
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous introduction of various palladium active sites into MOF via one-pot synthesis: Pd@[Cu3−xPdx(BTC)2]n†
Wenhua
Zhang
a,
Zhihao
Chen
b,
Majd
Al-Naji
b,
Penghu
Guo
c,
Stefan
Cwik
a,
Olesia
Halbherr
a,
Yuemin
Wang
d,
Martin
Muhler
c,
Nicole
Wilde
b,
Roger
Gläser
b and
Roland A.
Fischer
*ef
aInorganic Chemistry II—Organometallics and Materials Chemistry, Ruhr-University Bochum, Universitätsstraße 150, 44801 Bochum, Germany
bInstitute of Chemical Technology, Universität Leipzig, Linnéstraße 3, 04103 Leipzig, Germany
cLaboratory of Industrial Chemistry, Ruhr-University Bochum, Universitätsstraße 150, 44801 Bochum, Germany
dInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), 76344 Karlsruhe, Germany
eChair of Inorganic and Metal-Organic Chemistry, Department of Chemistry, Technical University Munich, Lichtenbergstraße 4, 85748 Garching, Germany
fCatalysis Research Centre, Technical University Munich, Ernst-Otto-Fischer Strasse 1, 85748 Garching, Germany. E-mail: roland.fischer@tum.de
First published on 22nd August 2016
Abstract
Simultaneous incorporation of palladium within Pd–Pd and/or Pd–Cu paddlewheels as framework-nodes and Pd nanoparticle (NP) dispersion into MOF have been achieved for the first time via one-pot synthesis. In particular, the framework substitution of Cu2+ by Pd2+ as well as the pore loading with PdNPs have been confirmed and characterized by XPS. The obtained solids featuring such multiple Pd-sites show enhanced catalytic activity in the aqueous-phase hydrogenation of p-nitrophenol (PNP) with NaBH4 to p-aminophenol (PAP).
High structural and compositional design ability of metal–organic frameworks (MOFs) and hence, the ability of tailoring their properties made MOFs to be among the most topical materials over last decades.1–7 Along with linker(s) modification8–10 and guest(s) inclusion,11,12 variation of the metal center(s) is also a powerful tool to fine tune MOFs functionalities. In fact, both control over coordinatively unsaturated metal sites (CUS) (e.g., via “defects-engineering”)13 and partial metal substitution (e.g., via “solid solution” approach) could considerably enhance MOFs activity, particularly in catalysis14–16 and selective gas sorption/separation.17,18
Combination of distinct metal-ions, which are closely related in coordination chemistry and have similar effective ionic radii (rion), such as Cu2+ (73 pm)/Zn2+ (74 pm) couple19 within single MOF, has been reported for several structural types.17,20,21 In fact, partial substitution of Cu2+ by Zn2+ and some other metals of 3d-row, namely, Co, Fe and Mn, in HKUST-1 ([Cu3(BTC)2]n, BTC = benzene-1,3,5-tricarboxylate) has been recently reported.18,22 In a later case, doping with a second metal caused enhanced selective sorption of O2vs. N2.18,23 What is more challenging, however, is to introduce more distinct metals of 4d-24 or 5d-row25 as framework-nodes. In particular, metals of the platinum group are highly attractive as catalytic/strong sorption centers. Moreover, square planar coordination is preferred such as Cu2+.26–28,29 However, due to kinetic reasons, these metal ions are difficult to be crystallized within 3D structures28,29 and therefore, are almost neglected in MOF field.30 To the best of our knowledge, studies on integration of palladium as a framework node into a MOF are thus far in its infancy.30–33 Reports on MOFs featuring Pd–Pd and Pd–M paddlewheel nodes could not be found. Interestingly, bimetallic Pd/M-paddlewheel complexes have been lately found to be promising economical catalysts in the intramolecular benzylic C–H amination.34 Moreover, porous metal–organic polyhedrals (MOPs) constructed with Pd2+–M2+ (M = Cu, Ni, Zn) paddlewheel nodes has been very recently described.29 Considering its structural peculiarities (i.e., paddlewheel SBUs and CUSs), choosing HKUST-1 as a matrix for palladium in-framework incorporation via solid solution approach would be of interest.35 However, a rigorous exclusion of reducing conditions during MOF synthesis is difficult. Even technical activation may cause reduction at some metal sites due to decarboxylation.36,37 Therefore, the standard protocol for HKUST-1 synthesis is, intentionally, not changed at this stage.
