
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving selectivity in catalytic hydrodefluorination by limiting SNV reactivity†
Juliane
Krüger
a,
Christian
Ehm
*b and
Dieter
Lentz
*a
aInstitut für Chemie und Biochemie, Anorganische Chemie, Freie Universität Berlin, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: dieter.lentz@fu-berlin.de
bDipartimento di Scienze Chimiche, Università di Napoli Federico II, Via Cintia, 80126 Napoli, Italy. E-mail: christian.ehm@unina.it
First published on 18th August 2016
Abstract
Catalytic hydrodefluorination of perfluoroallylbenzene with Cp2TiH in THF is unselective and yields a variety of previously unknown compounds, predominantly activated in the allylic position. Several different mechanisms have been examined in detail using solvent corrected (THF) DFT(M06-2X) calculations for the archetypal perfluorinated olefin perfluoropropene and perfluoroallylbenzene: (a) single electron transfer, (b) hydrometallation/fluoride elimination, (c) σ-bond metathesis (allylic or vinylic), and (d) nucleophilic vinylic substitution (SNV, w/o Ti–F contacts in the TS). SNV is shown to be a competitive mechanism to hydrometallation and proceeds via ionic species from which F-elimination is facile and unselective leading to low selectivity in polar solvents. Subsequent experiments show that selectivity can be increased in a non-polar solvent.
Introduction
Fluoroorganic compounds have received increasing attention because of their unique properties especially in the fields of pharmaceuticals, agricultural chemistry and materials science.1–11 However, the carbon–fluorine bond is the strongest and the most unreactive bond found in organic molecules and its use as an active functional group is a significant chemical challenge.12,13 A growing number of scientific publications have focused on transition metal-catalyzed C–F bond activation and C–F bond functionalization.14–18,19–25 Several reviews on the current state of the art in early and late transition metal catalyzed C–F bond activation have been published.14–17,26–30 For the simplest kind of C–F bond activation, hydrodefluorination (HDF), a feasible and promising strategy is to use transition metal hydrido complexes.29,31–48 Several systems capable of catalytic HDF are known, often using expensive late transition metal compounds.28,36,42,43,47–53 Only a few examples of group 4 catalyzed processes are known; Rosenthal et al. and Crimmin et al. described a zirconium based catalytic system using alanes as hydride sources.35,47 Recently we published an updated catalytic cycle for an earlier reported HDF employing a commercially available and air-stable precatalyst (titanocene difluoride, 1a) and a comparatively inexpensive hydride source (diphenylsilane, 2).54 The active species, a titanocene(III) hydrido complex (1), is generated in trace amounts in situ from 1a and 2 as catalyst regeneration with 2 is endergonic; the resting state is the trimeric titanocene(III) fluoride. E/Z regioselectivity in catalytic olefinic HDF using 1 appears to be substrate dependent but often unselective while vinylic C–F activation is always strongly preferred over allylic C–F activation.46 Based on experimental selectivities a hydrometallation/elimination mechanism (HM/E) and σ-bond metathesis (SBM) were proposed to be competing mechanisms but the reason for the pronounced substrate effect on regioselectivity remained elusive. Some group 4d0 systems have been studied theoretically. The groups of Jones and Eisenstein described the defluorination of perfluoropropene using tetravalent Cp*2ZrH2 and Cp*2ZrHF complexes.55 The first vinylic C–F bond exchange occurs via an internal insertion of the olefin followed by β-F elimination (HM/E mechanism).In the following, we will give new experimental and computational insights into the mechanism and regioselectivity of HDF reactions of catalyst 1 with hexafluoropropene (3) and perfluoroallylbenzene (4) and show that the solvent choice has a marked influence on HDF regioselectivity.
Results and discussion
In earlier experiments with substrate 3 and catalyst 1 we observed the following experimental trends: (a) temperature changes (−25 to +25 °C) do not influence the observed product selectivity,56 (b) steric and electronic changes in the catalyst influence its activity but barely influence the selectivity (E/Z 0.67 to 1.07 for 3), (c) changing the substrate has tremendous influence on both activity (TOF) as well as the selectivity (e.g. 1,1,1,3,3-pentafluoropropene (5) and 1,1,2-trifluorovinyl-ferrocene (6) in Table 1) and (d) the choice of silane for the regeneration reaction has no influence on the selectivity but does influence the activity.46 The low selectivity observed with 1 differs notably from selective HDF of 3 using Zr(IV).55Mechanistic studies on the HDF of fluoroolefins
Solvent corrected (PCM:THF) mechanistic DFT studies were performed on the M06-2X/TZ//M06-2X/DZ level of theory, see the Experimental section for details.57In the following we will first analyze HDF pathways for 1 and the archetypical olefin 3 before presenting results for substrate 4.
Several different mechanisms are possible and all have low activation barriers; TS formation proceeds directly from the reactants as the SOMO blocks the vacant coordination site of 1 rendering the formation of all adducts (or contact pairs) of 3 and 1 endergonic.58,59 HM/E (type A TS), SNV60 (type B TS), 4-center-SBM, 6-center-SBM (type C TS) as well as SET were considered to play a role.
Hydrometallation/elimination pathway
