
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Geometric isomerism in coordination cages based on tris-chelate vertices: a tool to control both assembly and host/guest chemistry
Alexander J.
Metherell
and
Michael D.
Ward
*
Department of Chemistry, University of Sheffield, Sheffield S3 7HF, UK. E-mail: m.d.ward@sheffield.ac.uk
First published on 1st September 2016
Abstract
This ‘Perspective’ article summarises recent work from the authors’ research group on the exploitation of the simple fac/mer geometric isomerism of octahedral metal tris-chelates as a tool to control the chemistry of coordination cages based on bis(pyrazolyl-pyridine) ligands, in two different respects. Firstly this geometric isomerism plays a major role in controlling the guest binding properties of cages because a fac tris-chelate arrangement of pyrazolyl-pyridine chelates around a metal ion vertex results in formation of a convergent set of inwardly-directed C–H protons in a region of high positive electrostatic potential close to a metal cation. This collection of δ+ protons therefore provides a charge-assisted hydrogen-bond donor site, which interacts with the electron-rich regions of guest molecules that are of the correct size and shape to occupy the cage cavity, and the strength of this hydrogen-bonding interaction plays a major role in guest recognition in non-aqueous solvents. Secondly the ability to prepare mononuclear complexes with either a fac or mer arrangement of ligands provides an entry into the controlled, stepwise assembly of heterometallic cages based on a combination of kinetically inert and kinetically labile metal ions at different sites. This has allowed introduction of useful physical properties such as redox activity or luminescence, commonly associated with inert metal ions which are not amenable to participation in thermodynamic self-assembly processes, to be incorporated in a predictable way into the superstructures of coordination cages at specific sites.
 Alexander J. Metherell | Alex Metherell was born in Bristol and grew up in Maidenhead. After graduating from the University of Sheffield with an MChem in 2011, he remained in Sheffield to study for a PhD (2014) under the supervision of Prof. Michael Ward. Alex is currently undertaking postdoctoral studies within the same group, and his research so far has been based on the synthesis of heterometallic coordination cages and their subsequent applications in host–guest chemistry. In his spare time, Alex enjoys taking walks around the Peak District. |
 Michael D. Ward | Mike Ward did his BA at Cambridge, studying Natural Sciences (1983–1986). He remained in Cambridge for his PhD (1986–1989) with Ed Constable, studying some early examples of self-assembled helicate complexes. After a post-doc with Jean-Pierre Sauvage in Strasbourg, he was appointed to a lectureship at Bristol in 1990. He moved to Sheffield in 2003 and is currently the Head of Department. Mike's interests are all based around the coordination chemistry of transition metal and lanthanide ions and their multinuclear assemblies. Awards for his research include the RSC Corday Morgan medal for 1999; Sir Edward Frankland Fellowship for 2000–2001; the RSC ‘Chemistry of the Transition Metals’ award for 2005; and the RSC ‘Supramolecular Chemistry’ Award for 2016. He has been Chair of the Editorial Board of ‘RSC Advances’ since the journal started in 2011. |
Introduction
From its humble beginning with Pedersen's discovery of the first crown ether in 1960,1 the domain of supramolecular chemistry is one that has grown in many directions, with chemists’ ability to manipulate non-covalent interactions underpinning developments in research fields from synthetic biology to materials science. Within the domain of coordination chemistry, an area of particular interest over the last three decades has been the development of coordination cages: hollow, pseudo-spherical metal/ligand assemblies of sometimes remarkable complexity which are formed from simple metal ion and bridging ligand building blocks.2 These are of interest for both their structural and functional properties. Coordination cages provide some of the most elaborate structures that can be formed from simple building blocks by self-assembly methods: so the study of their assembly and the principles of symmetry which underpin this, and their structural characterisation, have been of long-standing interest. Functional behaviour can arise from binding guests in the central cavity, with the entrapment of guests in the microenvironment inside a cage cavity resulting in useful behaviour from highly effective catalysis of reactions of bound guests, to sensing and drug delivery.3The majority of coordination cages are homonuclear, homoleptic assemblies: that is, they contain one type of labile metal ion and one type of bridging ligand, with the structure dictated by the coordination preferences of the metal ion, and the disposition of donor sites in the ligand and its geometric flexibility.2 This naturally limits both the structural control available during the self-assembly process and, potentially, the functional properties of the resulting cage. Many potentially useful properties of transition metal ions that may be exploited to confer useful properties on a cage – in particular, reversible redox activity and photophysical activity – are associated with second and third row metal ions whose kinetic inertness makes them difficult to use in traditional self-assembly processes, with a few notable exceptions.4 The majority of coordination cages reported thus far are only ‘functional’ due to the structural attributes of the cage (size, shape, guest binding abilities and so on), with the exact nature of the metal ion being of little importance other than to direct the assembly of the ligands and support the overall cage structure.
As part of our exploration of coordination cage chemistry we have been interested in developing new routes to heterometallic cages, with a view to exploiting the different types of property associated with different metal ions to provide new forms of functional behaviour to cages. This synthetic control has relied on being able to treat different vertices of the cages in a different way during a stepwise assembly process as a consequence of their fac (‘facial’) or mer (‘meridional’) geometric isomerism. When an octahedrally-coordinated metal ion has three ligands of each of two different types – i.e. an MA3B3 complex – this can lead to two geometric isomers. The fac isomer arises when the three A ligands lie on one triangular face of the octahedron and the three B ligands are on the other, such that every ligand A is trans to a ligand B and there is a threefold rotation axis. The mer isomer arises when the three A ligands and the three B ligands are each disposed around an ‘equator’ such that one axis has two ligands A trans to one another; another has two ligands B trans to one another; and the third has one each of A and B trans to one another. This has no threefold rotation axis. These are sketched in Fig. 1 for the cases when A and B are monodentate ligands, as well as the case (in this work) when chelating bidentate ligands A–B are used.
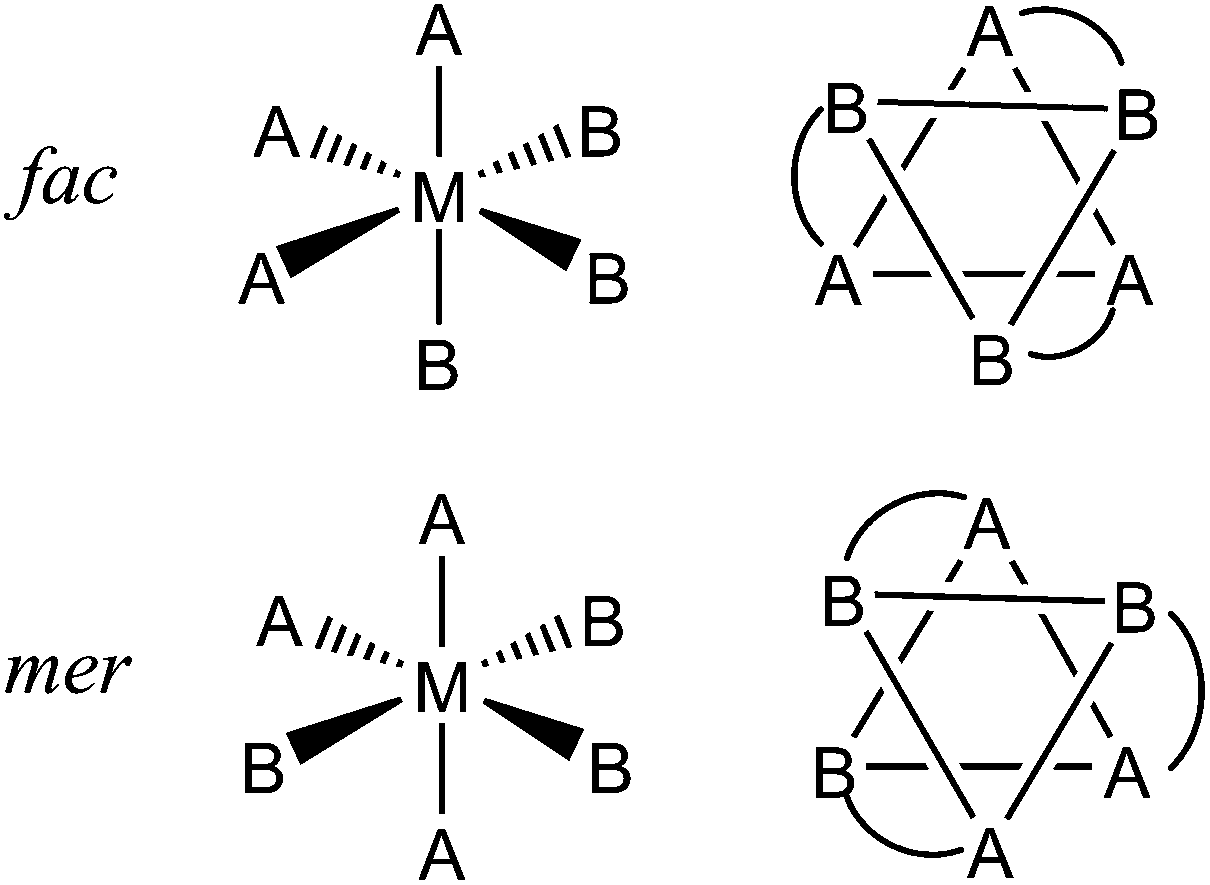 | ||
| Fig. 1 Top: Arrangement of the two ligand types in fac-MA3B3 complexes. Bottom: Arrangement of the two ligand types in mer-MA3B3 complexes. The sketches on the right are alternative depictions of the same species viewed through the centre of an octahedral face, with chelate links in the A–B ligands also included. | ||
This variation in the arrangement of donor atoms around otherwise similar metal tris-chelate vertices allows them to be differentiated between, and therefore provides an entry into a general stepwise assembly methodology. In addition, we have found that the occurrence of either or both of fac and mer tris-chelate vertices in the cages has an unexpectedly profound effect on the cages’ host guest chemistry.
In this review we describe how this simple variation in geometric isomerism between otherwise similar metal centres in the cages underpins both the control of heteronuclear cage assembly, and the ability of some of the cages to act as hosts for both anionic and neutral guests.
Background: occurrence of geometric fac/mer isomerism in coordination cages
Our research into coordination cage chemistry has centred mostly on the use of bis-bidentate ligands which consist of two chelating pyrazolyl-pyridine termini connected to a central aromatic spacer via flexible methylene hinges (Fig. 2).2c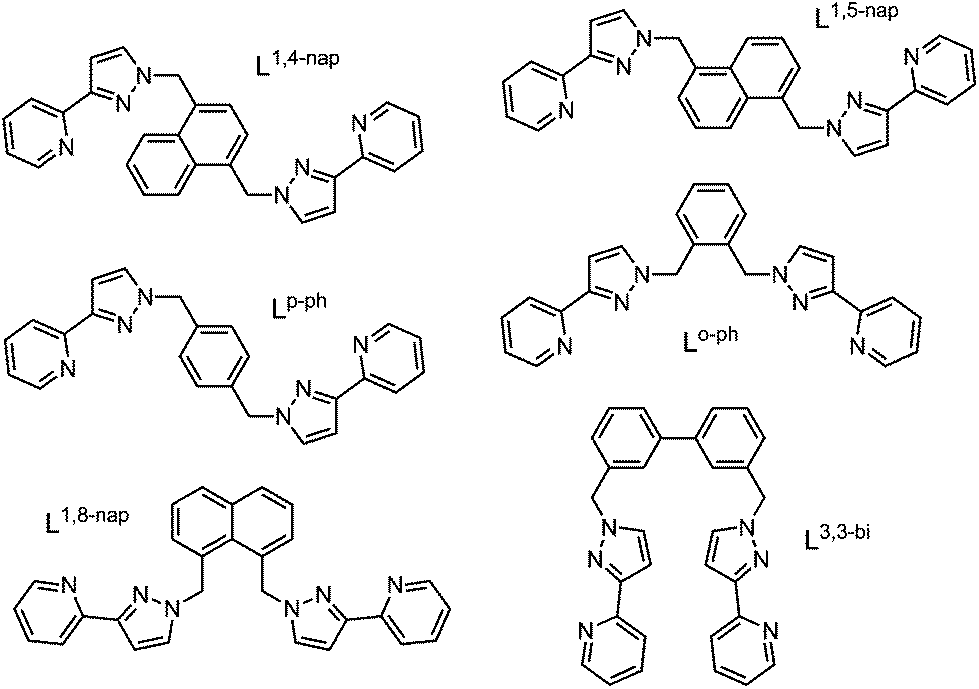 | ||
| Fig. 2 Structures of the bis(pyrazolyl-pyridine) ligands that are referred to in this article. | ||
Combination of these ligands, which possess four donor atoms, with kinetically labile octahedral transition metal dications, results in structures with a 2M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3L ratio, such that all donor atoms are used and all metal ions are coordinatively saturated (the ‘principle of maximum site occupancy’). This ratio has been expressed in numerous ways in polyhedral cages which have a 2:3 ratio of vertices to edges, with a metal ion occupying each vertex and a bridging ligand occupying each edge. Each vertex must be at the conjunction of three edges such that each metal ion interacts with three ligand termini and is therefore octahedral.
:3L ratio, such that all donor atoms are used and all metal ions are coordinatively saturated (the ‘principle of maximum site occupancy’). This ratio has been expressed in numerous ways in polyhedral cages which have a 2:3 ratio of vertices to edges, with a metal ion occupying each vertex and a bridging ligand occupying each edge. Each vertex must be at the conjunction of three edges such that each metal ion interacts with three ligand termini and is therefore octahedral.
The range of polyhedral shapes available to the cage family is controlled by these basic structural principles, and we have characterised an extensive family of such cages from M4L6 tetrahedra5a,b through to a M16L24 tetra-capped truncated tetrahedron, our largest cage reported to date;5c the syntheses and structures of this family of cages were reviewed a few years ago.2c The cages generally exhibit extensive aromatic stacking around the periphery between electron-poor (pyrazolyl-pyridine unit coordinated to a 2+ metal ion) and electron-rich (central aromatic spacer) ligand fragments which form alternating stacks; an example of inter-ligand stacking in a tetrahedral cage is in Fig. 3a.6
 | ||
| Fig. 3 (a) Structure of part of the complex cation of [Cd4(L3,3-bi)6](BF4)8, emphasising the inter-ligand aromatic stacking regions with arrows indicating the direction of the donor–acceptor interactions; (b) close-up view of the interaction of the bound anion with the convergent set of CH protons around the fac tris-chelate metal vertex (ref. 6). | ||
This stacking appears to contribute substantially to the cages’ stability, helping to overcome the entropy penalty associated with formation of such large assemblies. The few cages which do not exhibit aromatic stacking in the solid state do not persist in solution.7 This stacking is facilitated by the flexibility of the ligands – arising from the methylene groups which act as ‘hinges’ – which permits them to adjust their conformations to maximise the stacking.
A key structural feature of all of these cages, and a consequence of the ligands’ flexibility, is the presence of a combination of fac and/or mer tris chelate metal centres at different positions in the metal array, that can arise during cage formation because the two donor atoms of the pyrazolyl-pyridine units are inequivalent.8 When labile metal ions are used for cage assembly there is no control over this, with each cage type adopting different numbers of fac and/or mer tris-chelate metal vertices as required by that particular self-assembly process. Thus, the ortho-phenylene spaced ligand Lo-ph (Fig. 2) affords [M4(Lo-ph)6]8+ tetrahedral cages in which all metal centres have a fac tris-chelate coordination geometry (Fig. 4a), and in addition all metal ions have the same optical configuration resulting in T symmetry.5a,b With the related ligand L3,3-bi, tetrahedral [M4(L3,3-bi)6]8+ cages also form, but with one fac and three mer tris-chelate vertices to give C3 symmetric assemblies (Fig. 3 and 4b).6,9 The octanuclear cubic cages [M8(L1,5-nap)12]16+ contain a mixture of six mer and two fac vertices (Fig. 4c),7a whereas the truncated tetrahedral dodecanuclear cages [M12(L1,5-nap)18]24+ contain only mer tris-chelate metal vertices.10
 | ||
| Fig. 4 Schematic diagrams of (a) M4(Lo-ph)6, (b) M4(L3,3-bi)6 and (c) M8(L1,5-nap)12 cages, indicating the coordination geometry (fac or mer) at each position. The colours indicate how the metal ions can be divided into distinct sets in our stepwise synthetic strategy. | ||
Fig. 4 illustrates examples of the disposition of fac and mer tris-(pyrazolyl-pyridine) metal fragments at the vertices of different cages. These metal complex units are relatively structurally invariant between different cages, apart from minor differences in M–N distances arising from metal ions of different radii, with the structural variety of cages arising from the various ways in which different numbers of these fac or mer metal complex units are connected by different spacers.
Within the general class of (M2L3)n cages based on octahedral ions connected by bis-bidentate edge-bridging ligands, the presence of both geometric isomers of metal vertices in the same assembly is rare.11 It is far more common that the bridging ligands are relatively rigid, lacking the high conformational flexibility that ours possess from the methylene spacers, which inevitably results in fac tris-chelate metal centres dominating. Typical examples include the cages of Raymond,12 Nitschke,13 and others.14 Notable exceptions to this include a Co10L15 pentagonal prism15a and an Fe12L12 icosahedral capsule15b prepared by Nitschke and co-workers, both of which contain exclusively mer tris-chelate metal centres.
The occurrence of either or both types of vertex isomer in our cages has turned out to have two fundamentally important consequences. Firstly, it underpins the ability of the cages to act as hosts for hydrogen-bond accepting guests in organic solvents, because the arrangement of ligands around the fac tris-chelate vertices creates a hydrogen-bond donor pocket at which the guests bind (Fig. 3b). Secondly, it provides a basis for the synthetic control necessary to incorporate different metal ions at different sites in the cage superstructures, allowing some types of useful functional behaviour associated with kinetically inert metal ions to be incorporated into the cages. Whilst these two phenomena are distinct they have the same origin and are therefore interrelated. No other family of cages to our knowledge has provided a means for this simple geometric isomerism and its consequences to be explored in detail. The effects of the geometric isomerism of the cage vertices on both guest binding and control of self-assembly, which form the basis of this review, will now be discussed in turn.
Effect of geometric isomerism of vertices on host/guest chemistry
The presence of fac tris-chelate vertices at some positions in some of the cages turned out to play a crucial role in the ability of the cages to bind both anionic and neutral guests. We noticed from our early work with tetrahedral M4L6 cages based on Lo-ph and L3,3-bi that the small cavities contain anions which form a network of H-bonding interactions with inwardly-directed CH protons on the internal surface of the cationic cage.2a With the [M4(Lo-ph)6]8+ cages the anions are located symmetrically in the centre of the cavity,5a,b but in the [M4(L3,3-bi)6]8+ cages the anion is clearly off-centre and displaced towards the fac tris-chelate vertex (Fig. 3b).6,9 A closer look shows that this permits a set of CH⋯X hydrogen bonds (where X is an electronegative atom from the anion, such as O from perchlorate or F from tetrafluoroborate or hexafluorophosphate) principally with the convergent set of methylene groups that form an H-bond donor pocket as a consequence of the fac tris-chelate geometry at that vertex.We did not attach much significance to this until we made, much more recently, a systematic study of the host/guest chemistry of the cubic cage [Co8(L1,5-nap)12]16+.16,17 Structural characterisation of the cage revealed that it has two fac tris-chelate vertices at either end of a long diagonal, with the other six vertices being mer tris-chelates. Fig. 5 shows the disposition of the two fac tris-chelate binding sites, as well as the convergent arrangement of CH protons in these positions that facilitates substrate binding. The threefold axis through the two fac tris-chelate units, and the inversion centre, result in molecular S6 symmetry which is preserved in solution according to the 1H NMR spectra.7a Notably, the solvent molecule guests in the cavity (water or methanol) are located in the two binding pockets at the fac tris-chelate vertices,7a,16,17 just like the anions in the cavities of the [M4(L3,3-bi)6]8+ cages (Fig. 3).6,9
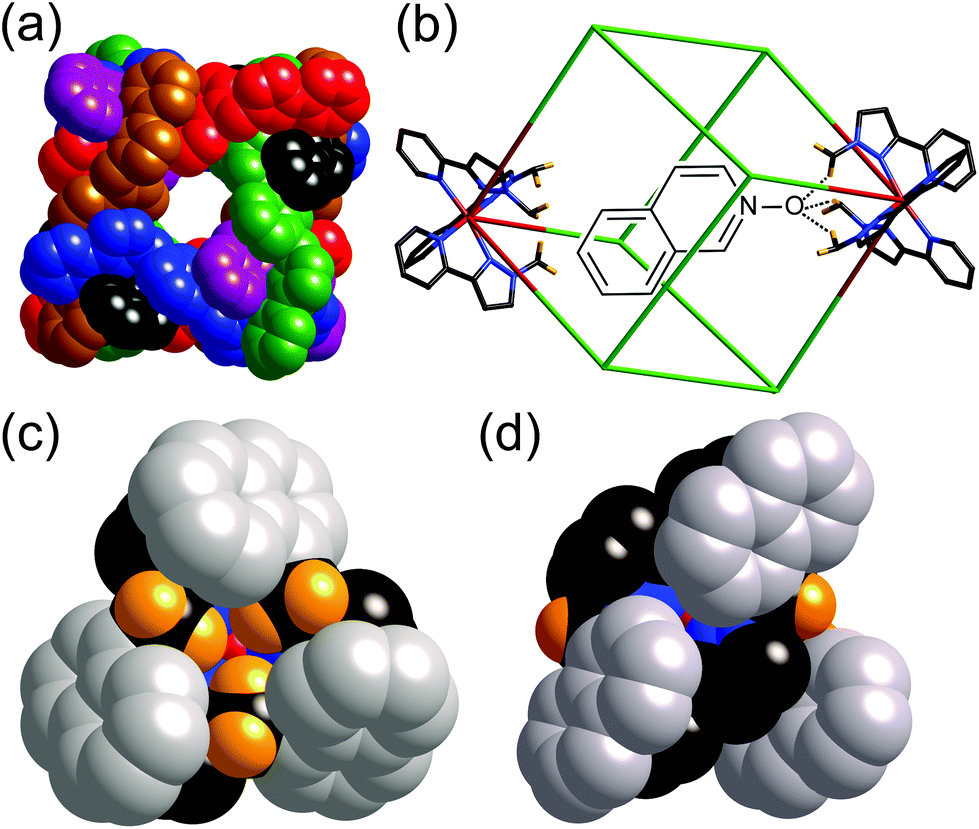 | ||
| Fig. 5 (a) Space-filling view of the cage complex cation of [Co8(L1,5-nap)12](BPh4)16, with the ligands coloured separately for clarity; (b) a view (host and guest not to scale) showing the disposition of the two fac tris-chelate sites at either end of a long diagonal, and the hydrogen-bonding interaction with a hydrogen-bond accepting guest in the cavity; (c) a space-filling view of the environment around a fac tris-chelate vertex showing the convergent set of CH protons at the hydrogen-bonding site; and (d) a space-filling view of the environment around a mer tris-chelate vertex showing the absence of such a hydrogen-bonding pocket. Reprinted with permission from ref.17; copyright 2013, American Chemical Society. | ||
The central cavity has a volume of ca. 400 Å3 and is readily accessible through portals in the centres of the faces of the cube (Fig. 5a). According to the Rebek ‘55% rule’ the optimal volume for a bound guest should be ca. 220 Å3,18 which means that a wide range of small molecules could be plausible guest candidates. Accordingly we used 1H NMR spectroscopy to screen a wide variety of potential guests in CD3CN solution. By virtue of the paramagnetism associated with the high-spin Co(II) centres the 1H NMR spectra of the cages are dispersed over the range +100 to −100 ppm,7a,16,17 which minimises overlap and makes changes in individual signals associated with guest binding easy to detect.
After screening many potential guests that showed no sign of binding, we identified that coumarin bound in the cavity of [Co8(L1,5-nap)12]16+ with K = 80 M−1.16 Binding was in slow exchange on the NMR timescale such that separate signals for empty and bound cage could be seen, with the former decreasing in intensity and the latter increasing in intensity during the titration; integration of these allowed the K value to be determined taking into account the concentrations of cage and total guest. The 1H NMR signals for bound coumarin were shifted substantially upfield due to the paramagnetism of the host, with the single biggest Δδ for a coumarin proton on binding being −12 ppm. Molecular modelling indicated that coumarin could fit nicely in the cavity in an orientation such that the carbonyl O-atom, a reasonable H-bond acceptor, was directed towards the convergent set of CH protons around one of the fac tris-chelate vertices: this oxygen atom occupies the same position as the oxygen atom of water or methanol solvent molecule guests in the ‘empty’ cage. Starting the calculation with the coumarin guest in a range of different orientations always resulted in this minimum-energy structure arising; this is illustrated in Fig. 5b for a different (but approximately isosteric) guest isoquinoline-N-oxide.17
So we had a hypothesis that coumarin binds in the cavity of [Co8(L1,5-nap)12]16+ not just because of a shape/size match, but because the electronegative carbonyl O atom acts as an H-bond acceptor, interacting with the interior surface of the cage at the fac tris-chelate sites where several CH groups converge, and also where the surface is relatively highly charged due to its proximity to a partially-exposed metal ion (Fig. 5c). In agreement with this, isosteric guests such as isoquinoline-N-oxide in which the H-bond accepting O atom is more electron rich than that of coumarin (its H-bond acceptor β parameter19 is higher), bind more strongly.17
Subsequent structural characterisation of a wide range of cage/guest complexes has shown that, invariably, an H-bond acceptor atom on the guest lies in the fac tris-chelate binding pocket;20 two examples are shown in Fig. 6 and include one of a series of alkyl phosphonates used as chemical warfare agent simulants,20b and the cyclic ketone cycloundecanone.20c The extent to which this weak hydrogen-bonding interaction contributes to binding is highly solvent dependent. In MeCN, this H-bonding interaction between cage and guest is stronger than the interactions that the guest can form with solvent, so it contributes positively up to ca. 10 kJ mol−1 in the case of isoquinoline-N-oxide.17 In water an H-bonding guest could form stronger hydrogen bonds with the solvent than it can with the cage surface so this interaction does not drive binding thermodynamics, which are dominated by the hydrophobic effect associated with the non-polar part of the guest surface area.20a However once the guest is desolvated and the hydrophobic effect has resulted in its uptake by the cage, the interaction with the fac tris-chelate vertex still serves to orient the guest in the cavity.
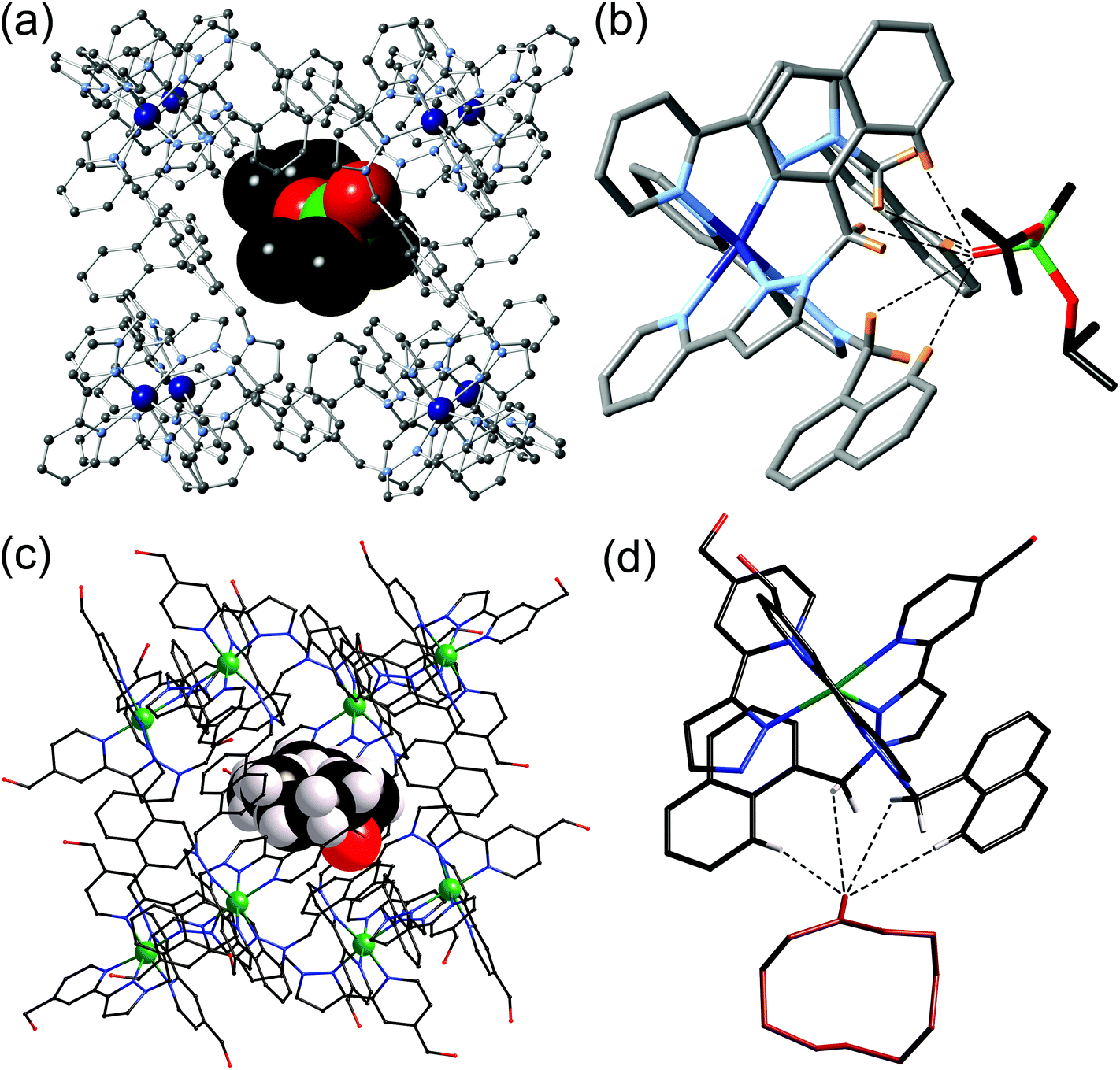 | ||
| Fig. 6 (a) Crystal structure of the [Co8(L1,5-nap)12]16+ host with a molecule of di(isopropyl)methylphosphonate as guest; (b) an expansion of (a) showing the hydrogen-bonding interactions between the guest and the CH groups around the fac tris-chelate vertex of the cage (CH⋯O distances shown by dashed lines are in the range 2.55–2.83 Å); (c) crystal structure of the [Co8(L1,5-nap)12]16+ host with a molecule of cycloundecanone as guest; (d) an expansion of (c) showing the hydrogen-bonding interactions between the guest and the CH groups around the fac tris-chelate vertex of the cage (CH⋯O distances shown by dashed lines are in the range 2.54–3.06 Å). Parts (c) and (d) are reprinted with permission from ref. 20c; copyright 2014, the American Chemical Society. | ||
We have not yet extended detailed investigations of guest binding to other cages. However we note that the larger cages [M16(Lp-ph)24]32+ contain four fac tris-chelate vertices, disposed in a large tetrahedron around the central cavity which has a volume of ca. 1300 Å3,5c so the possibility for large guests with multiple H-bond acceptor groups to bind, anchored at two or more sites by interactions with the cage interior surface, is appealing.
Initial studies on geometric isomers of mononuclear complexes
Given the importance of the fac tris-(pyrazolyl-pyridine) vertices of the cages in facilitating guest binding, we were interested to examine this further using isolated mononuclear complex units outside of the cage environment. In order to provide reasonable mononuclear analogues of the vertices, we chose the mer and fac isomers of mononuclear [M(LBn)3]2+ where the ligand LBn is essentially half of the ligand L1,5-nap (Fig. 7). Preparation of these as their pure fac or mer isomers requires a kinetically inert metal ion, as use of labile Co(II) afforded – unsurprisingly – a statistical mixture of the two isomers. Given the requirement for a kinetically inert but synthetically tractable octahedral metal ion with a 2+ charge and similar M–N distances to those provided in the Co(II) cages, Ru(II) was the obvious choice.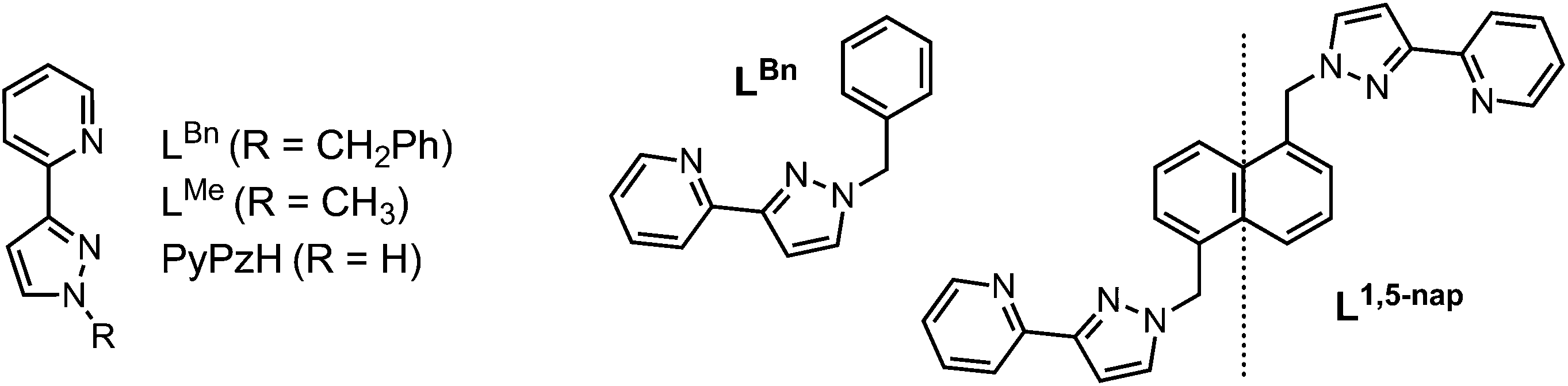 | ||
| Fig. 7 Left: Structure of the ligands used in the mononuclear complex studies. Right: LBn and its relationship to L1,5-nap. | ||
It is well documented that post-synthetic separation of mer and fac isomers is generally not a trivial process;8 following preparation of [Ru(LBn)3](PF6)2 as the expected 3:1 mixture of mer and fac isomers, this became increasingly obvious. Reported methods of such isomeric separation by Piguet21 and Fletcher22 by chromatography or fractional crystallisation were unsuccessful. Consequently, a novel method of separation was devised starting from the complex [Ru(PyPzH)3]2+, which contains three pyrazole NH groups which may be functionalised (see Fig. 7 for ligand structure).
[Ru(PyPzH)3](PF6)2 was prepared as 3:1 mer:fac statistical mixture of isomers, as was evident from the presence by four ligand environments with equal intensity in the 1H NMR spectrum. Separation is based on the differing ability of the two isomers to form adducts with Cu(I) salts:23 the three pyrazolyl N1 donors, which lie together on one face of the fac isomer, can coordinate to a Cu(I) ion in a chelating tridentate face-capping manner, which allows the fac isomer of [Ru(PyPzH)3](PF6)2 to be precipitated selectively. The mer isomer of [Ru(PyPzH)3](PF6)2, in which the three pyrazolyl N1 donors do not converge, cannot coordinate to a Cu(I) ion in this way so remains in solution. Thus, the as-isolated 3:1 mer:fac statistical mixture could be quantitatively separated into pure mer and fac isomers.24
With separation of these isomers achieved, alkylation of the reactive pyrazolyl NH groups with benzyl bromide was achieved with retention of isomeric integrity to yield the complexes mer- and fac-[Ru(LBn)3](PF6)2 as mononuclear models for the coordination cage vertices. The crystal structure of fac-[Ru(LBn)3](PF6)2 highlights the set of convergent protons from the three methylene groups which define the pocket where H-bonding guests bind in the cages. An acetone solvent molecule neatly demonstrates this viewpoint, with the carbonyl oxygen interacting with this binding pocket (Fig. 8).24
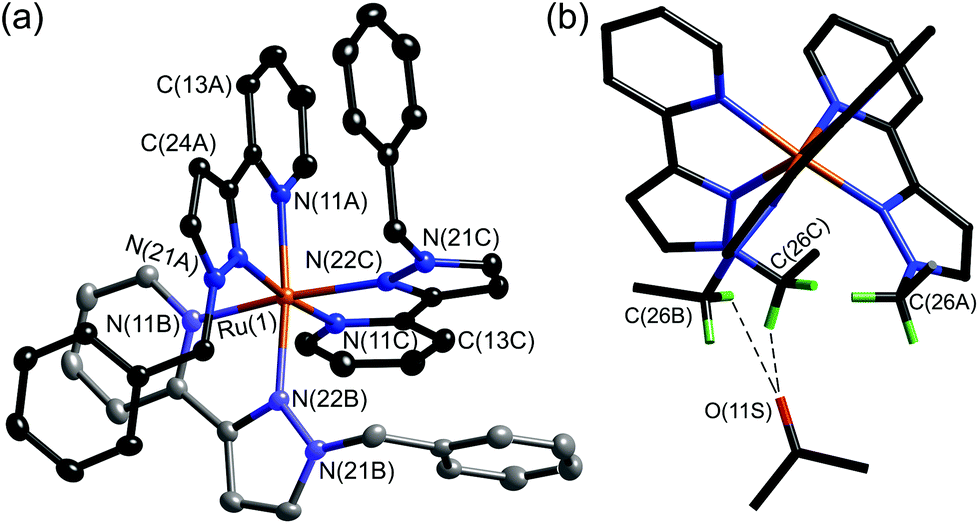 | ||
| Fig. 8 (a) Structure of the complex cation of fac-[Ru(LBn)3](PF6)2·2Me2CO. (b) Alternative view of the complex cation with the phenyl rings not shown, emphasising the convergent arrangement of methylene protons to form an hydrogen-bond donor site, and the presence of a hydrogen-bonded molecule of acetone at this site; the two shortest CH⋯O interactions are shown by dashed lines (ref. 24). | ||
The different abilities of these two geometric isomers of [Ru(LBn)3](PF6)2 to act as H-bond donors was demonstrated by titration of the hydrogen-bond acceptor isoquinoline-N-oxide into a solution of each isomer, which revealed a stronger interaction of isoquinoline-N-oxide with fac-[Ru(LBn)3](PF6)2 than with mer-[Ru(LBn)3](PF6)2. The absolute value of the binding 1:1 constant between isoquinoline-N-oxide and fac-[Ru(LBn)3](PF6)2 is much lower (ca. 1 M−1) than in a cage complex host, because the local environment is different and also possibly because the hexafluorophosphate anions may compete for this binding site in solution which does not seem to happen in the cages where the anions remain surface-bound.20a However the preference for binding to the fac compared to the mer isomer is clear (Fig. 9). In addition, during the titration of isoquinoline-N-oxide with fac-[Ru(LBn)3](PF6)2, one of the diastereotopic methylene proton signals was observed to shift, whereas the other did not, which provides good evidence for the N-oxide group of the guest interacting with the inwardly directed methylene protons of the host, but not to a significant extent with the outwardly directed protons (Fig. 10).
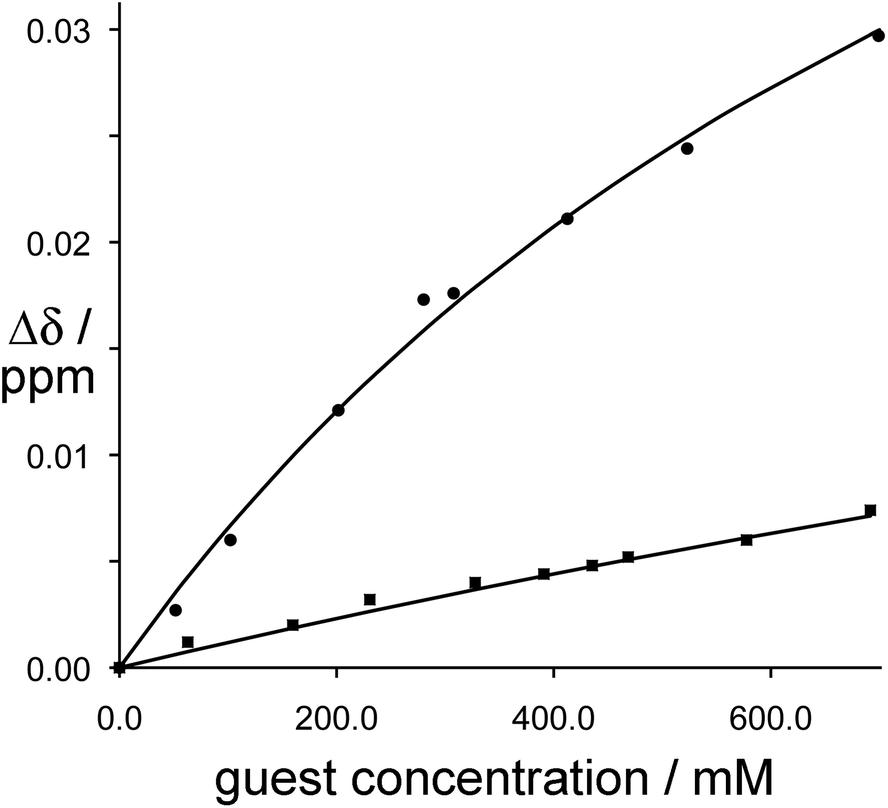 | ||
| Fig. 9
1H NMR binding curves showing the shift of the methylene proton signals in fac-[Ru(LBn)3](PF6)2 (circles, upper line) and mer-[Ru(LBn)3](PF6)2 (squares, lower line) during titration with isoquinoline-N-oxide. For fac-[Ru(LBn)3](PF6)2 the data fit a 1:1 binding isotherm with K ≈ 1 M−1; for mer-[Ru(LBn)3](PF6)2 the binding constant is too weak to quantify (ref. 24). | ||
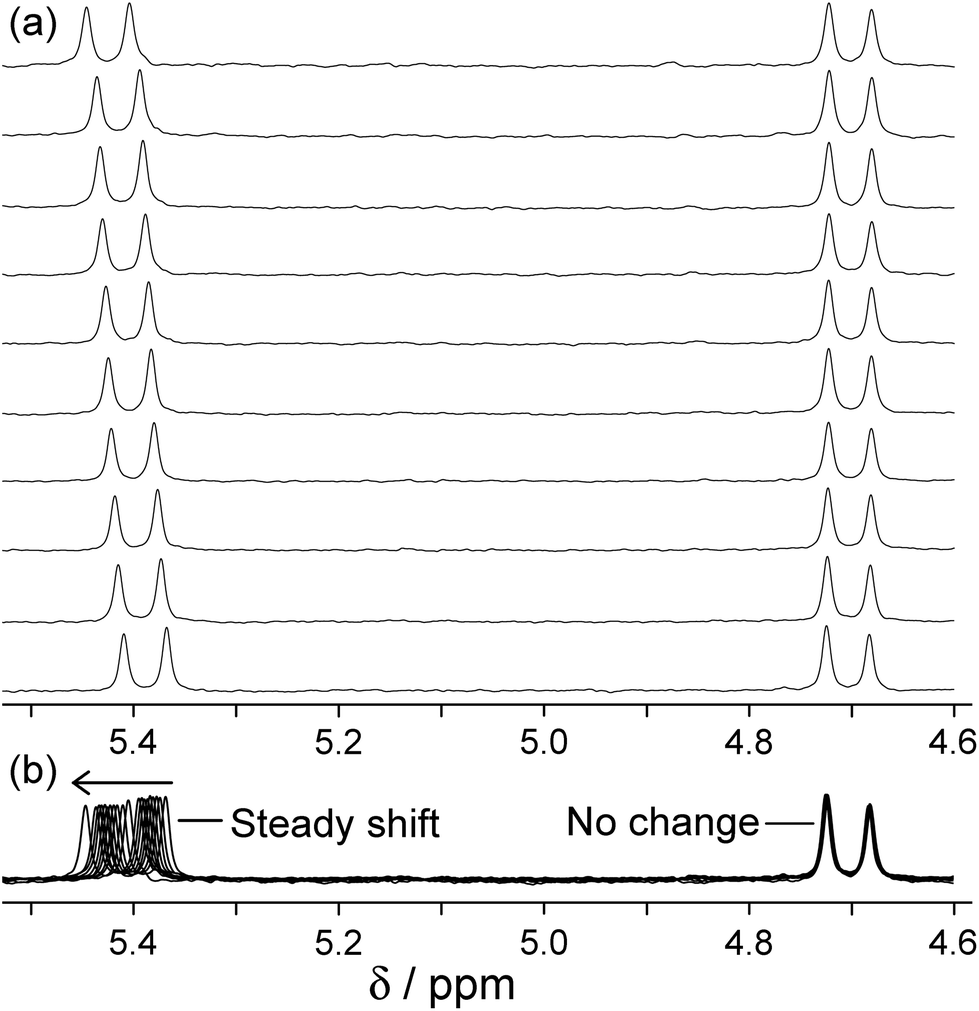 | ||
| Fig. 10 Changes in the 1H NMR chemical shift of the two inequivalent methylene proton signals of fac-[Ru(LBn)3](PF6)2 (0.23 mM) during titration with isoquinoline-N-oxide (up to 0.7 M) in CD3CN. (a) Stacked plots; (b) overlaid plots to emphasise how one signal moves but the other does not (ref. 24). | ||
General methodologies for preparing heteronuclear assemblies: basic principles and some examples from other groups
Having found a route to pure fac or mer Ru(II) complexes with the pyrazolyl-pyridine chelate, with post-coordination alkylation of the pyrazolyl NH groups providing a route to extending the ligands with different substituents, we realised that we now had an entry into stepwise assembly of heterometallic cages. Before this is discussed in the next section, we briefly summarise here other work on heterometallic coordination ages.There exist two general methods for the formation of heterometallic supramolecular assemblies. The first is a thermodynamic approach, in which a ligand with orthogonal binding sites discriminates between two (or more) different types of metal based on the matching of metal ion and ligand types using the simple principles of hard/soft acid and base (HSAB) theory. Thus, a ligand possessing both hard and soft binding sites can selectively bind a hard and a soft metal ion at each site, respectively. Normally both metal ion types are labile such that the assembly occurs in one step, with the hard metal/hard ligand and soft metal/soft ligand combinations providing the orthogonal interactions that direct the self-assembly. For example, Raymond has used a ligand with soft phosphine termini and hard catechol termini which bind to Pd(II) and Ti(IV), respectively (Fig. 11);25 we have used an unsymmetrical ligand with catechol and pyrazolyl-pyridine termini, which respectively bind to Ti(IV) and Zn(II) ions to form a tetrameric hetero-octanuclear cyclic helicate.26 This general method has been used by many groups including those of Wang,27 Shionoya28 and Stang,29 and has been well-covered in many reviews.2e,i,30
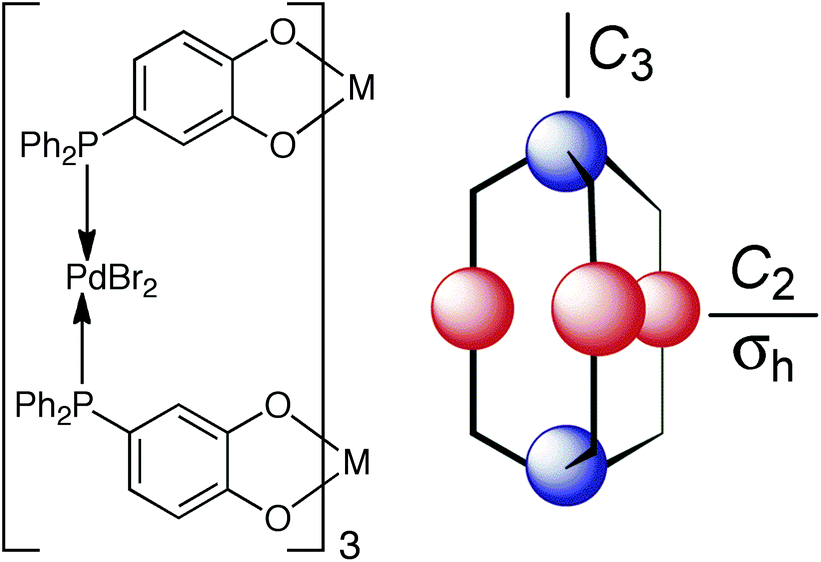 | ||
| Fig. 11 An example of heterometallic self-assembly using the thermodynamic control approach: Raymond's phosphino-catechol ligand and the pentanuclear complex it forms on reaction with Ti(IV) and Pd(II) ions (blue and red spheres represent Ti(IV) and Pd(II), respectively). Reprinted with permission from ref. 25; copyright 2001, the American Chemical Society. | ||
This thermodynamic approach may also be used when the two orthogonal metal/ligand interactions are based on differing coordination numbers or geometric preferences rather than on hard/soft differences.31 For example, Nitschke and co-workers reported a heterometallic [Fe8Pt6L24] cubic cage, which formed due to the differing geometric coordination preferences of each type of metal ion.31a The ligand, possessing a pyridine terminus and a diimine terminus, selectively coordinates to Pt(II) via the pyridine donor to form square-planar coordination (face centres), and to octahedral Fe(II) ions using the chelating bidentate site to give octahedral coordination (vertices), enabling the synthesis of a heterometallic assembly with complete control of metal site selectivity. Again, this requires both metal ions to be kinetically labile, and in this case coordination of monodentate pyridyl donors to Pt(II) is a sufficiently labile process for a conventional thermodynamic self-assembly to occur.
The second approach used to form heterometallic assemblies – which is the one we have adapted for our use – is the use of kinetically inert subcomponents, which is sometimes known as the ‘metalloligand’ or ‘complexes as ligands’ approach.32,33 In this strategy a kinetically inert mononuclear unit with pendant binding sites is used as a pre-formed building block; combining these with additional labile ions in a separate step then completes the assembly of heteronuclear assemblies in which different types of metal ion (inert and labile) are incorporated at specific sites with complete positional control. For example, the synthesis of a cage with octahedral tris-chelate metal ions at the vertices of a cube and square planar ions with four monodentate ligands at the face centres may be achieved by preforming a metalloporphyrin component (Fig. 12).33 A related alternative is to electrochemically ‘lock’ a system by converting a labile oxidation state to an inert one after the self-assembly process has taken place [e.g. Co(II) to Co(III)], but this method is less robust due to the structural rearrangements which may occur upon oxidation of the metal, for example changes in metal–ligand bond distances.34
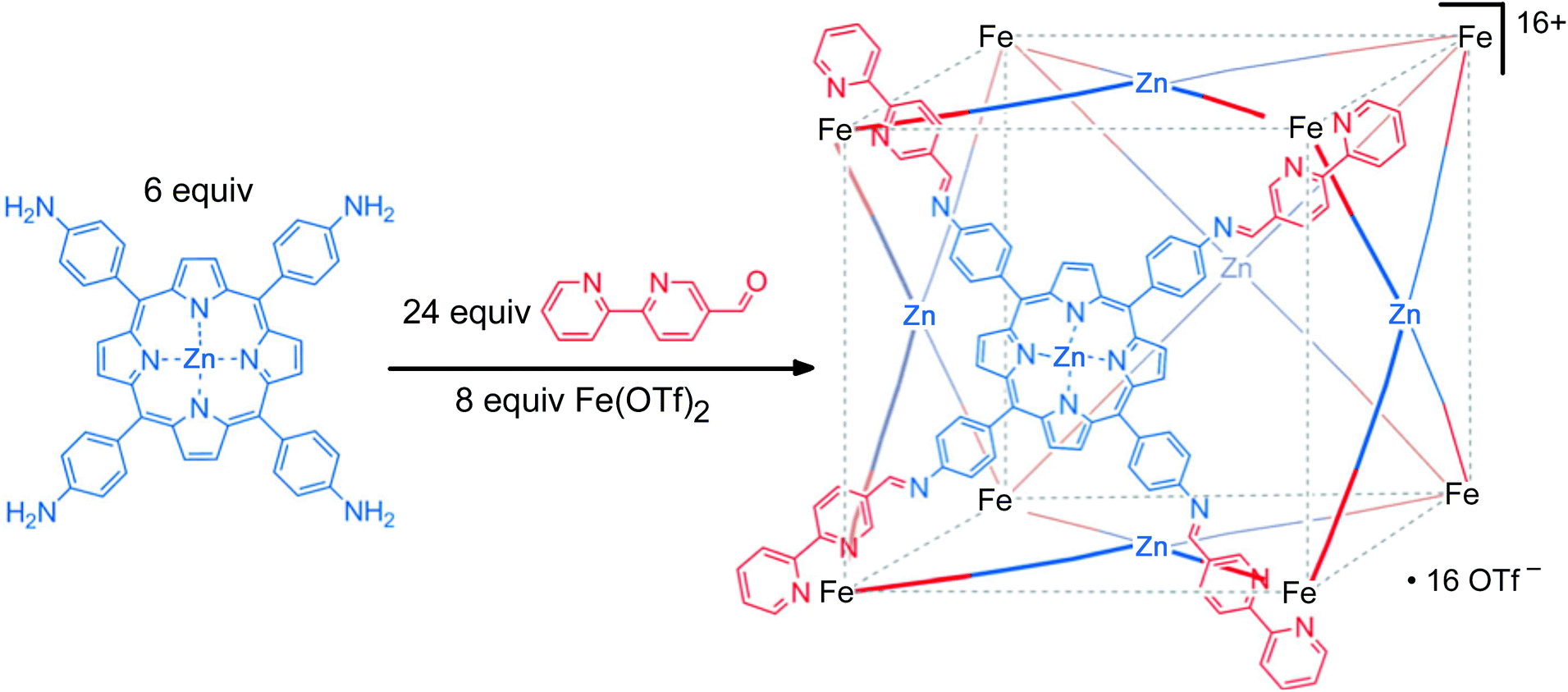 | ||
| Fig. 12 An example of heterometallic self-assembly using the kinetic control approach: a preformed metalloporphyrin subcomponent is used as the basis of synthesising a heterometallic cube (see ref. 33a). | ||
Stepwise self-assembly of heterometallic cages based on bis(pyrazolyl-pyridine) ligands
The preparation of pure fac-[Ru(PyPzH)3](PF6)2 described above, and its elaboration into fac-[Ru(LBn)3](PF6)2 by alkylation with benzyl bromide,24 provided the basis for our route into stepwise assembly of heterometallic versions of some of our coordination cages in which kinetically inert and kinetically labile metal ions are incorporated at pre-determined sites in the cage superstructure.Heterometallic octanuclear cubic cages
We targeted in the first instance the octanuclear cubic cage [M8(L1,5-nap)12]16+ for two reasons. Firstly, we know that it has particularly extensive and interesting host/guest chemistry: being able to include redox metal ions such as Ru(II) in the cage might, for example, allow guest binding to be modulated by a redox change in the host. Secondly, the combination of two fac and six mer tris-chelate vertices that this cage contains7a turned out to be particularly synthetically convenient, as explained below.The eight metal vertices of the [M8(L1,5-nap)12]16+ cube can be split into two subsets of four (Ma and Mb, Fig. 13). When a kinetically inert metal ion with pendant binding sites is used as a building block and combined with labile metal ions in the next step, it must follow that ‘inert’ and ‘labile’ ions strictly alternate around the periphery. For [M8(L1,5-nap)12]16+ we find that each subset of ions – Ma and Mb – contains one fac and three mer tris-chelate units. This is a consequence of the fact that the two fac vertices lie at opposite ends of a long diagonal, such that each subset of four metal vertices can only contain one of the two fac vertices. Thus, if we can prepare [Ru(L1,5-nap)3]2+ (mononuclear complex with three pendant binding sites) as a 1:3 mixture of fac and mer isomers, we will have one subset of four metal ions Ma as the required combination of geometric isomers, plus all twelve ligands needed for the cage. Combination of this with labile ions Mb, such as Co(II) or Cd(II), in the second step will result in assembly of the pendant twelve binding sites around the four Mb ions to complete assembly of the cage, with the necessary 1:3 fac:mer ratio of Mb ions arising naturally from the assembly process. What makes this particularly convenient is that a 1:3 mixture of fac and mer isomers of [Ru(L1,5-nap)3]2+ is exactly what is expected to arise on statistical grounds8 during its synthesis from a source of Ru(II) and three equivalents of L1,5-nap, which means that no separation of isomers or isomer-specific synthetic routes should be required.
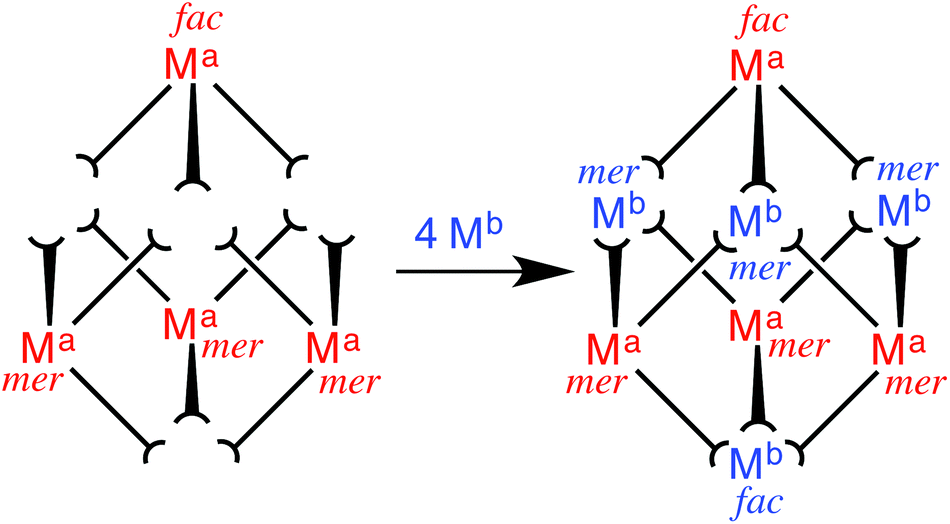 | ||
| Fig. 13 Sketch outlining the stepwise synthetic strategy used to prepare the heterometallic cubic cage complexes: viz. the combination of pre-formed, kinetically inert [(Ma)(L1,5-nap)3]2+ (Ma = Ru, Os) with additional labile ions (Mb)2+ (Mb = Co, Cd) in a 4:4 ratio to give octanuclear [(Ma)4(Mb)4(L1,5-nap)12]16+ (ref. 35). | ||
Reaction of RuCl2(dmso)4 with excess L1,5-nap yielded [Ru(L1,5-nap)3]2+ in the required 3:1 mer:fac ratio, as determined by 1H NMR spectroscopy.35 Each L1,5-nap ligand is coordinated to Ru(II) through one pyrazolyl-pyridine terminus with the second one pendant and available for propagation of the cage. Although bulky ligands sometimes give a higher than expected proportion of the mer isomer for steric reasons,8 that was not the case here. This kinetically inert subcomponent, which possesses three pendant pyridyl-pyrazole units, was then combined with Cd(ClO4)2 to complete assembly of the cage. The labile Cd(II) ions direct the assembly of the pendant binding groups from the pre-formed Ru(II) complexes in a reversible self-assembly step, and following crystallisation from nitromethane the mixed-metal [Ru4Cd4(L1,5-nap)12](ClO4)16 cage was isolated.35
The identity of the cage was confirmed by solution and solid-state studies. High-resolution mass spectrometry confirmed the Ru4Cd4 formulation, and the synthetic route used necessarily means that these two metal ion types should alternate around the cage. This was confirmed crystallographically (Fig. 14): the heteronuclear cage [Ru4Cd4(L1,5-nap)12](ClO4)16 is basically isostructural with the homonuclear analogues reported previously,7a,16,17,20 but significantly different M–N bond distances around Ru(II) and Cd(II) allowed unambiguous assignment of the metal ions. In addition, deliberate mis-assignment of the Ru metal centres as Cd (and vice versa) resulted in significantly worsened thermal displacement parameters for those atoms during the refinement, confirming the correct assignment.35
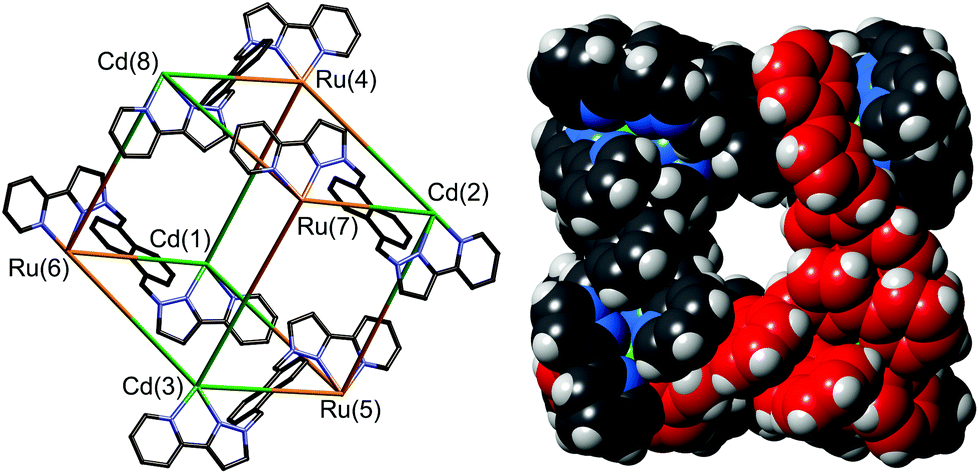 | ||
| Fig. 14 Left: A view of [Ru4Cd4(L1,5-nap)12](ClO4)16 emphasizing the approximately cubic array of metal ions with four of the bridging ligands included (ref. 35). Right: Space-filling view of the complex cation of [Os4Cd4(L1,5-nap)12](ClO4)16, with one of the pre-formed fac-[Os(L1,5-nap)3]2+ units highlighted in red (ref. 36). | ||
We could perform an identical type of synthesis using Os(II) as the kinetically inert component, allowing stepwise formation of heterometallic [Os4M4(L1,5-nap)12]16+ (M = Co, Cd) cages.36 In these cases the metal ions at the Ma and Mb sites could not be distinguished crystallographically, as disorder in the cage orientation in the crystal means that all metal sites had to be refined as a 50:50 mixture of Os(II) and the labile metal ion. However, as before, the high-resolution ES mass spectra confirmed the cage formulation with four of each type of metal ion; and the synthetic route necessarily results in the metal ions alternating around the periphery, as was established for the Ru4Cd4 cage.
Incorporation of Ru(II) or Os(II) tris-diimine units into the octanuclear cages brings new behaviour to the cages in the form of redox activity associated with reversible M(II)/M(III) couples for both Ru and Os. For both the Ru4Cd4 and Os4Cd4 cages, cyclic voltammetry revealed a single chemically reversible redox process associated with all four metal-centred redox processes occurring simultaneously: unsurprisingly, the Ru(II) or Os(II) ions within a cage are too far apart to show any electronic coupling which would allow the four one-electron process to occur at different potentials and be individually resolved. For the Ru4Cd4 cage the redox process occurred at +0.96 V vs. the ferrocene/ferrocenium couple (Fc/Fc+);35 for the Os4Cd4 cage it was at +0.46 V vs. Fc/Fc+ (Fig. 15).36 The considerably greater ease of oxidation of Os(II) compared to Ru(II) in the same coordination environment is normal and arises from the smaller effective nuclear charge experienced by the 5d electrons of Os(II) compared to the 4d electrons of Ru(II). In both cases, these redox processes are (i) at similar potentials to those observed for the mononuclear Ru(II) or Os(II) subcomponent complexes in the same coordination environment, and (ii) fully chemically reversible, meaning that the cage can be switched in a single step between 16+ and 20+ forms without any decomposition. This was further confirmed by the observations that (i) aerial oxidation of the {OsII4CdII4}16+ cage to the {OsIII4CdII4}20+ form resulted in no significant change in diffusion coefficient by DOSY NMR studies, and (ii) overlaid UV/Vis absorption spectra recorded during gradual oxidation of {OsII4CdII4}16+ cage to {OsIII4CdII4}20+ showed a clear isosbestic point.36
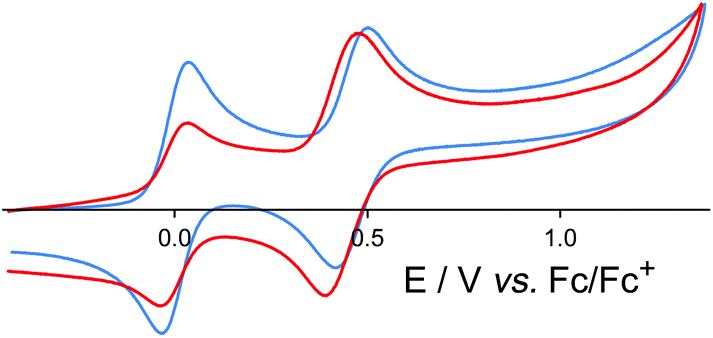 | ||
| Fig. 15 Cyclic voltammograms in MeCN of [Os(L1,5-nap)3](PF6)2 (blue trace) and [Os4Cd4(L1,5-nap)12](ClO4)16 (red trace) at a scan rate of 0.1 V s−1. In both cases the reversible wave at 0.0 V is internal ferrocene added as a reference (ref. 36). | ||
Given that we have demonstrated how binding of electron-rich guests in organic solvents involves a substantial contribution from charge-assisted hydrogen-bonding to the internal surface of the cage,17 a reversible redox swing could affect the strength of the host–guest interaction and may provide a mechanism for controlling uptake and release of bound guests that operates in a complementary way to changing the charge on the guest by protonation/deprotonation.20d
In addition to the redox properties imparted to the cages, the Os4Cd4 cage was found to have potentially valuable photophysical properties arising from a long-lived 3MLCT excited state that is a good photo-electron donor. The absorption spectrum of the mononuclear ‘vertex unit’ [Os(L1,5-nap)3](PF6)2 (as-synthesised 1:3 mix of fac and mer isomers) showed features typical of an Os(II) tris-diimine complex (Fig. 16), with spin-allowed 1MLCT absorptions at around 400 nm, plus a weaker spin-forbidden 3MLCT absorption manifold which provides a low-energy absorption tail in the 500–600 nm region.
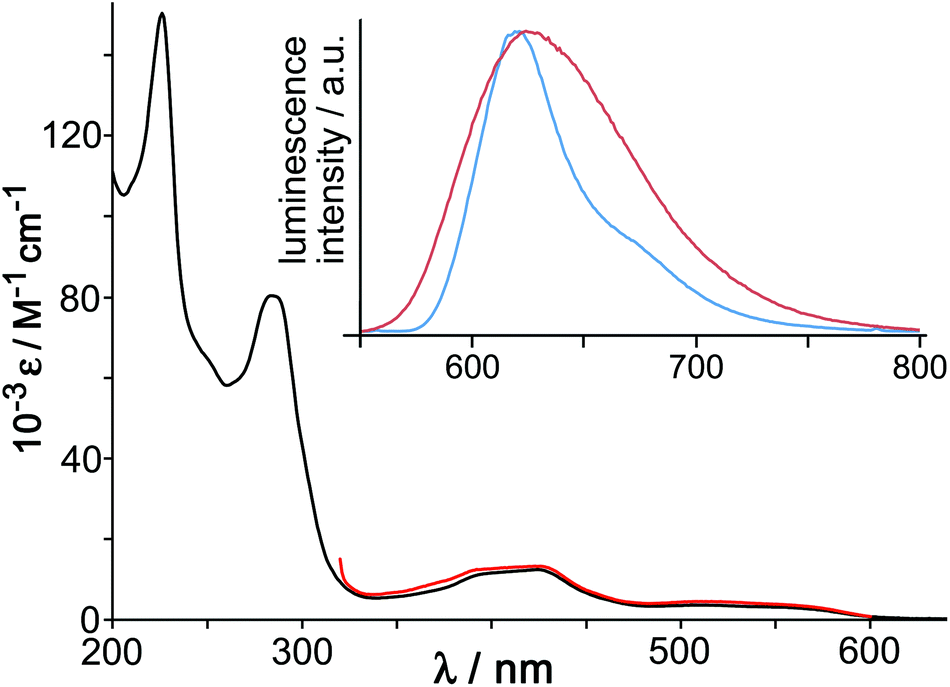 | ||
| Fig. 16 Main figure: UV/Vis absorption spectrum of [Os(L1,5-nap)3](PF6)2 in MeCN (black) with, overlaid in red, the excitation spectrum for the Os-based emission at 620 nm. Inset: luminescence spectra of [Os(L1,5-nap)3](PF6)2 in MeCN fluid solution at room temperature (red trace) and of [Os(L1,5-nap)3]Cl2 in EtOH/MeOH (4:1, v/v) glass at 77 K (blue trace) (ref. 36). | ||
[Os(L1,5-nap)3](PF6)2 showed a typical broad, structureless luminescence band with a maximum at 625 nm in MeCN (Fig. 16, inset) and two lifetime components of 200 ns (minor component) and 72 ns (major component), consistent with the presence of a mixture of fac and mer isomers.36 At 77 K the emission spectrum of [Os(L1,5-nap)3]2+ is sharpened and the maximum slightly blue-shifted with the highest energy feature at 620 nm (Fig. 16, inset). The 3MLCT energy content is therefore 16100 cm−1, which is relatively high for an Os(II) tris-diimine unit;37,38 and this, together with the ease with which the Os(III) state can be formed by oxidation, makes the photo-excited state of [Os(L1,5-nap)3](PF6)2 a better electron donor than the well-known [Ru(bipy)3]2+ unit that has been widely used in photocatalysis.38
The photophysical properties of the mononuclear [Os(L1,5-nap)3](PF6)2 unit were essentially unchanged when these units are incorporated into the cage [Os4Cd4(L1,5-nap)12](ClO4)16. Significantly, the strong naphthalene-based fluorescence characteristic of the free ligand, and which we also saw in the cage complex [Cd8(L1,5-nap)12](ClO4)16,7a is completely quenched; this implies the presence of (naphthyl) → Os(II) energy-transfer from the stacked ligand array to the Os(II) ions at the vertices of the cage.
This cage accordingly has the potential be a good excited-state photoelectron donor to electron-deficient guests that occupy the central cavity. A particularly exciting possibility is that the array of four long-lived chromophores surrounding a single bound guest may provide the possibility of multiple photoinduced electron transfer events to a single guest in a supramolecular assembly. Such studies are in progress.
A heterometallic Ru4Cd12 cage
The stepwise synthetic methodology described above was then taken a significant step further, with preparation of a hexadecanuclear cage5c,39,40 containing not just two different types of metal ion at different vertices of the metal polyhedron, but also containing a mixture of two different types of ligand along different edges. For this we focussed on the [M16L24]32+ cages, which can be formed with either Lp-ph or L1,4-nap along the edges, and any of several octahedral dications at the vertices.5c,39,40 As this is the largest of the cages in this family identified so far it presents a particularly appealing challenge; and the arrangement of fac and mer tris chelate units in this type of cage lends itself particularly well to a stepwise assembly approach.The arrangement of metal ion geometries is such that there are four triangular {M3(μ-L)3}6+ cyclic helical units, in which all three metal ions have a mer geometry. These are connected by fac-{ML3}2+ units, each of which links to three different {M3(μ-L)3}6+ units via its pendant binding sites. The result is a structure best described as a capped truncated tetrahedron, with the four fac-{ML3}2+ vertices being arrayed at the vertices of a large tetrahedron; although somewhat cumbersome to describe this is easy to visualise in Fig. 17.
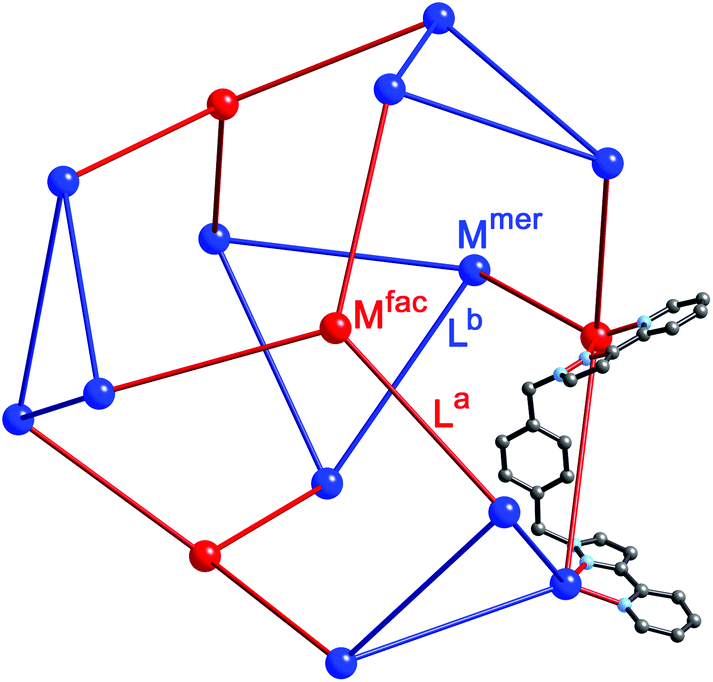 | ||
| Fig. 17 Representation of the core structure of [Cd16(Lp-ph)24](ClO4)32 (ref. 39) with one bridging ligand included. All metal sites are Cd(II) but the two different types of geometric isomer are colour-coded: fac tris-chelates are in red (Mfac, see main text) and mer tris-chelates are in blue (Mmer). Likewise the two ligand environments are La in red, and Lb in blue. The blue triangles are M3(μ-L)3 cyclic helicates subcomponents, four of which are connected by (red) ML3 units. | ||
A ‘retrosynthetic analysis’ of the cage allows us to identify how best to dissect the structure into component parts for a stepwise synthesis. The 16 metal ions can be conveniently split into a set of four (fac, isolated from one another; red in Fig. 16) and a set of 12 (mer, connected to one another in sets of three; blue in Fig. 16). The ligands likewise can be split into two types: those that connect a fac to a mer vertex (La, red), and those connecting two mer vertices in the cyclic helicate triangular panels (Lb, blue). There are 12 of each type of ligand. As with the mixed-metal cubes, our initial choice of metal ions is Ru(II) for the kinetically inert vertices, given the straightforward and well-established synthesis and purification of stable tris-chelate complexes as their pure fac and mer isomers;24 and diamagnetic Cd(II) for the kinetically labile vertices to facilitate 1H NMR analysis. The most logical solution is to use Ru(II) at the four Mfac sites and Cd(II) at the twelve Mmer sites. This will permit the three pendant binding sites associated with each (inert) Ru(II) unit to connect to three (labile) Cd(II) ions during the second assembly step. Around the triangular helical faces, three Cd(II) ions will end up adjacent to one another but as they are kinetically labile that is not an issue.
The strategy requires preparation of homoleptic fac-[Ru(La)3]2+ units in which La could be either Lp-ph or L1,4-nap. Each of these will necessarily bind to three Cd(II) ions in the subsequent assembly step. The remaining bridging ligands Lb, which connect the Cd(II) ions around the triangular cyclic helicate units, will need to be added separately to the reaction. There is no problem with them being chemically different from La, as long as both La and Lb support the same cage structure (as Lp-ph and L1,4-nap do).5c,40 This dissection of the structure therefore suggests that a cage containing two different metal ions at predictable positions, and two different ligand types at predictable positions, can be assembled from four pre-prepared fac-[Ru(Lp-ph)3]2+ units, twelve additional Cd(II) ions, and twelve additional ligands L1,4-nap to give the heteronuclear, heteroleptic cage [Ru4Cd12(Lp-ph)12(L1,4-nap)12]32+ (Fig. 18). In this assembly Mfac = Ru; Mmer = Cd; La = Lp-ph; Lb = L1,4-nap and there is no possibility for disorder of metal ions or ligand types between sites as long as the [Ru(Lp-ph)3]2+ units remain stable as the fac isomer.
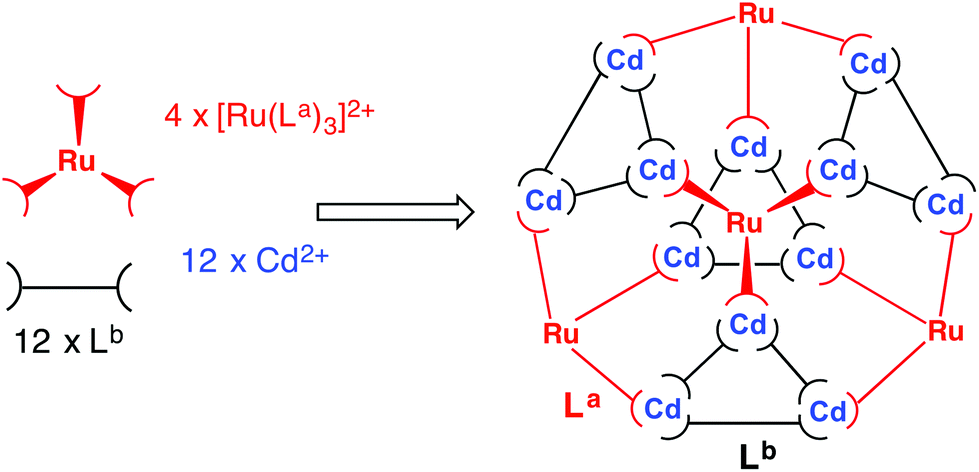 | ||
| Fig. 18 Sketch of the stepwise synthetic strategy used to prepare the heterometallic, mixed-ligand cage: viz. combination of pre-formed fac-[Ru(La)3]2+ (red), additional labile Cd2+ ions (blue), and free ligand (Lb, black) in a 4:12:12 ratio to give hexadecanuclear [Ru4Cd12(La)12(Lb)]32+ (ref. 43). | ||
The key to this is therefore preparation of the kinetically inert Ru(II) complex [Ru(Lp-ph)3][PF6]2 as the pure fac isomer. To avoid any tedious separation of isomers, we prepared isomerically pure fac-[Ru(Lp-ph)3][PF6]2 using the same method that we described above for pure fac-[Ru(LBn)3](PF6)2, which was used in the modelling of coordination cage vertices.24 Thus, fac-[Ru(PyPzH)3](PF6)2 was alkylated at the pyrazolyl NH positions using the previously-reported intermediate A,41 which consists of a pyrazolyl-pyridine terminus connected to a para-xylyl-bromide (Fig. 19), under conditions mild enough to prevent any fac/mer scrambling at the Ru(II) centre. This afforded pure fac-[Ru(Lp-ph)3](PF6)2; its crystal structure shows how all three pendant arms are arranged on the same face of the complex (Fig. 20).42
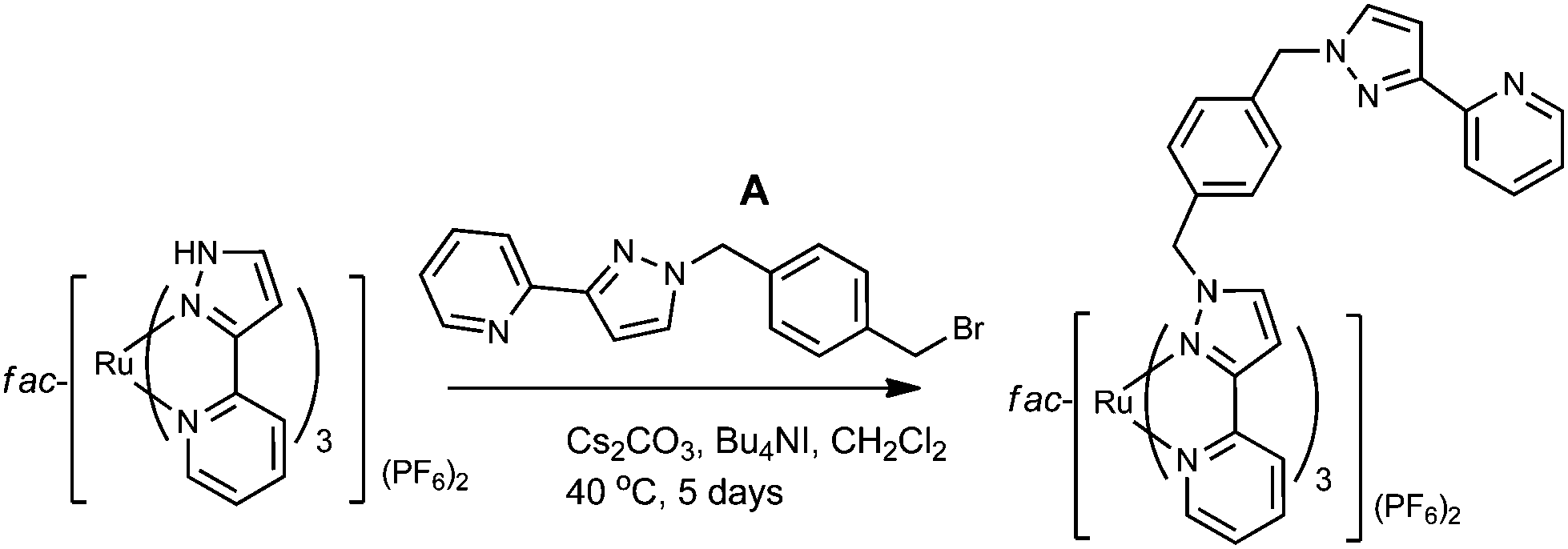 | ||
| Fig. 19 Preparation of fac-[Ru(Lp-ph)3](PF6)2 (ref. 42). | ||
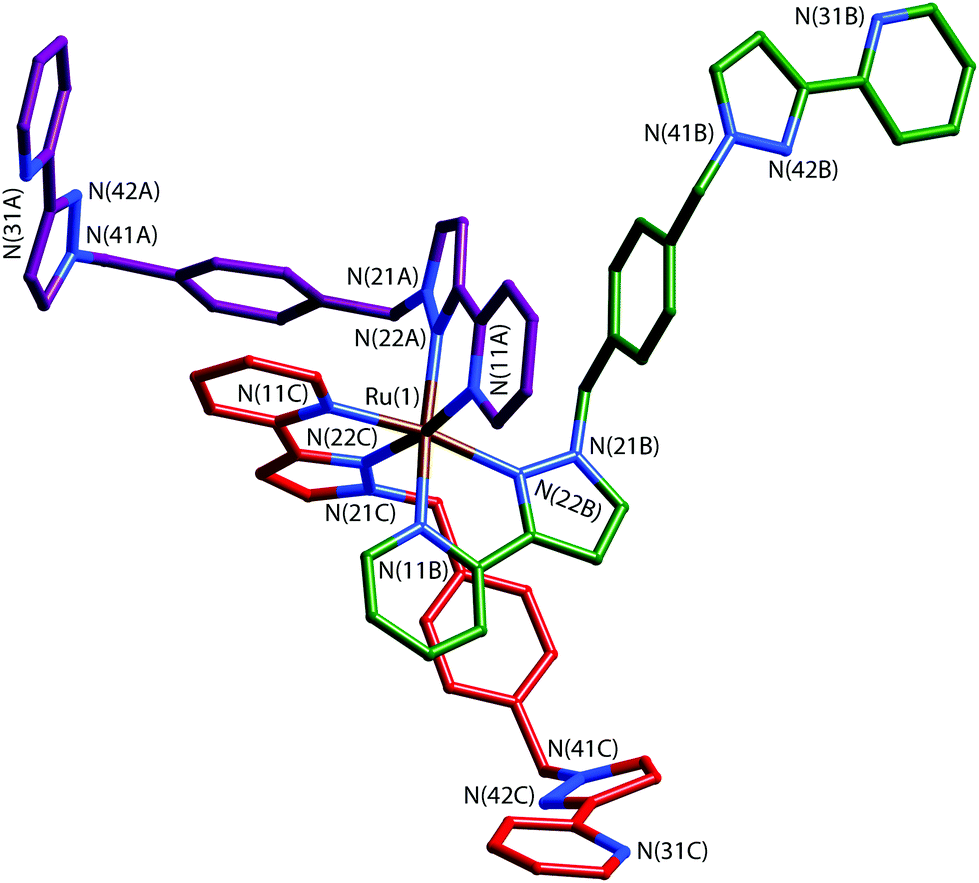 | ||
| Fig. 20 Structure of the complex cation of fac-[Ru(Lp-ph)3][PF6]2·acetone with the three ligands coloured differently for clarity (ref. 42). | ||
This is the key intermediate that makes the synthetic strategy proposed in Fig. 18 possible. The subcomponent fac-[Ru(Lp-ph)3][PF6]2 was combined with Cd(BF4)2 and L1,4-nap in a 1:3:3 ratio – all of the component parts necessary to complete assembly of the mixed metal, mixed-ligand cage – in nitromethane. Recrystallisation over several weeks afforded the product as X-ray quality yellow crystals, whose crystal structure revealed the desired cage complex [Ru4Cd12(Lp-ph)12(L1,4-nap)12](PF6)7(BF4)25 (Fig. 21 and 22).43
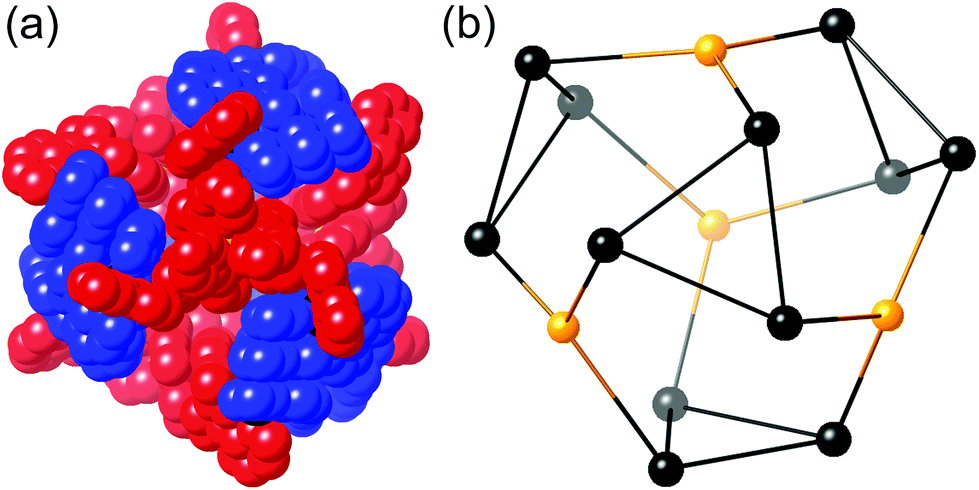 | ||
| Fig. 21 Two views of the crystal structure of [Ru4Cd12(Lp-ph)12(L1,4-nap)12](PF6)7(BF4)25. Left: The entire complex cation in spacefilling view (L1,4-nap shown in blue, Lp-ph shown in red); Right: Structure of the Ru4Cd12 core (Ru shown in yellow, Cd shown in black) (ref. 43). | ||
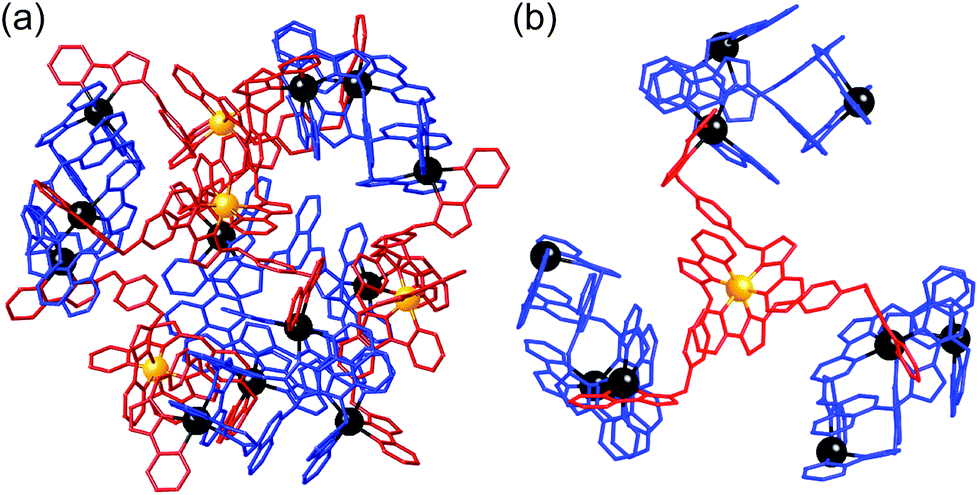 | ||
| Fig. 22 Left: View of the complete complex cation of [Ru4Cd12(Lp-ph)12(L1,4-nap)12](PF6)7(BF4)25, (same colouring scheme as Fig. 21). Right: Partial view of the complex, emphasising how each fac-[Ru(Lp-ph)3]2+ vertex is connected to three [Cd3(L1,4-nap)3]6+ cyclic helicate subunits (ref. 43). | ||
It is clear from the crystal structure that the core structure of the cage is the same as that of the previously reported [M16L24]32+ cages,5c,39,40 as required, with sixteen metal ions arrayed in a tetra-capped truncated tetrahedral array with M⋯M separations along the edges in the region of 10 Å (Fig. 21 and 22). The identities of the Ru(II) and Cd(II) ions are clear from their significantly different M–N distances (Ru–N, 2.0–2.1 Å; Cd–N, 2.3–2.4 Å). The different ions are in their allotted positions according to the synthetic scheme with the Ru(II) ions occupying the four fac sites and the Cd(II) ions occupying the twelve mer sites. The two different types of ligand are trivial to distinguish as they are chemically different, with the twelve Lp-ph ligands (containing a 1,4-phenylene spacer) spanning the Ru⋯Cd edges, and the twelve L1,4-nap ligands spanning the Cd⋯Cd edges around the Cd3 triangles. The other features of the structure (inter-ligand stacking around the cage periphery, disposition of anions both inside the cavity and around the cage surface) are the same as has been reported before with the parent {M16L24}32+ cages.5c,39,40
The cage is stable in solution, with high-resolution ESMS confirming the cage formulation and an 800 MHz 1H NMR spectrum confirming the expected symmetry, with two independent ligand types in a 1:1 ratio, each with no internal symmetry, corresponding to 42 independent 1H signals of equal intensity. A DOSY spectrum clearly confirmed the presence of a single species with a diffusion coefficient typical of a cage of this size but quite different from that of any mononuclear species such as fac-[Ru(Lp-ph)3][PF6]2.42 Unlike other cages based on Lp-ph as the bridging ligand which rearrange slowly (days/weeks) in solution,5c [Ru4Cd12(Lp-ph)12(L1,4-nap)12](PF6)7(BF4)25 is indefinitely stable, which we ascribe to the presence of the L1,4-nap ligands in the ligand set which provide an increased surface area for inter-ligand π-stacking compared to Lp-ph.40 In addition the presence of four kinetically inert Ru(II) centres will aid solution stability, as these are unlikely to undergo dissociation of a chelating ligand under mild conditions – which is the essential first step to rearrangement of a coordinatively saturated complex at room temperature.
It is clear from this that the ability to prepare kinetically inert Ru(II)-based mononuclear ‘complex ligands’ with the necessary isomeric purity provides a powerful tool for the rational synthesis of these cages. In principle the same methodology could be extended to other octahedral metal ions with desirable redox or photophysical properties [Os(II), Ir(III)…] to prepare new types of cage with specific types of useful, metal-based property incorporated into the metal superstructure at precisely controlled positions.
Stepwise self-assembly of an adamantoid Ru4Ag6 structure
Finally, we show how we have extended the work reported in the previous section to the preparation of a new cage type based on a mixture of metal ions with different coordination preferences; six-coordinate octahedral Ru(II), and four-coordinate tetrahedral Ag(I).Having prepared geometrically pure fac-[Ru(Lp-ph)3][PF6]2 we also tested the assembly of this complex with Ag(I) salts. The hope was that the use of a kinetically labile metal ion with a markedly different coordination geometry to Ru(II) would provide an entry into structurally novel assemblies. On the basis that three pendant bidentate sites are available for coordination from fac-[Ru(Lp-ph)3](PF6)2, and that Ag(I) ions are likely to prefer a four-coordinate geometry with two bidentate chelating ligands, we combined fac-[Ru(Lp-ph)3](PF6)2 with 1.5 equivalents of AgPF6 (following, again, the principle of maximum site occupancy).44 If each pendant ligand fragment from the fac-[Ru(Lp-ph)3](PF6)2 unit coordinates to a different Ag(I) ion, as is likely on steric grounds given the distance between the pendant pyrazolyl-pyridine units, we might expect a mixed-metal cage in which each [Ru(Lp-ph)3]2+ unit caps a triangular array of Ag(I) ions.
The crystalline product proved to be the decanuclear Ru4Ag6 cage [Ru4(Lp-ph)12Ag6](PF6)14 (Fig. 23 and 24). The cage core has an adamantane-like structure, with a Ru(II) tris-chelate unit at each of the four three-connected vertices which are arranged in an approximate tetrahedron. An Ag(I) bis-chelate unit occupies each of the six two-connected vertices. The structure can therefore be described as a tetrahedral array of Ru(II) ions with an Ag(I) ion lying in the centre of each Ru⋯Ru edge (Fig. 23), with every adjacent Ru(II)/Ag(I) pair connected by a bis-bidentate bridging ligand Lp-ph.
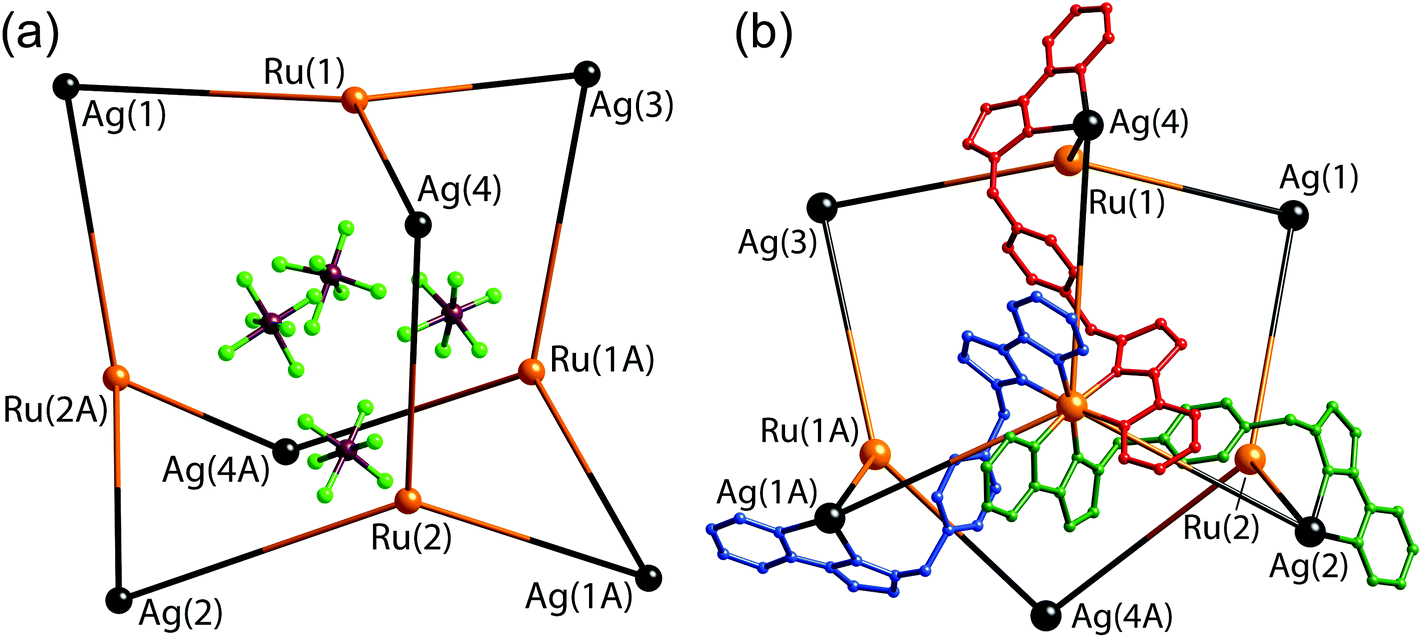 | ||
| Fig. 23 Two views of the structure of [Ru4(Lp-ph)12Ag6](PF6)14: (a) the adamantane-like arrangement of metal ions, with the four anions that lie within the cavity also shown; (b) the metal superstructure with three of the bridging ligands included (ref. 42). | ||
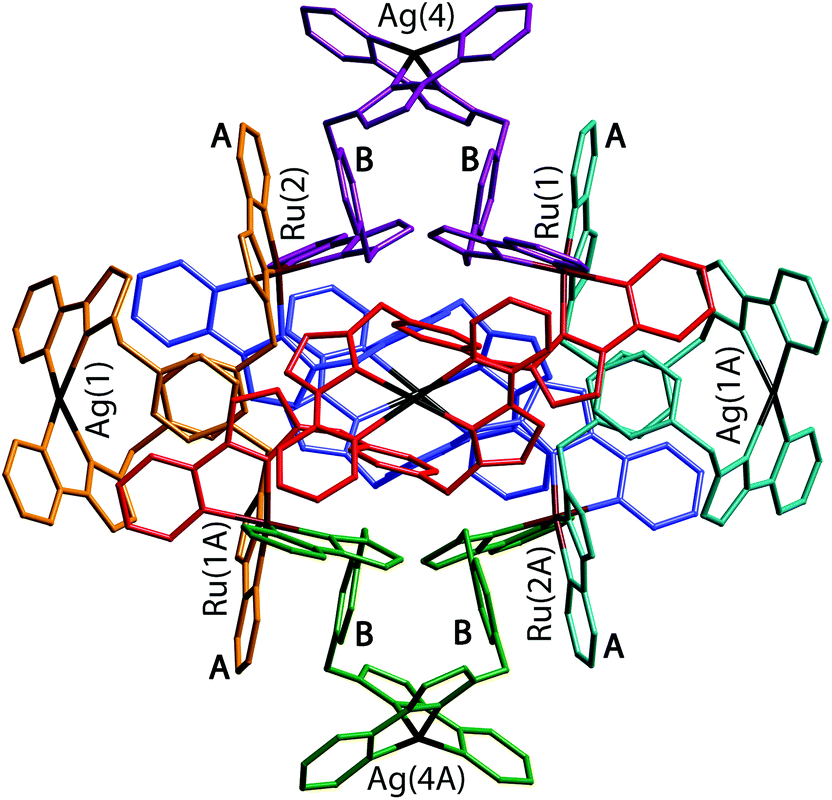 | ||
| Fig. 24 A view of the complete complex cation of [Ru4(Lp-ph)12Ag6](PF6)14. The two ligands coordinated to each Ag(I) have the same colour. Labels A and B denote the electron-deficient (pyrazolyl-pyridine) and electron-rich (phenyl) units involved in the pairwise π-stacking interactions (ref. 42). | ||
The flexibility of the ligands associated with the CH2 ‘hinges’ allows them to adopt a conformation which maximises inter-ligand π-stacking,2c with every central (electron-rich) phenyl ring of a bridging ligand (labelled ‘B’ in Fig. 24) lies parallel to, and overlapping with, an (electron deficient) pyrazolyl-pyridine unit of another ligand coordinated to a Ru(II) ion (labelled ‘A’ in Fig. 24), forming alternating stacks of electron-rich and electron-deficient components. In the view in Fig. 24 we can see four such A/B stacked pairs; there are twelve such interactions overall, involving every phenyl group.
The structural integrity of the complex in solution was confirmed by ES mass spectrometry and 1H NMR spectroscopy. That this species is a large assembly is confirmed by its DOSY spectrum which clearly shows that all of its 1H signals belong to a single species which has a much lower diffusion rate than fac-[Ru(Lp-ph)3][PF6]2.42
Assembly of this adamantanoid cage therefore is based on an obviously different type of synthetic control compared to the examples using purely octahedral metal ions, with its adamantane-like structure thus relies on two different types of geometric control at different metal sites. It requires a combination of metal that are three-connected for the vertices of the cage [each tris-chelate, Ru(II) ion is connected to three Ag(I) ions]; and two connected [each bis-chelate Ag(I) ion is connected to two Ru(II) ions] along the edges of the cage. As with the first two examples reported above however, the principle of using a pre-formed, kinetically inert subcomponent which is combined with a labile metal ion a separate step is still crucial.
Conclusions
Coordination cage chemistry has come a long way since the days of exploratory synthesis, when simply combining labile components and characterising the products that formed was the extent to which any planning was realistically achievable. As the ‘rules’ of self-assembly have become ever more clear to the supramolecular chemist, rational design of more complicated structures becomes increasingly feasible. The synthetic methodology that we have developed – based on the preparation of kinetically inert mononuclear subcomponents with a particular fac:mer isomeric ratio, to exploit the distribution of such sites in the cages – has clear benefits. Firstly, it is a useful synthetic tool which allows synthetic control that can be exploited in the rational synthesis of new types of cage. Secondly, it allows specific types of metal-based functionality (we have used the examples of redox activity and luminescence) to be included in the cage superstructure at specific sites, thus allowing cages to be used as active participants in functional behaviour rather than just passive ‘containers’. Finally – and this is specific to our cage family – this has potential importance for control of guest binding, as binding of electron-rich guests in the cage cavities involves specific hydrogen-bonding interactions with the cage interior surface at the fac tris-chelate sites.
Acknowledgements
We thank the funding agencies that have supported this work (EPSRC, Leverhulme Trust) and the numerous talented co-workers whose efforts made this possible; their names appear in the reference list.References
- C. J. Pedersen, Angew. Chem., Int. Ed. Engl., 1988, 27, 1021 CrossRef.
- (a) D. Fiedler, D. H. Leung, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2005, 38, 349 CrossRef CAS PubMed; (b) M. Fujita, M. Tominaga, A. Hori and B. Therrien, Acc. Chem. Res., 2005, 38, 369 CrossRef CAS PubMed; (c) M. D. Ward, Chem. Commun., 2009, 4487 RSC; (d) R. Chakrabarty, P. S. Mukherjee and P. J. Stang, Chem. Rev., 2011, 111, 6810 CrossRef CAS PubMed; (e) T. R. Cook and P. J. Stang, Chem. Rev., 2015, 115, 7001 CrossRef CAS PubMed; (f) M. D. Pluth, R. G. Bergman and K. N. Raymond, Acc. Chem. Res., 2009, 42, 1650 CrossRef CAS PubMed; (g) M. M. J. Smulders, I. A. Riddell, C. Browne and J. R. Nitschke, Chem. Soc. Rev., 2013, 42, 1728 RSC; (h) H. Amouri, C. Desmarets and J. Moussa, Chem. Rev., 2012, 112, 2015 CrossRef CAS PubMed; (i) L. Li, D. J. Fanna, N. D. Shepherd, L. F. Lindoy and F. Li, J. Inclusion Phenom. Macrocyclic Chem., 2015, 82, 3 CrossRef CAS; (j) L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201 CrossRef CAS PubMed; (k) H. Li, Z.-J. Yao, D. Liu and G.-X. Jin, Coord. Chem. Rev., 2015, 293, 139 CrossRef.
- M. D. Ward and P. R. Raithby, Chem. Soc. Rev., 2013, 42, 1619 RSC.
- (a) O. Chepelin, J. Ujma, X. Wu, A. M. Z. Slawin, M. B. Pitak, S. J. Coles, J. Michel, A. C. Jones, P. E. Barran and P. J. Lusby, J. Am. Chem. Soc., 2012, 134, 19334 CrossRef CAS PubMed; (b) K.-I. Yamashita, M. Kawano and M. Fujita, Chem. Commun., 2007, 4102 RSC; (c) S. Roche, C. Haslam, H. Adams, S. L. Heath and J. A. Thomas, Chem. Commun., 1998, 1681 RSC; (d) J. Yang, M. Bhadbade, W. A. Donald, H. Iranmanesh, E. G. Moore, H. Yan and J. E. Beves, Chem. Commun., 2015, 51, 4465 RSC.
- (a) J. S. Fleming, K. L. V. Mann, C. A. Carraz, E. Psillakis, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 1998, 37, 1279 CrossRef CAS; (b) R. L. Paul, Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4883 CrossRef CAS PubMed; (c) A. Stephenson, S. P. Argent, T. Riis-Johannessen, I. S. Tidmarsh and M. D. Ward, J. Am. Chem. Soc., 2011, 133, 858 CrossRef CAS PubMed.
- B. R. Hall, L. E. Manck, I. S. Tidmarsh, A. Stephenson, B. F. Taylor, E. J. Blaikie, D. A. Vander Griend and M. D. Ward, Dalton Trans., 2011, 40, 12132 RSC.
- (a) I. S. Tidmarsh, T. B. Faust, H. Adams, L. P. Harding, L. Russo, W. Clegg and M. D. Ward, J. Am. Chem. Soc., 2008, 130, 15167 CrossRef CAS PubMed; (b) A. M. Najar, C. Avci and M. D. Ward, Inorg. Chem. Commun., 2012, 15, 126 CrossRef CAS.
- S. L. Dabb and N. C. Fletcher, Dalton Trans., 2015, 44, 4406 RSC.
- R. L. Paul, S. P. Argent, J. C. Jeffery, L. P. Harding, J. M. Lynam and M. D. Ward, Dalton Trans., 2004, 3453 RSC.
- (a) Z. R. Bell, J. C. Jeffery, J. A. McCleverty and M. D. Ward, Angew. Chem., Int. Ed., 2002, 41, 2515 CrossRef CAS; (b) S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding, O. Mamula and M. D. Ward, Inorg. Chem., 2006, 45, 3905 CrossRef CAS PubMed.
- I. A. Riddell, Y. R. Hristova, J. K. Clegg, C. S. Wood, B. Breiner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 2723 CrossRef CAS PubMed.
- D. L. Caulder, C. Bruckner, R. E. Powers, S. Konig, T. N. Parac, J. A. Leary and K. N. Raymond, J. Am. Chem. Soc., 2001, 123, 8923 CrossRef CAS PubMed.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed.
- (a) C. R. K. Glasson, G. V. Meehan, J. K. Clegg, L. F. Lindoy, P. Turner, M. B. Duriska and R. Willis, Chem. Commun., 2008, 1190 RSC; (b) R. Custelcean, J. Bosano, P. V. Bonnesen, V. Kertesz and B. P. Hay, Angew. Chem., Int. Ed., 2009, 48, 4025 CrossRef CAS PubMed.
- (a) I. A. Riddell, M. M. J. Smulders, J. K. Clegg, Y. R. Hristova, B. Breiner, J. D. Thoburn and J. R. Nitschke, Nat. Chem., 2012, 4, 860 CrossRef CAS; (b) R. A. Bilbeisi, T. K. Ronson and J. R. Nitschke, Angew. Chem., Int. Ed., 2013, 52, 9027 CrossRef CAS PubMed.
- S. Turega, M. Whitehead, B. R. Hall, M. F. Haddow, C. A. Hunter and M. D. Ward, Chem. Commun., 2012, 48, 2752 RSC.
- S. Turega, M. Whitehead, B. R. Hall, A. J. H. M. Meijer, C. A. Hunter and M. D. Ward, Inorg. Chem., 2013, 52, 1122 CrossRef CAS PubMed.
- S. Mecozzi and J. Rebek, Chem. – Eur. J., 1998, 4, 1016 CrossRef CAS.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310 CrossRef CAS PubMed.
- (a) W. Cullen, M. C. Misuraca, C. A. Hunter, N. H. Williams and M. D. Ward, Nat. Chem., 2016, 8, 231 CrossRef CAS PubMed; (b) C. G. P. Taylor, J. R. Piper and M. D. Ward, Chem. Commun., 2016, 52, 6225 RSC; (c) S. Turega, W. Cullen, M. Whitehead, C. A. Hunter and M. D. Ward, J. Am. Chem. Soc., 2014, 136, 8475 CrossRef CAS PubMed; (d) W. Cullen, S. Turega, C. A. Hunter and M. D. Ward, Chem. Sci., 2015, 6, 625 RSC.
- T. Riis-Johannessen, N. Dupont, G. Canard, G. Bernardinelli, A. Hauser and C. Piguet, Dalton Trans., 2008, 3661 RSC.
- (a) N. C. Fletcher, R. T. Brown and A. P. Doherty, Inorg. Chem., 2006, 45, 6132 CrossRef CAS PubMed; (b) N. C. Fletcher, M. Nieuwenhuyzen and S. Rainey, J. Chem. Soc., Dalton Trans., 2001, 2641 RSC; (c) R. T. Brown, N. C. Fletcher, M. Nieuwenhuyzen and T. E. Keyes, Inorg. Chim. Acta, 2005, 358, 1079 CrossRef CAS.
- M. H. W. Lam, S. T. C. Cheung, K. M. Fung and W. T. Wong, Inorg. Chem., 1997, 36, 4618 CrossRef CAS PubMed.
- A. J. Metherell, W. Cullen, A. Stephenson, C. A. Hunter and M. D. Ward, Dalton Trans., 2014, 43, 71 RSC.
- X. K. Sun, D. W. Johnson, D. L. Caulder, K. N. Raymond and E. H. Wong, J. Am. Chem. Soc., 2001, 123, 2752 CrossRef CAS PubMed.
- A. J. Metherell and M. D. Ward, RSC Adv., 2013, 3, 14281 RSC.
- H. B. Wu and Q. M. Wang, Angew. Chem., Int. Ed., 2009, 48, 7343 CrossRef CAS PubMed.
- S. Hiraoka, Y. Sakata and M. Shionoya, J. Am. Chem. Soc., 2008, 130, 10058 CrossRef CAS PubMed.
- M. Wang, V. Vajpayee, S. Shanmugaraju, Y.-R. Zheng, Z. Zhao, H. Kim, P. S. Mukherjee, K.-W. Chi and P. J. Stang, Inorg. Chem., 2011, 50, 1506 CrossRef CAS PubMed.
- G. Kumar and R. Gupta, Chem. Soc. Rev., 2013, 42, 9403 RSC.
- (a) M. M. J. Smulders, A. Jimenez and J. R. Nitschke, Angew. Chem., Int. Ed., 2012, 51, 6681 CrossRef CAS PubMed; (b) M. L. Saha and M. Schmittel, J. Am. Chem. Soc., 2013, 135, 17743 CrossRef CAS PubMed; (c) F. Reichel, J. K. Clegg, K. Gloe, K. Gloe, J. J. Weigand, J. K. Reynolds, C.-G. Li, J. R. Aldrich-Wright, C. J. Kepert, L. F. Lindoy, H.-C. Yao and F. Li, Inorg. Chem., 2014, 53, 688 CrossRef CAS PubMed; (d) I. S. Tidmarsh, H. Fenton and M. D. Ward, Dalton Trans., 2010, 39, 3805 RSC.
- J. A. Thomas, Chem. Soc. Rev., 2007, 36, 856 RSC.
- (a) M. Otte, P. F. Kuijpers, O. Troeppner, I. Ivanovic-Burmazovic, J. N. H. Reek and B. de Bruin, Chem. – Eur. J., 2013, 19, 10170 CrossRef CAS PubMed; (b) P. Ballester, M. Claudel, S. Durot, L. Kocher, L. Schoepff and V. Heitz, Chem. – Eur. J., 2015, 21, 15339 CrossRef CAS PubMed; (c) W. Meng, B. Breiner, K. Rissanen, J. D. Thoburn, J. K. Clegg and J. R. Nitschke, Angew. Chem., Int. Ed., 2011, 50, 3479 CrossRef CAS PubMed; (d) T. Nakamura, H. Ube, M. Shiro and M. Shionoya, Angew. Chem., Int. Ed., 2013, 52, 720 CrossRef CAS PubMed; (e) K. Li, L.-Y. Zhang, C. Yan, S.-C. Wei, M. Pan, L. Zhang and C.-Y. Su, J. Am. Chem. Soc., 2014, 136, 4456 CrossRef CAS PubMed; (f) M. D. Wise, J. J. Holstein, P. Pattison, C. Besnard, E. Solari, R. Scopelliti, G. Bricogne and K. Severin, Chem. Sci., 2015, 6, 1004 RSC.
- (a) S. Rigault, C. Piguet, G. Bernardinelli and G. Hopfgartner, Angew. Chem., Int. Ed., 1998, 37, 169 CrossRef CAS; (b) P. R. Symmers, M. J. Burke, D. P. August, P. I. T. Thomson, G. S. Nichol, M. R. Warren, C. J. Campbell and P. J. Lusby, Chem. Sci., 2015, 6, 756 RSC.
- A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 6330 RSC.
- A. B. Wragg, A. J. Metherell, W. Cullen and M. D. Ward, Dalton Trans., 2015, 44, 17939 RSC.
- L. De Cola, V. Balzani, F. Barigelletti, L. Flamigni, P. Belser, A. Von Zelewsky, M. Frank and F. Vögtle, Inorg. Chem., 1993, 32, 5228 CrossRef CAS.
- C. T. Lin and N. Sutin, J. Phys. Chem., 1976, 80, 97 CrossRef CAS.
- S. P. Argent, H. Adams, T. Riis-Johannessen, J. C. Jeffery, L. P. Harding and M. D. Ward, J. Am. Chem. Soc., 2006, 128, 72 CrossRef CAS PubMed.
- A. Stephenson, D. Sykes and M. D. Ward, Dalton Trans., 2013, 42, 6756 RSC.
- S. P. Argent, H. Adams, L. P. Harding, T. Riis-Johannessen, J. C. Jeffery and M. D. Ward, New J. Chem., 2005, 29, 904 RSC.
- A. J. Metherell and M. D. Ward, Chem. Commun., 2014, 50, 10979 RSC.
- A. J. Metherell and M. D. Ward, Chem. Sci., 2016, 7, 910 RSC.
- J. M. Lehn and A. V. Eliseev, Science, 2001, 291, 2331 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |