
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Oxygen reduction reaction catalyzed by nickel complexes based on thiophosphorylated calix[4]resorcinols and immobilized in the membrane electrode assembly of fuel cells†
M. K.
Kadirov
*ab,
I. R.
Knyazeva
a,
I. R.
Nizameev
ac,
R. A.
Safiullin
b,
V. I.
Matveeva
a,
K. V.
Kholin
ab,
V. V.
Khrizanforova
a,
T. I.
Ismaev
b,
A. R.
Burilov
a,
Yu. H.
Budnikova
a and
O. G.
Sinyashin
a
aA.E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center, Russian Academy of Sciences, 8, Akad. Arbuzov str., Kazan 420088, Russia. E-mail: kadirovmarsil@gmail.com
bKazan National Research Technological University, 68, K. Marx str., Kazan 420015, Russia
cKazan National Research Technical University, 10, K. Marx str., Kazan 420111, Russia
First published on 30th September 2016
Abstract
The catalytic activity of the nickel complexes of thiophosphorylated calix[4]resorcinols for oxygen reduction in a polymer electrolyte membrane fuel cell (PEMFC) has been studied. The conformation of the macrocyclic ligand determines the morphology and catalytic properties of the resulting organometallic species.
Fuel cells are electrochemical devices that convert the chemical energy in fuels into the electrical energy directly, promising power generation with high efficiency and low environmental impact. However, the use of platinum-based PEMFC electrocatalysts remains a major issue for commercial introduction due to the scarcity of platinum and price pressures due to the increasing demand.1–3 Moreover, overpotentials as large as several hundred mV, which result from poor oxygen-reduction reaction (ORR) kinetics on carbon-supported Pt catalysts, limit the rate of energy conversion in PEMFCs.2 The energy associated with this voltage loss is converted to heat, which has to be removed through a complex heat management.4 A crucial target is to improve the ORR catalyst at the cathodic end of hydrogen- and direct methanol-based fuel cells.5
The ORR is the foundation of biological aerobic energy generation. Efficient and rapid reduction of oxygen is carried out without the use or with the partial use of noble metals in nature by cytochrome c oxidase enzymes to power the cellular proton pump during the final stage of respiration.4 Recently a series of papers connected with the use of the biomimetic properties of organometallic complexes in the ORR for fuel cell (FC) membrane electrode assembly (MEA) have been published.6
Calix[4]arene derivatives have been used in catalytic reactions over the last four decades because they tend to be highly stable towards hydrolysis, a factor which makes them highly useful as catalysts.7 The chemistry of heteroatom-containing, in particular, phosphorus-containing, macrocyclic compounds including calixarenes has undergone rapid development; this is primarily related to the search for new and effective ligands for various applications in science and engineering.8,9 It was reported8 that calix[4]resorcinols with four 2-thioxo-1,3,2-dioxaphosphorinane fragments included in aromatic substituents have been synthesized via a one-step condensation of resorcinol and its derivatives with a new para-thiophosphorylated benzaldehyde. It has been found that the diastereomeric ratio depends substantially on the reaction conditions, in particular the solvents and catalysts used. The macrocyclic products obtained are rctt- (Fig. 1a) and/or rccc-isomers (Fig. 1b), which were isolated and the structures determined by NMR and single crystal X-ray diffraction studies. It has been found that the diastereomeric ratio depends substantially on the reaction conditions, in particular the solvents and catalysts used. The main feature of these compounds is that a metal can be loosely tethered at an entrance to a receptor cavity, thereby favouring interactions between properly oriented, coordinated substrates and the inner part of the hollow molecule.10 The presence of additional coordination centers in the macromolecules represented by four P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S fragments with a hydrolytically stable tetracoordinate phosphorus atom enhances the complexation ability with various metal ions and extends the practical use of these compounds. We have designed membrane electrode assemblies with the NiII complexes of thiophosphorylated calix[4]resorcinols as molecular catalysts separately for the anode and cathode, and for both the anode and cathode sides simultaneously, and then tested them in H2/O2 fuel cells. Below we discuss these original results together with the results of other independent methods.