In plane approach of the catalyst towards the substrate C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond (hydride attack at C1) via 4-membered TSA3-5d (+8.5 kcal mol−1) leads to HM product 5d (−38.8 kcal mol−1).61 The barrier is purely entropic. 5d possesses no metal fluoride interactions and a staggered conformation (Fig. 1). HM is irreversible. IRC scans show that C–H bond formation is completed before Ti–C bond formation starts noticeably (see the ESI†). Rotamers 5a–5c have β-F dative interactions (Fig. 2). A β-H-agostic conformation could not be found. Interconversion between the resting states (RS) 5a–5cvia5d is easy as the energy difference between the RS is <4 kcal mol−1 and thus the ratio of the formed products depends only on ΔΔG‡ of the elimination TS (Curtin–Hammett).62
C double bond (hydride attack at C1) via 4-membered TSA3-5d (+8.5 kcal mol−1) leads to HM product 5d (−38.8 kcal mol−1).61 The barrier is purely entropic. 5d possesses no metal fluoride interactions and a staggered conformation (Fig. 1). HM is irreversible. IRC scans show that C–H bond formation is completed before Ti–C bond formation starts noticeably (see the ESI†). Rotamers 5a–5c have β-F dative interactions (Fig. 2). A β-H-agostic conformation could not be found. Interconversion between the resting states (RS) 5a–5cvia5d is easy as the energy difference between the RS is <4 kcal mol−1 and thus the ratio of the formed products depends only on ΔΔG‡ of the elimination TS (Curtin–Hammett).62
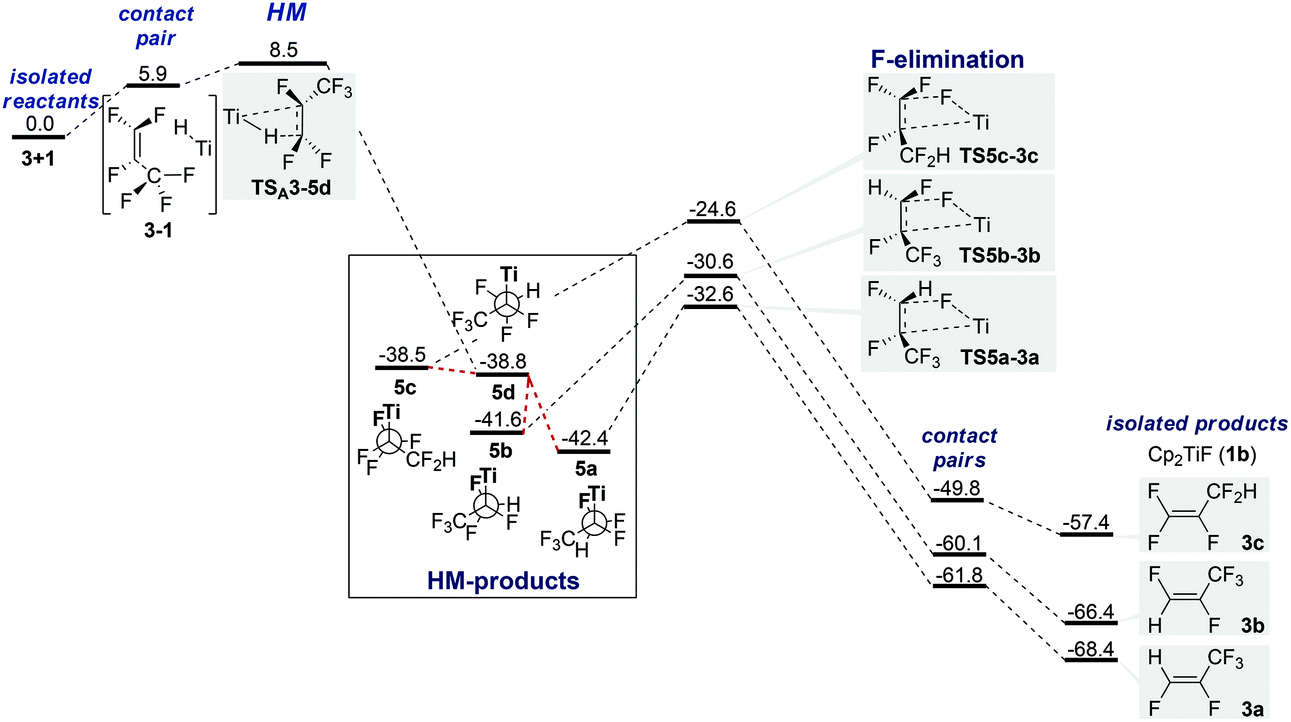 | ||
| Fig. 1 PES for HM/E pathway for HDF of 3 (ΔG in kcal mol−1, T = 298 K, 1 bar, in THF). | ||
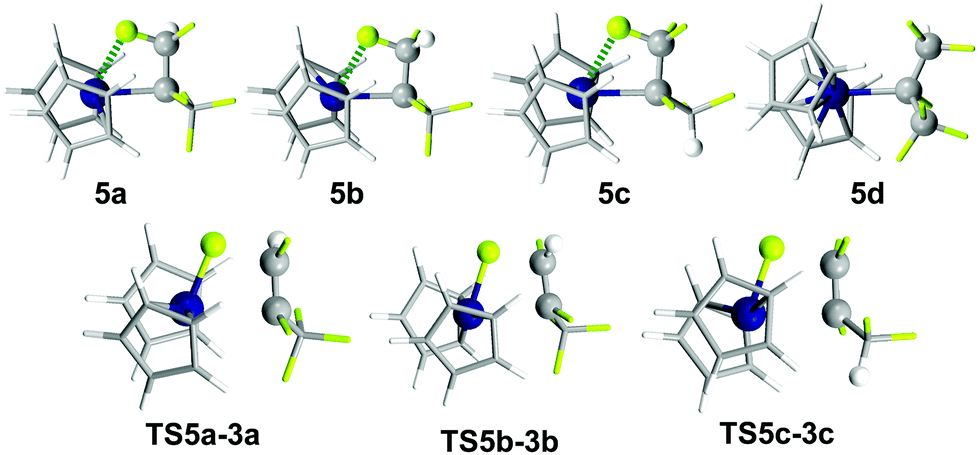 | ||
| Fig. 2 Resting states (RS) and elimination TS within the HM/E pathway. | ||
Barriers for F-elimination lie between 9.8 and 17.8 kcal mol−1 above the lowest RS 5a and lead via adduct complexes with H–F contacts to the isolated products. The elimination step is also irreversible and selectivity determining. Inspection of Table 2 and Fig. 1 shows that the β-fluorine elimination TS (TS5a-3a to TS5c-3c) are energetically closer to the RS (5a–d) than to the products (3a–c; 10–18 kcal mol−1 from RS, >30 kcal mol−1 from products).63 However, comparison of the geometries (Table 2 and Fig. 2) reveals that the elimination TS resemble the products much more than they resemble the RS. In particular, CC bond lengths in TS5a-3a to TS5c-3c are closer to the lengths in the isolated olefins 3a–c (+0.07 Å), i.e. formation of the olefin is almost complete already at the TS. Furthermore, C–FEl (>0.4 Å) and Ti–C bonds (>0.25 Å) are highly elongated, pointing to a large separation of titanocene fluoride and olefin in all TS (Table 2).
| C–H | CC |
C–FEl | F–Ti | C–Ti | ΔΔGa | |
|---|---|---|---|---|---|---|
| a With respect to the lowest ΔG in each of the three sets. | ||||||
| 5a | 1.090 | 1.501 | 1.427 | 2.292 | 2.324 | 0.0 |
| 5b | 1.092 | 1.509 | 1.421 | 2.297 | 2.321 | 0.8 |
| 5c | 1.092 | 1.513 | 1.401 | 2.349 | 2.329 | 3.9 |
| TS5a-3a | 1.083 | 1.392 | 1.824 | 2.050 | 2.585 | 0.0 |
| TS5b-3b | 1.085 | 1.395 | 1.831 | 2.056 | 2.578 | 2.0 |
| TS5c-3c | 1.093 | 1.396 | 1.873 | 2.040 | 2.577 | 8.0 |
| 3a | 1.081 | 1.323 | 0.0 | |||
| 3b | 1.084 | 1.324 | 2.0 | |||
| 3c | 1.090 | 1.325 | 11.0 | |||
| 1b | 1.863 | |||||
The large energy gain going from TS to the corresponding product (>30 kcal mol−1) varies by only 3 kcal mol−1 for the three different pathways (Fig. 1). The ΔΔG of the three products (0, 2.0 and 11.0 kcal) and H–F contact pairs (0.0, 1.7 and 12.0) almost match the corresponding TS ΔΔG‡ (0, 2.0 and 8.0 kcal mol−1).
The energy difference between 3a and 3b has been determined experimentally by Kaiser et al. (−2.85 kcal mol−1) and is in line with our DFT predictions.64
We tentatively conclude that barrier heights for elimination are dominated by product differences. Fluorinated sp2-carbons are destabilized as fluorine prefers a strong p-character in its bonds. TS5c-3c is the highest TS as 3c is destabilized by three fluorine substituents at the double bond.
Differences between 3a/3b and TS5a-3a/TS5b–3b, respectively, originate from the different destabilizations that occur if a CC double bond is destabilized by either cis- or trans-difluorination.65–67
σ-Bond metathesis and SNV
An approach of Ti–H to C1 with Ti–F interaction (SBM) results in either TSC3-3b (4-membered, +10.9 kcal mol−1) or TSC3-3c (6-membered, +10.5 kcal mol−1) and leads directly to product formation (Fig. 3).68 The activation barrier is again entirely entropic. IRC scans show that during SBM viaTSC3-3b and TSC3-3c C–H bond formation is completed before Ti–F elimination starts noticeably.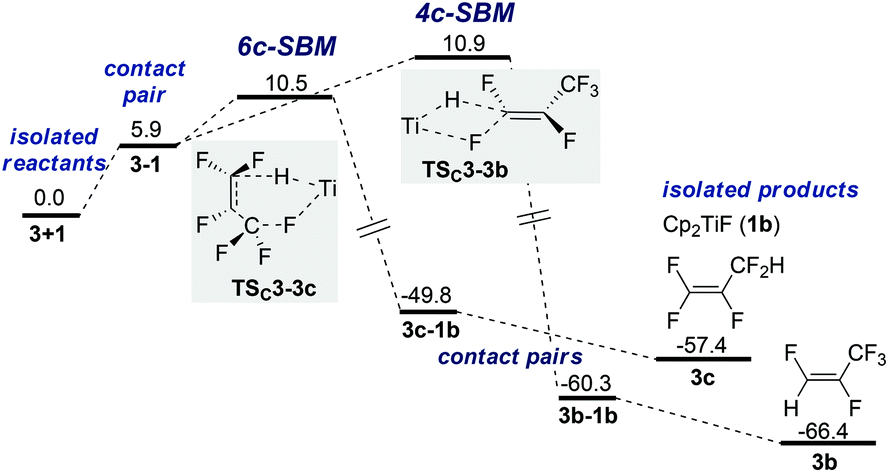 | ||
| Fig. 3 PES for SBM pathways for HDF of 3 (ΔG in kcal mol−1, T = 298 K, 1 bar, in THF). | ||
Nucleophilic attack of HTi at C1 (SNV) without Ti–F interaction viaTSB3-IP1 (+10.5 kcal mol−1) or with Ti–F interaction viaTSB3-IP2 (+12.7 kcal mol−1) results in the formation of contact ion pairs (CIP) IP1 and IP2 and the solvent separated ion pair (SIP) IP4, respectively (Fig. 4).69TSB3-IP1 is representative of several different, energetically close SNV TS.70IP4 is only 9.7 kcal mol−1 higher in free energy than IP2 and can recombine to the HM intermediate (5b)71 but can also decompose nearly barrierless.72 A hypothetical ‘IP3’ coming from TSC3-3b is unstable. Via rotation, IP1 leads equally to IP2 and unstable ‘IP3’, thus resulting in a statistical 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 3a and 3b. An SET mechanism should be excluded based on the low electron affinity of perfluorinated olefins compared to perfluorinated arenes (see also studies for 4 next page).
:1 ratio of 3a and 3b. An SET mechanism should be excluded based on the low electron affinity of perfluorinated olefins compared to perfluorinated arenes (see also studies for 4 next page).
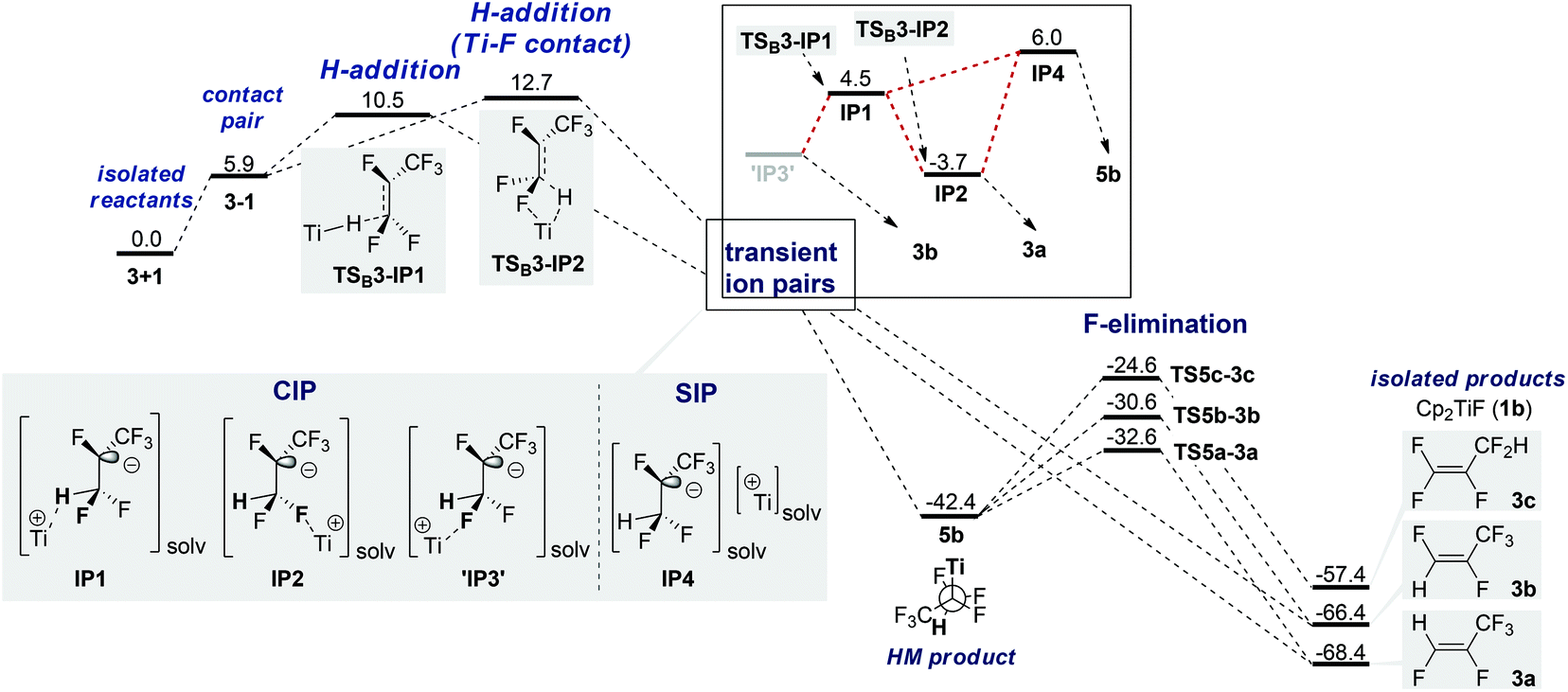 | ||
| Fig. 4 PES for SNV pathways for HDF of 3 (ΔG in kcal mol−1, T = 298 K, 1 bar, in THF). | ||
Comparison of 1st TS geometries for 3
First steps in the different possible HDF mechanisms are irreversible thus pathway competition is given by TS ΔΔG‡. All TS shown in Table 3 and Fig. 5 are energetically very early and purely entropic as a result of TS-formation out of two independent, yet almost unchanged, molecules (ΔG‡ 8.5–12.7 kcal mol−1 from the separated reactants, ΔH‡ −6.7 to −1.2 kcal mol−1). TSA3-5d and TSC3-3c show only minimal changes in geometry from the isolated reactants.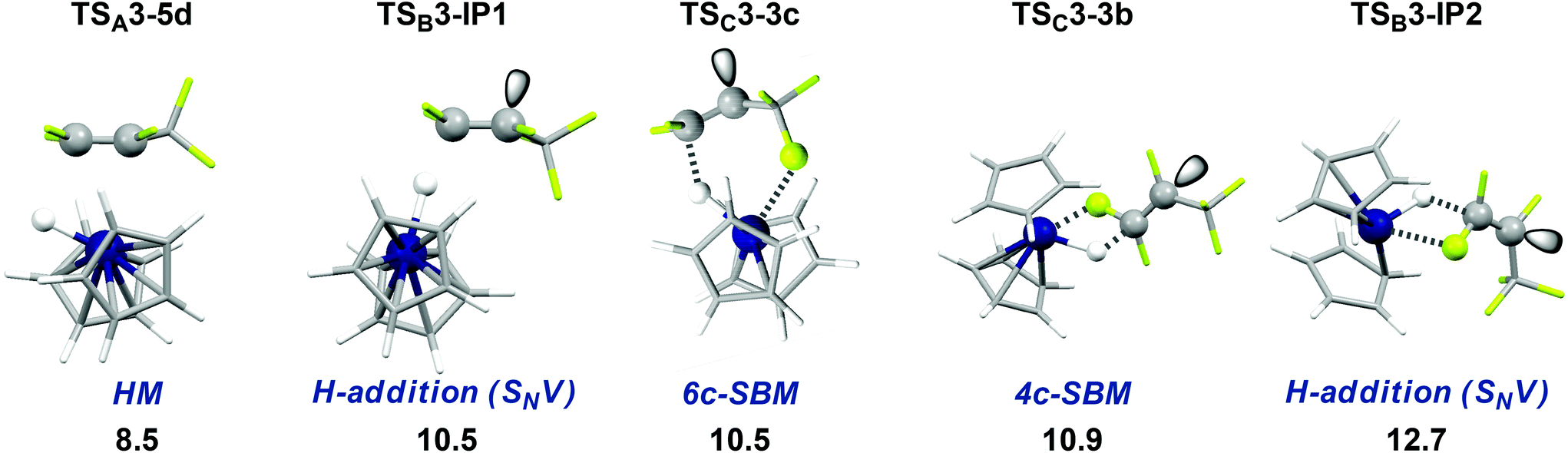 | ||
| Fig. 5 Comparison of the first TS (ΔG‡ in kcal mol−1). Gibbs free energies of activation from the isolated reactants in THF (T = 298 K, 1 bar). Only the lowest TS of type TSB3-IP1 is shown. | ||
| Ti–H | C–H | F–Ti/C–Ti | CC |
FCCF |
ΔH‡ | |
|---|---|---|---|---|---|---|
| 1 | 1.703 | |||||
| 3 | 1.326 | |||||
| 1–3 | 1.738 | 2.638 | 3.803 | 1.327 | 0 | −5.2 |
| TSB3-IP2 | 1.802 | 1.722 | 2.347 | 1.349 | 24 | −1.1 |
| TSB3-IP1 | 1.780 | 1.610 | — | 1.360 | 29 | −1.2 |
| TSC3-3b | 1.788 | 1.731 | 2.314 | 1.349 | 22 | −1.4 |
| TSC3-3c | 1.760 | 1.788 | 2.354 | 1.366 | 24 | −3.3 |
| TSA3-5d | 1.717 | 2.170 | 2.802 | 1.352 | 17 | −6.7 |
TS Gibbs free energies follow the trend in the distance of both reactants (shorter distance, higher TS energy) as exemplified by the HTi–C (1.61–2.17 Å), F–Ti (or C–Ti) distances (2.31–2.80 Å) and Ti–H bonds (stretched by max. 0.1 Å); all other bonds are not elongated more than 0.08 Å compared to the isolated reactants. For all TS beginning rehybridization is observed as the Fcis–CC–Fcis dihedral angle ranges between 17–29°; a full rehybridization from sp2 to sp3 would change the dihedral angle from 0 to 60°. Fig. 5 shows the beginning formation of a lone pair on C2 for the 1st TS. IRC scans show that for all pathways, H-addition is completed and the lone pair on C2 fully formed, before Ti–C bond formation or Ti–F elimination starts.
Pathway competition for 3
The analysis of the reaction mechanism by DFT calculations exhibits that three HDF pathways are active in the F/H-exchange of 3 with 1. With the exception of SNV with Ti–F contact (TSC3-IP2), all TS are within ±2 kcal mol−1, i.e. HM, SBM and SNV are competitive mechanisms.Each of these mechanisms has its preferred ratio of isomers (Table 4). Allylic products are predicted to be exclusively formed by 6c-SBM and not as assumed previously exclusively formed by HM.73
| Ratio (exp.) |
3a/3b/3c = 28:14:1 |
||||
|---|---|---|---|---|---|
| Mechanism | HM/E | 4c-SBM | 6c-SBM | SNV | SNV (Ti–F) |
| Product | 3a/3b/3c | 3b only | 3c only | 3a/3b | 3a only |
| Ratio (DFT) | 29:1:0 |
<1 | 1 | 1:1 |
0 |
| ΔG‡ (THF) | 8.5 | 10.9 | 10.5 | 10.5 | 12.7 |
Experimental selectivities correspond to energy differences (ΔG = RTln(k1/k2), T = 298.15 K) between the reaction pathways of 0.2 kcal mol−1 (3avs. 3b) and 2.3 kcal mol−1 (3avs. 3c). DFT predicts those differences to be 2.0 kcal mol−1via HM/E and 2.0 kcal mol−1via 6c-SBM, respectively. DFT predictions in THF are only qualitatively correct for the 3a/3b ratio via the energetically lowest HDF pathway HM/E. The only mechanism that is energetically close and yields a selectivity close to the experimentally observed one is SNV. All TS for HDF of 3 are very early, show only limited catalyst/substrate interactions and are basically entropy driven TS (see Fig. 6). Additionally, six different SNV TS were found for 3.
 | ||
| Fig. 6 Schematic overview of the plethora of possible HDF TS. Grey olefins differ in relative orientation wrt to the nearest black one, e.g. cis vs. trans C–F activation. | ||
It appears plausible that in all or some cases the TS found analysing the electronic energy PES is not identical with the one on the Gibbs free energy PES. This could affect the predicted ratio of 3a (via HM and SNV) and 3b (via SNV). Another possible explanation is that the functional M06-2X is not accurate enough or that despite out best efforts, we have not found the lowest TS of type TSB3-IP1.
Experimental studies on HDF of 4
In order to study the different reactivities of various C–F bonds within one molecule we chose perfluoroallylbenzene (4) as an ideal substrate in the sense that it is the smallest possible molecule comprising vinylic, allylic and aromatic C–F bonds. Additionally, it features an extended low lying π*-orbital and HDF regioselectivity could help to clarify the potential role of SET type chemistry. The 1st HDF of 4 takes places at the C3F5-moiety and allylic (4a,b) and vinylic (4c,d) products are formed in a 3:2 ratio (Scheme 1), in striking contrast to other reported olefins where only trace amounts of the allylic product are observed. E/Z mixtures were obtained for both constitutional isomers and formation of the E-isomers is always preferred.
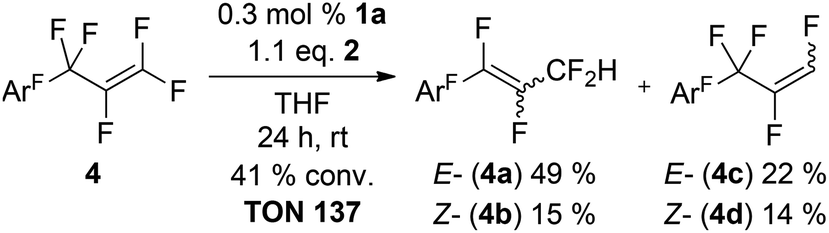 | ||
| Scheme 1 Competing catalytic HDF of 4 (ArF = C6F5). | ||
Increasing the amount of the catalyst or the reaction time or temperature leads to a second HDF step in the para position and formation of 4a′-d′ (Scheme 2).74
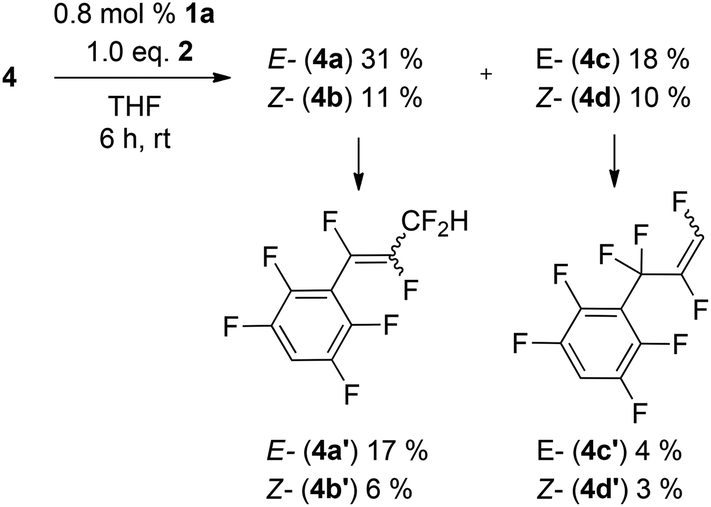 | ||
| Scheme 2 2nd generation HDF products. | ||
The eight new organofluorine compounds (4a,a′,b,b′,c,c′,d,d′) show distinct features in the 19F and 1H NMR spectra that allow for their unambiguous identification, see the Experimental section and ESI† for more details. The chemical shifts and coupling constants are only slightly affected by aromatic para H/F substitution, so that all resonances in the 19F NMR are doubled except those for the missing para-F atom. Fig. 7–9 show some illustrative cut outs of a 19F NMR spectra of a reaction mixture containing 1st and 2nd HDF products. Allylic HDF products show characteristic chemical shifts and coupling constants for the CF2H-group around −124 ppm (2JFH = 50.0 Hz for 4a and 2JFH = 51.3 Hz for 4b), additionally trans products have a characteristic large trans-F coupling constant (3JFF = 138.0 Hz for 4a and 3JFF = 136.0 Hz for 4c) and the vinylic HDF products show the typical gem-H/F coupling constants (2JFH = 71.5 Hz for 4c and 2JFH = 68.9 Hz for 4d). HDF for substrate 4, similar to 3 (E/Z = 0.67), proceeds very unselectively.
 | ||
| Fig. 7 19F NMR sequence of the para-F nuclei for 1st generation HDF products (−149.5 for 4a, −149.4 for 4b, −150.2 for 4c and −150.4 ppm for 4d). | ||
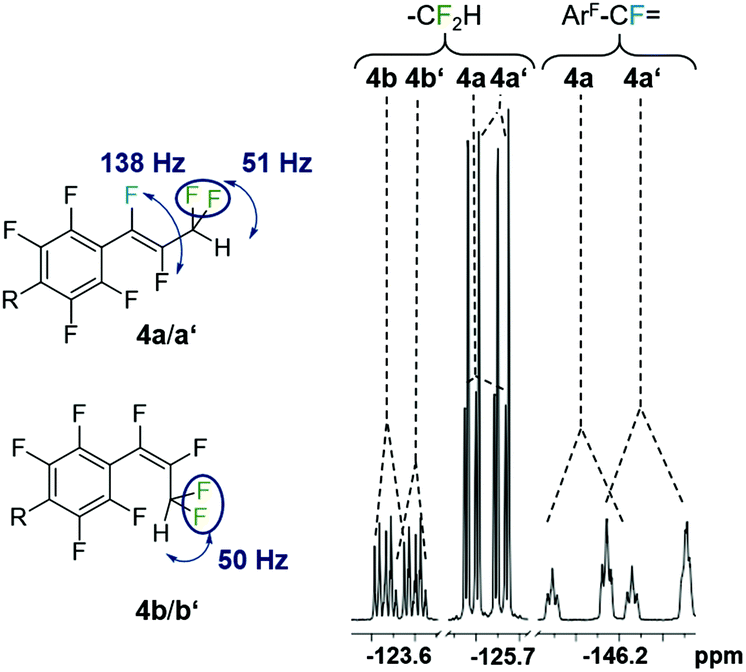 | ||
| Fig. 8 Characteristic 19F NMR resonances and coupling constants for 1st (4a,b with R = F) and 2nd generation allylic-HDF products (4a′,b′ with R = H). | ||
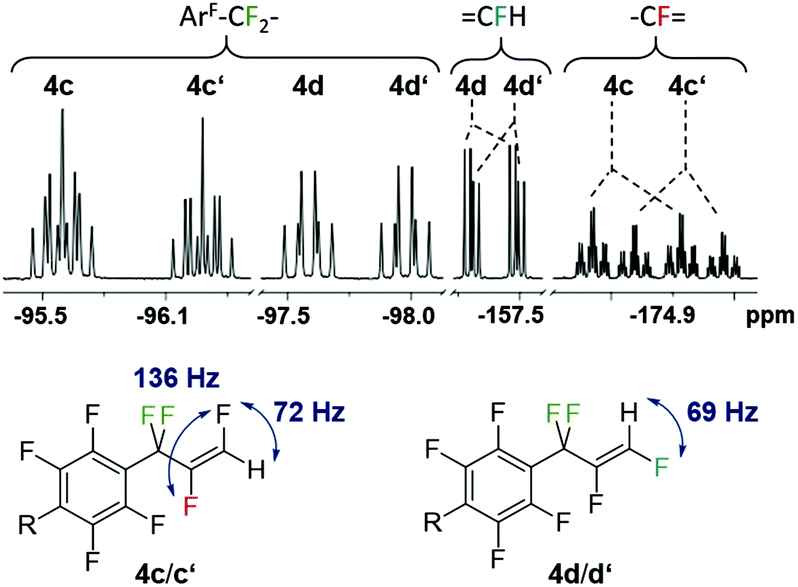 | ||
| Fig. 9 Characteristic 19F NMR resonances and coupling constants for 1st (4c,d with R = F) and 2nd generation vinylic-HDF products (4c′,d′ with R = H). | ||
DFT studies for 4
We assumed that the same mechanisms as just discussed for 3 could play a role for HDF of 4. All TS were followed by IRC scans and lead to similar resting states (HM and SNV pathways) or directly to the products (6c-SBM). Attack of 1 on C1, with or without a TiF contact in the TS, proceeds via SNV and leads to a stable ion pair, regardless of a cis or trans approach (TSB4-IP1, TSB4-IP2 and TSB4-IP3; Fig. 10).75 Both 6c-SBM TS (TSC4-4a and TSC4-4b) are favored over SNV (with Ti–F contact). HM viaTSA4-HM is predicted to be the overall preferred pathway. All activation barriers are approx. 1 kcal mol−1 lower than they are for 3. Within the HM pathway, the elimination TS leading to 4d is heavily preferred (see the ESI†). The TS leading to allylic products (4a and 4b) are 7 kcal, and the TS leading to 4c are ∼2.5 kcal mol−1 higher in energy.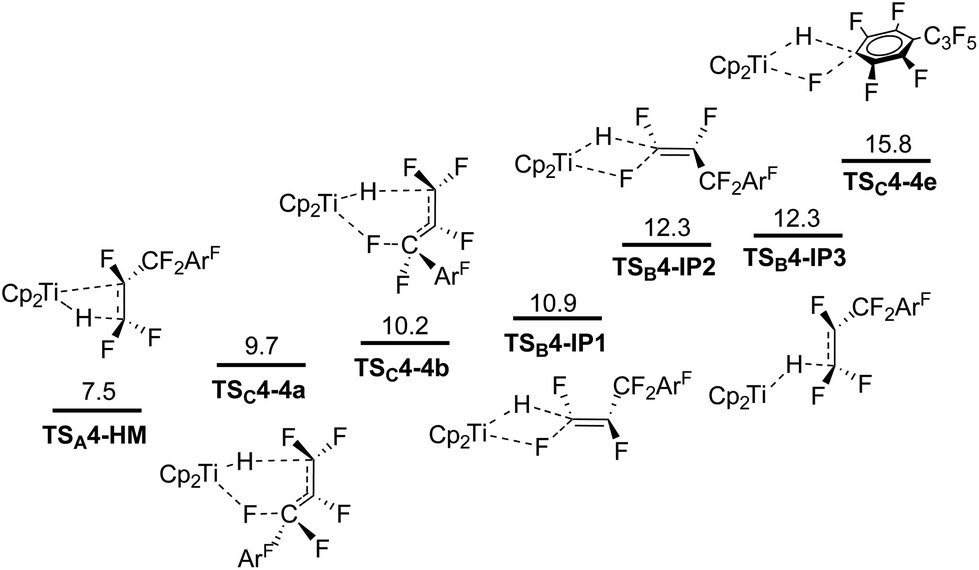 | ||
| Fig. 10 Pathway competition for HDF of 4 (ΔG in kcal mol−1, T = 298 K, 1 bar, in THF). | ||
DFT predictions are thus not in line with the experimentally observed selectivities as only 4d should be formed. The aromatic C–F activation barrier is predicted to be significantly higher (but accessible for 2nd generation HDF) than the allylic and vinylic ones. SET was shown to be a possible mechanism for HDF of fluorinated arenes using late transition metals. However, the experimental selectivity in the 1st HDF product of 4 hints that SET cannot play a role here. While the low lying π*-LUMO of 4 is spread out through the whole molecule (Fig. 11), the most stable isomer of radical anion 4˙− is 4A˙− (negative charge and SOMO concentrated on the arene ring), and not 4B˙− (calculated ΔG = +4.6 kcal mol−1, negative charge and SOMO concentrated on C1 and C2 of the former alkene, respectively). However, 1st generation aromatic HDF products are not observed.
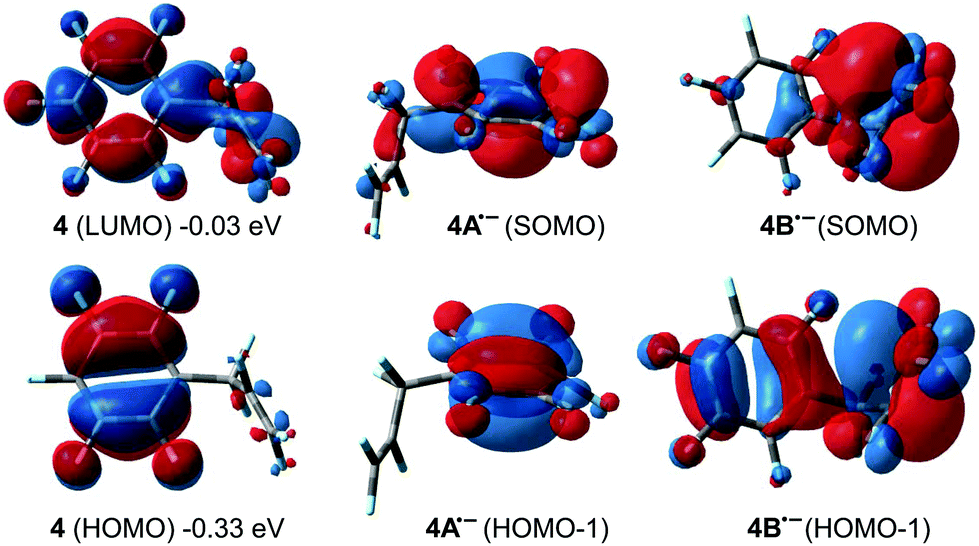 | ||
| Fig. 11 Visualization of LUMO and HOMO for 4 and SOMO/HOMO−1 for 4˙− and 4B˙− (isovalue = 0.03). | ||
Suppressing SNV reactivity
For both substrates (3 and 4), DFT is not able to correctly predict the product isomer ratios in THF. In both cases however, energetically low lying SNV TS, which could be responsible for the low selectivity, were found. In order to suppress this SNV reactivity via ion pairs we have changed the solvent from THF to toluene, both in the experiment and the calculations (Table 5). HDF of 3 in toluene exhibits an isomeric ratio of 3a to 3b to 3c of 9:1:1, which is now completely in line with DFT results.76 As expected the TS energy for TSB3-IP1 increases to +13.8 kcal mol−1 excluding this reaction pathway.77
| Ratio (exp.) |
3a/3b/3c = 9:1:1 |
||||
|---|---|---|---|---|---|
| Mechanism | HM/E | 4c-SBM | 6c-SBM | SNV | SNV (Ti–F) |
| Product | 3a/3b/3c | 3b only | 3c only | 3a/3b | 3a only |
| Ratio (DFT) | 12:1:0 |
>1 | 1 | 0:0 |
0 |
| ΔG‡ (tol.) | 6.6 | 9.4 | 7.4 | 13.8 | 13.4 |
A solvent change increases selectivity for 4 as well (Table 6). The initial test for other substrates, e.g.5, interestingly leads to multiple subsequent HDF steps.78 We are currently optimizing reaction conditions for these substrates.
As mentioned earlier, 5 and 6 give one HDF product in major amounts (Table 1) even in polar solvents.46 SNV should be disfavored for HDF of 5 and 6 because ion pairs are destabilized as Fc or H groups are more electron donating than F, explaining the increase in selectivity, compared to 3.79
Although solvent change (THF to toluene) lowers most barriers for activation, it decreases the TOF as it affects the rate limiting catalyst regeneration step, too.54
Similarly to 1, DFT calculations for HDF of 3 using Cp2ZrH2 in toluene predict the Z-isomer 3a but experimentally, Cp2*ZrH2 leads to the E-isomer 3b.55 This indicates that catalyst tuning should have an influence, and we subsequently tested the stoichiometric reaction of Cp*2TiH with 3, which similarly favors the formation of the E-isomer (E/Z = 2.7). Previously, we tested selectivity of HDF of 3 with different catalysts in a polar solvent but the results presented here indicate that those tests should be repeated in a non-polar solvent to avoid SNV reactivity.46
Conclusion
For the first time, a substrate (perfluoroallylbenzene) allowing conclusions for intramolecular competition of vinylic, allylic and aromatic was tested in HDF using 1. A yet unmatched TON was observed but the reaction proceeds unselectively. Vinylic and allylic attacks are preferred over aromatic for the first HDF step but aromatic HDF becomes dominant in the subsequent step. SET likely plays no role in this reactivity.DFT calculations provide previously unknown insight into Ti-catalyzed HDF reactions. In polar solvents, HM/E, 4-center-SBM, 6-center-SBM and SNV are competitive reaction pathways. The latter one proceeds unselectively, which likely is responsible for low selectivities. Non-polar solvents can suppress SNV reactivity leading to higher selectivity. Selectivity within the HM/E pathway is driven by product energy differences. 6c-SBM is preferred over SBM-4 reactivity.
We have recently shown that the barrier for regeneration is heavily dependent on the choice of silane. A combination of the right choice of silane and a non-polar solvent could therefore open routes to achieve highly selective HDF at low temperatures. This is a current focus in our laboratory.
Experimental section
All preparations were performed using standard Schlenk-type and vacuum-line techniques, or by working in an argon-filled glove box. Diglyme, THF and toluene were freshly distilled from sodium or potassium/benzophenone. 1a, 2, 4 (abcr) and 5 (Syn-Quest Labs) were obtained from commercially available sources and Cp*2TiH was synthesized analogous to a procedure reported for Cp′2TiH (Cp′ = (1,2,3,4-tetramethyl-5-phenyl)cyclopentadienyl) starting from Cp*2TiMe.802 and 4 were distilled from calcium hydride and 1a was sublimed prior to use. 3 (Solvay Fluor) was obtained free of charge and 3 and 5 were used as received.Catalytic reactions
Reaction conditions and substrates are listed in Table S1 of the ESI.† A single-necked flask equipped with a PTFE valve was charged with 1a, 2 and solvent. If no color change occurred the mixture was shortly heated with a heat gun (∼10 s) until its color changed to green. Substrate addition: the frozen mixture was degassed prior to condensing the gaseous substrates. 4 was added via a syringe and degassed afterwards. By opening the valve the reaction was stopped, accompanied by a change of color from green to yellow; fluorobenzene (standard) was added and stirred for 5 min and NMR spectra were recorded.Stoichiometric reaction
A single-necked flask equipped with a PTFE valve was charged with Cp*2TiH (20 mg, 0.06 mmol), dissolved in THF and an excess 3 was added. After a reaction time of one hour at room temperature the reaction was stopped by opening the vessel, fluorobenzene (standard) was added and stirred for 5 min and NMR spectra were recorded.Hydrodefluorination products were identified by NMR spectroscopy, using literature data for 3a,813b81 and 4e.82
), −149.5 (1F, m, para-Farene), −160.1 (2F, m, ortho-Farene), −165.1 (1F, 3JFF = 138.0 Hz, CF–) ppm. 4b: 1H (400 MHz, C6D6): δ = 6.60 (1H, td, 2JHF = 50.0 Hz, 3JHF = 34.5 Hz, CFH) ppm; 19F (376 MHz, C6D6): δ = −122.9 (1F, m, ArF–CF), −123.8 (2F, ddd, 2JFH = 50.1 Hz, 3JFF = 19.7 Hz, 4JFF = 10.2 Hz, –CF2H), −137.8 (2F, m, meta-Farene), −149.4 (1F, m, para-Farene), −157.5 (1F, CF–), −160.2 (2F, m, ortho-Farene) ppm. 4c: 1H (400 MHz, C6D6): δ = 7.82 (1H, ddt, 2JHF = 68.5 Hz, 3JHF = 4.4 Hz, 4JFH = 1.4 Hz, CFH) ppm; 19F (376 MHz, C6D6): δ = −95.6 (2F, m, ArF–CF2–), −141.2 (2F, m, meta-Farene), −150.2 (1F, ttt, 3JFF = 21.6 Hz, 4JFF = 5.3 Hz, 6JFF = 1.7 Hz, para-Farene), −160.3 (2F, m, ortho-Farene), −165.7 (1F, ddtt, 2JFH = 71.4 Hz, 3JFF = 136.0 Hz, 4JFF = 20.5 Hz, 6JFF = 2.3 Hz, CFH), −174.4 (1F, dtdt, 3JFF = 136.0 Hz, 3JFF = 18.1 Hz, 3JFH = 4.0 Hz, 5JFF = 3.6 Hz, –CF) ppm. 4d: 1H (400 MHz, C6D6): δ = 7.51 (1H, ddt, 2JHF = 68.6 Hz, 3JHF = 15.4 Hz, 4JFH = 1.2 Hz, CFH) ppm; 19F (376 MHz, C6D6): δ = −97.6 (2F, td, 4JFF = 25.8 Hz, 3JFF = 20.7 Hz, ArF–CF2–), −140.6 (2F, m, meta-Farene), −150.4 (1F, ttt, 3JFF = 21.9 Hz, 4JFF = 5.4 Hz, 6JFF = 1.7 Hz, para-Farene), −153.8 (1F, m, –CF), −157.1 (1F, dd, 2JFH = 68.9 Hz, 3JFF = 9.0 Hz, CFH), −159.9 (2F, m, ortho-Farene) ppm. 4a′: 1H (400 MHz, C6D6): δ = 7.10 (1H, tdd, 2JHF = 51.3 Hz, 3JHF = 15.6 Hz, 4JHF = 2.5 Hz, –CF2H) ppm; 19F (376 MHz, C6D6): δ = −125.5 (2F, ddd, 2JFH = 51.2 Hz, 3JFF = 18.9 Hz, 4JFF = 1.3 Hz, –CF2H), −137.0 (2F, m, meta-Farene), −146.0 (1F, dtt, 3JFF = 138.0 Hz, 4JFF = 8.3 Hz, 4JFF = 1.4 Hz, ArF–CF), −160.8 (2F, m, ortho-Farene), −165.3 (1F, 3JFF = 138.0 Hz, CF–) ppm. 4b′: 1H (400 MHz, C6D6): δ = 6.55 (1H, tdd, 2JHF = 50.3 Hz, 3JHF = 15.6 Hz, 4JHF = 2.5 Hz, –CF2H) ppm; 19F (376 MHz, C6D6): δ = −123.1(1F, m, ArF–CF), −123.7 (2F, ddd, 2JFH = 51.2 Hz, 3JFF = 21.2 Hz, 4JFF = 7.8 Hz, –CF2H), −137.1 (2F, m, meta-Farene), −154.8 (1F, m, CF–), −160.3 (2F, m, ortho-Farene) ppm. 4c′: 1H (400 MHz, C6D6): δ = 7.80 (1H, ddt, 2JHF = 68.5 Hz, 3JHF = 4.4 Hz, 4JFH = 1.4 Hz, CFH) ppm; 19F (376 MHz, C6D6): δ = −96.3 (2F, m, ArF–CF2–), −141.4 (2F, m, meta-Farene), −160.8 (2F, m, ortho-Farene), −166.0 (1F, ddtt, 2JFH = 71.4 Hz, 3JFF = 136.0 Hz, 4JFF = 20.5 Hz, 6JFF = 2.3 Hz, CFH), −174.9 (1F, dtdt, 3JFF = 136.0 Hz, 3JFF = 18.1 Hz, 3JFH = 4.0 Hz, 5JFF = 3.6 Hz, –CF) ppm. 4d′: 1H (400 MHz, C6D6): δ = 7.50 (1H, ddt, 2JHF = 68.6 Hz, 3JHF = 15.4 Hz, 4JFH = 1.2 Hz, CFH) ppm; 19F (376 MHz, C6D6): δ = −97.9 (2F, td, 4JFF = 25.8 Hz, 3JFF = 20.7 Hz, ArF–CF2–), −140.7 (2F, m, meta-Farene), −154.0 (1F, m, –CF), −157.2 (1F, dd, 2JFH = 68.9 Hz, 3JFF = 9.0 Hz, CFH), −160.1 (2F, m, ortho-Farene) ppm.
GC-MS (EI, eV): m/z = 279.8 M+ (4.43, 4.51, 4.58 min; 4a–d), 261.7 M+ (5.20, 5.24, 5.30; 4a′–d′).
Computational details
All structures were fully optimized at the M06-2X(PCM)83/6-31+(2d,p) level of theory using Gaussian 0957 coupled to an external optimizer (PQS; convergence settings: DMAX 0.1, TOLG 0.0003, TOLE 0.00004, TOLD 0.0018)84,85 instead of the internal Gaussian optimizer, using an ultrafine grid (Int(Grid = ultrafine)) and standard SCF convergence quality settings (Scf = tight) for Gaussian single point calculations. The nature of each stationary point was checked with an analytical second-derivative calculation (no imaginary frequency for minima, exactly one imaginary frequency for transition states, corresponding to the reaction coordinate) and the accuracy of the TS was confirmed with an IRC scan. S2 values for all doublet species are below 0.76. Solvent influence (THF, ε = 4.2457, toluene, ε = 2.3741) was modeled explicitly, using the polarizable continuum model (PCM) implemented in the Gaussian 09 software suite.Transition states were located using a suitable guess and the Berny algorithm (Opt = TS)86 or the Synchronous Transit-Guided Quasi-Newton (STQN) Method, developed by H. B. Schlegel and coworkers87,88 (Opt = QST2 or QST3) or a relaxed potential energy scan to arrive at a suitable transition state guess, followed by a quasi-Newton or eigenvector-following algorithm to complete the optimization.
Vibrational analysis data derived at this level of theory were used to calculate thermal corrections (enthalpy and entropy, 298 K, 1 bar) for all species considered. Final single-point (SP) energies were calculated at the M06-2X(PCM)89,90 level of theory employing triple-ζ Dunning basis sets (cc-pVTZ) from the EMSL basis set exchange library,91,92 to minimize BSSE contributions,93 including Grimmes’ dispersion corrections for M06-2X (zero option in dftd3 standalone program).94,95
Acknowledgements
The authors would like to thank Prof. P. H. M. Budzelaar (U-Manitoba, Winnipeg) for generous donation of CPU time and valuable discussions on the manuscript. We thank Dr. Darina Heinrich for supportive data evaluation and helpful discussions. Support from the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged.Notes and references
- D. O'Hagan, J. Fluorine Chem., 2010, 131, 1071 CrossRef.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC.
- I. S. R. Stenhagen, A. K. Kirjavainen, S. J. Forsback, C. G. Jørgensen, E. G. Robins, S. K. Luthra, O. Solin and V. Gouverneur, Chem. Commun., 2013, 49, 1386 RSC.
- M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426 CrossRef CAS PubMed.
- E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639 CrossRef CAS PubMed.
- J.-P. Bégué and D. Bonnet-Delpon, Bioorganic and medicinal chemistry of fluorine, John Wiley & Sons, Hoboken, N.J., 2008 Search PubMed.
- T. Nakajima and H. Groult, Fluorinated materials for energy conversion, Elsevier, Amsterdam, San Diego, CA, Oxford, 2005 Search PubMed.
- B. Ameduri and B. Boutevin, Well-Architectured Fluoropolymers: Synthesis, Properties and Applications, Elsevier Science, 2004 Search PubMed.
- A. McCulloch, J. Fluorine Chem., 2003, 123, 21 CrossRef CAS.
- M. P. Krafft and J. G. Riess, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 1185 CrossRef CAS.
- P. Kirsch, Modern Fluoroorganic Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2013 Search PubMed.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22 RSC.
- D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308 RSC.
- E. Clot, O. Eisenstein, N. Jasim, S. A. Macgregor, J. E. McGrady and R. N. Perutz, Acc. Chem. Res., 2011, 44, 333 CrossRef CAS PubMed.
- T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931 CrossRef CAS PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 52, 3328 CrossRef PubMed.
- T. A. Unzner and T. Magauer, Tetrahedron Lett., 2015, 56, 877 CrossRef CAS.
- J. Weaver and S. Senaweera, Tetrahedron, 2014, 70, 7413 CrossRef CAS.
- M. Ohashi, M. Shibata and S. Ogoshi, Angew. Chem., Int. Ed., 2014, 53, 13578 CrossRef CAS PubMed.
- C. L. Clark, J. J. Lockhart, P. E. Fanwick and S. C. Bart, Chem. Commun., 2015, 51, 14084 RSC.
- H. Luo, G. Wu, S. Xu, K. Wang, C. Wu, Y. Zhang and J. Wang, Chem. Commun., 2015, 51, 13321 RSC.
- S. I. Kalläne, M. Teltewskoi, T. Braun and B. Braun, Organometallics, 2015, 34, 1156 CrossRef.
- D. McKay, I. M. Riddlestone, S. A. Macgregor, M. F. Mahon and M. K. Whittlesey, ACS Catal., 2015, 5, 776 CrossRef CAS.
- B. Procacci, Y. Jiao, M. E. Evans, W. D. Jones, R. N. Perutz and A. C. Whitwood, J. Am. Chem. Soc., 2015, 137, 1258 CrossRef CAS PubMed.
- P. Tian, C. Feng and T.-P. Loh, Nat. Commun., 2015, 6, 7472 CrossRef PubMed.
- M. F. Kuehnel, D. Lentz and T. Braun, Angew. Chem., Int. Ed., 2013, 125, 3412 CrossRef.
- M. Klahn and U. Rosenthal, Organometallics, 2012, 31, 1235 CrossRef CAS.
- M. K. Whittlesey and E. Peris, ACS Catal., 2014, 4, 3152 CrossRef CAS.
- J.-Y. Hu and J.-L. Zhang, Top. Organomet. Chem., 2015, 52, 143 CrossRef CAS.
- N. A. LaBerge and J. A. Love, Top. Organomet. Chem., 2015, 52, 55 CrossRef CAS.
- T. Braun, D. Noveski, M. Ahijado and F. Wehmeier, Dalton Trans., 2007, 3820 RSC.
- T. Braun, F. Wehmeier and K. Altenhöner, Angew. Chem., Int. Ed., 2007, 46, 5321 CrossRef CAS PubMed.
- R. D. Rieth, W. W. Brennessel and W. D. Jones, Eur. J. Inorg. Chem., 2007, 2839 CrossRef CAS.
- R. J. Lindup, T. B. Marder, R. N. Perutz and A. C. Whitwood, Chem. Commun., 2007, 3664 RSC.
- U. Jäger-Fiedler, M. Klahn, P. Arndt, W. Baumann, A. Spannenberg, V. V. Burlakov and U. Rosenthal, J. Mol. Catal. A: Chem., 2007, 261, 184 CrossRef.
- S. P. Reade, M. F. Mahon and M. K. Whittlesey, J. Am. Chem. Soc., 2009, 131, 1847 CrossRef CAS PubMed.
- D. Breyer, T. Braun and A. Penner, Dalton Trans., 2010, 39, 7513 RSC.
- B. M. Kraft, E. Clot, O. Eisenstein, W. W. Brennessel and W. D. Jones, J. Fluorine Chem., 2010, 131, 1122 CrossRef CAS.
- M. F. Kühnel and D. Lentz, Angew. Chem., Int. Ed., 2010, 49, 2933 CrossRef PubMed.
- J. A. Panetier, S. A. Macgregor and M. K. Whittlesey, Angew. Chem., Int. Ed., 2011, 50, 2783 CrossRef CAS PubMed.
- T. F. Beltrán, M. Feliz, R. Llusar, J. A. Mata and V. S. Safont, Organometallics, 2011, 30, 290 CrossRef.
- D. Breyer, T. Braun and P. Kläring, Organometallics, 2012, 31, 1417 CrossRef CAS.
- J.-H. Zhan, H. Lv, Y. Yu and J.-L. Zhang, Adv. Synth. Catal., 2012, 354, 1529 CrossRef CAS.
- M. F. Kuehnel, T. Schlöder, S. Riedel, B. Nieto-Ortega, F. J. Ramírez, J. T. López Navarrete, J. Casado and D. Lentz, Angew. Chem., Int. Ed., 2012, 51, 2218 CrossRef CAS PubMed.
- P. Fischer, K. Götz, A. Eichhorn and U. Radius, Organometallics, 2012, 31, 1374 CrossRef CAS.
- M. F. Kuehnel, P. Holstein, M. Kliche, J. Krüger, S. Matthies, D. Nitsch, J. Schutt, M. Sparenberg and D. Lentz, Chem. – Eur. J., 2012, 18, 10701 CrossRef CAS PubMed.
- S. Yow, S. J. Gates, A. J. P. White and M. R. Crimmin, Angew. Chem., Int. Ed., 2012, 51, 12559 CrossRef CAS PubMed.
- H. Lv, Y.-B. Cai and J.-L. Zhang, Angew. Chem., Int. Ed., 2013, 52, 3203 CrossRef CAS PubMed.
- J. Li, T. Zheng, H. Sun and X. Li, Dalton Trans., 2013, 42, 13048 RSC.
- H. Lv, J.-H. Zhan, Y.-B. Cai, Y. Yu, B. Wang and J.-L. Zhang, J. Am. Chem. Soc., 2012, 134, 16216 CrossRef CAS PubMed.
- J. Vela, J. M. Smith, Y. Yu, N. A. Ketterer, C. J. Flaschenriem, R. J. Lachicotte and P. L. Holland, J. Am. Chem. Soc., 2005, 127, 7857 CrossRef CAS PubMed.
- A. D. Selmeczy, W. D. Jones, M. G. Partridge and R. N. Perutz, Organometallics, 1994, 13, 522 CrossRef CAS.
- A. A. Peterson and K. McNeill, Organometallics, 2006, 25, 4938 CrossRef CAS.
- C. Ehm, J. Krüger and D. Lentz, Chem. – Eur. J., 2016, 22, 9305 CrossRef CAS PubMed.
- E. Clot, C. Mégret, B. M. Kraft, O. Eisenstein and W. D. Jones, J. Am. Chem. Soc., 2004, 126, 5647 CrossRef CAS PubMed.
- M. F. Kühnel, Doctoral thesis, FU-Berlin, 2011.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2009.
- Rehybridization of the SOMO allowing participation of an empty coordination site at 1 can occur in the TS however.
- The SOMO is predominantly concentrated on the metal in all TS but some delocalization into the organic fragments occurs in some of them. See the ESI† for SOMO pictures.
- C. F. Bernasconi and Z. Rappoport, Acc. Chem. Res., 2009, 42, 993 CrossRef CAS PubMed.
- The barrier for hydride attack at C2 is +6.8 kcal mol−1 higher than TSA3–5d.
- D. Y. Curtin and M. C. Crew, J. Am. Chem. Soc., 1955, 77, 354 CrossRef CAS.
- Relative values are derived from absolute values from Fig. 1via subtraction TS-RS or TS-separated products.
- E. W. Kaiser and D. S. Pierce, J. Phys. Chem. A, 2015, 119, 9000 CrossRef CAS PubMed.
- N. C. Craig and E. A. Entemann, J. Am. Chem. Soc., 1961, 83, 3047 CrossRef CAS.
- R. Kanakaraju, K. Senthilkumar and P. Kolandaivel, J. Mol. Struct., 2002, 589–590, 95 CrossRef CAS.
- C. Ehm and D. Lentz, J. Phys. Chem. A, 2010, 114, 3609 CrossRef CAS PubMed.
- Attack at C2 has significantly higher barriers. See the ESI.†.
- SNV has not been reported as a possible mechanism for HDF using zirconocenes. The relatively strong Zr–H bond compared to Ti–H might be responsible for this (Cp*2ZrH2 81.0 ± 0.5 vs. Cp2TiH 71.7 kcal mol−1).
- We found at least six, differing in orientation of the Cp rings and olefin.
- This would represent a two step hydrometallation pathway but would lead back to a very selective mechanism (elimination from Ti-alkyl species) which is not in line with experimental observations.
- Despite multiple attempts we were not able to locate an elimination TS from IP2 but comparison of IRC scans of TSC3-3b (F−-elimination) and TSB3-IP2 (charge separation) indicates that subtle changes in the relative orientation of anions and cations in the SIP can lead to barrierless F-elimination.
- Earlier, we rendered a 6-membered SBM TS not feasible based on experimentally observed Ti(III)-halide angles but considering that the TS are very early, angles at Ti(III) should not be important and 6c-SBM is predicted to be more feasible than each of the two 4c-SBM.
- Additional information and enhanced spectra in the ESI.†.
- This implies that the anion 4H− is even more stable than the anion 3H−.
- DFT calculations were repeated in toluene. Experimental selectivities in toluene correspond to energy differences of 1.5 kcal mol−1 (3avs. 3b) and 1.5 kcal mol−1 (3avs. 3c). DFT predicts 1.9 and 0.8 kcal mol−1, respectively, assuming 3a and 3b stem from HM/E and 3c from 6c-SBM.
- The barrier for TSB3-IP2 is heavily affected by the choice of solvent which is not observed for the other TS.
- Something that was also observed for 3,3,3-trifluoropropene and trifluoroethylene in diglyme (see ref. 46).
- DFT correctly predicts preference for the E-isomer via HM/E (0.7 kcal mol−1vs. 1.2 kcal mol−1 experimentally) in THF.
- J. M. de Wolf, A. Meetsma and J. H. Teuben, Organometallics, 1995, 14, 5466 CrossRef CAS.
- H. Koroniak, K. W. Palmer, W. R. Dolbier and H.-Q. Zhang, Magn. Reson. Chem., 1993, 31, 748 CrossRef CAS.
- J.-j. Ma, W.-b. Yi, G.-p. Lu and C. Cai, Adv. Synth. Catal., 2015, 357, 3447 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 215 CrossRef CAS.
- J. Baker, J. Comput. Chem., 1986, 385 CrossRef CAS.
- J. Baker, PQS, 2.4, Parallel Quantum Solutions, Fayetteville, AR, 2001 Search PubMed.
- The Berny algorithm was never completely published, see the Gaussian documentation for details.
- C. Peng, P. Y. Ayala, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 1996, 17, 49 CrossRef CAS.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449 CrossRef CAS.
- R. Raucoules, T. de Bruin, P. Raybaud and C. Adamo, Organometallics, 2009, 28, 5358 CrossRef CAS.
- S. Tobisch and T. Ziegler, J. Am. Chem. Soc., 2004, 126, 9059 CrossRef CAS PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, R. Huenerbein and S. Ehrlich, ChemPhysChem, 2011, 12, 1258 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt02961b |
| This journal is © The Royal Society of Chemistry 2016 |