On the other hand, loading of Pd0 NPs onto MOF is widely investigated as gas adsorbents38–40 and catalysts used in hydrogenation38,41–44 and cross coupling42,45 reactions.46 It can be noted that one of the most common approaches to obtain Pd0@MOFs is using H2 to reduce Pd2+-precursor loading MOFs that are prepared via solution impregnation by soluble Pd2+ species. In some cases, trace amount of Pd2+ can be still observed after reduction.45 Actually, according to the concept of post-synthetic metal-ion exchange,47 Pd2+ substitution in the metal nodes of MOFs could probably occur under this impregnation. Interestingly, the discrimination between the two Pd-sites (extra-framework loading or in-framework incorporation) is disregarded to some extent in the current literature. Given both facts, herein, an appropriate one-pot synthesis has been selected. Both H2O and alcohols are found to be important components to reduce Pd2+ ions to Pd0 at elevated temperatures.48–50 Therefore, it is expected that utilizing a standard protocol for HKUST-1 synthesis (employing H2O and EtOH under solvothermal conditions) can simultaneously introduce both Pd2+-doped nodes and Pd0 NPs in one step.
Pd@[Cu3−xPdx(BTC)2]n (Cu/Pd-BTC_1–3) materials have been synthesized under solvothermal conditions by mixing corresponding metal salts (Pd(II) acetate and Cu(NO3)2·3H2O) with H3BTC directly in the starting reactions (see ESI†). All Cu/Pd-BTC_1–3 samples (both as-synthesized and dried phases) are crystalline solids and isostructural with the parent Cu-BTC (Fig. 1 and S1†), indicating the preserved structure integrity after the Pd-sites introduction. Recorded PXRD patterns and accordingly calculated cell parameters of Cu/Pd-BTC_1–3 match very well with the respective simulated data of the reported [Cu3(BTC)2]n single-crystal structure (Fig. S3 and Table S1†). Remarkably, intensity of the reflections at ca. 6.67°, 9.40° and 13.34° (2θ) assigned to the (200), (220) and (400) planes, respectively, varies (Fig. 1). This indicates certain changes of electronic density compared to the non-doped Cu-analogue (Cu-BTC). The main contribution to the reflection of the (200) and (220) planes is due to the metal-nodes (Fig. S4†). Hence, such difference in intensities should primarily stem from the partial substitution of Cu2+ by Pd2+ within the M2-paddlewheel units of MOFs, indicating Pd2+ framework incorporation. Considering the presence of Pd NPs in the discussed Cu/Pd-BTC_1–3, small reflections at 40.02° resulting from (111) planes of the face-centered cubic structure of Pd are observed (Fig. 1).51 This is probably due to the rather small particle size of these Pd NPs and therefore, they are difficult to be detected by PXRD technique. Finally, it should be pointed out that employing equal molar amounts of Cu- and Pd-salts or only Pd-precursor in the starting reaction mixtures, formation of only metal/metal–oxide products has been observed (Fig. S5 and S6†).
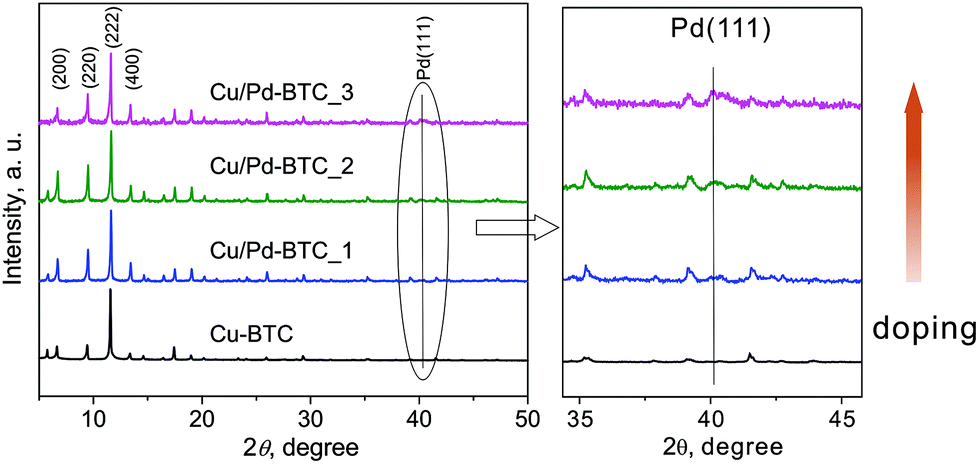 | ||
| Fig. 1 PXRD patterns of activated Pd@[Cu3−xPdx(BTC)2]n (Cu/Pd-BTC_1–3) in comparison with activated non-doped Cu-BTC. The vertical lines correspond to the peak position of (111) planes of the face-centered cubic (fcc) Pd-structure. | ||
AAS analysis of the activated Cu/Pd-BTC_1–3 solids confirms quantitative incorporation of palladium. Thus, the Pd-contents and Pd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu ratios in the final Cu/Pd-BTC_1–3 samples remain almost unchanged with respect to the taken feeding ratios of the metal-precursors (Table S2†). Furthermore, SEM-EDX elemental mapping demonstrates quite homogeneous Cu/Pd distribution within the obtained Cu/Pd-BTC solids (Fig. S8†). Incorporation of Pd2+ into the framework as well as the Pd0 NPs loading has been subsequently corroborated by high-resolution XPS. Fig. 2 shows the Pd 3d core-level spectra of the Cu/Pd-BTC_1 and Cu/Pd-BTC_3 (representative samples with relatively low and high Pd-contents). Three Pd 3d doublets (3d5/2 and 3d3/2) are resolved in the deconvoluted spectra. The doublet located at 337.9 and 343.2 eV (Pd2) is characteristic for Pd2+ species (Table S3†).52,53 In addition, the higher binding energies observed at 338.9 and 344.2 eV for Pd1 reveal the presence of an electronically modified Pd2+ species. Moreover, we could unambiguously rule out an assignment of both Pd2+ species to PdO NPs, as no typical O 1s peak at about 530 eV has been detected in the corresponding spectra, which showed only one O 1s peak at 531.8 eV, originating from the carboxylate (–COO) groups in the frameworks. Therefore, both the found Pd2+ species (Pd1 and Pd2) are very likely associated with the formation of Pd–Pd and Cu–Pd framework-nodes. The doublet at ca. 335.8 and 341.0 eV is attributed to metallic Pd0 species (Pd3), indicating the existence of Pd metallic NPs in these samples. Moreover, the amount of Pd NPs in the total Pd species (Pd2+ and Pd0) increases (from 33% for Cu/Pd-BTC_1 to 44% for Cu/Pd-BTC_3) along with the doping increase of Pd(II) acetate. TEM characterization has not been performed because of the beam sensitivity of HKUST-1, which does not allow characterization of the “pristine” Pd@[Cu3−xPdx(BTC)2]n sample as well.
:Cu ratios in the final Cu/Pd-BTC_1–3 samples remain almost unchanged with respect to the taken feeding ratios of the metal-precursors (Table S2†). Furthermore, SEM-EDX elemental mapping demonstrates quite homogeneous Cu/Pd distribution within the obtained Cu/Pd-BTC solids (Fig. S8†). Incorporation of Pd2+ into the framework as well as the Pd0 NPs loading has been subsequently corroborated by high-resolution XPS. Fig. 2 shows the Pd 3d core-level spectra of the Cu/Pd-BTC_1 and Cu/Pd-BTC_3 (representative samples with relatively low and high Pd-contents). Three Pd 3d doublets (3d5/2 and 3d3/2) are resolved in the deconvoluted spectra. The doublet located at 337.9 and 343.2 eV (Pd2) is characteristic for Pd2+ species (Table S3†).52,53 In addition, the higher binding energies observed at 338.9 and 344.2 eV for Pd1 reveal the presence of an electronically modified Pd2+ species. Moreover, we could unambiguously rule out an assignment of both Pd2+ species to PdO NPs, as no typical O 1s peak at about 530 eV has been detected in the corresponding spectra, which showed only one O 1s peak at 531.8 eV, originating from the carboxylate (–COO) groups in the frameworks. Therefore, both the found Pd2+ species (Pd1 and Pd2) are very likely associated with the formation of Pd–Pd and Cu–Pd framework-nodes. The doublet at ca. 335.8 and 341.0 eV is attributed to metallic Pd0 species (Pd3), indicating the existence of Pd metallic NPs in these samples. Moreover, the amount of Pd NPs in the total Pd species (Pd2+ and Pd0) increases (from 33% for Cu/Pd-BTC_1 to 44% for Cu/Pd-BTC_3) along with the doping increase of Pd(II) acetate. TEM characterization has not been performed because of the beam sensitivity of HKUST-1, which does not allow characterization of the “pristine” Pd@[Cu3−xPdx(BTC)2]n sample as well.
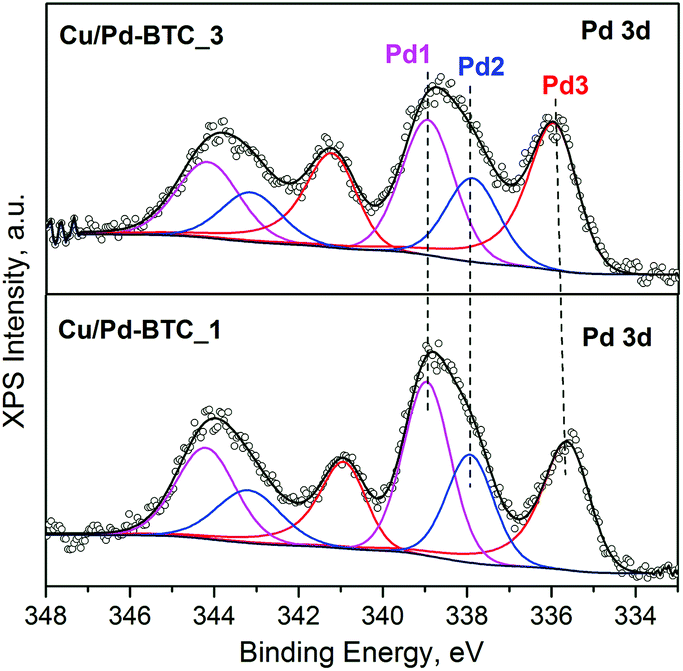 | ||
| Fig. 2 Deconvoluted XP spectra of Cu/Pd-BTC_1 and 3 in Pd 3d region. | ||
Furthermore, any vibrations stemming neither from the non-reacted H3BTC linker nor used metal-precursors (i.e., acetate-, nitrate-groups) could be revealed in the FT-IR spectra of all prepared Cu/Pd-BTC solids (Fig. S12†). Moreover, 1H-NMR spectra of the digested Cu/Pd-BTC_1–3 samples show the presence of only BTC-linker (Fig. S13†). Thus, absence of any resonances at about 1.9 ppm, stemming from the acetate protons, fully supports the IR results ruling out any residuals of the employed starting Pd-reactant. Hence, the presence of Pd2+-sites should originate from the in-framework nodes rather than Pd2+-precursor loading.
TGA suggests that thermal stability of the obtained Cu/Pd-BTC_1–3 solids is fully preserved (in comparison with the parent Cu-BTC)35 with the decomposition temperatures close to 300 °C (Fig. S14†). The N2 (77 K) sorption isotherms recorded for obtained samples reveal type I isotherm (Fig. S15†), confirming their permanent microporosity. Brunauer–Emmett–Teller (BET) surface area of Cu/Pd-BTC_1 and _2 increase a little in comparison with the parent Cu-BTC (prepared under similar conditions) (Table S4†), indicating that the presence of Pd2+ incorporation as metal nodes in the framework are dominant. When it turns to Cu/Pd-BTC_3, the increasing of Pd0 NPs results in a slight lower BET surface area in comparison with the others samples.
Both Pd2+-containing MOF ([Pd(2-pymo)2]n, 2-pymo = 2-pyrimidinolate)31–33 and Pd0@MOF,38,42,43,45 have been reported as good reusable catalysts in typical palladium-catalyzed reactions such as Suzuki C–C couplings and hydrogenation. Owing to both Pd0 NPs and potential Pd2+-CUS (that should be available after thermal treatment and specifically promote H2 splitting) in obtained Pd-containing Cu/Pd-BTC materials, it was of interest to test their catalytic activity in hydrogenation reactions. Herein, it should be mentioned that in order to investigate which Pd species (Pd2+ or Pd0) would play the important role on the catalysis as well, we prepared another isostructural analog Cu/Pd-BTC_4 sample employing PdCl2 and longer reaction time instead (see ESI for the preparation and characterization: Tables S2, S3 and Fig. S2, S9, S11, S16†). This sample contains dominant Pd0 (70%) rather than Pd2+ on the basis of the Pd 3d region XP spectra (Fig. S11 and Table S3†).
As a test reaction, the aqueous-phase hydrogenation of PNP to PAP using NaBH4 as a reducing agent has been chosen. It can be conducted at room temperature and ambient pressure within a reaction time of <10 min using a simple on-line UV-Vis spectroscopy to monitor the progress of the reaction.43,54,55 The concentration of PNP as a function of time as well as the initial reaction rate normalized to the amount of Pd in the catalyst for Cu/Pd-BTC_1–4 are depicted in Fig. 3. Before addition of the solid catalyst, i.e., within the first 0.5 min of the experiment, no PNP conversion was observed. Complete conversion of PNP to PAP was reached within 2 min of reaction time using Cu/Pd-BTC_1–4, whereas the conversion of PNP remained incomplete for the Pd-free Cu-BTC. This incomplete conversion is known for catalysts with low activity and results from unproductive NaBH4 decomposition to hydrogen.54 Evidently, the Pd-containing Cu-BTC catalysts are significantly more active than the Pd-free Cu-BTC. Interestingly, the initial reaction rate related to the Pd amount for Cu/Pd-BTC_1 and 2 (i.e., 4.0 × 10−3 and 2.0 × 10−3 dm−3 min−1) is significantly higher than that achieved over a metallic Pd catalyst supported on an Al-containing mesocellular silica foam (1.0 × 10−3 dm−3 min−1) under the same reaction conditions.56 Note that the activity of Cu/Pd-BTC_1 and 2 is so high that the difference in their catalytic activity cannot be distinguished under the reaction conditions used.
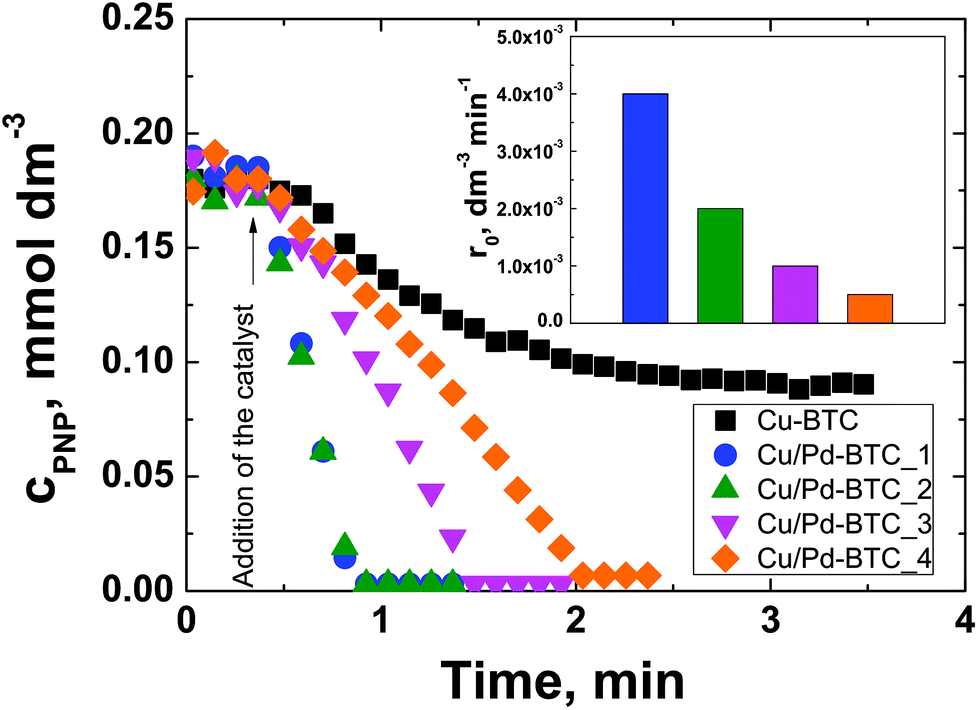 | ||
| Fig. 3 Concentration of PNP as a function of time for non-doped Cu-BTC and Pd containing Cu/Pd-BTC_1–4 as catalysts in the aqueous-phase hydrogenation of PNP with NaBH4 to PAP. The insert shows the initial reaction rate for Cu/Pd-BTC_1–4 normalized to the amount of Pd in the catalyst, respectively. Reaction conditions: cPNP = 0.18 mmol dm−3, cNaBH4 = 0.60 mmol dm−3, T = 298 K, stirring speed = 1300 min−1, mcatalyst = 5.0 mg, ambient pressure. | ||
Surprisingly, the normalized initial reaction rate decreases with increasing overall Pd content from 4 × 10−3 dm−3 min−1 for Cu/Pd-BTC_1 to 1.0 × 10−3 dm−3 min−1 for Cu/Pd-BTC_3, respectively (insert in Fig. 3). This can be explained by considering the decreasing concentration of Pd2+ species with increasing Pd content from Cu/Pd-BTC_1 to Cu/Pd-BTC_3, as observed by XPS (Table S3†). Particularly, Cu/Pd-BTC_4 containing the lowest concentration of Pd2+ species (70%) displays the lowest initial reaction rate in comparison with the other three Cu/Pd-BTC samples. It may, therefore, be concluded that Pd2+ species in the catalyst are catalytically active and they are dominantly responsible for the observed high catalytic activity. Moreover, it has been reported that Pd2+ can act as active metal sites in olefin hydrogenations.31–33 It can be noted that PXRD patterns of the Cu/Pd-BTC samples before and after reaction indicate that the structure of the framework remains unchanged (Fig. S17†). Finally, according to the ICP-OES analysis of the solution after reaction (Cu/Pd-BTC_2 as selective catalysts), leaching of palladium is negligible (i.e., 0.01 mg L−1), indicating the stability of the used MOF catalysts as well as the heterogeneous nature of the catalytic reaction.
In summary, via one-pot synthesis, we successfully obtained crystalline porous Pd@[Cu3−xPdx(BTC)2]n MOFs with various doping levels of Pd. On the basis of entire experimental data, structural incorporation of Pd2+ serving as frameworks-nodes (within Cu–Pd or/and Pd–Pd paddlewheels) as well as Pd0 NPs dispersion in the framework of HKUST-1 are concluded. To the best of our knowledge, it is the singular case of MOFs bearing both Pd2+-paddlewheels and Pd NPs known so far. Moreover, distinct Pd sites, especially Pd2+/M-CUS considerably enhances the catalytic activity of MOFs in the aqueous-phase hydrogenation of PNP to PAP with respect to the “Pd-free” Cu-BTC catalysts, which affords a perspective way to tune their catalytic activity. Furthermore, a synthesis of HKUST-1 analogs excluding Pd0 formation and featuring exclusive Pd2+/Cu2+ mixed-metal paddlewheels would lead to highly active catalysts.
W. Z. and Z. C. thank the China Scholarship Council (CSC) for PhD fellowships. W. Z. is also grateful to the Research SchoolPlus at Ruhr-University Bochum for the support of her PhD project. P. G. acknowledges the support of the EU innovative Training Network “DEFect NETwork materials science and engineering” (DEFNET).
Notes and references
- M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J.-H. Lee, J.-S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227–8231 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- E. D. Bloch, W. L. Queen, R. Krishna, J. M. Zadrozny, C. M. Brown and J. R. Long, Science, 2012, 335, 1606–1610 CrossRef CAS PubMed.
- A. Corma, H. García and F. X. Llabrés i Xamena, Chem. Rev., 2010, 110, 4606–4655 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974–5978 CrossRef CAS PubMed.
- A. A. Talin, A. Centrone, A. C. Ford, M. E. Foster, V. Stavila, P. Haney, R. A. Kinney, V. Szalai, F. El Gabaly, H. P. Yoon, F. Léonard and M. D. Allendorf, Science, 2014, 343, 66–69 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- H. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gándara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- H. Deng, C. J. Doonan, H. Furukawa, R. B. Ferreira, J. Towne, C. B. Knobler, B. Wang and O. M. Yaghi, Science, 2010, 327, 846–850 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- D. Yan, G. O. Lloyd, A. Delori, W. Jones and X. Duan, ChemPlusChem, 2012, 77, 1112–1118 CrossRef CAS.
- A. Carné-Sánchez, I. Imaz, M. Cano-Sarabia and D. Maspoch, Nat. Chem., 2013, 5, 203–211 CrossRef PubMed.
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed.
- F. Vermoortele, B. Bueken, G. Le Bars, B. Van de Voorde, M. Vandichel, K. Houthoofd, A. Vimont, M. Daturi, M. Waroquier, V. Van Speybroeck, C. Kirschhock and D. E. De Vos, J. Am. Chem. Soc., 2013, 135, 11465–11468 CrossRef CAS PubMed.
- O. Kozachuk, I. Luz, F. X. Llabrés i Xamena, H. Noei, M. Kauer, H. B. Albada, E. D. Bloch, B. Marler, Y. Wang, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2014, 53, 7058–7062 CrossRef CAS PubMed.
- C. K. Brozek and M. Dincă, J. Am. Chem. Soc., 2013, 135, 12886–12891 CrossRef CAS PubMed.
- G.-T. Vuong, M.-H. Pham and T.-O. Do, Dalton Trans., 2013, 42, 550–557 RSC.
- D. F. Sava Gallis, M. V. Parkes, J. A. Greathouse, X. Zhang and T. M. Nenoff, Chem. Mater., 2015, 27, 2018–2025 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Cryst., 1976, 32, 751–767 CrossRef.
- O. Kozachuk, K. Khaletskaya, M. Halbherr, A. Bétard, M. Meilikhov, R. W. Seidel, B. Jee, A. Pöppl and R. A. Fischer, Eur. J. Inorg. Chem., 2012, 2012, 1688–1695 CrossRef CAS.
- Y. Jiao, C. R. Morelock, N. C. Burtch, W. P. Mounfield, J. T. Hungerford and K. S. Walton, Ind. Eng. Chem. Res., 2015, 54, 12408–12414 CrossRef CAS.
- F. Gul-E-Noor, B. Jee, M. Mendt, D. Himsl, A. Pöppl, M. Hartmann, J. Haase, H. Krautscheid and M. Bertmer, J. Phys. Chem. C, 2012, 116, 20866–20873 CAS.
- M. V. Parkes, D. F. Sava Gallis, J. A. Greathouse and T. M. Nenoff, J. Phys. Chem. C, 2015, 119, 6556–6567 CAS.
- M. A. Gotthardt, R. Schoch, S. Wolf, M. Bauer and W. Kleist, Dalton Trans., 2015, 44, 2052–2056 RSC.
- Q. Zhang, L. Cao, B. Li and L. Chen, Chem. Sci., 2012, 3, 2708–2715 RSC.
- N. Y. Kozitsyna, S. E. Nefedov, F. M. Dolgushin, N. V. Cherkashina, M. N. Vargaftik and I. I. Moiseev, Inorg. Chim. Acta, 2006, 359, 2072–2086 CrossRef CAS.
- N. S. Akhmadullina, N. V. Cherkashina, N. Y. Kozitsyna, I. P. Stolarov, E. V. Perova, A. E. Gekhman, S. E. Nefedov, M. N. Vargaftik and I. I. Moiseev, Inorg. Chim. Acta, 2009, 362, 1943–1951 CrossRef CAS.
- G. Nickerl, U. Stoeck, U. Burkhardt, I. Senkovska and S. Kaskel, J. Mater. Chem. A, 2014, 2, 144–148 CAS.
- J. M. Teo, C. J. Coghlan, J. D. Evans, E. Tsivion, M. Head-Gordon, C. J. Sumby and C. J. Doonan, Chem. Commun., 2016, 52, 276–279 RSC.
- J. A. R. Navarro, E. Barea, J. M. Salas, N. Masciocchi, S. Galli, A. Sironi, C. O. Ania and J. B. Parra, Inorg. Chem., 2006, 45, 2397–2399 CrossRef CAS PubMed.
- F. X. Llabrés i Xamena, A. Abad, A. Corma and H. Garcia, J. Catal., 2007, 250, 294–298 CrossRef.
- S. Schuster, E. Klemm and M. Bauer, Chem. – Eur. J., 2012, 18, 15831–15837 CrossRef CAS PubMed.
- S. Opelt, V. Krug, J. Sonntag, M. Hunger and E. Klemm, Microporous Mesoporous Mater., 2012, 147, 327–333 CrossRef.
- X. Zhang, H. Xu, X. Liu, D. L. Phillips and C. Zhao, Chem. – Eur. J., 2016, 22, 7288–7297 CrossRef CAS PubMed.
- S. S. Y. Chui, S. M. F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- J. Szanyi, M. Daturi, G. Clet, D. R. Baer and C. H. F. Peden, Phys. Chem. Chem. Phys., 2012, 14, 4383–4390 RSC.
- P. St. Petkov, G. N. Vayssilov, J. Liu, O. Shekhah, Y. Wang, C. Wöll and T. Heine, ChemPhysChem, 2012, 13, 2025–2029 CrossRef PubMed.
- M. Sabo, A. Henschel, H. Frode, E. Klemm and S. Kaskel, J. Mater. Chem., 2007, 17, 3827–3832 RSC.
- C. Zlotea, R. Campesi, F. Cuevas, E. Leroy, P. Dibandjo, C. Volkringer, T. Loiseau, G. Férey and M. Latroche, J. Am. Chem. Soc., 2010, 132, 2991–2997 CrossRef CAS PubMed.
- G. Li, H. Kobayashi, J. M. Taylor, R. Ikeda, Y. Kubota, K. Kato, M. Takata, T. Yamamoto, S. Toh, S. Matsumura and H. Kitagawa, Nat. Mater., 2014, 13, 802–806 CrossRef CAS PubMed.
- S. Opelt, S. Türk, E. Dietzsch, A. Henschel, S. Kaskel and E. Klemm, Catal. Commun., 2008, 9, 1286–1290 CrossRef CAS.
- B. Gole, U. Sanyal, R. Banerjee and P. S. Mukherjee, Inorg. Chem., 2016, 55, 2345–2354 CrossRef CAS PubMed.
- C. Wang, H. Zhang, C. Feng, S. Gao, N. Shang and Z. Wang, Catal. Commun., 2015, 72, 29–32 CrossRef CAS.
- X. Jia, S. Wang and Y. Fan, J. Catal., 2015, 327, 54–57 CrossRef CAS.
- B. Yuan, Y. Pan, Y. Li, B. Yin and H. Jiang, Angew. Chem., Int. Ed., 2010, 49, 4054–4058 CrossRef CAS PubMed.
- M. Gulcan, M. Zahmakiran and S. Özkar, Appl. Catal., B, 2014, 147, 394–401 CrossRef CAS.
- P. Deria, J. E. Mondloch, O. Karagiaridi, W. Bury, J. T. Hupp and O. K. Farha, Chem. Soc. Rev., 2014, 43, 5896–5912 RSC.
- B. P. Fors, P. Krattiger, E. Strieter and S. L. Buchwald, Org. Lett., 2008, 10, 3505–3508 CrossRef CAS PubMed.
- T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632–3640, 10.1039/JR9650003632.
- W. A. Carole and T. J. Colacot, Chem. – Eur. J., 2016, 22, 7686–7695 CrossRef CAS PubMed.
- G. H. Jeong, S. H. Kim, M. Kim, D. Choi, J. H. Lee, J.-H. Kim and S.-W. Kim, Chem. Commun., 2011, 47, 12236–12238 RSC.
- H.-F. Wang, W. E. Kaden, R. Dowler, M. Sterrer and H.-J. Freund, Phys. Chem. Chem. Phys., 2012, 14, 11525–11533 RSC.
- R. Arrigo, M. E. Schuster, Z. Xie, Y. Yi, G. Wowsnick, L. L. Sun, K. E. Hermann, M. Friedrich, P. Kast, M. Hävecker, A. Knop-Gericke and R. Schlögl, ACS Catal., 2015, 5, 2740–2753 CrossRef CAS.
- M. Goepel, M. Al-Naji, P. With, G. Wagner, O. Oeckler, D. Enke and R. Gläser, Chem. Eng. Technol., 2014, 37, 551–554 CrossRef CAS.
- J. Sun, Y. Fu, G. He, X. Sun and X. Wang, Catal. Sci. Technol., 2014, 4, 1742–1748 CAS.
- M. Al-Naji, A. M. Balu, A. Roibu, M. Goepel, W.-D. Einicke, R. Luque and R. Glaser, Catal. Sci. Technol., 2015, 5, 2085–2091 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, IR, TGA, NMR, SEM-EDX, N2 sorption, elementals analysis and catalytic reaction details. See DOI: 10.1039/c6dt02893d |
| This journal is © The Royal Society of Chemistry 2016 |