S fragments with a hydrolytically stable tetracoordinate phosphorus atom enhances the complexation ability with various metal ions and extends the practical use of these compounds. We have designed membrane electrode assemblies with the NiII complexes of thiophosphorylated calix[4]resorcinols as molecular catalysts separately for the anode and cathode, and for both the anode and cathode sides simultaneously, and then tested them in H2/O2 fuel cells. Below we discuss these original results together with the results of other independent methods.
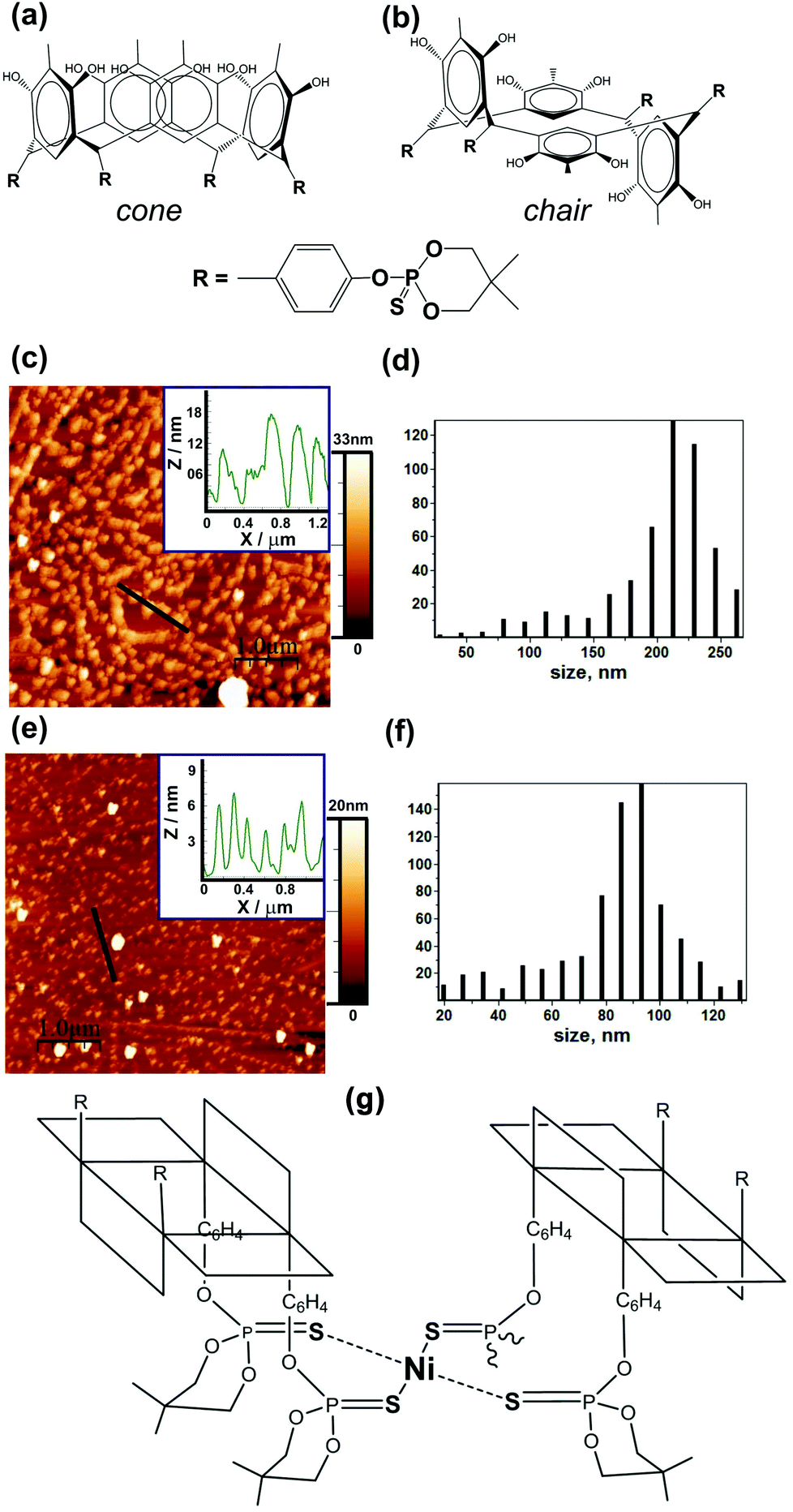 | ||
| Fig. 1 Thiophosphorylated calix[4]resorcinols in (a) cone (1) and (b) chair (2) conformations. AFM images of 1 (c) and 2 (e) on HOPG, inset: cross section along a black line. Particles lateral size distribution corresponding to 1 (d) and 2 (f). (g) Schematic representation of nickel ion coordination of a flat-square Ni–S4 coordination node formed in the Ni complex of calix[4]resorcinol in the chair conformation. An atomic force microscope MultiMode V has been used to reveal the morphology of the particles. The 250–350 kHz cantilevers (Veeco, USA) with silicone tips (tip curvature radius is 10–12 nm) have been used in all measurements. The microscopy images were obtained with 512 × 512 resolution. The scanning rate was 1 Hz. | ||
Some of us have improved11 the method of Pt and Pd lattice production on the graphite surface. This method allows controlling the morphological characteristics of synthesized noble metal nanolattices. These characteristics directly depend on the morphological characteristics of the original hemi- and pre-cylindrical micellar templates. AFM proved to be very informative and is widely used to study the morphology of both micellar self-associated templates and metal clusters. MEAs with the noble metal nanolattices as the catalytic surface on both anode and cathode sides in a H2/O2 fuel cell have been designed and satisfactory diagnostic curves have been obtained.
In order to find out which particles form organometallic catalysts immobilized on a carbon substrate, we deposited them on an atomically smooth surface of pyrolytic graphite. Nickel complexes of thiophosphorylated calix[4]resorcinol 1 or 2 were prepared in situ by mixing salts of nickel Ni(BF4)2 with a ligand of cone or chair conformation, respectively, in the ratio of metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand = 1:1 in DMF. The formation of particles (Fig. 1c) with an average lateral size of 220 nm (Fig. 1d) is typical of compound 1 with a cone conformation ligand. And particles (Fig. 1e) with a lateral size of 90 nm (Fig. 1f) are typical of compounds with two chair conformation ligands. The nickel ion in thiophosphorylated calix[4]resorcinol 1 is most probably coordinated near four sulfur atoms at the lower hydrophobic rim within one macromolecule. By means of these hydrophobic lower parts the complexes immobilize on a hydrophobic carbon substrate. The adjacent complexes immobilized in this way have been fastened on the upper hydrophilic surface of the complexes lying there with the hydrophilic upper rim down with a certain shift relative to the underlying complex. Complexes of the next layer also lie with some shifts relative to the neighbors on the underlying upper surface of the second layer with the hydrophobic rim down. Thus relatively large molecular aggregates of the polymer with vertical sizes of 10–18 nm are formed (Fig. 1c, inset). This corresponds to about 10–15 layers of the complex with the ligands in cone conformation. So a direct connection with the electrically conductive carbon substrate provides a relatively small amount of complex fragments and also the approach of oxygen molecules to the metal ion near the carbon substrate can be difficult. And perhaps this is one of the reasons why the nickel complex of the compound 1 does not function as a satisfactory oxygen reduction catalyst on the cathode side of the fuel cell, as shown in entry 1 of Table 1, demonstrating low currents and power densities with such a catalyst immobilized on the MEA. As can be seen in entry 3 of Table 1, the immobilization in MEA complex 2 with chair conformation of thiophosphorylated calix[4]resorcinols shows a power density of more than 5 times greater for the ORR in the fuel cell at the cathode. The chair conformation of the thiophosphorylated calix[4]resorcinols suggests that the metal ion can be coordinated in a complex by two sulfur atoms of the first thiophosphorylated calix[4]resorcinol and by two sulfur atoms of the second thiophosphorylated calix[4]resorcinol (Fig. 1h) forming flat-square coordinating node Ni–S4, which is parallel and close to the carbon substrate surface. Metal:ligand ratio = 1:1 supposes the coordination of other metal ions by other substituents in the trans position of thiophosphorylated calix[4]resorcinol already involved in the coordination of the first Ni ion. The nickel complexes of compound 2 are more uniformly distributed over the surface of the carbon substrate forming smaller aggregates, whose height is 4–6 nm according to the inset in Fig. 1e. The latter implies a possible arrangement of these complexes in two or three layers, where the shortest distance between the nearest nickel ions is about 4 nm. Such a large distance does not allow the capture of the oxygen molecule by two nickel ions of the immobilized complexes, which is a necessary condition for the four-electron reduction of oxygen with the formation of water molecules.12
:ligand = 1:1 in DMF. The formation of particles (Fig. 1c) with an average lateral size of 220 nm (Fig. 1d) is typical of compound 1 with a cone conformation ligand. And particles (Fig. 1e) with a lateral size of 90 nm (Fig. 1f) are typical of compounds with two chair conformation ligands. The nickel ion in thiophosphorylated calix[4]resorcinol 1 is most probably coordinated near four sulfur atoms at the lower hydrophobic rim within one macromolecule. By means of these hydrophobic lower parts the complexes immobilize on a hydrophobic carbon substrate. The adjacent complexes immobilized in this way have been fastened on the upper hydrophilic surface of the complexes lying there with the hydrophilic upper rim down with a certain shift relative to the underlying complex. Complexes of the next layer also lie with some shifts relative to the neighbors on the underlying upper surface of the second layer with the hydrophobic rim down. Thus relatively large molecular aggregates of the polymer with vertical sizes of 10–18 nm are formed (Fig. 1c, inset). This corresponds to about 10–15 layers of the complex with the ligands in cone conformation. So a direct connection with the electrically conductive carbon substrate provides a relatively small amount of complex fragments and also the approach of oxygen molecules to the metal ion near the carbon substrate can be difficult. And perhaps this is one of the reasons why the nickel complex of the compound 1 does not function as a satisfactory oxygen reduction catalyst on the cathode side of the fuel cell, as shown in entry 1 of Table 1, demonstrating low currents and power densities with such a catalyst immobilized on the MEA. As can be seen in entry 3 of Table 1, the immobilization in MEA complex 2 with chair conformation of thiophosphorylated calix[4]resorcinols shows a power density of more than 5 times greater for the ORR in the fuel cell at the cathode. The chair conformation of the thiophosphorylated calix[4]resorcinols suggests that the metal ion can be coordinated in a complex by two sulfur atoms of the first thiophosphorylated calix[4]resorcinol and by two sulfur atoms of the second thiophosphorylated calix[4]resorcinol (Fig. 1h) forming flat-square coordinating node Ni–S4, which is parallel and close to the carbon substrate surface. Metal:ligand ratio = 1:1 supposes the coordination of other metal ions by other substituents in the trans position of thiophosphorylated calix[4]resorcinol already involved in the coordination of the first Ni ion. The nickel complexes of compound 2 are more uniformly distributed over the surface of the carbon substrate forming smaller aggregates, whose height is 4–6 nm according to the inset in Fig. 1e. The latter implies a possible arrangement of these complexes in two or three layers, where the shortest distance between the nearest nickel ions is about 4 nm. Such a large distance does not allow the capture of the oxygen molecule by two nickel ions of the immobilized complexes, which is a necessary condition for the four-electron reduction of oxygen with the formation of water molecules.12
| Entry | Anode catalyst | Cathode catalyst | OCV (V) | Maximum current density [mA cm−2] | Maximum power density [mW cm−2] |
|---|---|---|---|---|---|
| a Operating conditions: T = 80 °C, flow rate of water-saturated H2 gas = 20 mL min−1, flow rate of water-saturated O2 gas = 20 mL min−1, humidity 100%, active area of MEA = 1 cm2, proton-conducting polymer electrolyte: Nafion®212, carbon black: Cabot Corporation Vulcan XC-72, gas diffusion layer: Sigracet® 25CC. | |||||
| 1 | Pt | 1 | 0.470 | 12.8 | 0.726 |
| 2 | 1 | Pt | — | — | — |
| 3 | Pt | 2 | 0.538 | 48.4 | 3.924 |
| 4 | 2 | Pt | 0.052 | 0.136 | 0.002 |
| 5 | Pt | Pt | 1 | 1280 | 324 |
Fig. 2a shows the diagnostic curves at 80 °C for the H2–O2 fuel cell with the cathode of the organometallic catalyst 2 and the Pt anode. The diagnostic characteristics of FC with the cathode of the organometallic catalyst 2 (entry 3 of the Table 1) are significantly better than the characteristics of FC with the cathode of the organometallic catalyst 1 (entry 1 of Table 1).
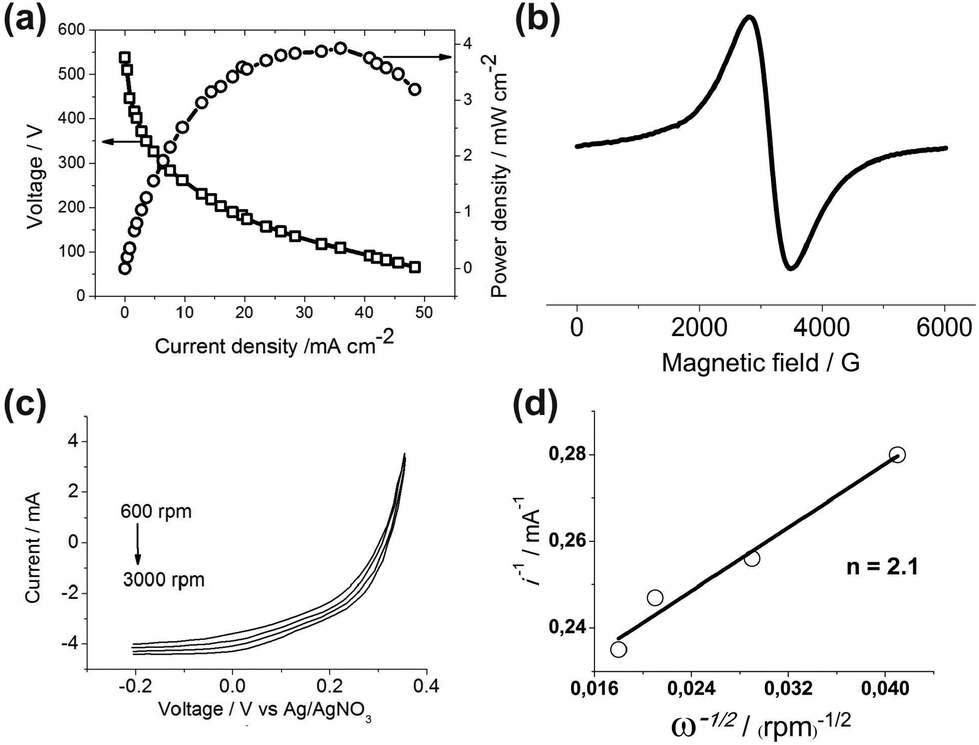 | ||
| Fig. 2 (a) Polarization and power density curves at 80 °C for the H2–O2 fuel cell with the cathode of the organometallic catalyst 2 and the Pt anode (entry 3 in Table 1). The catalytic ink was prepared according to the following formulation: 0.5 mg of organometallic catalyst and 5 mg of Vulcan XC-72 were added to 375 mL of isopropyl alcohol (IPA). The ink was first sonicated for 15 min then 46 μL of 5 wt% Nafion® solution (Aldrich) was added to the ink and sonicated again. The E-TEK (Pt20/C) catalytic ink was prepared according to the almost similar procedure: 20 mg of E-TEK were added to 8.4 mL of IPA, then the ink was sonicated for 15 min, and then 163 μL of 5 wt% Nafion® solution were added to the ink and sonicated again. The obtained inks were deposited on the carbon paper gas diffusion layer (GDL) Sigracet® 25CC. MEA was obtained by hot-pressing GDLs on either side of a Nafion® 212 membrane at 80 °C with a load of about 200 lbs for 10 minutes. (b) The EPR spectrum of 1 at a potential of −1.5 V in DMF vs. Ag/AgNO3. (c) Current–potential curves for the O2 reduction at rotating modified by the catalyst 2 glassy carbon disk electrode in 0.5 M H2SO4 saturated with O2. Pt counter electrode, Ag/AgNO3 reference electrode, scan rate = 50 mV s−1, disk area = 0.0314 cm2. To produce a modified adsorbed layer catalyst 20 μl of the ink is applied to the horizontal surface of a glassy carbon disk and dried at room temperature for at least 10 minutes. (d) Koutecky–Levich plot of the inverse of the rotating disk current measured at 0 V vs. Ag/AgNO3 as a function of the square root of the inverse of the rotation rate. The fitted line yields 2.1 electrons. The parameters for the Koutecky–Levich plot including the diffusion constant of O2, concentration of O2 in O2-saturated aqueous solution, and kinetic viscosity of the solution were taken from ref. 12. | ||
To find out other possible causes of such a significant difference in the catalytic abilities of the two isomeric forms of the organometallic catalyst the method of stationary electron spin resonance (ESR) was used. ESR proved to be a very productive method that can be applied in the direct registration of decomposition products in perfluorosulphonated membranes,13 investigation of the micelles of classical surfactants in bulk solutions,14 amphiphilic calix[4]arenes,15 and simultaneous studies of paramagnetic intermediates by electrochemistry and ESR.16 The ESR spin probe method17 and combination of the AFM and ESR methods allowed the study of the nature of surfactant surface aggregates more closely.18 The experiments on the electrochemical reduction of the Ni complexes of compounds 1 and 2 in DMF at a platinum working electrode (reference electrode: Ag/AgNO3) in a combined electrochemistry–ESR cell directly in the ESR X-band spectrometer cavity have been performed. Some differences in sample behavior have been revealed. Through reduction of the complex of 2 to a potential of −2.5 V no signal appears. In the case of the complex of 1 at the potential of −1.5 V a very intensive and wide ESR spectrum appears (Fig. 2b). Its magnetic-resonance parameters are: g = 2.19, ΔHpp = 680 Gauss. A very large integrated intensity of the spectrum cannot be explained by the formation of paramagnetic particles in the solution. Indeed, the working electrode surface has changed its color to black. The same electrode continued to give a similar spectrum in the absence of the solution, indicating the formation of the paramagnetic film. It is known from the literature19 that Ni(0) gives the spectrum, which corresponds to all of the acquired parameters. So it can be concluded that the film contains Ni(0) formed on the electrode. Thus, in the case of the Ni complex of compound 1 at the FC operating potentials, at least part of the nickel ions are reduced to the metal dropping out of the catalytic process. Let's return to Table 1, namely, to its entries 2 and 4, which show that complexes 1 and 2 immobilized in MEA hardly act as a hydrogen oxidation catalyst, having zero or near-zero current, voltage and power at the output of the corresponding FC, although some of us have recently found that the homogeneous nickel complexes of the thiophosphorylated calix[4]resorcinols showed high catalytic activity for hydrogen evolution from acidic solutions and stability in the electro-assisted catalysis at the surface of both glassy carbon and Hg-pool electrodes.20 The possible reason for this contradiction, in our opinion, is the blocking of the active sites of the hydrogen oxidation in the molecular catalysts immobilized in MEA.
To study the ORR mechanism the voltammograms of the O2 reduction at the rotating glassy carbon disk electrode modified by the catalyst 2 in 0.5 M H2SO4 aqueous solution saturated with O2 have been acquired (Fig. 2c). A fitted Koutecky–Levich line (Fig. 2d) of the inverse of the rotating disk current measured at 0 V versus Ag/AgNO3 as a function of the square root of the inverse of the rotation rate yields 2.1 electrons.
Earlier we have already mentioned the work12 which deals with a CuI complex of 3-ethynyl-phenanthroline covalently immobilized onto an azide-modified glassy carbon surface. The rate of O2 reduction is second-order, suggesting that two CuI species are necessary for the efficient 4-e reduction of O2. It has been proposed that a binding event occurs between two proximal Cu centers and a single O2 molecule to form a Cu2O2 intermediate species. A second O2-reduction pathway is due to site-isolated mononuclear Cu complexes, {CuI}, which reduce O2 by two electrons and two protons to H2O2 at more negative potentials. By analogy, it is logical to assume the presence of site-isolated mononuclear Ni complexes, {NiII} (Fig. 1h), where the nickel ion captures an oxygen molecule. The modified glassy carbon or organometallic fuel cell cathode reduces it by two electrons and two protons to H2O2 according to Fig. 3 and eqn (1)–(3).
| {NiII} + O2 → {NiIIO2} | (1) |
| {NiIIO2} + e− → {NiIIO2−} | (2) |
| {NiIIO2−} + 2H+ + e− → {NiII} + H2O2 | (3) |
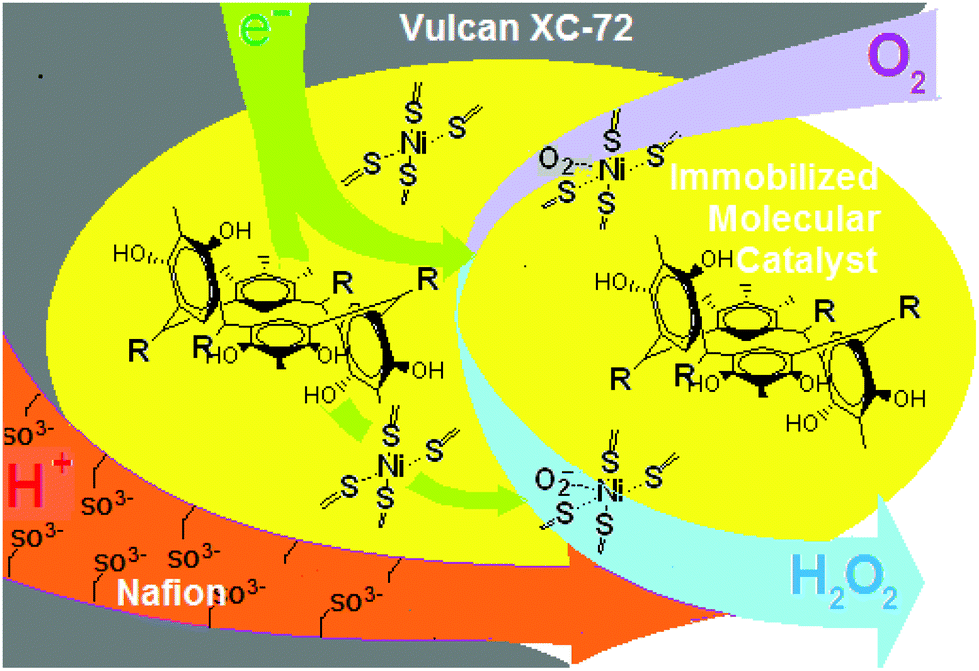 | ||
| Fig. 3 Schematic representation of the reaction of the oxygen reduction by the immobilized molecular catalyst of the thiophosphorylated calix[4]resorcinol in the chair conformation. | ||
Since the number of electrons transported in the ORR was found to be 2.1, this reaction is 95% and goes through the two-electron mechanism with the formation of hydrogen peroxide.
So the catalyst for FC with the use of a perspective class of supramolecular compounds such as calix[4]resorcinols has been first built. The specific role in the catalysis of the stereoisomers of the nickel complexes of thiophosphorylated calix[4]resorcinols in both chair and cone configurations has been shown. The choice of other metal ions (CoII, FeII), variations of substituents at both the upper and lower rims of calix[4]resorcinols and types of conformational isomers could be a powerful tool in the development of effective molecular calixarene catalysts not only for ORR, but also for hydrogen or other fuels oxidation.
Acknowledgements
This work was financially supported by the Russian Science Foundation No. 14-23-00016.Notes and references
- G. Brumfiel, Nature, 2003, 422, 104 CrossRef CAS PubMed; D. J. Berger, Science, 1999, 286, 49 CrossRef.
- Q. He, T. Mugadza, X. Kang, X. Zhu, S. Chen, J. Kerr and T. Nyokong, J. Power Sources, 2012, 216, 67 CrossRef CAS.
- V. S. Murthi, R. C. Urian and S. Mukerjee, J. Phys. Chem. B, 2004, 108, 11011 CrossRef CAS.
- J. Y. Yang, R. M. Bullock, W. G. Dougherty, W. S. Kassel, B. Twamley, D. L. DuBois and M. R. DuBois, Dalton Trans., 2010, 39, 3001 RSC.
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345 CrossRef CAS PubMed.
- T. Matsumoto, K. Kim and S. Ogo, Angew. Chem., Int. Ed., 2011, 50, 11202 CrossRef CAS PubMed; X. Zhu, J. B. Kerr, Q. He, G. Hwang, Z. Martin, K. Clark, A. Z. Weber and N. Zhao, ECS Trans., 2012, 45, 143 CrossRef; T. Matsumoto, K. Kim, H. Nakai, T. Hibino and S. Ogo, ChemCatChem, 2013, 5, 1368 CrossRef; P. D. Tran, A. Morozan, S. Archambault, J. Heidkamp, P. Chenevier, H. Dau, M. Fontecave, A. Martinent, B. Jousselme and V. Artero, Chem. Sci., 2015, 6, 2050 RSC; M. K. Kadirov, T. I. Ismaev, R. A. Safiullin, I. R. Nizameev, I. D. Strelnik, E. I. Musina, Yu. H. Budnikova, A. A. Karasik and O. G. Sinyashin, Phosphorus, Sulfur Silicon, 2016, 191 DOI:10.1080/10426507.2016.1212050.
- C. Weiser, C. B. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93 CrossRef.
- I. R. Knyazeva, V. I. Sokolova, M. Gruner, W. D. Habicher, V. V. Syakaev, V. V. Khrizanforova, B. M. Gabidullin, A. T. Gubaidullin, Y. H. Budnikova, A. R. Burilov and M. A. Pudovik, Tetrahedron Lett., 2013, 54, 3538 CrossRef CAS.
- I. R. Knyazeva, V. I. Sokolova, M. Gruner, W. D. Habicher, Ju. K. Voronina, A. R. Burilov and M. A. Pudovik, J. Inclusion Phenom. Macrocyclic Chem., 2013, 76, 231 CrossRef CAS; I. R. Knyazeva, V. I. Matveeva, V. V. Syakaev, B. M. