DOI:
10.1039/C6DT03005J
(Paper)
Dalton Trans., 2016,
45, 16441-16452
Intrinsic reactivity of a uranium metallacyclopropene toward unsaturated organic molecules†
Received
29th July 2016
, Accepted 12th September 2016
First published on 12th September 2016
Abstract
The uranium metallacyclopropene (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) reacts with various small unsaturated organic molecules. For example, replacement of bis(trimethylsilyl)acetylene occurs when complex 1 is exposed to alkynes, conjugated alkenes, nitriles and quinones. Reaction of 1 with internal phenyl(alkyl)acetylene PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CMe selectively yields the Cs symmetric uranium metallacyclopentadiene (η5-C5Me5)2U[η2-C(Ph)
CMe selectively yields the Cs symmetric uranium metallacyclopentadiene (η5-C5Me5)2U[η2-C(Ph)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)–C(Ph)C(Me)] (6) after the loss of bis(trimethylsilyl)acetylene, while treatment of 1 with phenyl(silyl)acetylenes (PhCCR, R = SiHMe2, SiMe3) gives the corresponding C2v symmetric isomers (η5-C5Me5)2U[η2-C(R)C(Ph)–C(Ph)C(R)] (R = SiHMe2 (7), SiMe3 (8)). Furthermore, while no deprotonation occurs between complex 1 and pyridine derivatives, cyclohexanone can be inserted into the uranium metallacyclopropene moiety of 1 to yield the five-membered, heterocyclic complex (η5-C5Me5)2U[OC(CH2)5(C2(SiMe3)2)] (14) in quantitative conversion. Density functional theory (DFT) studies have been performed to complement the experimental studies.
C(Me)–C(Ph)C(Me)] (6) after the loss of bis(trimethylsilyl)acetylene, while treatment of 1 with phenyl(silyl)acetylenes (PhCCR, R = SiHMe2, SiMe3) gives the corresponding C2v symmetric isomers (η5-C5Me5)2U[η2-C(R)C(Ph)–C(Ph)C(R)] (R = SiHMe2 (7), SiMe3 (8)). Furthermore, while no deprotonation occurs between complex 1 and pyridine derivatives, cyclohexanone can be inserted into the uranium metallacyclopropene moiety of 1 to yield the five-membered, heterocyclic complex (η5-C5Me5)2U[OC(CH2)5(C2(SiMe3)2)] (14) in quantitative conversion. Density functional theory (DFT) studies have been performed to complement the experimental studies.
Introduction
Metallacyclopropenes have various synthetic and catalytic applications,1–3e.g., metallacyclopropenes of group 4 metallocenes have been employed for the preparation of complex organic molecules or heterocyclic main group element compounds.1–3 Therefore group 4 metallacyclopropenes bearing the Cp′2M fragment (where Cp′ = substituted or unsubstituted η5-cyclopentadienyl) represent synthetically useful synthons liberating the coordinated alkyne under mild conditions and transferring the Cp′2M(II) fragment when reacted with unsaturated substrates.1,2 While group 4 chemistry is now well established, the corresponding actinide and lanthanide metallacycles have been neglected.1g,4 This is remarkable considering the recent advances in actinide mediated small molecule activation,1g,5 in which the influence of 6d and 5f orbitals on the reactivity of these species has been evaluated.6 In the course of our investigations, we have recently reported on stable actinide metallacyclopropenes [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2)7a and (η5-C5Me5)2U[η2-C2(SiMe3)2] (1).8 Interestingly, whereas the alkyne in the thorium metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) is strongly coordinated and reacts as a nucleophile towards hetero-unsaturated molecules or as a strong base inducing intermolecular C–H bond activation,7a,b replacement of the coordinated alkyne occurs when the uranium metallacyclopropene (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) reacts with unsaturated molecules such as alkynes, imines, bipy, carbodiimide, organic azides, and diazene derivatives.8 Encouraged by these remarkably different reactivities, we have now extended the substrate scope, and report herein on its reaction with pyridine derivatives, imines, (un)symmetrically substituted internal alkynes, conjugated alkenes, quinones, ketones and nitriles. These studies are also compared to those with related thorium metallacyclopropenes.9
Results and discussion
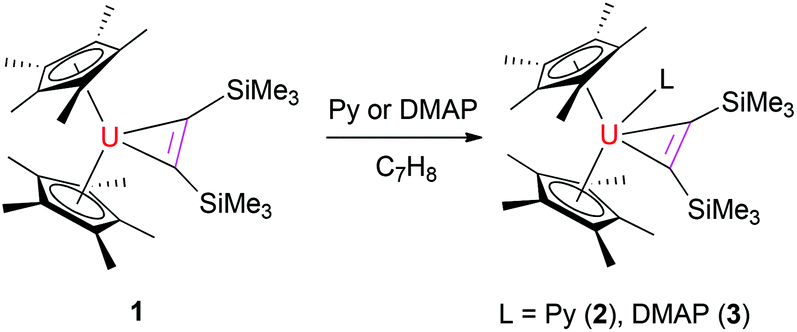
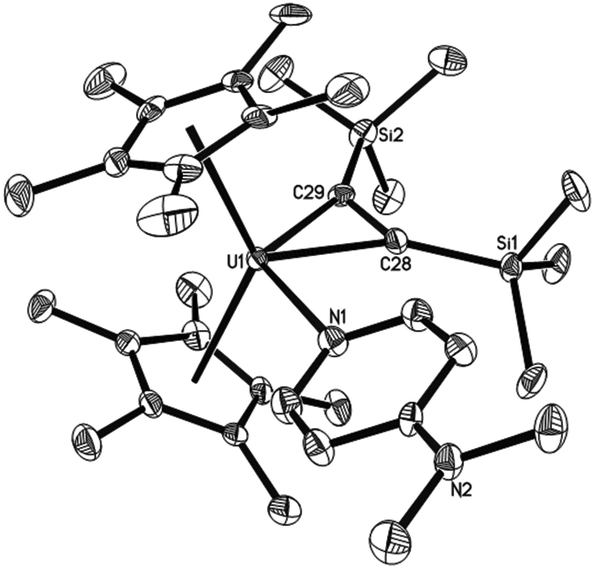
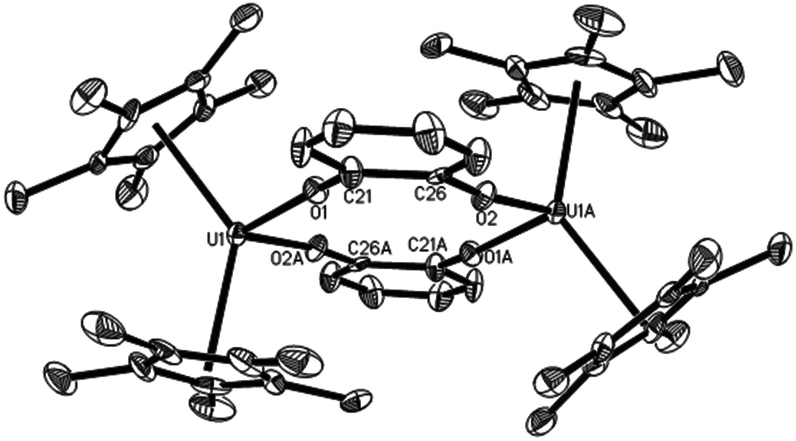
The reaction of the thorium metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) with pyridine derivatives induces C–H bond activation to give pyridyl alkenyl thorium compounds.7b,9 Nevertheless, similar to group 4 metallacyclopropene complexes,1,2 no deprotonation is observed between the uranium metallacyclopropene (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) and pyridine or 4-(dimethylamino)pyridine (DMAP) even when heated at 50 °C overnight, instead, the corresponding adducts (η5-C5Me5)2U[η2-C2(SiMe3)2](L) (L = Py (2), DMAP (3)) are formed in quantitative yields (Scheme 1). The molecular structures of 2 and 3 are shown in Fig. 1 and 2, and the selected bond distances and angles are listed in Table 1. In the U[η2-C2(SiMe3)2] fragment, the average U–C distances are 2.357(8) Å and 2.385(6) Å for 2 and 3, respectively, and the C–U–C angles are 33.0(3)° and 33.8(2)° for 2 and 3, respectively. These structural parameters are comparable to those found in the base-free complex 1 with an average U–C distance of 2.333(9) Å and the C–U–C angle of 33.3(3)°.8 The relatively long U–N distances of 2.625(8) Å (for 2) and 2.632(6) Å (for 3) are consistent with those of a datively coordinated nitrogen atom, which, however, are longer than that found in [η5-1,2,4-(Me3C)3C5H2]2UO(DMAP) (2.535(4) Å).10 Nevertheless, deprotonation occurs when complex 1 is exposed to di(naphthalen-1-yl)methanimine (1-C10H7)2CNH to form the alkenyl iminato (η5-C5Me5)2U[C(SiMe3)CH(SiMe3)][NC(1-C10H7)2] (4) (Scheme 2). The molecular structure of 4 is shown in Fig. 3, and the selected bond distances and angles are listed in Table 1. The C(21)–C(22) distance of 1.330(14) Å is in the typical range of a CC bond, whereas the U–C(21) distance of 2.436(9) Å is slightly longer than those in 2 and 3 (Table 1). The short U–N distance of 2.191(8) Å and the angle of U–N–C(29) of 177.8(6)° suggest some nitrogen π donation to the uranium atom. These structural parameters may be compared to those found in (η5-C5Me5)2U(NCPh2)2 with the U–N distances of 2.169(6)–2.185(5) Å and the U–N–C angles of 172.8(6)–176.5(5)°,8,11 and those in imidazolin-2-iminato uranium compounds with the U–N distances in the range of 2.118(8)–2.143(4) Å and the U–N–C angles of 169.5(5)–169.8(4)°.12
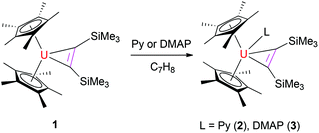 |
| | Scheme 1 Synthesis of complexes 2 and 3. | |
 |
| | Fig. 1 Molecular structure of 2 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Fig. 2 Molecular structure of 3 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Scheme 2 Synthesis of complex 4. | |
 |
| | Fig. 3 Molecular structure of 4 (thermal ellipsoids drawn at the 35% probability level). | |
Table 1 Selected distances (Å) and angles (°) for compounds 2–4, 6–8 and 11–14a
| Compound |
C(Cp)–Ub |
C(Cp)–Uc |
Cp(cent)–Ub |
U–X |
Cp(cent)–U–Cp(cent) |
X–U–X/Y |
|
Cp = cyclopentadienyl ring.
