingen, 1997 Search PubMed.
- A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, C34 Search PubMed; A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- P. Van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 194 CrossRef.
- C. Papatriantafyllopoulou, C. P. Raptopoulou, A. Terzis, E. Manessi-Zoupa and S. P. Perlepes, Z. Naturforsch., B: Chem. Sci., 2006, 61, 37 CrossRef CAS.
- J. Lopez-Carriga, G. T. Badcock and J. F. Harrison, J. Am. Chem. Soc., 1986, 108, 7241 CrossRef.
- A. B. P. Lever and A. Mantovani, Inorg. Chem., 1971, 10, 817 CrossRef CAS.
- (a) F.-R. Dai, D. C. Becht and Z. Wang, Chem. Commun., 2014, 5385 RSC; (b) B. Biswas, S. Khanra, T. Weyhermuller and P. Chaudhuri, Chem. Commun., 2007, 1059 RSC; (c) K. V. Shuvaev, S. S. Tandon, L. N. Dawe and L. K. Thompson, Chem. Commun., 2010, 4755 RSC; (d) H. Jiang, T. Sheng, S. Bai, S. Hu, X. Wang, R. Fu, P. Yu and X. Wu, Inorg. Chem., 2013, 52, 12305 CrossRef CAS PubMed; (e) K. Su, F. Jiang, J. Qian, Y. Gai, M. Wu, S. M. Bawaked, M. Mokhtar, S. A. AL-Thabaiti and M. Hong, Cryst. Growth Des., 2014, 14, 3116 CrossRef CAS.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes in C: The Art of Scientific Computing, Cambridge University Press, Cambridge, 2nd edn, 1992 Search PubMed.
- G.-Y. An, C.-M. Ji, A.-L. Cui and H.-Z. Kou, Inorg. Chem., 2011, 50, 1079 CrossRef CAS PubMed.
- (a) M. Evangelisti, F. Luis, L. J. De Jongh and M. Affronte, J. Mater. Chem., 2006, 16, 2534 RSC; (b) M. Evangelisti, Molecular Magnets, NanoScience and Technology, in Molecule-based Magnetic Coolers: Measurement, Design and Application, ed. J. Bartolomé, F. Luis and J. F. Fernandez, Springer-Verlag, Berlin, Heidelberg, 2014, pp. 365–387 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables with bond distances and angles, hydrogen bonding details, magnetism and heat capacity plots for 2·10H2O·26MeOH. CCDC 678537 and 1502969. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03511f |
| This journal is © The Royal Society of Chemistry 2016 |