
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Donor-substituted phosphanes – surprisingly weak Lewis donors for phosphenium cation stabilisation†
Ewan R.
Clark
*,
Andryj M.
Borys
and
Kyle
Pearce
School of Physical Sciences, University of Kent, Ingram Building, Canterbury, Kent CT2 7NH, UK. E-mail: e.r.clark@kent.ac.uk
First published on 21st September 2016
Abstract
Paradoxically, N- and O-donor substituted tri-arylphosphanes are shown to be weaker donors than PPh3 when binding the soft Lewis acid moiety [PPh2]+. This arises from internal solvation and rehybridisation at phosphorus, precluding chelation and increasing steric demand, in direct contrast to coordination modes observed for metal complexes.
Phosphacations in the form of highly electron poor, four coordinate P(V) cations have been shown to exhibit high Lewis acidity enabling C–F bond activation1 and their use as catalysts for hydrogenation via frustrated Lewis pair (FLP) chemistry has been demonstrated.2,3 In contrast, two coordinate, cationic P(III) (phosphenium) species are less well studied.4 These highly reactive species rapidly insert into C–X bonds.5,6 and in general require significant steric bulk and strongly π-donating substituents to render them sufficiently stable for isolation.4 Stability may also be imparted by quenching the Lewis acidity with a σ donor (Lewis basic) species,7 regenerating the three coordinate phosphorus centre, which for chelating ligands may permit higher coordination modes (Fig. 1).8–10 Phosphanes themselves are good donors for this and the stability of phosphane–phosphenium complexes is attributed to the thermodynamic favourability of the P–P bond.11 Interestingly, whilst tetracoordinate P(III) monocations are known with internal chelating ligands,12,13 there are very few examples for phosphorus donors or intramolecular chelates (Fig. 1). Burford's attempted syntheses using symmetrical diphosphane ligands instead gave either phosphane–phosphenium complexes with three coordinate phosphacation centres and a free, unbound donor centre or symmetrical bis-phosphenium species for alkyl bridged ligands,14 or rearrangement in the case of aryl bridged species.15
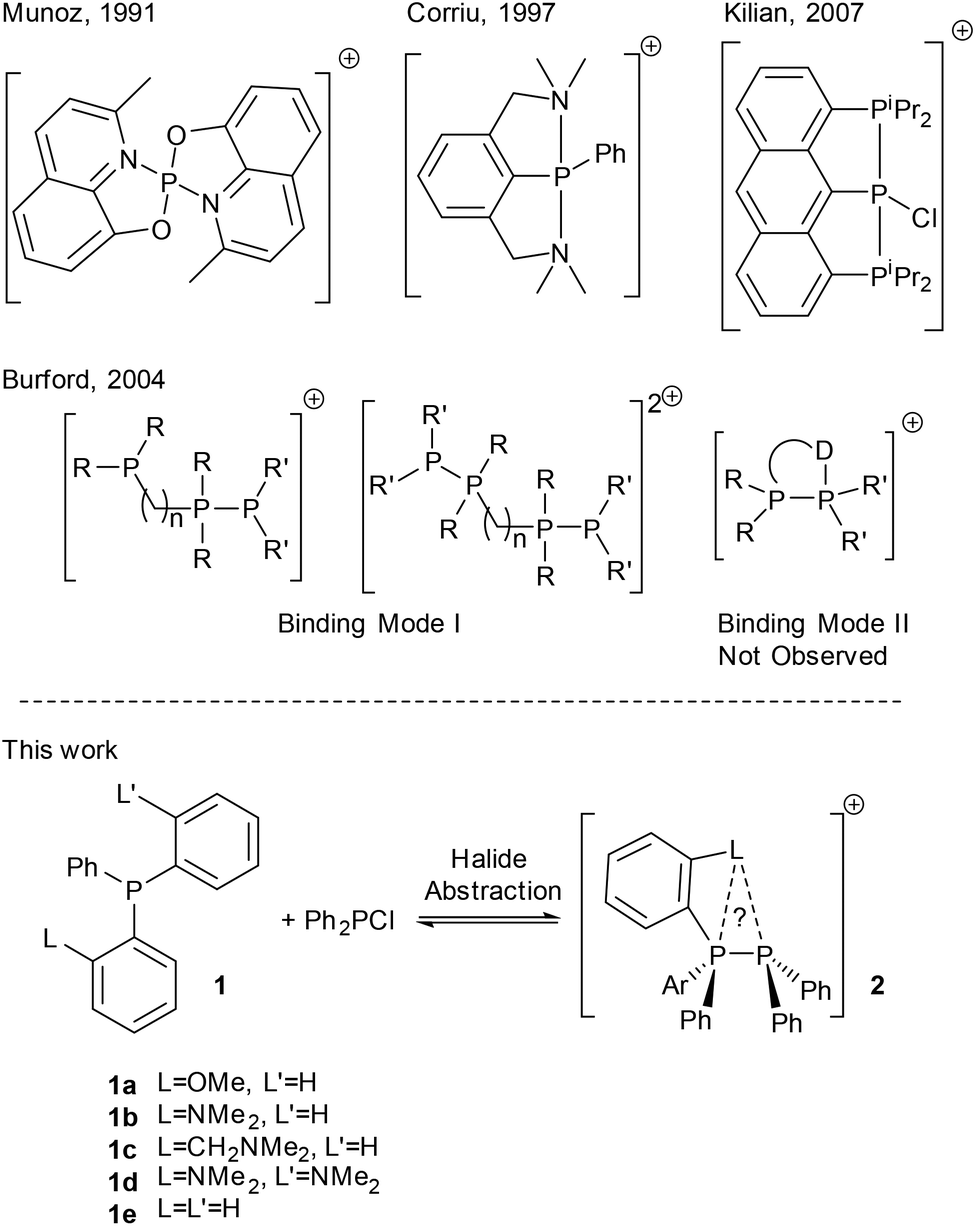 | ||
| Fig. 1 Previously reported and proposed phosphane–phosphenium coordination modes with chelating and multidentate ligands. | ||
We hypothesised that combination of a phosphane donor moiety with rigidly linked first row main-group donors would favour the formation of the elusive binding mode II by preorganisation towards binding and the increased stability of hypervalent bonding involving more electronegative elements. In this report, we describe the synthesis of a family of simple donor functionalised phosphane-derived phosphane–phosphenium salts; the effect of donor substitution on the overall donor strength and resultant cation stability is discussed.
Results and discussion
Initial synthesis and structure
No reaction is observed on combination of phosphanes 1a to 1e with Ph2PCl in DCM; addition of one equivalent of NaBArF lead in all cases to complete loss of 31P NMR signals for both starting materials and signals indicative of phosphane–phosphenium formation – see Table 1. In contrast to the broad, unresolved signals often seen for 2e salts,151JP–P coupling could be resolved in solution for 2b[BArF], 2c[BArF] and 2d[BArF] (Table 1).| BArF | OTf | GaCl4 | |
|---|---|---|---|
| a Not cleanly formed. | |||
| [2a] | 16.4, −6.4 | 17.1 −6.4 | 18.9, −5.8 |
| — | (355) | (307) | |
| [2b] | 13.7, −8.4 | 13.8, −8.9 | 13.8, −8.8 |
| (345) | (344) | (344) | |
| [2c] | 17.0, −6.1 | —, —a | 17.0, −6.5a |
| (335) | — | (332) | |
| [2d] | 12.8, −6.3 | 12.8, −6.8 | 12.8, −6.6 |
| (335) | (331) | (335) | |
| [2e] | 13.7, −10.1 | 15.3, −10.6 | 13, −1315 |
| — | (∼340) | (340) | |
The reaction with Me3SiOTf or GaCl3 likewise resulted in clean chloride abstraction and the quantitative formation of the desired triflate or [GaCl4]− salts except for 1c which gave complex mixtures for both reagents. Significant variation is seen for the 1JP–P coupling constants, especially for [2a] salts indicating varying degree of anion association. Attempted synthesis of the cheaper [AlCl4]− salts by halide abstraction with AlCl3 lead to complex behaviour with multiple species present in solution by 31P NMR, likely due to the more coordinating nature of the anion coupled with competition from the harder nitrogen donor centres. Both the 31P chemical shifts and coupling constants are comparable to those seen for 2e, implying that binding mode II is not adopted. This was confirmed upon successful isolation and characterisation of single crystals of [2c]BArF, revealing that it exhibits the unexpected mode III with short N(1)–P(1) contacts (Fig. 2).
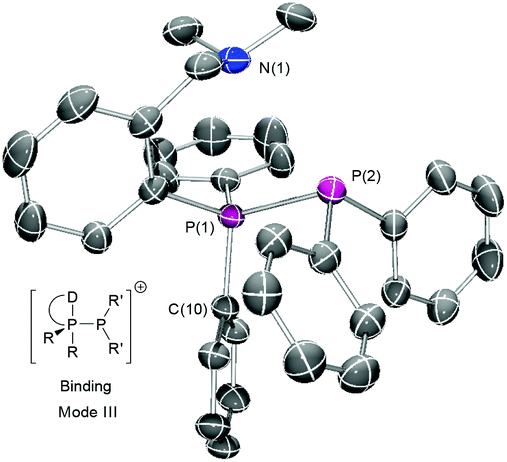 | ||
| Fig. 2 Crystal structure of [2c]BArF drawn with ellipsoids at 50% probability; hydrogens and disordered anion omitted for clarity. | ||
Compound [2c]BArF crystallises with a single ion pair in the asymmetric unit, with disordered CF3 units in the counteranion (Fig. 2). The P(1)–P(2) bond length is unexceptional but slightly long at 2.2477(9) Å (cf. 2.2302(13) Å for [2e]OTf),15 and there is a close contact between the donor group and the adjacent phosphorus centre (dN(1)–P(1) = 3.014(3) Å, less than the sum of the van der Waals radii (3.35 Å)16) with the nitrogen lone-pair clearly oriented towards the phosphorus centre. The donor phosphorus may therefore be described as either a monocapped tetrahedron or a highly distorted trigonal bipyramid – the sum of equatorial angles = 334.37° (cf. 328.4° for a tetrahedron) but the axial N(1)–P(1)–C(10) angle is 175.4(1)° suggestive of trigonal bipyramid geometry. This is therefore the rather unusual situation in which a P(III) centre is acting as a Lewis acid and a Lewis base simultaneously. Similar internal coordination has been seen for internally solvated phosphanes,17–20 and for P-alkylated phosphonium salts bearing rigid bearing 2-donor-1-napthyl fragments17,18 (the more flexible N,N-dimethylbenzylamino-substituted variants instead react with alkylating agents at the harder nitrogen centre). Despite the inequivalence of the P(1)-bound phenyl and N-methyl groups in the crystal structure, these give single resonances in the 1H NMR down to −50 °C, indicating a dynamic structure in solution. Notably, this differs significantly from the chelating P,N coordination mode observed for 1c with transition metal Lewis acids.21 The remaining donor-functionalised salts were found to decompose on exposure to nhexane or Et2O (see ESI†), implying that the additional donor moieties destabilise the cation with respect to the easily isolated [2e] and, with the coupling constant variation, that there is significant anion dependence on their stability.
Computational investigation and VT NMR studies
Computational methods have been used to explore and support the crystallographic observations of internal solvation (see ESI† for further details).Calculations were performed on full, rather than model structures, given the likely significant steric and electronic influence of the adjacent π systems. In light of this computational demand, we selected three cations – 2b,c,e – for modelling and also the hypothetical adducts 3b,c,e for comparison of donor strength in a neutral complex (Table 2). The optimised structures of 1b, and 2c were in agreement with experimental data,22 giving confidence in the model. The computed structures for 2b also exhibited a short N(1)–P(1) contact, again in contrast to the behaviour of the ligand 1b with transition metal Lewis acids, but similar to the more substituted analogues.18,19 Examination of the computed molecular orbitals show that the HOMOs of 1b, 1c and 1e all correspond to phosphorus centred lone pairs, with energies of −7.18 eV, −7.16 eV and −7.46 eV respectively; internal coordination therefore significantly raises the energies of the lone pairs (expected to increase donor strength) whilst also increasing positive charge at phosphorus. Furthermore, the HOMO−1 for 1b and 1c are in both cases P–N σ bonding interactions. For the cations, the donor-functionalised species are enthalpically stabilised relative to 2e, (Table 4) though only to a small degree. In terms of free energy, 2c is in fact slightly destabilised with respect to 2e, presumably reflecting the entropic cost of binding the otherwise freely rotating benzyl moiety.
|
|
||||||
|---|---|---|---|---|---|---|
| Bond lengths/Å | Fuzzy bond indices | |||||
| N(1)–P(1) | P(1)–P(2) | P(2)–Cl(1) | N(1)–P(1) | P(1)–P(2) | P(2)–Cl(1) | |
| 1b | 2.92104 | 0.1445 | ||||
| 1c | 2.89306 | 0.1912 | ||||
| 1e | ||||||
| Ph2PCl | 2.12225 | 1.3485 | ||||
| 2b | 2.92143 | 2.26457 | 0.1311 | 1.0217 | ||
| 2c | 2.99666 | 2.27408 | 0.1440 | 1.0167 | ||
| 2e | 2.25743 | 1.0459 | ||||
| 3b | 3.07198 | 3.21079 | 2.16476 | 0.0907 | 0.2718 | 1.2758 |
| 3c | 3.01817 | 2.31104 | 3.08682 | 0.1362 | 0.9761 | 0.3831 |
| 3e | 3.47776 | 2.15110 | 0.1847 | 1.3031 | ||
The calculated Mayer's Fuzzy bond indices show a significant degree of covalent bonding between the nitrogen and phosphorus centres for both 1b and 1c, which decreases in 2b and 2c compared to the free phosphanes. A slight increase in negative charge at the donor nitrogen and apical carbon centre is seen in the cationic complexes, coupled with an increased positive charge at P(1) (Table 3) relative to 2e. This suggests a decrease in P–N bonding, supported by P–N bond elongation on complex formation and attributed to steric repulsion. With this in mind, the increased exothermicity of P–P bonding with respect to phosphane exchange (Table 4) cannot arise from the naïve argument of electron donors increasing the electron density available at phosphorus as calculated charges show increased positive charge at phosphorus for 1b and 1c relative to 1e. Instead, the P–N bonding results in a rehydridisation at phosphorus and a change in the nature of the donor orbital. Ultimately, the calculated P–P bond order is (albeit slightly) lower for the internally coordinated salts than for 2e, and the P–P bonds longer. The increased stability likely therefore arises from the change in degree of P–N electrostatic interaction, whilst the decrease in covalency argues for weaker, more reactive P–P bonds.
Phosphane–phosphenium systems are highly susceptible to nucleophilic attack by stronger donor species,23,24 resulting in phosphane exchange. With the above in mind, we reacted one equivalent of Ph3P with [2c]BArF to obtain experimental support for the calculated stabilities. This gave immediate formation of a dynamic mixture as seen by 31P NMR (Fig. 3). Immediately upon addition of Ph3P, the 1JP–P coupling was lost and the broad peaks seen in the 31P NMR did not obviously correspond to any free components of the equilibrium. On cooling, however, the peaks sharpened and resolved into free 1c and [2e]BArF as predicted by the computed relative free energies. This was repeated with [2b]BArF formed in situ with the same result; likewise an identical spectrum was formed by the addition of 1b to [2e]BArF. From these combined computational and experimental results, we must conclude that the increased steric bulk of the ortho substituent coupled with rehybridisation at phosphorus counterintuitively lead to weaker donor systems for main group Lewis acids. This is in direct contrast to their behaviour as chelates in which the change in hybridisation does not occur.
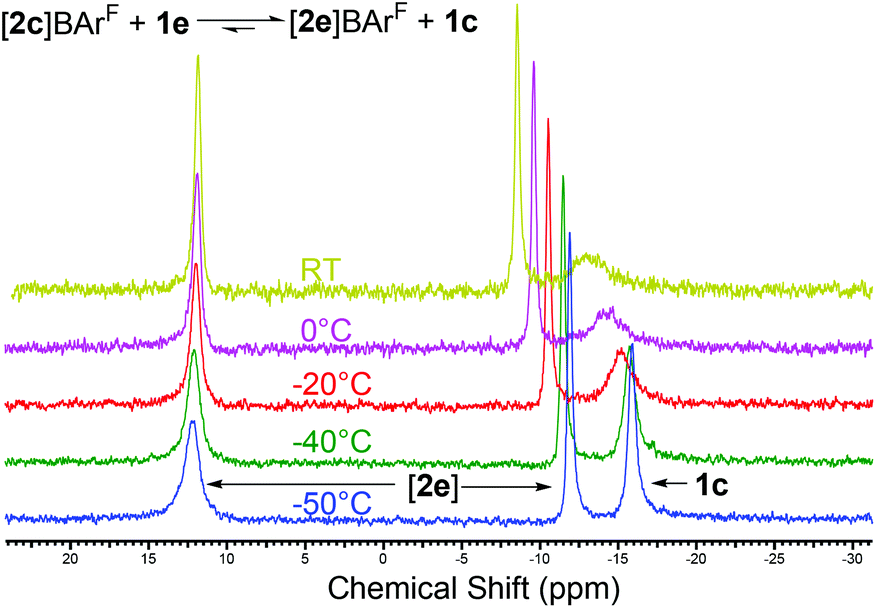 | ||
| Fig. 3 Variable temperature 31P NMR showing the dynamic exchange between [2c]BArF, [2e]BArF, and the free phosphanes. | ||
With respect to the neutral complexes 3, for 3b and 3e we were only able to locate a minimum corresponding to a weak interaction between donor and Ph2PCl and slight elongation of the P–Cl bond relative to free Ph2PCl. For 3c, no such weak complex could be found, but instead significant P–P bonding character and near scission of the P–Cl bond was found in both Fuzzy bond indices and bond lengths. The formation of the complexes, 3, is enthalpically favourable in all three cases, though the free energy change is positive for all cases, not unexpected given the entropic costs of binding. No evidence of adduct formation was seen by variable temperature NMR studies on 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of 1b or 1c and Ph2PCl. Repeating the experiment using the softer Ph2PI gave dramatically different results. Upon combination with one equivalent of 1b, the 31P NMR immediately changes – whilst there are still only two major resonances visible, they are broad and centred at δ 35.5 ppm and δ −11.8 ppm and do not correspond to any single known species. On cooling the signals continue to move and at −30 °C, 1JP–P coupling becomes resolved. By −50 °C, the 31P spectrum is essentially identical to that of [2b]BArF and is ascribed to the formation of [2b]I; on warming the spectra revert to those seen at room temperature. Similar results are observed for the reaction of 1c and 1e with Ph2PI, which converge upon the spectra for [2c]I and [2e]I. In no case were any signals which could be attributed to neutral adducts observed. From this we conclude that the barriers to interconversion (not calculated) are in all cases small such that the intermediate is too short lived to be observed on the NMR timescale. This would also explain the anomalous calculated structure of 3c, indicating narrow, shallow potential wells in the energy surface. The resonances observed are therefore simple weighted averages of the signals of 1, Ph2PI and 2 in fast exchange down to −60 °C.
:1 mixtures of 1b or 1c and Ph2PCl. Repeating the experiment using the softer Ph2PI gave dramatically different results. Upon combination with one equivalent of 1b, the 31P NMR immediately changes – whilst there are still only two major resonances visible, they are broad and centred at δ 35.5 ppm and δ −11.8 ppm and do not correspond to any single known species. On cooling the signals continue to move and at −30 °C, 1JP–P coupling becomes resolved. By −50 °C, the 31P spectrum is essentially identical to that of [2b]BArF and is ascribed to the formation of [2b]I; on warming the spectra revert to those seen at room temperature. Similar results are observed for the reaction of 1c and 1e with Ph2PI, which converge upon the spectra for [2c]I and [2e]I. In no case were any signals which could be attributed to neutral adducts observed. From this we conclude that the barriers to interconversion (not calculated) are in all cases small such that the intermediate is too short lived to be observed on the NMR timescale. This would also explain the anomalous calculated structure of 3c, indicating narrow, shallow potential wells in the energy surface. The resonances observed are therefore simple weighted averages of the signals of 1, Ph2PI and 2 in fast exchange down to −60 °C.
Conclusions
Herein we report simultaneous inter- and intra-molecular phosphorus Lewis donor–acceptor complex formation when internally solvated triarylphosphanes react with the soft main group Lewis acid [Ph2P]+, wherein the donor centre also acts as a Lewis acid. This internal solvation results in higher energy lone pairs at phosphorus, arising from rehybridisation of the phosphorus centre due to hypervalent bonding. This does not translate directly to stronger donor–acceptor bonding in phosphane–phosphenium salts, however, due to competing unfavourable steric interactions and increased positive charge at phosphorus. The existence of this binding mode has implications for the utility of such (conventionally) chelating phosphines in main group cation chemistry.Acknowledgements
The authors gratefully acknowledge the University of Kent (ERC, ABS) and the Royal Society of Chemistry Undergraduate Research Bursary (KP) for funding. The authors would also like to thank Drs Simon Holder and Barry Blight for generous donation of NaBArF. Furthermore, the authors would like to acknowledge the invaluable use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out this work.Notes and references
- C. B. Caputo, L. J. Hounjet, R. Dobrovetsky and D. W. Stephan, Science, 2013, 341, 1374–1377 CrossRef CAS PubMed.
- T. vom Stein, M. Peréz, R. Dobrovetsky, D. Winkelhaus, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 10178–10182 CrossRef CAS PubMed.
- M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298–7301 CrossRef CAS PubMed.
- A. H. Cowley and R. A. Kemp, Chem. Rev., 1985, 85, 367–382 CrossRef CAS.
- T. Hatakeyama, S. Hashimoto and M. Nakamura, Org. Lett., 2011, 13, 2130–2133 CrossRef CAS PubMed.
- C. Tsang, C. A. Rohrick, T. S. Saini, B. O. Patrick and D. P. Gates, Organometallics, 2002, 21, 1008–1010 CrossRef CAS.
- N. Burford and P. J. Ragogna, J. Chem. Soc., Dalton Trans., 2002, 4307–4315, 10.1039/B208715D.
- L. Lamandé and A. Munoz, Tetrahedron Lett., 1991, 32, 75–78 CrossRef.
- S. E. Pipko, Y. V. Balitzky, A. D. Sinitsa and Y. G. Gololobov, Tetrahedron Lett., 1994, 35, 165–168 CrossRef CAS.
- F. Carré, C. Chuit, R. J. P. Corriu, A. Mehdi and C. Reyé, J. Organomet. Chem., 1997, 529, 59–68 CrossRef.
- B. D. Ellis, P. J. Ragogna and C. L. B. Macdonald, Inorg. Chem., 2004, 43, 7857–7867 CrossRef CAS PubMed.
- H. H. Karsch, E. Witt and F. E. Hahn, Angew. Chem., Int. Ed. Engl., 1996, 35, 2242–2244 CrossRef CAS.
- P. Kilian and A. M. Z. Slawin, Dalton Trans., 2007, 3289–3296 RSC.
- N. Burford, D. E. Herbert, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2004, 126, 17067–17073 CrossRef CAS PubMed.
- N. Burford, P. J. Ragogna, R. McDonald and M. J. Ferguson, J. Am. Chem. Soc., 2003, 125, 14404–14410 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–446 CrossRef CAS.
- C. Chuit, R. J. P. Corriu, P. Monforte, C. Reyé, J. Declercq and A. Dubourg, J. Organomet. Chem., 1996, 511, 171–175 CrossRef CAS.
- C. Chuit, R. J. P. Corriu, P. Monforte, C. Reyé, J. Declercq and A. Dubourg, Angew. Chem., 1993, 105, 1529–1531 CrossRef CAS.
- R. Mitea, A. Covaci, C. Silvestru and A. Silvestru, Rev. Roum. Chim., 2013, 58, 265–273 CAS.
- P. Wawrzyniak, A. L. Fuller, A. M. Z. Slawin and P. Kilian, Inorg. Chem., 2009, 48, 2500–2506 CrossRef CAS PubMed.
- G. S. Ananthnag, N. Edukondalu, J. T. Mague and M. S. Balakrishna, Polyhedron, 2013, 62, 203–207 CrossRef CAS.
- P. Suomalainen, S. Jääskeläinen, M. Haukka, R. H. Laitinen, J. Pursiainen and T. A. Pakkanen, Eur. J. Inorg. Chem., 2000, 2000, 2607–2613 CrossRef.
- N. Burford, T. S. Cameron, P. J. Ragogna, E. Ocando-Mavarez, M. Gee, R. McDonald and R. E. Wasylishen, J. Am. Chem. Soc., 2001, 123, 7947–7948 CrossRef CAS PubMed.
- J. Slattery, C. Fish, M. Green, T. Hooper, J. Jeffery, R. Kilby, J. Lynam, J. McGrady, D. Pantazis, C. Russell and C. Willans, Chem. – Eur. J., 2007, 13, 6967–6974 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and X-ray diffraction data. CCDC 1429292. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03478k |
| This journal is © The Royal Society of Chemistry 2016 |