Average value.
Range.
The angle of C–U–C.
|
|
2
|
2.794(9) |
2.754(9) to 2.830(9) |
2.520(9) |
C26: 2.369(8), C27: 2.346(8) |
133.4(3) |
33.0(3)d |
| N1: 2.625(8) |
|
3
|
2.831(6) |
2.792(6) to 2.876(6) |
2.555(6) |
C28: 2.387(6), C29: 2.382(6) |
133.4(2) |
33.8(2)d |
| N1: 2.632(6) |
|
4
|
2.787(9) |
2.746(8) to 2.813(9) |
2.514(8) |
C21: 2.436(9), N1: 2.191(8) |
134.8(3) |
99.0(3) |
|
6
|
2.748(8) |
2.714(8) to 2.774(8) |
2.471(8) |
C22: 2.399(9), C25: 2.365(8) |
141.7(3) |
78.4(3) |
|
7
|
2.752(11) |
2.734(10) to 2.777(11) |
2.476(10) |
C21: 2.400(11), C24: 2.382(11) |
137.0(4) |
79.7(4) |
|
8
|
2.764(7) |
2.755(7) to 2.780(7) |
2.497(7) |
C14: 2.370(8), C14A: 2.370(8) |
136.9(3) |
85.6(3) |
|
11
|
2.766(10) |
2.715(10) to 2.827(10) |
2.495(10) |
N1: 2.447(10), N2: 2.364(11) |
131.8(4) |
N1–U–N2: 54.5(4) |
| N3: 2.270(11) |
N1–U–N3: 120.2(5) |
| N2–U–N3: 67.8(4) |
|
12
|
2.714(8) |
2.684(8) to 2.749(8) |
2.434(8) |
O1: 2.191(5), O2: 2.202(5) |
139.2(2) |
73.1(2) |
|
13
|
2.750 (11) |
2.655(11) to 2.805(10) |
2.476(10) |
O1: 2.130(6), O2A: 2.127(6) |
133.3(2) |
98.3(2) |
|
14
|
2.810(14) |
2.734(14) to 2.886(11) |
2.556(12) |
O1: 2.062(8), C21: 2.512(12) |
130.4(3) |
67.9(3) |
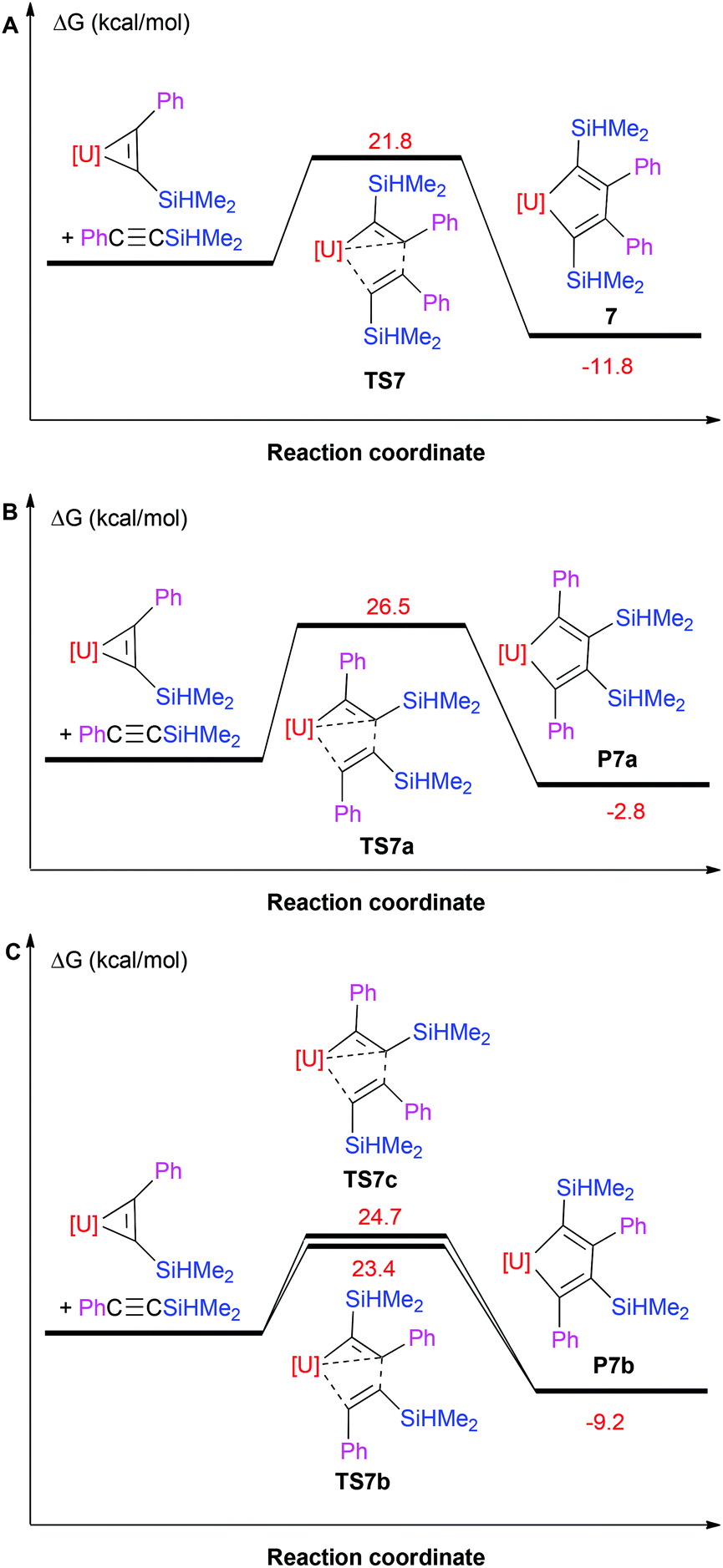
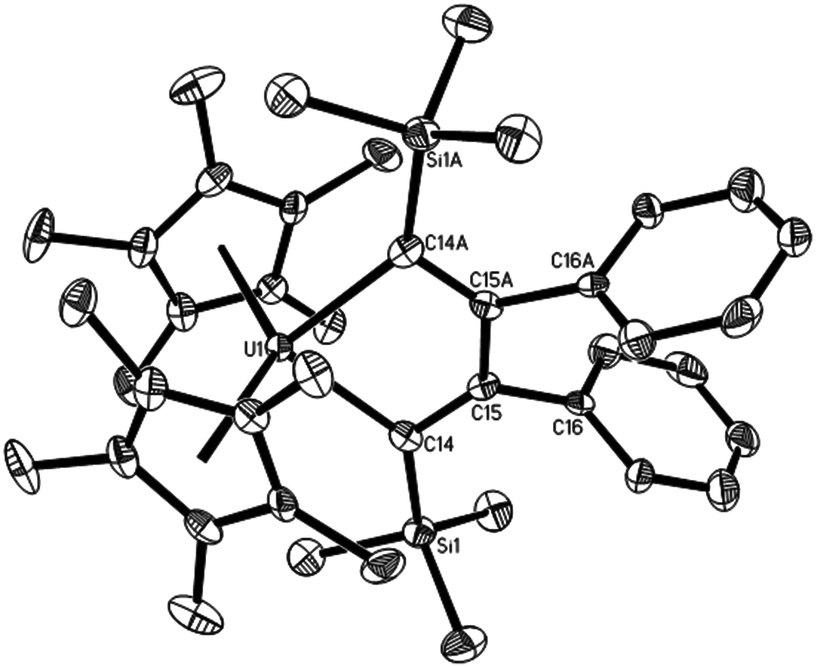
However, in contrast to the thorium metallacyclopropenes,7a,9 the coordinated bis(trimethylsilyl)acetylene in 1 can be exchanged with the internal alkynes. Mixing the uranium metallacyclopropene 1 with internal alkynes PhCCR (where R = Ph, Me) in toluene at ambient temperature forms the corresponding metallacyclopentadienes (η5-C5Me5)2U[η2-C(Ph)C(R)–C(Ph)C(R)] (R = Ph (5),8 Me (6)) in quantitative conversions (Scheme 3). Our previous DFT computations suggest that one molecule of PhCCR initially reacts with 1 to displace bis(trimethylsilyl)acetylene and to form the corresponding metallacyclopropenes (η5-C5Me5)2U(η2-C2Ph(R)), followed by a second insertion of PhCCR to yield the thermodynamically preferred metallacyclopentadienes (Scheme 3),8 which is presumably a consequence of the more open coordination sphere in the metallacyclopropene intermediates (η5-C5Me5)2U(η2-C2Ph(R)). Similar to the formation of thorium metallacyclopentadienes,7c the C–C bond formation is selective, i.e, the methyl-end of PhCCMe couples with the phenyl-substituted terminus of a second acetylene, leading to the Cs-symmetric U[η2-C(Ph)C(Me)–C(Ph)C(Me)] fragment. DFT studies confirm that the formation of this Cs-symmetric U[η2-C(Ph)C(Me)–C(Ph)C(Me)] fragment is thermodynamically more favourable (ΔG(298 K) = −17.7 kcal mol−1) than the C2v-symmetric isomer U[η2-C(Ph)C(Me)–C(Me)C(Ph)] (P6b; ΔG(298 K) = −16.4 kcal mol−1) or U[η2-C(Me)C(Ph)–C(Ph)C(Me)] (P6a; (ΔG(298 K) = −15.2 kcal mol−1) and also proceeds with the lower activation barrier ΔG‡(298 K) = 21.3 kcal mol−1 (Fig. 4). This selectivity in the C–C bond formation observed for complex 6 may be rationalized by the Mulliken charges in the free alkyne PhCCMe, the uranium metallacyclopropene intermediate (η5-C5Me5)2U[η2-C2Ph(Me)] and the transition state TS6 (Fig. 5). The more negatively charged end of the internal alkyne coordinates to the electropositive U(IV) atom and therefore electronic effects prevail over steric effects. Moreover, the formation of 6 may also proceed by two different reaction pathways, i.e., via transition state TS6 or TS6c (Fig. 4C), but the insertion viaTS6 (ΔG‡(298 K) = 21.3 kcal mol−1) is computed to be energetically more favourable than that proceeding viaTS6c (ΔG‡(298 K) = 21.9 kcal mol−1), which is consistent with the electronic arguments developed above. When phenyl(silyl)acetylene PhCCSiHMe2 or PhCCSiMe3 is added to compound 1, the metallacyclopentadienes (η5-C5Me5)2U[η2-C(R)C(Ph)–C(Ph)C(R)] (R = SiHMe2 (7), SiMe3 (8)) are isolated exclusively, but the selectivity in the C–C bond formation changes (Scheme 3), that is, the phenyl-substituted terminus of PhCCR couples with the phenyl-substituted one of a second acetylene to give a C2v-symmetric U[η2-C(R)C(Ph)–C(Ph)C(R)] moiety. Our DFT investigations also reproduce this change in selectivity. The C2v-symmetric isomer (η5-C5Me5)2U[η2-C(SiHMe2)C(Ph)–C(Ph)C(SiHMe2)] is energetically more favorable (7; ΔG(298 K) = −11.8 kcal mol−1) than the C2v-symmetric (P7a; ΔG(298 K) = −2.8 kcal mol−1) and Cs-symmetric isomers (P7b; ΔG(298 K) = −9.2 kcal mol−1), and it also forms with the lowest barrier of activation ΔG‡(298 K) = 21.8 kcal mol−1 (Fig. 6). As discussed above, the selectivity of the C–C bond formation to give complex 7 can also be explained by the Mulliken charges computed for the free alkyne PhCCSiHMe2, the intermediate (η5-C5Me5)2U[η2-C2Ph(SiHMe2)] and the transition state TS7 (Fig. 5). However, in contrast to the uranium metallacyclopentadiene 5,8 no thermal degradation is observed for complexes 6–8, in line with the previous observations establishing that the substituents on the acetylene significantly influenced the reactivity of the actinide metallacyles.7c The molecular structures of 6–8 are shown in Fig. 7–9, and the selected bond distances and angles are provided in Table 1. Furthermore, the U–C distances of 2.365(8)–2.400(11) Å are comparable to those of the U–C(sp2) σ-bonds found in complexes 1–4 (2.315(9)–2.436(9) Å). The C–C distances within the metallacyclopentadiene fragments are 1.344(10), 1.503(11) and 1.363(11) Å for 6, 1.372(15), 1.510(15) and 1.352(15) Å for 7 and 1.374(9), 1.558(12) and 1.374(9) Å for 8, and therefore are very close to those previously reported for related actinide metallacyclopentadiene compounds,4,7ce.g., (η5-C5Me5)2U(η2-C4Ph4) (1.365(3), 1.509(4) and 1.365(3) Å)4e and (η5-C5Me5)2Th(η2-C4Me4) (1.354(4), 1.521(6) and 1.354(4) Å).4n
 |
| | Scheme 3 Synthesis of complexes 5–9. | |
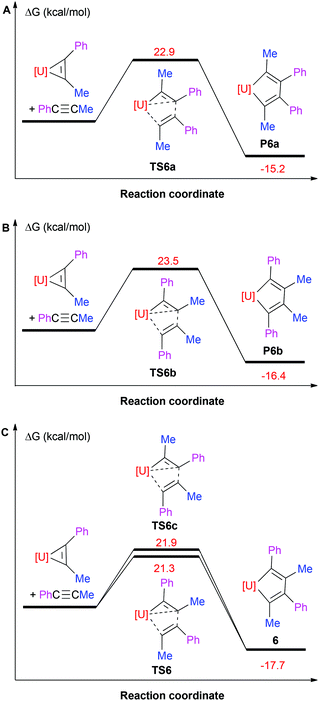 |
| | Fig. 4 Free energy profile (kcal mol−1) for the reaction of (η5-C5Me5)2U[η2-C(Ph)C(Me)] + PhCCMe (U was treated with ECP60MWB). [U] = (η5-C5Me5)2U. | |
 |
| | Fig. 5 Mulliken charges of the free alkynes, their respective uranium metallacyclopropenes and transition state complexes. [U] = (η5-C5Me5)2U. | |
 |
| | Fig. 6 Free energy profile (kcal mol−1) for the reaction of (η5-C5Me5)2U[η2-C(Ph)C(SiHMe2)] + PhCCSiHMe2 (U was treated with ECP80MWB). [U] = (η5-C5Me5)2U. | |
 |
| | Fig. 7 Molecular structure of 6 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Fig. 8 Molecular structure of 7 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Fig. 9 Molecular structure of 8 (thermal ellipsoids drawn at the 35% probability level). | |
The coordinated bis(trimethylsilyl)acetylene in 1 may also be replaced with conjugated alkynes or olefins. For example, reaction of 1 with 1 equiv. of 1,4-diphenylbutadiyne (PhCCCCPh) or 1,4-diphenylbutadiene (PhCHCHCHCHPh) yields the uranium metallacyclopentatriene (η5-C5Me5)2U(η4-C4Ph2) (9) (Scheme 3) and the metallacyclopentene (η5-C5Me5)2U[η2-CH(Ph)CHCHCH(Ph)] (10) (Scheme 4), respectively, in quantitative conversions. However, no reaction occurs when 1 is exposed to olefins such as RCHCHR (R = H, Ph, Me) even when heated at 100 °C for one week.
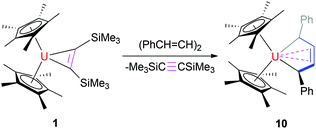 |
| | Scheme 4 Synthesis of complex 10. | |
The bis(trimethylsilyl)acetylene moiety in 1 can also be replaced with hetero-unsaturated organic molecules. For example, complex 1 reacts with three equivalents of the nitrile C6H11CN to yield the C–C and N–C coupling product (η5-C5Me5)2U[η3-NC(C6H11)C(C6H11)NC(C6H11)N] (11) (Scheme 5). This contrasts the reaction of the related thorium metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) with PhCN,7a,9 for which an insertion product was isolated. In analogy to the reactivity of group 4 metallacyclopropene (η5-C5Me5)2M[η2-C2(SiMe3)2] (M = Ti, Zr),2h,l we propose that C6H11CN initially replaces the bis(trimethylsilyl)acetylene fragment to give a metal η2-nitrile intermediate,13 which immediately couples with a second molecule of C6H11CN to give a five-membered metallaheterocycle,2h,l that further reacts with a third molecule of C6H11CN to afford 11 (Scheme 5). Fig. 10 shows the molecular structure of 11 and the selected bond distances and angles are provided in Table 1. These structural parameters suggest some degree of electron delocalization with the N(1)–C(21)–N(2)–C(28)–C(35)–N(3) fragment. The U–N distances are 2.447(10) Å for N(1) and 2.364(11) Å for N(2) and 2.270(11) Å for N(3), which are longer than that found in 4 (2.191(8) Å). Addition of 9,10-phenanthrenequinone (9,10-C14H8O2) to 1 forms the monomeric uranium quinonate (η5-C5Me5)2U(9,10-O2C14H8) (12)14 concomitant with free bis(trimethylsilyl)acetylene (Scheme 6), whereas the less sterically encumbered o-benzoquinone affords the dimeric quinonate [(η5-C5Me5)2U]2(μ-o-O2C6H4)2 (13) (Scheme 6). The molecular structures of 12 and 13 are shown in Fig. 11 and 12, and the selected bond distances and angles are listed in Table 1. The average U–O distance is 2.191(5) Å for 12, which is larger than that found in 13 (2.127(6) Å). Nevertheless, in contrast to the reaction with quinones, but similar to the reactivity of the thorium metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) towards ketones (for details see the ESI†), insertion of 1 equiv. of cyclohexanone ((CH2)5CO) into the uranium metallacyclopropene moiety of 1 is observed at ambient temperature to exclusively yield the five-membered uranium heterocycle (η5-C5Me5)2U[OC(CH2)5(C2(SiMe3)2)] (14) (Scheme 6). The molecular structure of 14 is shown in Fig. 13, and the selected bond distances and angles are compiled in Table 1. The U–O distance is 2.062(8) Å, which is comparable to those in 12 and 13 (Table 1), whereas the U–C(21) distance is 2.512(12) Å, which is significantly longer than those of the U–C(sp2) σ-bonds found in compounds 1–4 (2.315(9)–2.436(9) Å).
 |
| | Scheme 5 Synthesis of complex 11. | |
 |
| | Fig. 10 Molecular structure of 11 (thermal ellipsoids drawn at the 35% probability level). | |
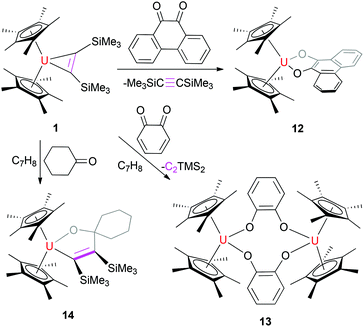 |
| | Scheme 6 Synthesis of complexes 12–14. | |
 |
| | Fig. 11 Molecular structure of 12 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Fig. 12 Molecular structure of 13 (thermal ellipsoids drawn at the 35% probability level). | |
 |
| | Fig. 13 Molecular structure of 14 (thermal ellipsoids drawn at the 35% probability level). | |
Conclusions
While the coordinated alkyne in the thorium metallacyclopropenes is inert towards alkyne exchange, it can react as a nucleophile towards hetero-unsaturated molecules or as a strong base inducing inter- or intramolecular C–H bond activations.7,9 In contrast, addition of pyridine derivatives to the uranium complex (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) forms the corresponding Lewis-base adducts (η5-C5Me5)2U[η2-C2(SiMe3)2](L) (L = Py (2), DMAP (3)) without C–H bond activations. The reactivity difference observed for uranium relative to thorium can be rationalized by the more covalent bonds between the (η5-C5Me5)2U2+ and [η2-C2(SiMe3)2]2− fragments and is a consequence of the enhanced 5f orbitals contributing to the bonding in the uranium metallacyclopropene U–(η2-CC) moiety.8 Furthermore, in contrast to the thorium metallacyclopropenes,7,9 replacement of the coordinated alkyne occurs when complex 1 is exposed to alkynes, conjugated alkenes, nitriles and quinones. These distinct reactivity patterns are similar to those of the more covalent group 4 metallacyclopropene complexes.1,2 Nevertheless, thorium and uranium metallacyclopropenes exhibit similar reactivity patterns when exposed to ketones, which are inserted into the actinide metallacyclopropene moieties to yield the five-membered heterocyclic compounds. Further investigations regarding the intrinsic reactivity of actinide metallacyclopropenes and of uranium metallacycles 9 and 10 are currently in progress.
Experimental
General methods
All reactions and product manipulations were carried out under an atmosphere of dry dinitrogen with rigid exclusion of air and moisture using standard Schlenk or cannula techniques, or in a glove box. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) was prepared according to literature methods.8 All other chemicals were purchased from Aldrich Chemical Co. and Beijing Chemical Co. and used as received unless otherwise noted. Infrared spectra were recorded in KBr pellets on an Avatar 360 Fourier transform spectrometer. 1H and 13C{1H} NMR spectra were recorded at 25 °C on a Bruker AV 400 spectrometer at 400 and 100 MHz, respectively. All chemical shifts are reported in δ units with reference to the residual protons of the deuterated solvents, which served as internal standards, for proton and carbon chemical shifts. Melting points were measured on X-6 melting point apparatus and were uncorrected. Elemental analyses were performed on a Vario EL elemental analyzer.
Syntheses
Preparation of (η5-C5Me5)2U[η2-C2(SiMe3)2](Py) (2).
A toluene (5 mL) solution of pyridine (32 mg, 0.40 mmol) was added to a toluene (10 mL) solution of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) with stirring at room temperature. After this solution was stirred at room temperature for one hour, the solvent was evaporated. The residue was dried in a vacuum at 50 °C overnight to give 2 as a brown solid in quantitative yield (Found: C, 52.30; H, 7.02; N, 1.86. C33H53NSi2U requires C, 52.29; H, 7.05; N, 1.85%). M.p.: 93–95 °C (dec.). 1H NMR (C6D6): δ 25.95 (br s, 1H, py), 14.96 (s, 9H, SiCH3), 6.38 (s, 1H, py), −2.07 (s, 1H, py), −3.27 (s, 30H, CpCH3), −3.94 (s, 9H, SiCH3), −6.56 (s, 1H, py), −10.67 (br s, 1H, py) ppm. 13C{1H} NMR (C6D6): δ 133.4 (ring C), 125.6 (py C), 99.7 (py C), 79.8 (py C), 60.4 (SiCH3), −54.7 (SiCH3), −66.8 (CpCH3) ppm; carbons of UCSiMe3 were not observed. IR (KBr, cm−1): 2962 (s), 2895 (s), 1598 (m), 1570 (m), 1433 (s), 1402 (s), 1242 (s), 1082 (s), 1018 (s), 839 (s). Brown crystals of 2 suitable for X-ray structure analysis were grown from an n-hexane solution at room temperature.
Preparation of (η5-C5Me5)2U[η2-C2(SiMe3)2](DMAP) (3).
This compound was prepared as a brown solid in quantitative yield from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and DMAP (49 mg, 0.40 mmol) in toluene (15 mL) at room temperature and dried in a vacuum at 50 °C by a similar procedure to that in the synthesis of 2 (Found: C, 52.46; H, 7.32; N, 3.48. C35H58N2Si2U requires C, 52.48; H, 7.30; N, 3.50%). M.p.: 107–109 °C (dec.). 1H NMR (C6D6): δ 16.13 (br s, 9H, SiCH3), 14.52 (br s, 9H, SiCH3), −0.50 (s, 1H, py), −2.94 (s, 6H, NCH3), −3.36 (s, 30H, CpCH3), −3.52 (s, 1H, py) ppm; two protons were not observed. 13C{1H} NMR (C6D6): δ 128.5 (py C), 128.1 (py C), 127.9 (py C), 113.3 (ring C), 62.3 (SiCH3), 36.1 (NCH3), −65.9 (CpCH3) ppm; carbons of UCSiMe3 were not observed. IR (KBr, cm−1): 2960 (s), 1608 (s), 1527 (s), 1438 (s), 1384 (s), 1228 (s), 1062 (s), 1004 (s), 839 (s), 804 (s). Brown crystals of 3 suitable for X-ray structure analysis were grown from an n-hexane solution at room temperature.
Preparation of (η5-C5Me5)2U[C(SiMe3)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CH(SiMe3)][NC(1-C10H7)2] (4).
Method A. A toluene (5 mL) solution of (1-C10H7)2CNH (113 mg, 0.4 mmol) was added to a toluene (10 mL) solution of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) with stirring at room temperature. After the solution was stirred at room temperature overnight, the solvent was removed. The residue was extracted with benzene (10 mL × 3) and filtered. The volume of the filtrate was reduced to 5 mL, and brown crystals of 4 were isolated when this solution was kept at room temperature for one week. Yield: 338 mg (88%) (Found: C, 61.26; H, 6.60, N, 1.47. C49H63NSi2U requires C, 61.29; H, 6.61; N, 1.46%). M.p.: 177–179 °C (dec.). 1H NMR (C6D6): δ 19.01 (s, 9H, SiCH3), 15.05 (br s, 4H, aryl), 9.23 (s, 2H, aryl), 7.60 (m, 2H, aryl), 4.59 (s, 1H, CCH), 1.23 (s, 2H, aryl), 0.89 (s, 2H, aryl), 0.32 (s, 2H, aryl), −1.80 (s, 30H, CpCH3), −9.78 (s, 9H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 233.8 (UC), 142.1 (ring C), 132.1 (aryl C), 129.3 (aryl C), 128.5 (aryl C), 127.9 (aryl C), 125.6 (aryl C), 124.4 (aryl C), 44.9 (NC), 19.9 (SiCH3), 15.0 (SiCH3), −33.5 (CH) −51.6 (CpCH3) ppm. IR (KBr, cm−1): 2958 (s), 2899 (s), 1595 (s), 1579 (s), 1558 (s), 1402 (s), 1259 (s), 1246 (s), 1095 (s), 1018 (s), 840 (s), 775 (s).
CH(SiMe3)][NC(1-C10H7)2] (4).
Method A. A toluene (5 mL) solution of (1-C10H7)2CNH (113 mg, 0.4 mmol) was added to a toluene (10 mL) solution of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) with stirring at room temperature. After the solution was stirred at room temperature overnight, the solvent was removed. The residue was extracted with benzene (10 mL × 3) and filtered. The volume of the filtrate was reduced to 5 mL, and brown crystals of 4 were isolated when this solution was kept at room temperature for one week. Yield: 338 mg (88%) (Found: C, 61.26; H, 6.60, N, 1.47. C49H63NSi2U requires C, 61.29; H, 6.61; N, 1.46%). M.p.: 177–179 °C (dec.). 1H NMR (C6D6): δ 19.01 (s, 9H, SiCH3), 15.05 (br s, 4H, aryl), 9.23 (s, 2H, aryl), 7.60 (m, 2H, aryl), 4.59 (s, 1H, CCH), 1.23 (s, 2H, aryl), 0.89 (s, 2H, aryl), 0.32 (s, 2H, aryl), −1.80 (s, 30H, CpCH3), −9.78 (s, 9H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 233.8 (UC), 142.1 (ring C), 132.1 (aryl C), 129.3 (aryl C), 128.5 (aryl C), 127.9 (aryl C), 125.6 (aryl C), 124.4 (aryl C), 44.9 (NC), 19.9 (SiCH3), 15.0 (SiCH3), −33.5 (CH) −51.6 (CpCH3) ppm. IR (KBr, cm−1): 2958 (s), 2899 (s), 1595 (s), 1579 (s), 1558 (s), 1402 (s), 1259 (s), 1246 (s), 1095 (s), 1018 (s), 840 (s), 775 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of (1-C10H7)2CNH (5.6 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 4 were observed by 1H NMR spectroscopy (100% conversion).
Preparation of (η5-C5Me5)2U[η2-C(Ph)C(Me)C(Ph)C(Me)]·0.5C6H6 (6·0.5C6H6).
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and PhCCMe (93 mg, 0.8 mmol) in toluene (15 mL) at room temperature and recrystallized from a benzene solution by a similar procedure as that in the synthesis of 4. Yield: 234 mg (75%) (Found: C, 63.13; H, 6.32. C41H49U requires C, 63.15; H, 6.33%). M.p.: 103–105 °C (dec.). 1H NMR (C6D6): δ 7.15 (s, 3H, C6H6), 6.14 (s, 4H, phenyl), 5.58 (s, 3H, CH3), 2.94 (s, 15H, CpCH3), 2.72 (s, 1H, phenyl), 1.28 (s, 15H, CpCH3), −1.39 (s, 2H, phenyl), −6.00 (s, 3H, CH3), −13.65 (s, 2H, phenyl), −20.74 (s, 1H, phenyl) ppm. 13C{1H} NMR (C6D6): δ 278.4 (UCPh), 260.2 (UCCH3), 197.4 (ring C), 188.4 (ring C), 130.3 (phenyl C), 129.6 (phenyl C), 129.3 (phenyl C), 128.0 (C6H6), 124.7 (phenyl C), 122.9 (phenyl C), 122.5 (phenyl C), 122.4 (phenyl C), 121.9 (phenyl C), 112.2 (CC(Ph)), 104.5 (CC(Me)), 91.2 (UCCH3), 69.7 (CCCH3), −41.2 (CpCH3), −45.3 (CpCH3). IR (KBr, cm−1): 2960 (s), 2922 (s), 1438 (s), 1402 (s), 1259 (s), 1074 (s), 1018 (s), 798 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of PhCCMe (4.6 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 6 and those of Me3SiCCSiMe3 (1H NMR (C6D6): δ 0.15 (s, 18H, SiCH3) ppm) were observed by 1H NMR spectroscopy (100% conversion).
Reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) with PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) CMe. NMR Scale. A C6D6 (0.2 mL) solution of PhCCMe (2.3 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.3 mL). Resonances of 6 along with those of unreacted 1 and Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (50% conversion based on 1).
CMe. NMR Scale. A C6D6 (0.2 mL) solution of PhCCMe (2.3 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.3 mL). Resonances of 6 along with those of unreacted 1 and Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (50% conversion based on 1).
Preparation of (η5-C5Me5)2U[η2-C(SiHMe2)C(Ph)C(Ph)C(SiHMe2)] (7).
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and PhCCSiHMe2 (128 mg, 0.8 mmol) in toluene (15 mL) at room temperature and recrystallized from an n-hexane solution by a similar procedure as that in the synthesis of 4. Yield: 279 mg (84%) (Found: C, 57.98; H, 6.61. C40H54Si2U requires C, 57.95; H, 6.57%). M.p.: 118–120 °C. 1H NMR (C6D6): δ 6.74 (d, 4H, J = 7.4 Hz, phenyl), 6.29 (t, 4H, J = 7.2 Hz, phenyl), 5.27 (t, 2H, J = 6.9 Hz, phenyl), 3.66 (s, 30H, CpCH3), −6.10 (s, 12H, SiCH3), −30.20 (s, 2H, SiH) ppm. 13C{1H} NMR (C6D6): δ 266.9 (UCSi), 231.4 (ring C), 128.5 (phenyl C), 127.9 (phenyl C), 124.3 (phenyl C), 123.4 (phenyl C), 105.1 (CPh), −1.5 (SiCH3), −41.4 (CpCH3) ppm. IR (KBr, cm−1): 2960 (s), 2902 (s), 2083 (s), 1593 (m), 1408 (s), 1259 (s), 1070 (s), 1018 (s), 937 (s), 887 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of PhCCSiHMe2 (6.4 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 7 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) with PhCCSiHMe2. NMR Scale. A C6D6 (0.2 mL) solution of PhCCSiHMe2 (3.2 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.3 mL). Resonances of 7 along with those of unreacted 1 and Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (50% conversion based on 1).
Preparation of (η5-C5Me5)2U[η2-C(SiMe3)C(Ph)C(Ph)C(SiMe3)] (8).
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and PhCCSiMe3 (140 mg, 0.8 mmol) in toluene (15 mL) at room temperature and recrystallized from an n-hexane solution by a similar procedure as that in the synthesis of 4. Yield: 281 mg (82%) (Found: C, 58.81; H, 6.87. C42H58Si2U requires C, 58.85; H, 6.82%). M.p.: 104–106 °C (dec.). 1H NMR (C6D6): δ 7.28 (d, 4H, J = 7.6 Hz, phenyl), 6.81 (t, 4H, J = 7.4 Hz, phenyl), 5.83 (t, 2H, J = 7.0 Hz, phenyl), 4.21 (s, 12H, SiCH3), 3.86 (s, 30H, CpCH3), −4.78 (s, 6H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 217.4 (UCSi), 146.5 (ring C), 132.2 (phenyl C), 127.0 (phenyl C), 125.9 (phenyl C), 123.9 (phenyl C), 110.7 (CPh), 0.0 (SiCH3), −39.7 (CpCH3), −65.1 (SiCH3) ppm. IR (KBr, cm−1): 2949 (m), 2906 (s), 1595 (m), 1487 (m), 1438 (s), 1400 (s), 1259 (s), 1236 (s), 1070 (s), 1018 (s), 935 (s), 827(s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of PhCCSiMe3 (7.0 mg, 0.04 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 8 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1) with PhCCSiMe3. NMR Scale. A C6D6 (0.2 mL) solution of PhCCSiMe3 (3.5 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.3 mL). Resonances of 8 along with those of unreacted 1 and Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (50% conversion based on 1).
Preparation of (η5-C5Me5)2U[η4-C4Ph2] (9).
Method A. This compound was prepared as brown microcrystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and PhCC–CCPh (81 mg, 0.4 mmol) in toluene (15 mL) at room temperature and recrystallized from a benzene solution by a similar procedure as that in the synthesis of 4. Yield: 219 mg (77%) (Found: C, 60.81; H, 5.71. C36H40U requires C, 60.84; H, 5.67%). M.p.: 136–138 °C (dec.). 1H NMR (C6D6): δ 6.69 (t, 2H, J = 7.0 Hz, phenyl), 5.18 (t, 4H, J = 6.8 Hz, phenyl), 2.09 (m, 4H, phenyl), −0.95 (s, 30H, CpCH3) ppm. 13C{1H} NMR (C6D6): δ 237.7 (UCPh), 190.6 (PhCC), 157.1 (ring C), 137.6 (phenyl C), 136.8 (phenyl C), 128.5 (phenyl C), 117.0 (phenyl C), −51.4 (CpCH3) ppm. IR (KBr, cm−1): 2962 (s), 2905 (s), 1612 (m), 1586 (m), 1439 (s), 1403 (s), 1383 (s), 1260 (s), 1068 (s), 1020 (s), 799 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of PhCC–CCPh (4.0 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 9 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Preparation of (η5-C5Me5)2U[η2-CH(Ph)CHCHCH(Ph)] (10).
Method A. This compound was prepared as brown microcrystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and PhCHCHCHCHPh (83 mg, 0.4 mmol) in toluene (15 mL) at 70 °C and recrystallized from a toluene solution by a similar procedure as that in the synthesis of 4. Yield: 234 mg (82%) (Found: C, 60.51; H, 6.18. C36H44U requires C, 60.49; H, 6.20%). M.p.: 153–155 °C (dec.). 1H NMR (C6D6): δ 47.19 (s, 2H, CHC), 12.58 (s, 15H, CpCH3), −0.21 (d, 4H, J = 8.9 Hz, phenyl), −0.32 (s, 15H, CpCH3), −10.91 (d, 2H, J = 11.7 Hz, phenyl), −20.32 (d, 4H, J = 9.2 Hz, phenyl), −171.11 (s, 2H, PhCH) ppm. 13C{1H} NMR (C6D6): δ 463.2 (UC), 219.8 (CHC), 169.8 (ring C), 155.4 (phenyl C), 155.1 (phenyl C), 127.9 (phenyl C), 74.3 (phenyl C), −11.5 (CpCH3), −49.8 (CpCH3) ppm. IR (KBr, cm−1): 2962 (s), 2910 (s), 1492 (m), 1438 (s), 1402 (s), 1384 (s), 1259 (s), 1072 (s), 1020 (s), 800 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of PhCHCHCHCHPh (4.1 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 10 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion) after this solution was kept at 70 °C for two days.
Preparation of (η5-C5Me5)2U[η3-NC(C6H11)C(C6H11)NC(C6H11)N]·0.5C6H6 (11·0.5C6H6).
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and C6H11CN (131 mg, 1.20 mmol) in toluene (15 mL) at room temperature and recrystallized from a benzene solution by a similar procedure as that in the synthesis of 4. Yield: 273 mg (78%) (Found: C, 60.36; H, 7.58; N, 4.82. C44H66N3U requires C, 60.39; H, 7.60; N, 4.80%). M.p.: 235–237 °C (dec.). 1H NMR (C6D6): δ 16.51 (m, 1H, CH2), 14.55 (m, 2H, CH2), 14.01 (m, 2H, CH2), 7.15 (s, 3H, C6H6), 7.04 (m, 2H, CH2), 6.09 (m, 2H, CH2), 5.76 (m, 2H, CH2), 4.89 (m, 1H, CH2), 4.70 (m, 1H, CH2), 3.70 (m, 2H, CH2), 2.98 (m, 2H, CH2), 2.63 (m, 1H, CH2), 1.88 (s, 30H, CpCH3), 0.20 (m, 2H, CH2), −1.23 (m, 1H, CH2), −3.39 (m, 1H, CH2), −4.84 (m, 2H, CH2), −5.20 (m, 2H, CH2), −6.71 (m, 1H, CH2), −13.12 (m, 2H, CH2), −13.50 (m, 1H, CH2), −19.15 (m, 2H, CH2), −83.76 (s, 1H, CH2) ppm. 13C{1H} NMR (C6D6): δ 195.3 (ring C), 166.6 (CN), 164.9 (CN), 128.0 (C6H6), 107.0 (CN), 64.2 (CH), 64.1 (CH), 63.3 (CH), 34.1 (CH2), 31.9 (CH2), 31.7 (CH2), 29.1 (CH2), 27.3 (CH2), 27.2 (CH2), 26.6 (CH2), 18.6 (CH2), 11.5 (CH2), −40.8 (CpCH3) ppm. IR (KBr, cm−1): 2962 (s), 2922 (s), 1617 (m), 1560 (m), 1400 (s), 1386 (s), 1259 (s), 1089 (s), 1016 (s), 798 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of C6H11CN (6.6 mg, 0.06 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 11 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Preparation of (η5-C5Me5)2U(9,10-O2C14H8) (12)14.
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and 9,10-phenanthrenequinone (85 mg, 0.40 mmol) in toluene (15 mL) at room temperature and recrystallized from a toluene solution by a similar procedure as that in the synthesis of 4. Yield: 258 mg (90%) (Found: C, 57.01; H, 5.31. C34H38O2U: C, 56.98; H, 5.34%). M.p.: >300 °C (dec.). 1H NMR (C6D6): δ 3.76 (s, 30H, CpCH3), 2.82 (d, 2H, J = 8.2 Hz, phenyl), 2.04 (t, 2H, J = 9.3 Hz, phenyl), −1.58 (d, 2H, J = 9.0 Hz, phenyl), −25.39 (d, 2H, J = 7.7 Hz, phenyl) ppm. 13C{1H} NMR (C6D6): δ 132.4 (ring C), 125.6 (aryl C), 115.6 (aryl C), 115.2 (aryl C), 113.7 (aryl C), 109.5 (aryl C), 97.0 (aryl C), 95.4 (aryl C), −27.3 (CpCH3) ppm. IR (KBr, cm−1): 2960 (s), 2906 (s), 1595 (m), 1517 (s), 1477 (s), 1409 (s), 1365 (s), 1109 (s), 1028 (s), 920 (s), 756 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of 9,10-phenanthrenequinone (4.2 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 12 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Preparation of [(η5-C5Me5)2U]2(μ-o-O2C6H4)2·C6H6 (13·C6H6).
Method A. This compound was prepared as orange crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and o-benzoquinone (43 mg, 0.40 mmol) in toluene (15 mL) at room temperature and recrystallized from a benzene solution by a similar procedure as that in the synthesis of 4. Yield: 220 mg (84%) (Found: C, 53.08; H, 5.71. C58H74O4U2 requires C, 53.13; H, 5.69%). M.p.: >300 °C (dec.). 1H NMR (C6D6): δ 7.15 (s, 6H, C6H6), 2.11 (s, 4H, phenyl), 1.78 (s, 60H, CpCH3), −12.33 (s, 4H, phenyl) ppm. 13C{1H} NMR (C6D6): δ 147.1 (ring C), 128.0 (C6H6), 125.6 (phenyl C), 108.4 (phenyl C), 108.2 (phenyl C), −36.5 (CpCH3) ppm. IR (KBr, cm−1): 2962 (m), 1581 (m), 1479 (s), 1402 (s), 1263 (s), 1224 (s), 1097 (s), 1018 (s), 904 (s), 825 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of o-benzoquinone (2.2 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 13 and those of Me3SiCCSiMe3 were observed by 1H NMR spectroscopy (100% conversion).
Preparation of (η5-C5Me5)2U[OC(CH2)5(C2(SiMe3)2)] (14).
Method A. This compound was prepared as brown crystals from the reaction of (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 272 mg, 0.40 mmol) and cyclohexanone (40 mg, 0.40 mmol) in toluene (15 mL) at room temperature and recrystallized from an n-hexane solution by a similar procedure as that in the synthesis of 4. Yield: 261 mg (84%) (Found: C, 52.53; H, 7.51. C34H58OSi2U requires C, 52.55; H, 7.52%). M.p.: 115–117 °C (dec.). 1H NMR (C6D6): δ 57.94 (d, 2H, J = 16.3 Hz, CH2), 48.10 (t, 2H, J = 11.9 Hz, CH2), 37.06 (q, 2H, J = 13.1 Hz, CH2), 24.90 (d, 1H, J = 10.1 Hz, CH2), 23.00 (d, 2H, J = 13.8 Hz, CH2), 18.20 (d, 1H, J = 9.0 Hz, CH2), 5.93 (s, 9H, SiCH3), −0.27 (s, 30H, CpCH3), −22.08 (s, 6H, SiCH3), −38.94 (s, 3H, SiCH3) ppm. 13C{1H} NMR (C6D6): δ 198.5 (UC), 143.3 (CH2), 142.5 (UCC), 141.5 (CH2), 118.7 (CO), 112.3 (ring C), 56.7 (CH2), 55.4 (CH2), 54.2 (CH2), 4.1 (SiCH3), −59.3 (CpCH3) ppm. IR (KBr, cm−1): 2958 (s), 2927 (s), 2854 (s), 1436 (s), 1377 (s), 1247 (s), 1076 (s), 1018 (s), 840 (s).
Method B. NMR Scale. A C6D6 (0.3 mL) solution of cyclohexanone (2.0 mg, 0.02 mmol) was slowly added to a J. Young NMR tube charged with (η5-C5Me5)2U[η2-C2(SiMe3)2] (1; 14 mg, 0.02 mmol) and C6D6 (0.2 mL). Resonances of 14 were observed by 1H NMR spectroscopy (100% conversion).
X-ray crystallography
Single-crystal X-ray diffraction measurements were carried out on a Bruker Smart APEX II CCD diffractometer at 100(2) K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program.15 All structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELXL program package.16 All the hydrogen atoms were geometrically fixed using the riding model. Disordered solvents in the voids of 15 were modelled or removed by using the SQUEEZE program.17 The crystal data and experimental data for 2–4, 6–8 and 11–16 are summarized in the ESI.† Selected bond lengths and angles are listed in Table 1.
Computational methods
All calculations were carried out with the Gaussian 09 program (G09),18 employing the B3PW91 functional, plus a polarizable continuum model (PCM) (denoted as B3PW91-PCM), with the standard 6-31G(d) basis set for C, H and Si elements and a quasi-relativistic 5f-in-valence effective-core potential (ECP60MWB) or 5f-in-core effective-core potential (ECP80MWB) treatment for the core region of U and the corresponding optimized segmented basis set for the valence shells of U,19 to fully optimize the structures of reactants, complexes, transition states, intermediates, and products, and also to mimic the experimental toluene-solvent conditions (dielectric constant ε = 2.379). All stationary points were subsequently characterized by vibrational analyses, from which their respective zero-point (vibrational) energies (ZPE) were extracted and used in the relative energy determinations; in addition, frequency calculations were also performed to ensure that the reactant, complex, intermediate, product and transition state structures resided at minima and 1st order saddle points, respectively, on their potential energy hypersurfaces.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21472013, 21573021, 21672024), and the Deutsche Forschungsgemeinschaft (DFG) through the Heisenberg program (WA 2513/6).
Notes and references
- For selected reviews, see:
(a) S. L. Buchwald and R. B. Nielsen, Chem. Rev., 1988, 88, 1047–1058 CrossRef CAS;
(b) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884–900 CrossRef CAS;
(c) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and V. B. Shur, Eur. J. Inorg. Chem., 2004, 4739–4749 CrossRef CAS;
(d) U. Rosenthal, Angew. Chem., Int. Ed., 2004, 43, 3882–3887 CrossRef CAS PubMed;
(e) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2005, 24, 456–471 CrossRef CAS;
(f) U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719–728 RSC;
(g) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303–415 CrossRef CAS;
(h) K. D. J. Parker and M. D. Fryzuk, Organometallics, 2015, 34, 2037–2047 CrossRef CAS.
- Selected papers on group 4 metallacyclopropenes, see:
(a) M. D. Walter, C. D. Sofield and R. A. Andersen, Organometallics, 2008, 27, 2959–2970 CrossRef CAS;
(b) T. Beweries, C. Fischer, S. Peitz, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg, D. Heller and U. Rosenthal, J. Am. Chem. Soc., 2009, 131, 4463–4469 CrossRef CAS PubMed;
(c) K. Kaleta, M. Ruhmann, O. Theilmann, T. Beweries, S. Roy, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2011, 133, 5463–5473 CrossRef CAS PubMed;
(d) M. Haehnel, M. Ruhmann, O. Theilmann, S. Roy, T. Beweries, P. Arndt, A. Spannenberg, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2012, 134, 15979–15991 CrossRef CAS PubMed;
(e) X. You, S. Yu and Y. Liu, Organometallics, 2013, 32, 5273–5276 CrossRef CAS;
(f) M. Haehnel, S. Hansen, K. Schubert, P. Arndt, A. Spannenberg, H. Jiao and U. Rosenthal, J. Am. Chem. Soc., 2013, 135, 17556–17565 CrossRef CAS PubMed;
(g) S. K. Podiyanachari, G. Kehr, C. Mück-Lichtenfeld, C. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 17444–17456 CrossRef PubMed;
(h) L. Becker, P. Arndt, H. Jiao, A. Spannenberg and U. Rosenthal, Angew. Chem., Int. Ed., 2013, 52, 11396–11400 CrossRef CAS PubMed;
(i) Ò. Àrias, A. R. Petrov, T. Bannenberg, K. Altenburger, P. Arndt, P. G. Jones, U. Rosenthal and M. Tamm, Organometallics, 2014, 33, 1774–1786 CrossRef;
(j) D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Organometallics, 2014, 33, 2760–2769 CrossRef CAS PubMed;
(k) L. Becker, P. Arndt, A. Spannenberg and U. Rosenthal, Chem. – Eur. J., 2014, 20, 12595–12600 CrossRef CAS PubMed;
(l) L. Becker, P. Arndt, A. Spannenberg, H. Jiao and U. Rosenthal, Angew. Chem., Int. Ed., 2015, 54, 5523–5526 CrossRef CAS PubMed.
- For selected reviews, see:
(a) E.-I. Negishi and T. Takahashi, Acc. Chem. Res., 1994, 27, 124–130 CrossRef CAS;
(b)
T. Takahashi and Y. Li, Zirconacyclopentadienes in Organic Synthesis, in Titanium and Zirconium in Organic Synthesis; Wiley-VCH, Weinheim, Germany, 2002, pp. 50–85 Search PubMed;
(c) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787–3802 CrossRef CAS PubMed;
(d) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC;
(e) S. Roy, U. Rosenthal and E. D. Jemmis, Acc. Chem. Res., 2014, 47, 2917–2930 CrossRef CAS PubMed.
- Selected actinide and lanthanide metallacycles, see:
(a) P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170–180 CrossRef CAS;
(b) P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650–6667 CrossRef CAS;
(c) J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS;
(d) J. M. Manriquez, P. J. Fagan and T. J. Marks, J. Am. Chem. Soc., 1978, 100, 3939–3941 CrossRef CAS;
(e) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC;
(f) L. Andrews, G. Kushto and C. J. Marsden, Chem. – Eur. J., 2006, 12, 8324–8335 CrossRef CAS PubMed;
(g) M. Foyentin, G. Folcher and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1987, 494–495 RSC;
(h) B. Fang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Dalton Trans., 2015, 44, 7927–7934 RSC;
(i) B. Fang, L. Zhang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Organometallics, 2015, 34, 5669–5681 CrossRef CAS;
(j) L. Xu, Y.-C. Wang, J. Wei, Y. Wang, Z. Wang, W.-X. Zhang and Z. Xi, Chem. – Eur. J., 2015, 21, 6686–6689 CrossRef CAS PubMed;
(k) L. Xu, J. Wei, W.-X. Zhang and Z. Xi, Chem. – Eur. J., 2015, 21, 15860–15866 CrossRef CAS PubMed;
(l) L. Xu, Y. Wang, Y.-C. Wang, Z. Wang, W.-X. Zhang and Z. Xi, Organometallics, 2016, 35, 5–8 CrossRef CAS;
(m) J. K. Pagano, J. M. Dorhout, R. Waterman, K. R. Czerwinski and J. L. Kiplinger, Chem. Commun., 2015, 51, 17379–17381 RSC;
(n) J. K. Pagano, J. M. Dorhout, K. R. Czerwinski, D. E. Morris, B. L. Scott, R. Waterman and J. L. Kiplinger, Organometallics, 2016, 35, 617–620 CrossRef CAS.
- For selected reviews, see:
(a) O. P. Lam and K. Meyer, Polyhedron, 2012, 32, 1–9 CrossRef CAS;
(b) O. P. Lam and K. Meyer, Angew. Chem., Int. Ed., 2011, 50, 9542–9544 CrossRef CAS PubMed;
(c) O. P. Lam, C. Anthon and K. Meyer, Dalton Trans., 2009, 9677–9691 RSC;
(d) K. Meyer and S. C. Bart, Adv. Inorg. Chem., 2008, 60, 1–30 CrossRef CAS;
(e) I. Castro-Rodríguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC;
(f) A. R. Fox, S. C. Bart, K. Meyer and C. C. Cummins, Nature, 2008, 455, 341–349 CrossRef CAS PubMed;
(g) M. S. Eisen, Top. Organomet. Chem., 2010, 31, 157–184 CrossRef CAS;
(h) T. Andrea and M. S. Eisen, Chem. Soc. Rev., 2008, 37, 550–567 RSC;
(i) E. Barnea and M. S. Eisen, Coord. Chem. Rev., 2006, 250, 855–899 CrossRef CAS;
(j) K. R. D. Johnson and P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947–1960 RSC;
(k) O. T. Summerscales and F. G. N. Cloke, Struct. Bonding, 2008, 127, 87–117 CrossRef CAS;
(l) P. L. Arnold, Chem. Commun., 2011, 47, 9005–9010 RSC;
(m) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS PubMed;
(n) M. Ephritikhine, Organometallics, 2013, 32, 2464–2488 CrossRef CAS;
(o) M. Ephritikhine, Dalton Trans., 2006, 2501–2516 RSC;
(p) T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC;
(q) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC;
(r) T. W. Hayton, Nat. Chem., 2013, 5, 451–452 CrossRef CAS PubMed;
(s) S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- Selected papers about the bonding of organoactinide complexes, see:
(a) T. Cantat, C. R. Graves, K. C. Jantunen, C. J. Burns, B. L. Scott, E. J. Schelter, D. E. Morris, P. J. Hay and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 17537–17551 CrossRef CAS PubMed;
(b) N. Barros, D. Maynau, L. Maron, O. Eisenstein, G. Zi and R. A. Andersen, Organometallics, 2007, 26, 5059–5065 CrossRef CAS;
(c) A. Yahia and L. Maron, Organometallics, 2009, 28, 672–679 CrossRef CAS;
(d) W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 40, 9662–9664 RSC;
(e) J. R. Walensky, R. L. Martin, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10007–10012 CrossRef CAS PubMed;
(f) L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589–10592 CrossRef CAS PubMed;
(g) B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Sci., 2014, 5, 2489–2497 RSC;
(h) N. L. Bell, L. Maron and P. L. Arnold, J. Am. Chem. Soc., 2015, 137, 10492–10495 CrossRef CAS PubMed;
(i) D. E. Smiles, G. Wu, P. Hrobárik and T. W. Hayton, J. Am. Chem. Soc., 2016, 138, 814–825 CrossRef CAS PubMed;
(j) K. P. Browne, K. A. Maerzke, N. E. Travia, D. E. Morris, B. L. Scott, N. J. Henson, P. Yang, J. L. Kiplinger and J. M. Veauthier, Inorg. Chem., 2016, 55, 4941–4950 CrossRef CAS PubMed;
(k) P. Yang, E. Zhou, G. Hou, G. Zi, W. Ding and M. D. Walter, Chem. – Eur. J., 2016, 22, 13845–13849 CrossRef CAS PubMed.
-
(a) B. Fang, W. Ren, G. Hou, G. Zi, D.-C. Fang, L. Maron and M. D. Walter, J. Am. Chem. Soc., 2014, 136, 17249–17261 CrossRef CAS PubMed;
(b) B. Fang, L. Zhang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Chem. Sci., 2015, 6, 4897–4906 RSC;
(c) B. Fang, G. Hou, G. Zi, W. Ding and M. D. Walter, Organometallics, 2016, 35, 1384–1391 CrossRef CAS.
- L. Zhang, G. Hou, G. Zi, W. Ding and M. D. Walter, J. Am. Chem. Soc., 2016, 138, 5130–5142 CrossRef CAS PubMed.
- For comparison, the reactivity of the thorium metallacyclopropenes is outlined in the ESI.†.
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- J. L. Kiplinger, D. E. Morris, B. L. Scott and C. J. Burns, Organometallics, 2002, 21, 3073–3075 CrossRef CAS.
- I. S. R. Karmel, N. Fridman, M. Tamm and M. S. Eisen, J. Am. Chem. Soc., 2014, 136, 17180–17192 CrossRef CAS PubMed.
- For selected well-characterized η2-nitrile metal complexes, see:
(a) T. C. Wright, G. Wilkinson, M. Motevalli and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 1986, 2017–2019 RSC;
(b) P. A. Chetcuti, C. B. Knobler and M. F. Hawthorne, Organometallics, 1988, 7, 650–660 CrossRef CAS.
- Complex 12 could also be prepared in 66% yield from [(η5-C5Me5)2 UCl]3 and 9,10-C14H8O2 in the presence of sodium amalgam, see ref. 4a.
-
G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- SQUEEZE: P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
-
(a) W. Küchle, M. Dolg, H. Stoll and H. Preuss, J. Chem. Phys., 1994, 100, 7535–7542 CrossRef;
(b) X. Cao, M. Dolg and H. Stoll, J. Chem. Phys., 2003, 118, 487–496 CrossRef CAS;
(c) X. Cao and M. Dolg, J. Mol. Struct.: THEOCHEM, 2004, 673, 203–209 CrossRef CAS;
(d) A. Moritz, X. Cao and M. Dolg, Theor. Chem. Acc., 2007, 118, 845–854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Reactivity of thorium metallacyclopropenes, molecular structures of 15 and 16, additional experiments and crystal parameters and Cartesian coordinates of all stationary points optimized at the B3PW91-PCM level. CCDC 1496477–1496481, 1496483–1496486 and 1496488–1496490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt03005j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence