
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic routes to iron chalcogenide nanoparticles and thin films
Peter D.
Matthews†
 a,
Masood
Akhtar†
a,
M. Azad
Malik
b,
Neerish
Revaprasadu
c and
Paul
O'Brien
*ab
a,
Masood
Akhtar†
a,
M. Azad
Malik
b,
Neerish
Revaprasadu
c and
Paul
O'Brien
*ab
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
bSchool of Materials, University of Manchester, Oxford Road, Manchester, M13 9Pl, UK. E-mail: paul.o'brien@manchester.ac.uk
cDepartment of Chemistry, University of Zululand, Private Bag X1001, Kwadlangezwa, 3886 South Africa
First published on 14th November 2016
Abstract
Iron chalcogenides are earth abundant, cheap and environmentally benign materials that have seen extensive research directed toward a range of applications, most notably for photovoltaics. The most common forms of materials for these applications are either nanoparticles or thin films. This perspective seeks to summarise the key synthetic routes to these materials by highlighting the key aspects that lead to control over phase and morphology.
 Peter D. Matthews | Peter D. Matthews studied for his MSci at the University of Cambridge (2008–2012) and completed his PhD (2012–2016) there with Prof. Dominic S. Wright and Prof. Ali Alavi FRS, working on polyoxotitanate cages and heteroatom doped graphites. He has moved to the University of Manchester to take up an EPSRC Doctoral Prize with Prof. Paul O'Brien FRS. |
 Masood Akhtar | Masood Akhtar completed his PhD at the University of Manchester in 2013 with Prof. Paul O'Brien FRS. He is a post-doctoral fellow at the University of Zululand and Visiting Researcher at the University of Manchester (2014–). He obtained his M.Sc. degree from the University of Azad Jammu and Kashmir and served as a lecturer in chemistry in the education department of Azad Kashmir. His research interest is the deposition of thin films and synthesis of nanoparticles for various applications. |
 M. Azad Malik | Mohammad Azad Malik completed his PhD at the University of London in 1990 and since then has been working with Prof. Paul O'Brien initially at Queen Mary University of London (1990–1995), then Imperial College (1995–2000), and currently the University of Manchester (2000–). He has been involved in various projects and has a wide range of experience in single-source molecular precursors for II/VI, III/V, III/VI, and IV/VI semiconductors, and the colloidal synthesis of nanoparticles. |
 Neerish Revaprasadu | Neerish Revaprasadu is a professor of Chemistry and SARChI Chair holder in Nanotechnology at the University of Zululand, South Africa. He obtained his BSc(Hons.) from the University of Natal in 1993 and PhD from Imperial College in 2000. He started as a Senior Lecturer at the University of Zululand in 2000, was promoted to associate professor in 2004 and full professor in 2008. His research interest is the synthesis and processing of semiconductor nanomaterials. He was elected Member of South African Academy of Science (ASSAF) in 2014. |
 Paul O'Brien | Paul O'Brien FRS FREng CBE is Chair of Inorganic Materials Chemistry in the Schools of Chemistry and Materials at the University of Manchester; he was Research Dean 2000–03, Head of the School of Chemistry 2002–09 and Head of the School of Materials 2011–15. He has held academic positions at Chelsea, Queen Mary and Imperial Colleges in the University of London. He was awarded the Kroll, Sir Colin Humphreys and Platinum Medals of the IoM3 and the 1st Peter Day Award and the Longstaff prize from the RSC. His research centres on developing new chemical processes for thin films and nanoparticles; especially of chalcogenides. In May 2013 he was elected a Fellow of the Royal Society London. In 2016 he was awarded a CBE and elected as a Fellow of the Royal Academy of Engineering. |
1. Introduction
Iron chalcogenides are earth abundant, cheap and environmentally benign materials that have seen extensive research across a range of applications. These include hydrogen evolution, photovoltaics, Li-ion batteries, high temperature superconductors, supercapacitors and memory devices.1–7 For these applications nanoparticles and thin films offer a large degree of flexibility as the size/thickness and morphology can be tuned during their formation. Unlike iron oxide nanoparticles and thin films, which have long been studied, the chalcogenide counterparts have historically received less attention, though this has changed in recent years.The most studied applications for iron chalcogenides are as photovoltaics or supercapacitors, with considerable research directed towards the magnetic properties of these materials. Iron chalcogenides have the potential to act as a photoabsorber layer within a photovoltaic device; this requires a very precisely defined morphology in order to maximise current flow whilst minimising hole-electron recombination at defect sites. Only pyrite (FeS2) demonstrates photoactivity, and contamination with secondary phases is prone to reduce the efficiency of devices.
Magnetic nanocrystals have a broad remit of applications, ranging from use as contrast agents in magnetic resonance imaging (MRI)8,9 to magnetic data storage10 and even paleomagnetism.11 Thus the variation of magnetic behaviour with particle morphology and size is an important area of study.
Iron sulfide has seven phases, whilst iron selenide and iron telluride both have three, which makes these systems quite complex. Most applications require a high degree of phase purity – secondary phases can hinder or reduce the efficiency of a device. Thus it is important that synthetic routes demonstrate the ability to control the phase of the obtained material, as well as the morphology.
The purpose of this perspective is not to summarise every single reaction in the literature, but to highlight the important aspects of those that lead to phase and/or shape/size control. It is through careful control of these variables that iron chalcogenides will be able to fulfil their exciting potential for applications.
2. Iron sulfide
There are seven major phases of iron sulfide, which indicates the complexity of the system. The phases are: iron sulfide (FeS), greigite (Fe3S4), pyrrhotite (Fe1−xS), troilite (FeS) mackinawite (Fe1+xS), marcasite (orthorhombic FeS2) and pyrite (cubic FeS2) and these are shown in Fig. 1.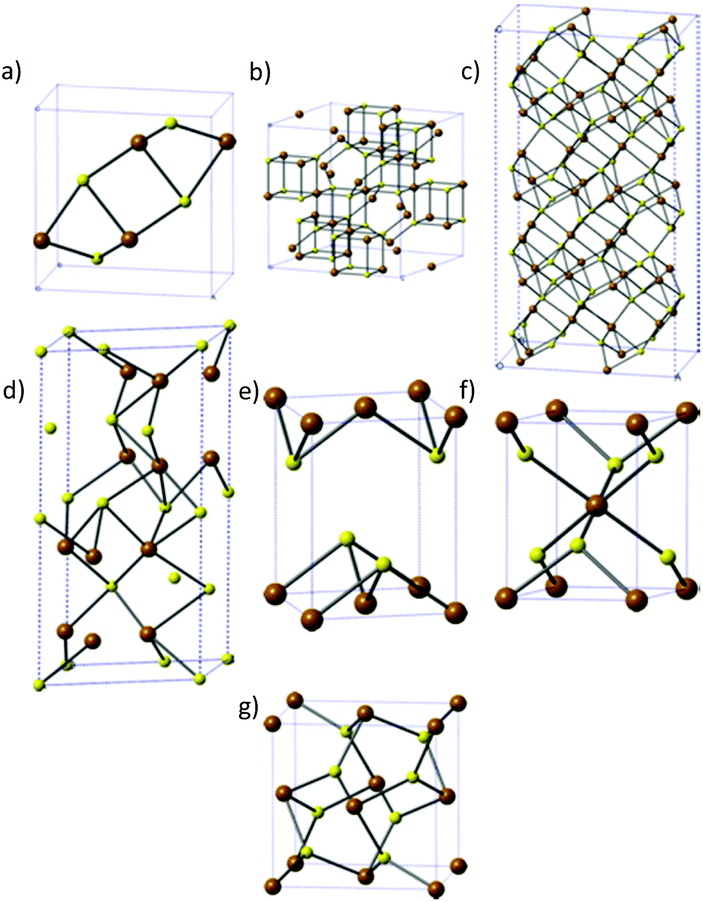 | ||
| Fig. 1 The different phases of iron sulfide. (a) FeS, (b) greigite, (c) pyrrhotite, (d) troilite, (e) mackinawite, (f) marcasite and (g) pyrite. Brown = Fe, yellow = S. | ||
Pyrite is the key phase for photovoltaic applications, with an appropriate band gap (0.95 eV), high absorption coefficient (>105 cm−1) a good carrier diffusion length (100–1000 nm) and an extremely high natural abundance.12–14 On the other hand, FeS and pyrrhotite (Fe1−xS) have been proposed as the preferred phases for Li-ion batteries,15 and troilite and greigite are the premier candidates for use in supercapacitors.16 It is apparent that the performance of each device here is phase dependent, and so it is clear that phase control is a clear requirement during the synthesis of iron sulfides.
2.1 Synthesis of nanoparticles
Iron sulfide nanoparticles, like most others have been synthesised via a number of different routes, the two most common of which are the hot-injection method or a solvothermal route.The former use a variety of iron sources in differing oxidation states, including FeCl2,17 [Fe(CO)5],18 [Fe(acac)2]19 or [Fe(acac)3]20 (acac = acectylacetonate). It appears that a degree of shape control, as well as the phase of the nanoparticles can be achieved by the choice of iron source.
FeCl2 favours the formation of pyrite (FeS2), with the iron oxidation state increasing from Fe(II) to Fe(IV), owing to the oxidizing environment created by the sulfur. It should be noted that Kirkeminde et al. successfully made FeS nanowires using FeCl2,21 though the majority of syntheses resulted in pyrite. Li et al., found that they could control both the size and shape of their products by varying the concentration of FeCl2 in their reaction. Low concentrations of FeCl2 in oleylamine (OA) resulted in the formation of ∼250 nm nanocubes, whilst higher concentrations resulted in the formation of ∼10 nm nanodendrites.22
FeCl2 has also been utilized by Steinhagen17 and Shukla23 in OA to generate cube-shaped nanoparticles, whilst Puthussery et al. found that they were able to make more stable colloidal suspensions by exchanging the OA ligands for octadexylxanthate.24 Macpherson et al. have produced a highly interesting study in which they were able to exert a high degree of shape control with FeCl2 (at the expense of monodispersity) through tuning the chemical potential of sulfur.2 They made use of a three step process: initial nucleation in a sulfur rich environment followed by 2 growth periods in near stoichiometric conditions for FeS2 (Fig. 2). This level of control was driven by theoretical predictions that the {100} face is the lowest energy face in S poor conditions, whilst the {210} and {111} faces are favoured with increasing S concentration.25,26
 | ||
| Fig. 2 TEM images and conditions employed by Macpherson et al.2 during their synthesis of pyrite nanocubes. Reprinted with permission from ref. 2, ©2012 American Chemical Society. | ||
[Fe(CO)5] has been used in conjunction with elemental sulfur in OA to generate hexagonally shaped nanoplates of pyrite,18 though its high toxicity makes it an undesirable reagent for large scale use.
Beal et al. used [Fe(acac)2] to synthesize both greigite (Fe3S4) and pyrrhotite (Fe1−xS) nanoparticles, though these were polydisperse and offered limited shape control.19,27 Other groups have used [Fe(acac)3] which resulted in carbon coated nanosheets of troilite (FeS). They found that the use of 1-dodecanethiol (1-DDT) gave more regular shapes than the usual OA/S mixture.20
The second major hot-injection technique is to use a single-source precursor that features preformed Fe–S bonds. The first examples of this in the iron sulfide field featured the decomposition in OA of [NnBu4]2[Fe4S4(SPh)4]28 and [{Fe(N-MeIm)6}S8] (N-MeIm = N-methylimidazole).29 The former resulted in the formation of pyrrhotite at 180 °C and greigite at 200 °C, demonstrating a good degree of phase control.28 The latter gave a multi-faceted morphology with greigite formed in a burst-nucleation when [Fe(N-MeIm)6]S8 was injected at 300 °C, followed by rapid cooling. The greigite was converted to pyrrhotite if the reaction was not immediately cooled to room temperature.29
Giovanni et al. investigated the use of [Fe2S2(CO)6] as a single source precursor (SSP), the thermolysis of which in OA led to the formation of pyrrhotite nanohexagons.30
A major class of SSPs that have been investigated are iron dialkyldithiocarbamates [Fe(S2CNR2)x] (x = 2 or 3, Fig. 3a) and iron O-alkyxanthates [Fe(S2COR)x] (x = 2 or 3, Fig. 3b). Hexagonal two-dimensional pyrrhotite (Fe1−xS) and greigite (Fe3S4) nanosheets were synthesized by thermolysing [Fe(S2CNEt2)2(phen)] (phen = 1,10-phenanthroline) and [Fe(S2CNEt2)3] respectively, both in OA.31 The influence of the OA as a capping ligand was investigated by the introduction of non-coordinating octadecene (ODE). For [Fe(S2CNEt2)2(phen)] this resulted in less defined, quasi-hexagonal shapes of Fe1−xS, which indicates that the oleylamine ligand controls the growth of the nanosheets along the {100} and {110} faces. Therefore, it is important to note that the choice of solvent system plays a key role in the shape of the obtained nanoparticles.
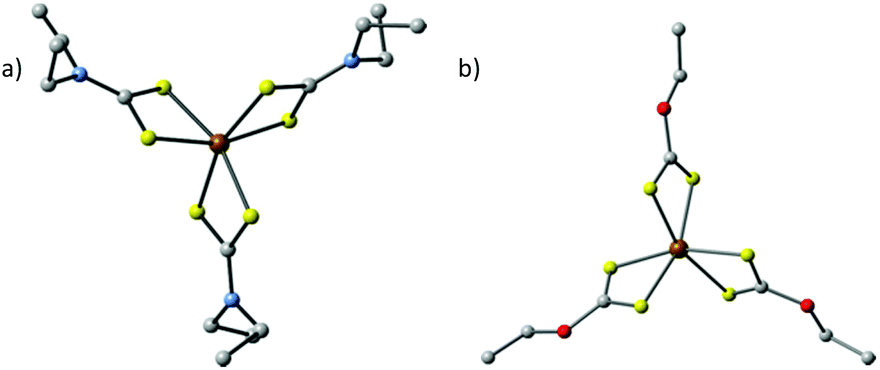 | ||
| Fig. 3 Two of the major classes of single source precursors that have been explored for the synthesis of iron sulfide nanoparticles and thin films. (an) Iron(III) diethyldithiocarbamate [Fe(S2CNEt2)3] and (b) iron(III) O-ethylxanthate [Fe(S2COEt2)3]. In both cases the ethyl group might be exchanged for other alkyl groups. Brown = Fe, yellow = S, grey = C, blue = N and red = O. Hydrogens omitted for clarity. | ||
A range of symmetrical and asymmetrical Fe(III) dithiocarbamates ([Fe(S2CNRR′)3] where R = Et, R′ = iPr; R,R′ = Hex; R = Me, R′ = Et; and R,R′ = Et) were used by Akhtar et al.32 They found that the precursors with symmetrical long chain alkyl groups gave pure greigite phase at lower thermolysis temperature but a mixture of greigite and pyrrhotite at higher temperatures. Symmetrical short chain alkyl groups give the pure greigite (Fe3S4) phase at both 230 and 300 °C. The unsymmetrical alkyl groups gave mixed phase (greigite and pyrrhotite) iron sulfide nanocrystals at all temperatures.
In a similar manner, O'Brien and co-workers made use of a series of tris(O-alkylxanthato)iron(III) complexes [(Fe(S2COR)3), R = Me, Et, and iBu, Fig. 3b] in oleylamine.33 These systems proved to be complex, with the O-methylxanthate giving a mixture of greigite and pyrrhotite. The O-ethylxanthate complexes gave pure greigite at low temperature, but a mixture of greigite, pyrrhotite and pyrite at high temperatures. This behaviour is also exhibited by the O-iso-butylxanthates. These nanocrystals showed random shapes with a wide polydispersity. The size range could be controlled somewhat by choice of solvent: 14–139 nm in length and 12–65 nm in width nanocrystals were synthesized in oleylamine whereas smaller nanocrystals 12–31 nm length 7–26 nm width were obtained from hexadecylamine.
The same group used an Fe(III) complex of 1,1,5,5-tetra-iso-propyl-2-thiobiuret [Fe(SON(CNiPr2)2)3] as a single source precursor for the synthesis of iron sulfide nanoparticles, by thermolysis in hot oleylamine (OA), octadecene (ODE), or 1-dodecanethiol (1-DDT).34 Several combinations of different injection solvents and capping agents were used in the reaction mixture to control the shape and the phase of the material. The thermolysis of the iron complex in OA or OA/1-DDT produced crystalline Fe7S8 nanoparticles with different morphologies (spherical, hexagonal plates and nanowires) depending on the growth temperature and precursor concentration. This system is more susceptible to solvent change than others, with the introduction of ODE resulting in an amorphous material.34
Kar,35,36 Nath37 and Xuefeng38 all reported the synthesis of nanowires from various iron salts and sulfur sources, with the constant being ethylenediamine as the solvent. Kar et al. found that the solvothermal reaction of [Fe(NO3)3·9H2O], [FeSO4·7H2O] or FeCl3 with thiourea or Na2S resulted in the formation of pyrite nanowires, though the Na2S reactions gave substoichiometric FeS2−x.35,36 Xuefeng et al. carried out a similar process with [FeSO4·7H2O] and Na2S3 as the precursors. They found a solvent-based morphology dependency that resulted in pyrite nanowires in ethylenediamine but pyrite nanoparticles in benzene (Fig. 4).38 This indicates that the solvent can be chosen to target the desired the morphology.
 | ||
| Fig. 4 The solvothermal reaction of FeSO4·7H2O and Na2S3 gives nanorods in ethylenediamine and nanoparticles in benzene, demonstrating the solvent dependency of the morphology. Reprinted with permission from ref. 38, ©2001 Elsevier. | ||
Nath et al. reacted [FeCl2·4H2O] with thioacetamide in ethylenediamine to generate a slurry that was annealed in an argon atmosphere to give Fe7S8 nanowires at 200 °C and Fe1−xS at 300 °C.
Cao39 and Zhang40 both successfully synthesized Fe3S4 nanoparticles, the former using FeSO4 and L-cysteine in water,39 whilst the latter used [FeCl3·6H2O] with thiourea in an ethyleneglycol–water mixture.40
Finally, Chen41 and Wadia42 both used a SSP in the form of iron tris-diethyldithiocarbamate [Fe(S2CNEt2)3] in water for Chen, resulting in pyrite nanocubes.41 Wadia on the other hand used iron tris-diethyldithiophosphate [Fe(S2P(OEt)3)] in hexadecyltrimethylammonium bromide to generate pyrite nanocubes.42
Morrish and co-workers also made use of Fe2O3 nanorods which they converted to FeS2 through plasma assisted sulfurization. For this preparation, nanorods of Fe2O3 (∼150 nm sized) were prepared by chemical bath deposition method using FeCl3 and NaNO3 on FTO glass plates. The Fe2O3 nanorods were converted to FeS2 by sulfurization using a mixture of 10% H2S: 90% Ar gas. Iron sulfide prepared by this method contained both marcasite and pyrite phases, which was confirmed by Raman spectroscopy measurement. The prolonged sulfurization of Fe2O3 nanorods increased the percentage of pyrite without completely eradicating the marcasite phase.45
Bauer et al. produce greigite nanorods through the vapour–solid interaction of Fe vapour and ZnS solid in an ultra-high vacuum environment.46
2.2 Synthesis of thin films
Iron sulfide thin films have been deposited by a number of methods, which includes the sintering of iron sulfide nanoparticle inks,24 sulfurization of iron oxides to FeS2,47 ion beam and reactive sputtering (FeS2),48 sulfurization of iron (FeS2),49,50 flash evaporation (FeS2),51 vacuum thermal evaporation (FeS2),52 vapour transport (FeS2),53 chemical spray pyrolysis (FeS2),14 high-energy mechanical milling combined with mechanochemical processing for FeS and FeS2,54 sulfur-reducing bacteria for Fe1−xS and Fe3S4,55,56 the decomposition of single-source precursors for FeS2,57 and other atmospheric- or low-pressure metal–organic chemical vapour deposition (AP- or LP-MOCVD) methods.58–60The two most commonly used types of CVD in this area are low-pressure (LP-) and aerosol assisted (AA-). Low pressure can improve film uniformity, whilst AA-CVD involves the formation of aerosols, allowing the use of less-volatile precursors. These two methods can be further broken down into multi-component precursor solutions and single-source precursors.
LP-CVD is more amenable to multi-component systems than single-source precursors, with Schleich60 and Thomas58 reporting the use of [Fe(CO)5] and tert-butyl disulfide to generate pyrite thin films. Schleich et al. also noted that it as possible to kinetically trap marcasite at lower temperatures (200 °C) in their system.60 Chi and co-workers used a related precursor [Fe2(CO)6S2] to make a mixture of Fe1−xS and Fe7S8 at 300 °C and FeS at 600 °C.66
In 2000 O'Brien et al. discovered that their iron(III) dithiocarbamates would not produce films under LP-conditions. However, they generated pyrite thin films via aerosol assisted- (AA-) CVD using [Fe(S2CNRR′)3] (R = Me, R′ = iPr; R,R′ = nBu).57 Takahashi used [FeCl3] and thioacetamide in an atmospheric pressure CVD apparatus to make pyrite at 500 °C,67 though this route has not been widely adopted.
Ramasamy used the iron thioburets [Fe{SON(CNR2)2}3] (R = iPr, Et, Me) that were successful in the synthesis of nanoparticles34 in an AA-CVD reaction to give an interesting mixture of films. The iPr complex gave hexagonal troilite FeS films with a small amount of tetragonal pyrrhotites at 300 °C, whereas only troilite was deposited above 350 °C. The ethyl compounds deposited a mixture of hexagonal troilite and cubic pyrite films at all temperatures (Fig. 5), whereas the methyl complexes produced very thin films of troilite at all temperatures.68
 | ||
| Fig. 5 SEM images of troilite thin films produced from the aerosol assisted chemical vapour deposition of [Fe{SON(CNEt2)2}3] at (a) 300 °C, (b) 350 °C, (c) 400 °C (inset 45° tilt image of film) and (d) 450 °C. Reprinted with permission from ref. 68, ©2010 American Chemical Society. | ||
The idea of using Fe(III) dithiocarbamates has been further expanded upon by Akhtar,69 Khalid70 and Mlowe71 to encompass short- and long-chain, asymmetrical and cyclic amine groups with mixed success. The asymmetrical groups gave mixed phase pyrite/marcasite films, whilst the use of dihexyldithiocarbamates led to a mixture of pyrite and pyrrhotite. Shorter chain, diethyldithiocarbamates on the other hand gave mixed pyrite/marcasite films, but at temperatures above 400 °C this turned into pure pyrrhotite.69 Khalid et al. used the same diethyldithiocarbamate complex as Akhtar, but exchanged the solvent for THF instead of toluene, resulting in the formation of clean pyrite films and thus indicating the importance of solvent choice during AA-CVD reactions.70 The use of heterocyclic amines in the form of tris-(piperidinedithiocarbamato)iron(III) and tris-(tetrahydroquinolinedithiocarbamato)iron(III) was trialled by Mlowe et al., but this appears to offer no significant advantage over the simpler systems, only resulting in the formation of a complex, mixed phase film.71
3. Iron selenide
Fewer phases of iron selenide are known than its sulfide counterpart, with three different phases: a tetragonal phase α-FeSe with PbO-structure (Fig. 6a), a NiAs-type β-phase (achavalite, hexagonal Fe7Se8 and monoclinic Fe3Se4, Fig. 6b) and a FeSe2 phase that has the orthorhombic marcasite structure (ferroselite, Fig. 6c). The hexagonal Fe7Se8 and monoclinic Fe3Se4 phases have attracted the most interest owing to their favourable magnetic properties.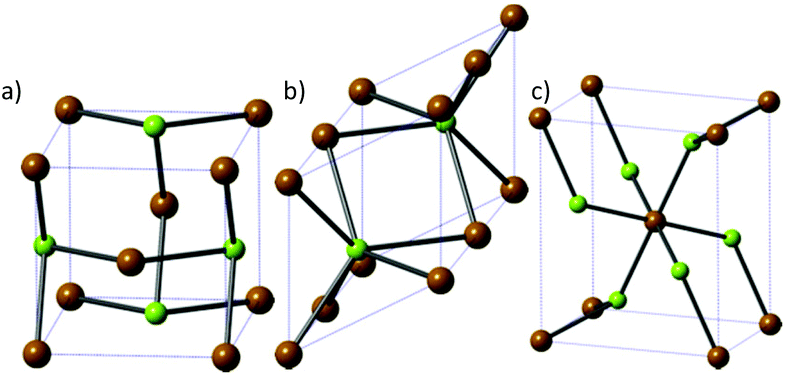 | ||
| Fig. 6 The three major phases of iron selenide: (a) α-FeSe, (b) achavalite and (c) ferroselite (FeSe2). Brown = Fe, green = Se. | ||
Iron selenide has garnered a lot of attention, due to its semiconductor, photoabsorption, and magnetic properties. It is a prime candidate for photovoltaics with a band gap of ∼1 eV and an absorption coefficient >105 cm−1.72–74 Iron selenide has also been shown to demonstrate high temperature superconductivity, which is a very exciting result.75,76
3.1 Synthesis of nanoparticles
Iron selenide nanoparticles have been synthesised by a number of different routes. Amongst these, the ubiquitous hot-injection method takes precedence. Chen et al. made PbO-type nanoflakes from the simple reaction of FeCl2 in a mixture of oleylamine (OA), oleic acid and trioctylphosphine selenide (TOPSe).77 TOPSe (and the corresponding telluride, TOPTe) are often described as a mixture of the elemental chalcogen in TOP (or other phosphine), though there is no ‘free’ chalcogen in the final solution. Instead, the phosphine is oxidised to the corresponding chalcogenide, though for tellurium there is an equilibrium between tellurium and the phosphine telluride.78 Zhang et al. also made use of a mixed precursor system, reacting [Fe(acac)3] and Se powder in OA generating ‘nanocacti’. Interestingly, they found that they could change the particles morphology to nanosheets by adding oleic acid into the reaction mixture (Fig. 7).79 | ||
| Fig. 7 SEM images of Fe3Se4 (a) ‘nanocacti’, (b) nanosheets, and (c) nanoplatelets synthesized by Zhang et al.,79 with the morphology dictated by the amount of oleic acid present in the reaction. Reprinted with permission from ref. 79, ©2011 American Chemical Society. | ||
Akhtar et al. decomposed the Fe(III) single source precursors tris(N,N-diethyl-N′-naphthoylselenoureato)iron(III) [Fe(napC-(O)NC(Se)NEt2)3] (nap = napthyl, Fig. 8a) in oleylamine (OA) at 190, 240 and 290 °C to make mixed phase iron selenide nanoparticles.80 The same authors found that they could make pure phase FeSe2 by switching to the Fe(II) complexes bis(tetraalklyldiselenoimidodiphosphinato)iron(II) where the alkyl is either an iso-propyl or a phenyl ([Fe{(SePR2)2N}2] R = iPr, Ph, Fig. 8b). Decomposition of these SSPs in OA resulted in plate-like crystallites of ferroselite (FeSe2).81
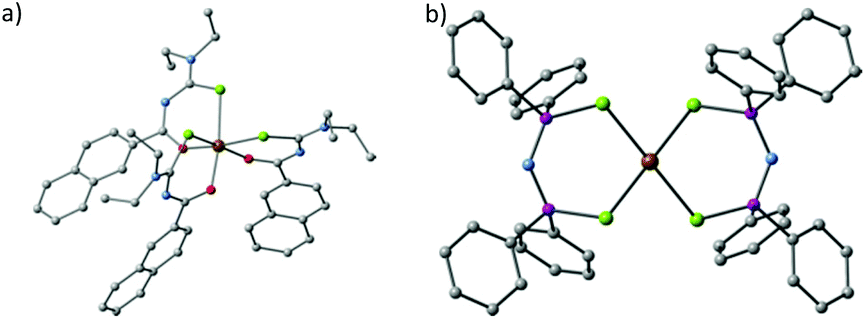 | ||
| Fig. 8 The crystal structures of (a) tris(N,N-diethyl-N′-naphthoylselenoureato)iron(III) [Fe(napC-(O)NC(Se)NEt2)3] (nap = napthyl) and (b) bis(tetraphenyldiselenoimidodiphosphinato)iron(II) [Fe{(SePPh2)2N}2], which have been used by Akhtar et al. to generate iron selenide nanoparticles and thin films.80,81 | ||
Iron selenide nanoparticles have also been synthesised by a variety of other routes. These include mechanochemical ball milling of Fe and Se powders, and though this method might be beautifully simple, it resulted in a mixture of FeSe2, FeSe, Fe7Se8 and Fe3O4.82
Liu et al. synthesised FeSe2 nanorods by the hydrothermal co-reduction method using hydrazine as the reductant. An aqueous solution of [FeCl3·6H2O], [Na2SeO3], in distilled water was heated in Teflon-lined stainless steel autoclave to 140 °C for 12 hours. After cooling to room temperature the black product was filtered off, dried and revealed to be the orthorhombic phase of FeSe2. The reaction was found to be dependent on the concentration of hydrazine, with the reaction only producing pure phase FeSe2 in 1.5 M aqueous hydrazine.83
PbO-type nanocrystals of FeSe have also been synthesized by the solid state reaction of Fe and Se. The elements were ground, cold-pressed into discs and heated to 700 °C under static vacuum. The samples were then reground at room temperature before being sintered again at 700 °C and then annealed at 400 °C.76 This method resulted in phase pure material, but represents poor potential for scalability, hence the interest in solution-based processing.
3.2 Synthesis of thin films
There are very few examples of iron selenide thin films, with the majority of synthetic routes focussing on CVD74,80,81,84,85 though more novel routes such as electrolytic bath deposition86 and pulsed laser deposition87 have also been explored.Wu et al. generated clean FeSe films from the toxic low-pressure- (LP-) CVD reaction of [Fe(CO)5] and H2Se.74,84 This process generated clean FeSe films that demonstrated good electrical properties, but both precursors are not the safest to use, and so interest in other routes to FeSe films remains.
Akhtar and co-workers used the Fe(III) selenoureato and the Fe(II) selenoimidophosphines (Fig. 8) that they used for iron selenide nanoparticle synthesis to deposit thin films through an AA-CVD process. The selenoimidophosphine precursors decomposed to form a mixture of Fe7Se8 and FeSe2,81 whilst the selenoureato gave FeSe thin films, but only at the relatively high temperature of 625 °C.80
A more complex precursor was chosen by Hussain et al.: 1-acetyl-3-(4-ferrocenylphenyl) selenourea, a substituted ferrocene derivative. This compound was dissolved in toluene and used in an AA-CVD process, but resulted in a very complicated mixture of different phases, indicating that simpler compounds with an easier decomposition route might be more appropriate.85
Chemical bath deposition is a process that has received considerable attention for materials such as zinc oxide, zinc sulfide and cadmium sulfide,88–91 but little research has focused on its suitability for iron sulfide deposition. Thanikaikarasan et al. have carried out aqueous electrolytic bath depositions using FeSO4 and SeO2, which resulted in the formation of FeSe films.86 One major advantage of this technique is that the average thickness of the deposited layers can be controlled through the applied plating current and the deposition time.
4. Iron telluride
Iron telluride is the least studied of the iron chalcogenides. There are three iron telluride structures: NiAs-type hexagonal FeTe (Fig. 9a), tetragonal Fe1.125Te (Fig. 9b) and orthorhombic frohbergite (FeTe2, Fig. 9c).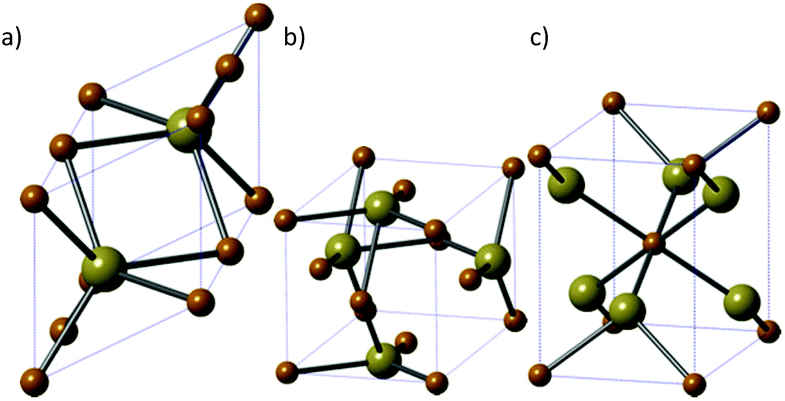 | ||
| Fig. 9 The three major phases of iron telluride. (a) NiAs-type hexagonal FeTe, (b) tetragonal Fe1.125Te and (c) orthorhombic frohbergite (FeTe2). Brown = Fe, beige = Te. | ||
Research in iron telluride has focussed on its potential to be a high temperature superconductor and its magnetic properties.92 There are therefore comparatively few examples of the synthesis of nanoparticles or thin films of this material.
Iron telluride has most often been prepared directly by mixing the elements in sealed tubes at high temperatures and high pressures.93–95 More recently, new synthetic methods and metalorganic chemical vapour deposition (MOCVD) and pulsed laser deposition routes have been used for the synthesis of iron telluride.96,97
4.1 Synthesis of nanoparticles
Zhang and co-workers reported an aqueous route to prepare nanocrystalline orthorhombic FeTe2 through a reaction between an aqueous alkaline of Te powder and KOH, and [Na2{Fe(EDTA)}]. An aqueous solution of tellurium was used to avoid handling H2Te and K2Te2.98Liu et al. extended their hydrothermal reduction synthesis of FeSe2 nanocrystals to include FeTe2, through the reaction of [FeCl3·6H2O] and [Na2TeO3] using hydrazine as the reducing agent.83
Another aqueous route by Roy et al. soaked Te nanorods, synthesized from the reduction of TeO2 by hydrazine,99 with FeCl3, to result in FeTe nanorods through a galvanic reaction. They discovered an interesting application in the FeTe rod's ability to detect glucose.100
Oyler and others used the traditional hot-injection route to make iron telluride nanoparticles from hexadecylamine (HDA), trioctylphosphine oxide (TOPO), trioctylphosphine telluride (TOPTe) and [Fe(CO)5]. The Fe and Te ratio should be 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to form pure FeTe and for FeTe2 a larger amount of Te is required. FeTe products are two-dimensional single crystals nanosheets with thickness of 2–3 nm and edge length ranging from 200 nm to several micrometres. FeTe2 formed as a mixture of nanosheets and one-dimensional sheet-derived nanostructures.73 This method reveals a strong ability to control the obtained phase of iron telluride, and so represents a good step forward in this field.
:1 to form pure FeTe and for FeTe2 a larger amount of Te is required. FeTe products are two-dimensional single crystals nanosheets with thickness of 2–3 nm and edge length ranging from 200 nm to several micrometres. FeTe2 formed as a mixture of nanosheets and one-dimensional sheet-derived nanostructures.73 This method reveals a strong ability to control the obtained phase of iron telluride, and so represents a good step forward in this field.
4.2 Synthesis of thin films
There are not many examples of iron telluride thin film synthesis, but Bochmann reported the synthesis of the iron–tellurium complex [Fe{tBu2P(Te)NR}2] (R = iPr, cyclohexyl) which they used for the gas-phase deposition of FeTe2 films.96,101 Additionally, Steigerwald has demonstrated a LP-CVD route to iron telluride thin films via the decomposition of [{Cp(Et3P)(CO)Fe}2Te] and [{Cp(Et3P)(CO)FeTe}2]. The former gave films of pure FeTe, whilst the latter gave pure films of FeTe2, demonstrating another great degree of control.102There are a couple of examples of iron telluride cages that, like the iron sulfide cubane clusters,28 might make good options for the formation of iron telluride nanocrystals/thin films. For example, Steigerwald has reported the synthesis of [(Et3P)4Fe4Te4],103 and Roof has also synthesised both [(Ph4P)2{Fe5Te4(CO)14}] and [(Ph4P)2{Fe8Te10(CO)20}].104 All three of these compounds have an Fe–Te core and so resembles an interesting target for future research.
5. Conclusions
This short perspective has sought to summarise the key synthetic routes to a relatively unexplored class of compounds. Iron sulfide represents a good candidate for thin film photovoltaics, and the synthetic routes to such a promising material must be improved if it is to be commercialised. The other iron chalcogenides, the selenides and particularly the tellurides, have received very little attention and the door remains wide open for interesting and novel research in this area.Acknowledgements
The authors thank the EPSRC (Doctoral Prize for P. D. M.) and the Royal Society DFID Africa Capacity Building Initiative for financial support.Notes and references
- D. Jasion, J. M. Barforoush, Q. Qiao, Y. Zhu, S. Ren and K. C. Leonard, ACS Catal., 2015, 5, 6653–6657 CrossRef CAS.
- H. A. Macpherson and C. R. Stoldt, ACS Nano, 2012, 6, 8940–8949 CrossRef CAS PubMed.
- J. W. Choi, G. Cheruvally, H. J. Ahn, K. W. Kim and J. H. Ahn, J. Power Sources, 2006, 163, 158–165 CrossRef CAS.
- Z. P. Yin, K. Haule and G. Kotliar, Nat. Mater., 2011, 10, 932–935 CrossRef CAS PubMed.
- M.-R. Gao, Y.-F. Xu, J. Jiang and S.-H. Yu, Chem. Soc. Rev., 2013, 42, 2986–3017 RSC.
- Y.-X. Wang, J. Yang, S.-L. Chou, H. K. Liu, W. Zhang, D. Zhao and S. X. Dou, Nat. Commun., 2015, 6, 8689 CrossRef CAS PubMed.
- M. Barawi, I. J. Ferrer, E. Flores, S. Yoda, J. R. Ares and C. Sánchez, J. Phys. Chem. C, 2016, 120, 9547–9552 CAS.
- D. A. J. Herman, P. Ferguson, S. Cheong, I. F. Hermans, B. J. Ruck, K. M. Allan, S. Prabakar, J. L. Spencer, C. D. Lendrum and R. D. Tilley, Chem. Commun., 2011, 47, 9221–9223 RSC.
- S. Cheong, P. Ferguson, K. W. Feindel, I. F. Hermans, P. T. Callaghan, C. Meyer, A. Slocombe, C. H. Su, F. Y. Cheng, C. S. Yeh, B. Ingham, M. F. Toney and R. D. Tilley, Angew. Chem., Int. Ed., 2011, 50, 4206–4209 CrossRef CAS PubMed.
- S. Sun, C. B. Murray, D. Weller, L. Folks and A. Moser, Science, 2000, 287, 1989–1992 CrossRef CAS PubMed.
- M. J. Dekkers, H. F. Passier and M. A. A. Schoonen, Geophys. J. Int., 2000, 141, 809–819 CrossRef.
- P. P. Altermatt, T. Kiesewetter, K. Ellmer and H. Tributsch, Sol. Energy Mater. Sol. Cells, 2002, 71, 181–195 CrossRef CAS.
- A. Ennaoui and H. Tributsch, Sol. Energy Mater., 1986, 14, 461–474 CrossRef CAS.
- G. Smestad, A. Da Silva, H. Tributsch, S. Fiechter, M. Kunst, N. Meziani and M. Birkholz, Sol. Energy Mater., 1989, 18, 299–313 CrossRef CAS.
- B. Wu, H. Song, J. Zhou and X. Chen, Chem. Commun., 2011, 47, 8653–8655 RSC.
- X. Rui, H. Tan and Q. Yan, Nanoscale, 2014, 6, 9889–9924 RSC.
- C. Steinhagen, T. B. Harvey, C. J. Stolle, J. Harris and B. A. Korgel, J. Phys. Chem. Lett., 2012, 3, 2352–2356 CrossRef CAS PubMed.
- A. Kirkeminde, B. A. Ruzicka, R. Wang, S. Puna, H. Zhao and S. Ren, ACS Appl. Mater. Interfaces, 2012, 4, 1174–1177 CAS.
- J. H. L. Beal, S. Prabakar, N. Gaston, G. B. Teh, P. G. Etchegoin, G. Williams and R. D. Tilley, Chem. Mater., 2011, 23, 2514–2517 CrossRef CAS.
- C. Xu, Y. Zeng, X. Rui, N. Xiao, J. Zhu, W. Zhang, J. Chen, W. Liu, H. Tan, H. H. Hng and Q. Yan, ACS Nano, 2012, 6, 4713–4721 CrossRef CAS PubMed.
- A. Kirkeminde, P. Gingrich, M. Gong, H. Cui and S. Ren, Nanotechnology, 2014, 25, 205603 CrossRef PubMed.
- W. Li, M. Doblinger, A. Vaneski, A. L. Rogach, F. Jackel, J. Feldmann, M. Döblinger, A. Vaneski, A. L. Rogach, F. Jäckel and J. Feldmann, J. Mater. Chem., 2011, 21, 17946–17952 RSC.
- S. Shukla, G. Xing, H. Ge, R. R. Prabhakar, S. Mathew, Z. Su, V. Nalla, T. Venkatesan, N. Mathews, T. Sritharan, T. C. Sum and Q. Xiong, ACS Nano, 2016, 10, 4431–4440 CrossRef CAS PubMed.
- J. Puthussery, S. Seefeld, N. Berry, M. Gibbs and M. Law, J. Am. Chem. Soc., 2011, 133, 716–719 CrossRef CAS PubMed.
- A. S. Barnard and S. P. Russo, J. Mater. Chem., 2009, 19, 3389–3394 RSC.
- D. R. Alfonso, J. Phys. Chem. C, 2010, 114, 8971–8980 CAS.
- J. H. L. Beal, P. G. Etchegoin and R. D. Tilley, J. Solid State Chem., 2012, 189, 57–62 CrossRef CAS.
- P. V. Vanitha and P. O'Brien, J. Am. Chem. Soc., 2008, 130, 17256–17257 CrossRef CAS PubMed.
- J. H. L. Beal, P. G. Etchegoin and R. D. Tilley, J. Phys. Chem. C, 2010, 114, 3817–3821 CAS.
- C. Di Giovanni, W.-A. Wang, S. Nowak, J.-M. Grenèche, H. Lecoq, L. Mouton, M. Giraud and C. Tard, ACS Catal., 2014, 4, 681–687 CrossRef CAS.
- W. Han and M. Gao, Cryst. Growth Des., 2008, 8, 1023–1030 CAS.
- M. Akhtar, J. Akhter, M. A. Malik, P. O'Brien, F. Tuna, J. Raftery and M. Helliwell, J. Mater. Chem., 2011, 21, 9737–9745 RSC.
- M. Akhtar, M. A. Malik, F. Tuna and P. O'Brien, J. Mater. Chem. A, 2013, 1, 8766–8774 CAS.
- A. L. Abdelhady, M. A. Malik, P. O'Brien and F. Tuna, J. Phys. Chem. C, 2012, 116, 2253–2259 CAS.
- S. Kar and S. Chaudhuri, Mater. Lett., 2005, 59, 289–292 CrossRef CAS.
- S. Kar and S. Chaudhuri, Chem. Phys. Lett., 2004, 398, 22–26 CrossRef CAS.
- M. Nath, A. Choudhury, A. Kundu and C. N. R. Rao, Adv. Mater., 2003, 15, 2098–2101 CrossRef CAS.
- Q. Xuefeng, X. Yi and Q. Yitai, Mater. Lett., 2001, 48, 109–111 CrossRef CAS.
- F. Cao, W. Hu, L. Zhou, W. Shi, S. Song, Y. Lei, S. Wang and H. Zhang, Dalton Trans., 2009, 9246–9252 RSC.
- Z. J. Zhang and X. Y. Chen, J. Alloys Compd., 2009, 488, 339–345 CrossRef CAS.
- X. Chen, Z. Wang, X. Wang, J. Wan, J. Liu and Y. Qian, Inorg. Chem., 2005, 44, 951–954 CrossRef CAS PubMed.
- C. Wadia, Y. Wu, S. Gul, S. K. Volkman, J. Guo and A. P. Alivisatos, Chem. Mater., 2009, 21, 2568–2570 CrossRef CAS.
- M. Cabán-Acevedo, M. S. Faber, Y. Tan, R. J. Hamers and S. Jin, Nano Lett., 2012, 12, 1977–1982 CrossRef PubMed.
- D. R. Cummins, H. B. Russell, J. B. Jasinski, M. Menon and M. K. Sunkara, Nano Lett., 2013, 13, 2423–2430 CrossRef CAS PubMed.
- R. Morrish, R. Silverstein and C. A. Wolden, J. Am. Chem. Soc., 2012, 134, 17854–17857 CrossRef CAS PubMed.
- E. Bauer, K. L. Man, A. Pavlovska, A. Locatelli, T. O. Mentes, M. A. Nino and M. S. Altman, J. Mater. Chem. A, 2014, 2, 1903–1913 CAS.
- B. Ouertani, J. Ouerfelli, M. Saadoun, B. Bessaïs, H. Ezzaouia and J. C. Bernède, Mater. Charact., 2005, 54, 431–437 CrossRef CAS.
- M. Birkholz, D. Lichtenberger, C. Höpfner and S. Fiechter, Sol. Energy Mater. Sol. Cells, 1992, 27, 243–251 CrossRef CAS.
- S. Bausch, B. Sailer, H. Keppner, G. Willeke, E. Bucher and G. Frommeyer, Appl. Phys. Lett., 1990, 57, 25–27 CrossRef CAS.
- Y. Hu, Z. Zheng, H. Jia, Y. Tang and L. Zhang, J. Phys. Chem. C, 2008, 112, 13037–13042 CAS.
- I. J. Ferrer and C. Sánchez, J. Appl. Phys., 1991, 70, 2641–2647 CrossRef CAS.
- B. Rezig, H. Dahman and M. Kenzari, Renewable Energy, 1992, 2, 125–128 CrossRef CAS.
- A. Ennaoui, S. Fiechter, C. Pettenkofer, N. Alonso-Vante, K. Buker, M. Bronold, C. Hopfner and H. Tributsh, Sol. Energy Mater. Sol. Cells, 1993, 29, 289–370 CrossRef CAS.
- P. P. Chin, J. Ding, J. B. Yi and B. H. Liu, J. Alloys Compd., 2005, 390, 255–260 CrossRef CAS.
- J. H. P. Watson, D. C. Ellwood, A. K. Soper and J. Charnock, J. Magn. Magn. Mater., 1999, 203, 69–72 CrossRef CAS.
- J. H. P. Watson, B. A. Cressey, A. P. Roberts, D. C. Ellwood, J. M. Charnock and A. K. Soper, J. Magn. Magn. Mater., 2000, 214, 13–30 CrossRef CAS.
- P. O'Brien, D. J. Otway and J. H. Park, Mater. Res. Soc. Symp. Proc., 2000, 606, 133–138 CrossRef.
- B. Thomas, C. Höpfner, K. Ellmer, S. Fiechter and H. Tributsch, J. Cryst. Growth, 1995, 146, 630–635 CrossRef CAS.
- B. Thomas and K. Ellmer, J. Mater. Sci.: Mater. Electron., 1998, 9, 61–64 CrossRef CAS.
- D. M. Schleich and H. S. W. Chang, J. Cryst. Growth, 1991, 112, 737–744 CrossRef CAS.
- D. B. Mitzi, Adv. Mater., 2009, 21, 3141–3158 CrossRef CAS.
- Y. Bi, Y. Yuan, C. L. Exstrom, S. A. Darveau and J. Huang, Nano Lett., 2011, 11, 4953–4957 CrossRef CAS PubMed.
- S. Seefeld, M. Limpinsel, Y. Liu, N. Farhi, A. Weber, Y. Zhang, N. Berry, Y. J. Kwon, C. L. Perkins, J. C. Hemminger, R. Wu and M. Law, J. Am. Chem. Soc., 2013, 135, 4412–4424 CrossRef CAS PubMed.
- W. Li, T. Dittrich, F. Jäckel and J. Feldmann, Small, 2014, 10, 1194–1201 CrossRef CAS PubMed.
- E. Strauss, D. Golodnitsky, K. Freedman, A. Milner and E. Peled, J. Power Sources, 2003, 115, 323–331 CrossRef CAS.
- K. Chi, S. Shyu, J. Wu, C. Wu and S. Chuang, Inorg. Chim. Acta, 2001, 334, 276–282 Search PubMed.
- N. Takahashi, T. Sawada, T. Nakamura, T. Nakamura and Y. Momose, J. Mater. Sci. Lett., 2000, 19, 2223–2224 CrossRef CAS.
- K. Ramasamy, M. A. Malik, M. Helliwell, F. Tuna and P. O'Brien, Inorg. Chem., 2010, 49, 8495–8503 CrossRef CAS PubMed.
- M. Akhtar, A. L. Abdelhady, M. Azad Malik and P. O'Brien, J. Cryst. Growth, 2012, 346, 106–112 CrossRef CAS.
- S. Khalid, E. Ahmed, M. Azad Malik, D. J. Lewis, S. Abu Bakar, Y. Khan and P. O'Brien, New J. Chem., 2015, 39, 1013–1021 RSC.
- S. Mlowe, D. J. Lewis, M. A. Malik, J. Raftery, E. B. Mubofu, P. O'Brien and N. Revaprasadu, Dalton Trans., 2016, 45, 2647–2655 RSC.
- G. Li, B. Zhang, J. Rao, D. Herranz Gonzalez, G. R. Blake, R. A. De Groot and T. T. M. Palstra, Chem. Mater., 2015, 27, 8220–8229 CrossRef CAS.
- K. D. Oyler, X. Ke, I. T. Sines, P. Schiffer and R. E. Schaak, Chem. Mater., 2009, 21, 3655–3661 CrossRef CAS.
- X. J. Wu, Z. Z. Zhang, J. Y. Zhang, B. H. Li, Z. G. Ju, Y. M. Lu, B. S. Li and D. Z. Shen, J. Appl. Phys., 2008, 103, 1–6 Search PubMed.
- L. Sun, X.-J. Chen, J. Guo, P. Gao, Q.-Z. Huang, H. Wang, M. Fang, X. Chen, G. Chen, Q. Wu, C. Zhang, D. Gu, X. Dong, L. Wang, K. Yang, A. Li, X. Dai, H. Mao and Z. Zhao, Nature, 2012, 483, 67–69 CrossRef CAS PubMed.
- F.-C. Hsu, J. Luo, K.-W. Yeh, T.-K. Chen, T.-W. Huang, P. M. Wu, Y.-C. Lee, Y.-L. Huang, Y.-Y. Chu, D.-C. Yan and M. Wu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14262–14264 CrossRef CAS PubMed.
- L. Chen, H. Zhan, X. Yang, Z. Sun, J. Zhang, D. Xu, C. Liang, M. Wu and J. Fang, CrystEngComm, 2010, 12, 4386–4391 RSC.
- R. García-Rodríguez, M. P. Hendricks, B. M. Cossairt, H. Liu and J. S. Owen, Chem. Mater., 2013, 25, 1233–1249 CrossRef.
- H. Zhang, G. Long, D. Li, R. Sabirianov and H. Zeng, Chem. Mater., 2011, 23, 3769–3774 CrossRef CAS.
- M. Akhtar, J. Akhtar, M. A. Malik, F. Tuna, M. Helliwell and P. O'Brien, J. Mater. Chem., 2012, 22, 14970–14975 RSC.
- M. Akhtar, M. A. Malik, J. Raftery and P. O'Brien, J. Mater. Chem. A, 2014, 2, 20612–20620 CAS.
- C. E. M. Campos, J. C. De Lima, T. A. Grandi, K. D. Machado, V. Drago and P. S. Pizani, J. Magn. Magn. Mater., 2004, 270, 89–98 CrossRef CAS.
- A. Liu, X. Chen, Z. Zhang, Y. Jiang and C. Shi, Solid State Commun., 2006, 138, 538–541 CrossRef CAS.
- X. J. Wu, D. Z. Shen, Z. Z. Zhang, J. Y. Zhang, K. W. Liu, B. H. Li, Y. M. Lu, B. Yao, D. X. Zhao, B. S. Li, C. X. Shan, X. W. Fan, H. J. Liu and C. L. Yang, Appl. Phys. Lett., 2007, 90, 88–91 Search PubMed.
- R. A. Hussain, A. Badshah, A. Younis, M. D. Khan and J. Akhtar, Thin Solid Films, 2014, 567, 58–63 CrossRef CAS.
- S. Thanikaikarasan, T. Mahalingam, K. Sundaram, A. Kathalingam, Y. Deak Kim and T. Kim, Vacuum, 2009, 83, 1066–1072 CrossRef CAS.
- Y. Han, W. Y. Li, L. X. Cao, S. Zhang, B. Xu and B. R. Zhao, J. Phys.: Condens. Matter., 2009, 21, 235702 CrossRef CAS PubMed.
- B. Cao and W. Cai, J. Phys. Chem. C, 2008, 112, 680–685 CAS.
- P. O'Brien and J. McAleese, J. Mater. Chem., 1998, 8, 2309–2314 RSC.
- P. O'Brien, T. Saeed and J. Knowles, J. Mater. Chem., 1996, 6, 1135–1139 RSC.
- A. Bayer, D. S. Boyle, M. R. Heinrich, D. J. Otway, O. Robbe and P. O'Brien, Green Chem., 2000, 2, 79–86 RSC.
- Y. Mizuguchi, F. Tomioka, S. Tsuda, T. Yamaguchi and Y. Takano, Physica C, 2009, 469, 1027–1029 CrossRef CAS.
- T. Bither, C. T. Prewitt, J. L. Gillson, P. E. Bierstedt, R. B. Flippen and H. S. Young, Solid State Commun., 1966, 4, 533–535 CrossRef CAS.
- T. Bither, R. J. Bouchard, W. H. Cloud, P. C. Dokohue and W. J. Siemons, Inorg. Chem., 1968, 7, 2208–2220 CrossRef CAS.
- Y. Mizuguchi, F. Tomioka, S. Tsuda, T. Yamaguchi and Y. Takano, Appl. Phys. Lett., 2009, 94, 7–10 CrossRef.
- M. Bochmann, Chem. Vap. Deposition, 1996, 8, 85–96 CrossRef.
- Y. Han, W. Y. Li, L. X. Cao, X. Y. Wang, B. Xu, B. R. Zhao, Y. Q. Guo and J. L. Yang, Phys. Rev. Lett., 2010, 104, 17003 CrossRef CAS PubMed.
- W. Zhang, Y. Cheng, J. Zhan, W. Yu, L. Yang, L. Chen and Y. Qian, Mater. Sci. Eng., B, 2001, 79, 244–246 CrossRef.
- Z. H. Lin, Z. Yang and H. T. Chang, Cryst. Growth Des., 2008, 8, 351–357 CAS.
- P. Roy, Z.-H. Lin, C.-T. Liang and H.-T. Chang, Chem. Commun., 2012, 48, 4079–4081 RSC.
- X. Song and M. Bochmann, J. Chem. Soc., Dalton Trans., 1997, 2689–2692 RSC.
- M. L. Steigerwald, Chem. Mater., 1989, 1, 52–57 CrossRef CAS.
- M. L. Steigerwald, T. Siegrist, S. M. Stuczynski and Y. U. Kwon, J. Am. Chem. Soc., 1992, 114, 3155–3156 CrossRef CAS.
- L. C. Roof, W. T. Pennington and J. W. Kolis, Angew. Chem., Int. Ed. Engl., 1992, 31, 913–915 CrossRef.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |