
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe synthesis and structure of a dianionic species with a bond between pentacoordinated tin atoms: bonding properties of the tin–tin bond†
Satoru
Tsukada
a,
Nathan J.
O'Brien
a,
Naokazu
Kano
*a,
Takayuki
Kawashima
*a,
Jing-Dong
Guo
b and
Shigeru
Nagase
b
aDepartment of Chemistry, Graduate School of Science, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: kano@chem.s.u-tokyo.ac.jp; takayuki@chem.s.u-tokyo.ac.jp
bFukui Institute for Fundamental Chemistry, Kyoto University, Takano-Nishihiraki-cho, Sakyo-ku, Kyoto 606-8103, Japan
First published on 11th November 2016
Abstract
The first dianionic compound bearing a bond between two pentacoordinated tin atoms, a distannate, was synthesized in a stable form by using two sets of an electron-withdrawing C,O-bidentate ligand on each tin atom. The structure of the distannate was determined by NMR spectroscopy and X-ray crystallographic analysis. The Sn–Sn bond of the distannate was shown to be a single bond featuring high s-character. The 1J(Sn–Sn) coupling constant was larger than that of Sn(sp3)–Sn(sp3) bonds found in most hexaorganodistannanes. This bond feature was also supported by computational studies. The Sn–Sn bond was cleaved by treatment with hydrochloric acid, which shows a different reactivity to the homonuclear bonds of pentacoordinated disilicates and digermanates.
Introduction
Homonuclear E–E bonds between heavier group 14 elements (E = silicon, germanium and tin), which are analogues of C–C bonds, have been extensively studied as the fundamental structural units of group 14 element compounds.1–11 For example, catenation of a Sn–Sn bond forms polystannanes,12–15 which have attracted much attention because of their potential application to semiconducting materials,13 photoresist technology,14 conductive films,14 and third-order optical nonlinear materials.12,16 The Sn–Sn bonds found in some distannanes are cleaved by several reagents and oxidized by air.7,11,17,18 They can also be cleaved by transition metals via oxidative addition and transmetallation.19 Additionally, Sn–Sn bond cleavage has also been applied to generate stannyl radicals due to their small bond dissociation energy (Δ(Sn–Sn) = 187 kJ mol−1).20 Some Sn–Sn bonded compounds have found use as a source of stannyl radicals in organic synthesis.21–25 The stability of a Sn–Sn bonded compound generally depends on the coordination number and charge of the tin atoms that form the bond. Thus, the Sn–Sn bonded compounds consisting of tin atoms with different coordination numbers and charges are expected to show divergent bond-cleavage reactivity. In contrast to general organotin compounds with a coordination number of four for the tin atom, there are mainly two types of organotin compounds with a coordination number of five for the tin atom. One is the neutral pentacoordinated organotin compounds featuring intramolecular coordination to the tin atom and the other is the anionic ate-type tin compounds. Actually, there have been some neutral compounds bearing a bond between two pentacoordinated tin atoms by taking advantage of intramolecular coordination. Following the first report of this type of compound, which used a pair of acetate bridging ligands,26 some neutral distannanes 1 with two acyloxy ligands were investigated (Chart 1).27–31 Alternatively, the formation of neutral pentacoordinated tin–tin bonds in compounds 2, 3, and 4 was achieved by intramolecular coordination of nitrogen atoms.32–35 Moreover, a few stannylenes formed a dimer through an interaction between tin atoms in the solid state and intramolecular P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O→Sn coordination made the tin atoms pentacoordinated.36 However, there is no report of a dianionic species containing a bond between two pentacoordinated ate-type tin atoms, a distannate, as far as we know. A reason for this is simply not ascribed to the instability of the ate-type structure of each tin atom as several stable pentacoordinated ate-type tin compounds, which show a trigonal bipyramidal (TBP) structure around the tin atom, have been synthesized by taking advantage of electron-withdrawing bidentate ligands.7,37–40
O→Sn coordination made the tin atoms pentacoordinated.36 However, there is no report of a dianionic species containing a bond between two pentacoordinated ate-type tin atoms, a distannate, as far as we know. A reason for this is simply not ascribed to the instability of the ate-type structure of each tin atom as several stable pentacoordinated ate-type tin compounds, which show a trigonal bipyramidal (TBP) structure around the tin atom, have been synthesized by taking advantage of electron-withdrawing bidentate ligands.7,37–40
 | ||
| Chart 1 | ||
We previously reported the synthesis of disilicate 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41,42 and digermanate 6,43 which are dianionic species containing a bond between two pentacoordinated ate-type silicon and germanium atoms, respectively. A tin analogue of 5 and 6 could be a challenging synthetic target considering the much smaller bond energy of the Sn–Sn bond in comparison to the Si–Si and Ge–Ge bonds.41,43 Disilicate 5 and digermanate 6 were successfully stabilized by taking advantage of four sets of an electron-withdrawing bidentate ligand. It was also presumed that these ligands could overcome the difficulties of stabilization of a distannate. Herein we report the synthesis and crystal structure of pentacoordinated distannate 7 bearing two bidentate ligands, –C6H4–2-C(CF3)2O–,44 on each tin atom. We also investigated the properties of the tin–tin bond by using 119Sn NMR spectroscopy and density functional theory (DFT) calculations.
41,42 and digermanate 6,43 which are dianionic species containing a bond between two pentacoordinated ate-type silicon and germanium atoms, respectively. A tin analogue of 5 and 6 could be a challenging synthetic target considering the much smaller bond energy of the Sn–Sn bond in comparison to the Si–Si and Ge–Ge bonds.41,43 Disilicate 5 and digermanate 6 were successfully stabilized by taking advantage of four sets of an electron-withdrawing bidentate ligand. It was also presumed that these ligands could overcome the difficulties of stabilization of a distannate. Herein we report the synthesis and crystal structure of pentacoordinated distannate 7 bearing two bidentate ligands, –C6H4–2-C(CF3)2O–,44 on each tin atom. We also investigated the properties of the tin–tin bond by using 119Sn NMR spectroscopy and density functional theory (DFT) calculations.
Experimental
General procedures
Solvents were dried and purified before use by using an MBraun MB-SPS solvent purification system. The reaction was carried out under an argon atmosphere. All NMR spectra were recorded on a Bruker Ultrashield™ 300 spectrometer. The 1H NMR (300 MHz) and 13C{1H} NMR (75 MHz) spectra were recorded using tetramethylsilane (TMS) as an external standard. The 19F NMR (282 MHz) and 119Sn NMR (112 MHz) spectra were recorded using Freon-11 and tetramethylstannane as external standards, respectively. The melting point was recorded with a Yanaco MP-500D micro melting point apparatus and was uncorrected. Mass spectral data were obtained on Thermo Scientific Exactive (ESI) and JOEL JMS-700P (FAB) instruments. Elemental analysis was performed by the Microanalytical Laboratory of the Department of Chemistry, Faculty of Science, the University of Tokyo.Synthesis of distannate 7
To a solution of fluorostannate 837 (0.86 g, 1.0 mmol) (dried at 182 °C under reduced pressure before use) in THF (2 mL) was added a THF solution of lithium naphthalenide (0.47 M, 2.6 mL, 1.2 mmol) at −78 °C under argon. The mixture was stirred for 3 hours, then warmed to room temperature and stirred for a further 14 hours. The reaction mixture was concentrated under reduced pressure and the resulting residue was washed with Et2O to give a grey solid. The solid was dissolved in THF, filtered through a bed of Celite® and concentrated under reduced pressure to give a colorless solid. The solid was recrystallized from THF/n-hexane to give the distannate 7 (0.40 g, 48%) as a colorless solid; m.p. 226–230 °C. 1H NMR (300 MHz, THF-d8) δ 1.05 (t, 3JHH = 7.4 Hz, 24H), 1.49 (sext, 3JHH = 7.4 Hz, 16H), 1.73–1.82 (m, 16H), 3.35–3.41 (m, 16H), 6.98 (dt, 3JHH = 7.5 Hz, JHSn = 1.0 Hz, 4H), 7.11 (dt, 3JHH = 7.2 Hz, JHSn = 1.0 Hz, 4H), 7.35 (d, 3JHH = 7.5 Hz, 4H), 8.14 (dt, 3JHH = 6.9 Hz, JHSn = 27 Hz, 4H). 13C{1H} NMR (75 MHz, THF-d8) δ 13.77 (s), 20.32 (s), 24.44 (s), 59.09 (s), 83.74 (sept, 2JCF = 27 Hz), 125.36 (s, JCSn = 79 Hz (satellite peaks)), 125.79 (q, 1JCF = 288 Hz), 126.39 (q, 1JCF = 289 Hz), 126.49 (s, JCSn = 83 Hz (satellite peaks)), 127.36 (s), 137.27 (s, JCSn = 46 Hz (satellite peaks)), 141.00 (s, 1JCSn = 50 Hz, 2JCSn = 14 Hz (satellite peaks)), 148.74 (s). 19F NMR (282 MHz, THF-d8) δ −77.15 (d, 4JFF = 7.1 Hz, 12F), −77.52 (d, 4JFF = 7.9 Hz, 12F). 119Sn NMR (112 MHz, THF-d8) δ −94.9 (JSnSn = 15532 Hz). LRMS (ESI, negative) m/z 1448 [M − (n-Bu)4N]−. Anal. Calcd for C68H88F24N2O4Sn2·2H2O: C, 47.30; H, 5.30; N, 1.62. Found: C, 47.17; H, 5.45; N, 1.71.
Reaction of distannate 7 with 1 M aqueous hydrochloric acid
To a solution of distannate 7 (100 mg, 60 μmol) in THF (3 mL) was added 1 M HCl (1.18 mL, 1.18 mmol) and the whole mixture was stirred at room temperature for 24 hours. The reaction mixture was diluted with H2O and the aqueous layer was extracted with CH2Cl2 (×2). The combined organics were washed with H2O, dried over MgSO4 and concentrated under reduced pressure to give a colorless solid. The solid was recrystallized from CH2Cl2/n-hexane to give the chlorostannate 9 (97 mg, 93%) as a colorless solid; m.p. 158–161 °C. 1H NMR (300 MHz, acetone-d6) δ 0.97 (t, 3JHH = 7.4 Hz, 12H), 1.42 (sext, 3JHH = 7.4 Hz, 8H), 1.76–1.87 (m, 8H), 3.40–3.46 (m, 8H), 7.45–7.59 (m, 4H), 7.71–7.82 (m, 2H), 7.98–8.24 (m, 2H). 13C{1H} NMR (75 MHz, acetone-d6) δ 13.8 (s), 20.3 (t, 2JCN = 1 Hz), 24.4 (s), 59.4 (t, 1JCN = 3 Hz), 81.1 (sept, 2JCF = 28 Hz), 125.9 (q, 1JCF = 289 Hz), 127.2 (s, JCSn = 86 Hz (satellite peaks)), 129.9 (s, JCSn = 79 Hz (satellite peaks)), 130.1 (s, JCSn = 16 Hz (satellite peaks)), 136.3 (s, JCSn = 56 Hz (satellite peaks)), 141.1 (s, JCSn = 92 Hz (satellite peaks)), 141.2 (s). 19F NMR (282 MHz, acetone-d6) δ −75.72 (s, 12F). 119Sn NMR (112 MHz, acetone-d6) δ −127.4 (s). LRMS (ESI, negative) m/z 638 [M − (n-Bu)4N]−. Anal. Calcd for C34H44ClF12NO2Sn: C, 46.36; H, 5.04; N, 1.59. Found: C, 46.44; H, 5.26; N, 1.56%.Results and discussion
Disilicate 541 and digermanate 643 were synthesized by the reductive coupling of tetracoordinated spiro-silane and germane with lithium, but the analogous spirostannane has not been reported due to the high electrophilicity of the tin atom. However, pentacoordinated fluorostannate 8, bearing two sets of the electron-withdrawing C,O-bidentate ligand, is known to react with nucleophiles and was used for the synthesis of stannates.37 It is expected that a single electron reduction of 8 would give lithium fluoride elimination and generate the corresponding tin radical anion. Dimerization of the radical anion would yield the distannate similarly to the formation of the previously reported disilicates 541 and digermanates 6.43 Actually, reductive coupling of tetrabutylammonium fluorostannate 837 with lithium naphthalenide in THF gave distannate 7 in 48% yield as a dihydrate (Scheme 1). Distannate 7 was characterized by multinuclear NMR spectroscopy, elemental analysis, and single-crystal X-ray diffraction analysis. The pentacoordinated fluorostannate 8 was used to generate the corresponding tin radical anion in this reaction.
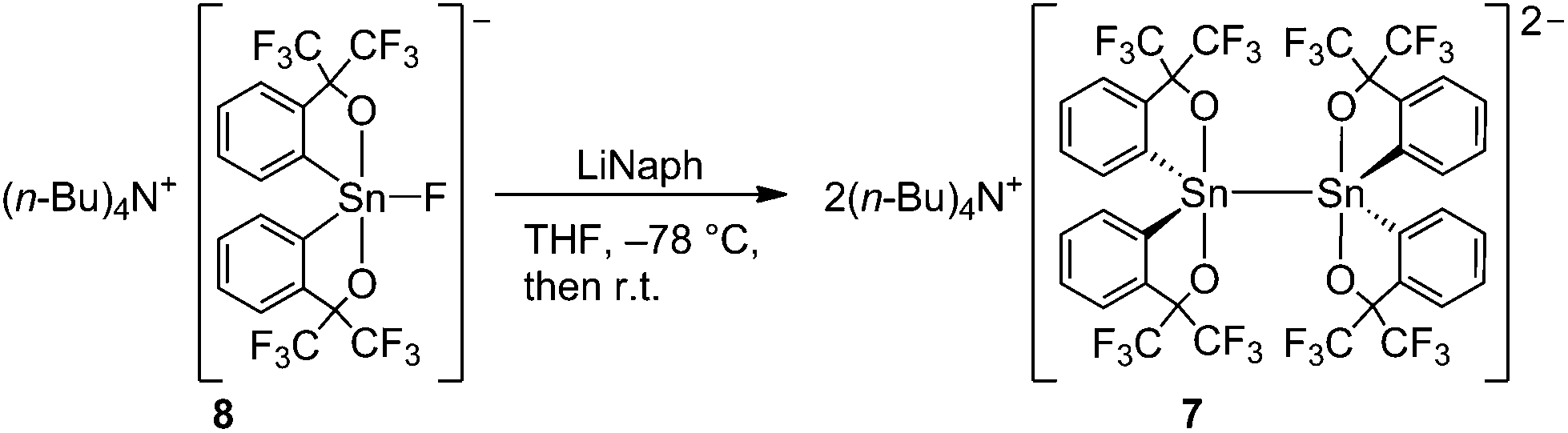 | ||
| Scheme 1 Synthesis of distannate 7 from fluorostannate 8. | ||
The crystal structure of 7 was determined by single-crystal X-ray diffraction analysis (Fig. 1). The tin-containing moiety was found to be a dianion and was accompanied by two separated tetrabutylammonium ions. The dianion has two pentacoordinated tin atoms, which are bonded to each other, showing its identity as a distannate. Distannate 7 shows a trigonal bipyramidal geometry around each crystallographically equivalent tin atom with two oxygen atoms at the apical positions and another tin and two carbon atoms at the equatorial positions. In other words, the Sn–Sn bond forms an equatorial bond on each tin atom.
 | ||
| Fig. 1 ORTEP drawing of distannate 7 with thermal ellipsoids at 50% probability level. Hydrogen atoms and water molecules are omitted for clarity. Color codes: light grey, C; red, O; yellow-green, F; orange, Sn; blue, N. Selected bond lengths (Å) and angle (deg.): Sn1–Sn1* 2.7522(14), Sn1–O1 2.166(9), Sn1–O2 2,162(9), Sn1–C1 2.155(13), Sn1–C10 2.104(11), O1–Sn1–O2 166.0(4). | ||
The Sn1–Sn1* bond length (2.7522(14) Å) is similar to that of Ph3Sn–SnPh3 (2.770(4) Å)45 and the sum of the covalent radii (2.78 Å)46 indicates single bond character. Moreover, among the pentacoordinated Sn–Sn bonded compounds, the Sn1–Sn1* bond length of 7 is between the values of neutral distannanes 1 bearing bridging carboxylato ligands (2.692(3)–2.721 Å)27–31 and distannanes 2, 3, and 4 bearing chelating amine or imine ligands (2.8014(13)–2.831 Å).32–35 The Sn–O1 and Sn–O2 apical bond lengths (2.166(9) and 2.162(9) Å) are longer than the sum of the Sn–O covalent radii (2.05 Å),46 which is in accordance with the apical bonds of pentacoordinated species having a tendency of being long. Nevertheless, the apical Sn–O bond lengths are slightly longer than those of the tetrabutylammonium fluorostannate bearing the same bidentate ligands (2.101(6) and 2.110(6) Å).37 The O1–Sn1–O2 angle (166.0(4)°) of 7 is considerably narrower than that of fluorostannate 8 (178.7(3)°). The Sn1–O1 and Sn1*–O2* bonds are staggered relative to each other, as exhibited by the O1–Sn1–Sn1*–O2* torsion angle (58.6(4)°), to avoid the steric repulsion of the bulky bidentate ligands. The torsion angle of 7 is smaller than the O–Si–Si–O and O–Ge–Ge–O torsion angles of disilicate 5 (66.64(5)°) and digermanate 6 (60.79(19)°), respectively.41,43 This reflects a decrease in steric repulsion between the bulky bidentate ligands on the group 14 element atoms due to the longer bond lengths between the two tin atoms.
To investigate the temperature dependence of the structure of 7, variable temperature 19F NMR measurements were carried out over a range of 30–65 °C in THF-d8 (Fig. S9†). There was no change in the spectra, indicating no pseudorotation around the Sn atom in the temperature range. In the 119Sn-NMR spectrum of 7 in THF-d8, one singlet was observed at δSn −94.9 ppm accompanied by satellite peaks due to the coupling between 119Sn and 117Sn nuclei. The 119Sn chemical shift of 7 is reasonable for an anionic pentacoordinated tin nucleus coordinated by electron-withdrawing ligands as it is similar to other pentacoordinated tin allyl ate complexes, e.g. (n-Et4N)+[(1,2-C6H4C(CF3)2O–)2Sn(CH2CHCH2)]− (δSn −105.0 ppm).38 The 119Sn–117Sn coupling constant (1J(Sn–Sn) = 15532 Hz) of 7 is similar to those of neutral pentacoordinated distannanes, e.g. Ph4Sn2(OOCCH3)21 [1J(Sn–Sn) = 16120 Hz],31 but is much larger than those of distannanes, such as Ph3Sn–SnPh3 (1J(Sn–Sn) = 4281 Hz) and (t-Bu)(i-Pr)2Sn–Sn(t-Bu)(i-Pr)2 (1J(Sn–Sn) = 764 Hz).47 The small coupling constant of (t-Bu)(i-Pr)2Sn–Sn(t-Bu)(i-Pr)2 is due to the bulky ligands on the tin atoms, which decreases the s-character of the Sn–Sn bond. Thus, the large coupling constant 1J(Sn–Sn) of 7 indicates high s-character of the Sn–Sn bond.
To investigate the molecular orbital and the bonding properties of the distannate, DFT calculations on the dianion were carried out using the Gaussian 09 program.48 Geometry optimization was performed with the dispersion-corrected B3PW91-D3BJ method49,50 by using the [4333111/433111/43] basis set augmented by two d polarization functions (d exponents 0.253 and 0.078)51 and diffusion function (sp exponent 0.0174) for Sn52 and the 6-31+G(d) basis set for other atoms. The natural bond orbital analysis was performed using the NBO program.53 The optimized structure of the dianion of 7 reproduced the dianion of the crystal structure (see the ESI†). The highest occupied molecular orbital (HOMO) was mainly contributed by the bonding σ orbital of the Sn–Sn bond and the lone pairs of the oxygen atoms (Fig. 2). The lowest unoccupied molecular orbital (LUMO) was contributed not by the σ* orbital of the Sn–Sn bond, but mainly by the π* orbitals of the benzene rings. These molecular orbital features are almost the same as 5 and 6.41,43 The Wiberg bond index54 of the Sn–Sn bond is 0.843, indicating that the Sn–Sn bond of distannate 7 is a single bond and corresponds to the crystallographic analysis. Additionally, the natural bond orbital analysis shows sp2.35 hybridization of the tin atoms that constitute the Sn–Sn bond of the distannate. The high s-character of the Sn–Sn bond is consistent with the larger 1J(Sn–Sn) coupling constant of the distannate than that of the Sn(sp3)–Sn(sp3) bonds in most distannanes. Charge distributions of the dianion of 7 were estimated by the natural population analysis. The tin atoms are positively charged (qSn, +1.581), while the oxygen and carbon atoms bound to the tin atoms are negatively charged (qO, −0.865, qC, −0.430). The localization of the negative charges on the bidentate ligands avoids the electrostatic repulsion between the two anionic halves of the distannate. The quantity of positive charge on the tin atoms was found to be larger than those of the silicon 5 (qSi, +1.393)41 and the germanium 6 (qGe, +1.30)43 atoms. This is a result of the high polarizability of the tin atom in comparison to the silicon and the germanium atoms.
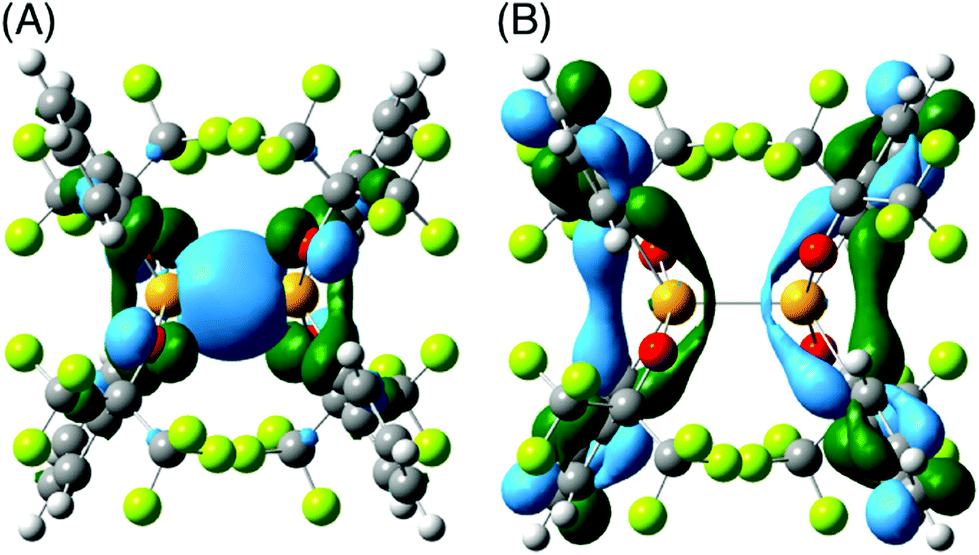 | ||
| Fig. 2 Molecular orbitals of the dianion part of 7 (isovalue, 0.03). (A) HOMO, and (B) LUMO. | ||
Mass spectroscopic analysis showed an ion peak of m/z 1448, assigned to the dianion part with a tetrabutylammonium ion of distannate 7, indicating its stability in the gas phase. Distannate 7 was also stable to air and moisture and could be handled without precautions. However, the reaction of 7 with 20 equivalents of 1 M aqueous hydrochloric acid caused its degradation. Analysis of the 1H and 19F NMR spectra (Fig. S10 and S11†) of the reaction mixture shows the formation of an intermediate, thought to be the mono- or diprotonated distannate. This intermediate was shown to have a 119Sn NMR chemical shift at δSn −88.6 ppm (Fig. S12†), which is different from the literature value of chlorostannate (δSn −127.6 ppm).37 Further reaction of this intermediate cleaved the Sn–Sn bond and gave clean conversion to chlorostannate 9 (Scheme 2). The reactivity of 7 is different from that of 5 and 6, both of which underwent protonation on oxygen atoms bonded to silicon and germanium atoms without homonuclear bond cleavage, upon treatment with hydrochloric acid.41–43 This result shows that the Sn–Sn bond of the distannate is weaker than the Si–Si and Ge–Ge bonds of the disilicate and digermanate, respectively, under the same conditions.
 | ||
| Scheme 2 Reaction of distannate 7 with 1 M aqueous HCl. | ||
Conclusions
In conclusion, we successfully synthesized the first example of a distannate, a dianionic compound bearing a bond between two pentacoordinated tin atoms, from the pentacoordinated fluorostannate. The Sn–Sn bond of the distannate was found to be of a similar length to common distannanes and was a single bond comprised of two sp2 hybrid tin atoms. Computational analysis indicated that the negative charges of the distannate are localized over the bidentate ligands. The distannate was stable towards both air and moisture, but the Sn–Sn bond was cleaved by treatment with hydrochloric acid. The unique reactivity of the distannate can be pointed out as a distinct difference in the stability of the homonuclear bonds of pentacoordinated group 14 elements, compared with those of the Si–Si and Ge–Ge systems. Utilization of this unique bonding property and structure could lead to the development of a new material, which might supersede polystannanes.Acknowledgements
We thank Prof. Yohsuke Yamamoto for his advice on this project. This work was partially supported by the Izumi Science and Technology Foundation and the Grant-in Aid for Challenging Exploratory Research (No. 25620024) from MEXT.Notes and references
- H. K. Sharma and K. H. Pannell, Chem. Rev., 1995, 95, 1351–1374 CrossRef CAS.
- E. Lukevics and O. Pudova, Main Group Met. Chem., 1998, 21, 123–183 CAS.
- M. Suginome and Y. Ito, J. Organomet. Chem., 2003, 685, 218–229 CrossRef CAS.
- K. M. Baines and W. G. Stibbs, Coord. Chem. Rev., 1995, 145, 157–200 CrossRef CAS.
- M. L. Amadoruge and C. S. Weinert, Chem. Rev., 2008, 108, 4253–4294 CrossRef CAS PubMed.
- A. K. Sawyer, in Organotin Componds, ed. A. K. Sawyer, Marcel Dekker, 1972, Vol. 3, pp. 823–879 Search PubMed.
- S. Patai, The chemistry of organic germanium, tin and lead compounds, Wiley, 1995 Search PubMed.
- M. Kaupp, B. Metz and H. Stoll, Angew. Chem., Int. Ed., 2000, 39, 4607–4609 CrossRef CAS.
- M. Kaupp and S. Riedel, Inorg. Chim. Acta, 2004, 357, 1865–1872 CrossRef CAS.
- S. T. Liddle, Molecular metal-metal bonds: compounds, synthesis, properties, Wiley-VCH, 2015 Search PubMed.
- G. S. Wilkinson, F. G. A. Stone and E. W. Abel, Comprehensive organometallic chemistry: the synthesis, reactions, and structures of organometallic compounds, Pergamon Press, 1982 Search PubMed, vol. 2.
- K. Takeda and K. Shiraishi, Chem. Phys. Lett., 1992, 195, 121–126 CrossRef CAS.
- T. Imori, V. Lu, H. Cai and T. D. Tilley, J. Am. Chem. Soc., 1995, 117, 9931–9940 CrossRef CAS.
- P. Braunstein and X. Morise, Chem. Rev., 2000, 100, 3541–3552 CrossRef CAS PubMed.
- M. Trummer, F. Choffat, P. Smith and W. Caseri, Macromol. Rapid Commun., 2012, 33, 448–460 CrossRef CAS PubMed.
- R. Priestley, A. D. Walser, R. Dorsinville, W. K. Zou, D. Y. Xu and N.-L. Yang, Opt. Commun., 1996, 131, 347–350 CrossRef CAS.
- W. V. Farrar and H. A. Skinner, J. Organomet. Chem., 1963, 1, 434–436 CrossRef.
- W. P. Neumann and J. Pedain, Justus Liebigs Ann. Chem., 1964, 672, 34–43 CrossRef CAS.
- J. Tsuji, Transition metal reagents and catalysts: innovations in organic synthesis, John Wiley, 2002 Search PubMed.
- Y.-R. Luo, Comprehensive handbook of chemical bond energies, CRC Press, 2007 Search PubMed.
- G. Stork and P. M. Sher, J. Am. Chem. Soc., 1983, 105, 6765–6766 CrossRef CAS.
- J. E. Baldwin, D. R. Kelly and C. B. Ziegler, J. Chem. Soc., Chem. Commun., 1984, 133–134 RSC.
- G. E. Keck and J. H. Byers, J. Org. Chem., 1985, 50, 5442–5444 CrossRef CAS.
- B. B. Snider and B. O. Buckman, J. Org. Chem., 1992, 57, 4883–4888 CrossRef CAS.
- M. Harendza, K. Leßmann and W. P. Neumann, Synlett, 1993, 283–285 CrossRef CAS.
- A. K. Sawyer and H. G. Kuivila, J. Am. Chem. Soc., 1960, 82, 5958–5959 CrossRef CAS.
- G. Bandoli, D. A. Clemente and C. Panattoni, J. Chem. Soc. D, 1971, 311–312 RSC.
- R. Faggiani, J. P. Johnson, I. D. Brown and T. Birchall, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 3742–3743 CrossRef.
- R. Faggiani, J. P. Johnson, I. D. Brown and T. Birchall, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1979, 35, 1227–1229 CrossRef.
- T. Birchall and J. P. Johnson, Can. J. Chem., 1982, 60, 934–938 CrossRef CAS.
- S. Adams, M. Dräger and B. Mathiasch, J. Organomet. Chem., 1987, 326, 173–186 CrossRef CAS.
- K. Jurkschat, A. Tzschach, C. Mügge, J. Piret-Meunier, M. Van Meerssche, G. Van Binst, C. Wynants, M. Gielen and R. Willem, Organometallics, 1988, 7, 593–603 CrossRef CAS.
- L. Rupnicki, Z. Urbanczyk-Lipkowska, A. Stepien, P. Cmoch, Z. Pianowski and K. Stalinski, J. Organomet. Chem., 2005, 690, 3690–3696 CrossRef CAS.
- J. Turek, Z. Padělková, Z. Černošek, M. Erben, A. Lyčka, M. S. Nechaev, I. Císařová and A. Růžička, J. Organomet. Chem., 2009, 694, 3000–3007 CrossRef CAS.
- J. Turek, Z. Padělková, M. S. Nechaev and A. Růžička, J. Organomet. Chem., 2010, 695, 1843–1847 CrossRef CAS.
- M. Henn, V. Deáky, S. Krabbe, M. Schürmann, M. H. Prosenc, S. Herres-Pawlis, B. Mahieu and K. Jurkschat, Z. Anorg. Allg. Chem., 2011, 637, 211–223 CAS.
- K.-y. Akiba, Y. Ito, F. Kondo, N. Ohashi, A. Sakaguchi, S. Kojima and Y. Yamamoto, Chem. Lett., 1992, 21, 1563–1566 CrossRef.
- S. Kojima, F. Kondoh, J.-Y. Kawakami, Y. Yamamoto and K.-y. Akiba, Heteroat. Chem., 2001, 12, 424–430 CrossRef CAS.
- Y. Yamamoto, A. Sakaguchi, N. Ohashi and K.-y. Akiba, J. Organomet. Chem., 1996, 506, 259–263 CrossRef CAS.
- M. Saito, S. Imaizumi, T. Tajima, K. Ishimura and S. Nagase, J. Am. Chem. Soc., 2007, 129, 10974–10975 CrossRef CAS PubMed.
- N. Kano, H. Miyake, K. Sasaki, T. Kawashima, N. Mizorogi and S. Nagase, Nat. Chem., 2010, 2, 112–116 CrossRef CAS PubMed.
- N. Kano, K. Sasaki, H. Miyake and T. Kawashima, Organometallics, 2014, 33, 2358–2362 CrossRef CAS.
- N. Kano, S. Tsukada, Y. Shibata, T. Kawashima, H. Sato, J.-D. Guo and S. Nagase, Organometallics, 2015, 34, 56–62 CrossRef CAS.
- E. F. Perozzi, R. S. Michalak, G. D. Figuly, W. H. Stevenson, D. Dess, M. R. Ross and J. C. Martin, J. Org. Chem., 1981, 46, 1049–1053 CrossRef CAS.
- H. Preut, H.-J. Haupt and F. Huber, Z. Anorg. Allg. Chem., 1973, 396, 81–89 CrossRef CAS.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- T. N. Mitchell and G. Walter, J. Chem. Soc., Perkin Trans. 2, 1977, 1842–1847 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision E. 01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS; (c) J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244–13249 CrossRef.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Huzinaga, J. Andzelm, M. Klobukowski, E. Radzio-Andzerm, Y. Sakai and H. Tatewaki, GAUSSIAN Basis Sets for Molecular Calculations, Elsevier, Amsterdam, 1984 Search PubMed.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem. A, 2001, 105, 8111–8116 CrossRef CAS.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, Version 3.1 Search PubMed.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectral charts of 7 and 9, summary of the optimized structures, and Cartesian coordinates of the dianion part of 7. CCDC 1501994. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03503e |
| This journal is © The Royal Society of Chemistry 2016 |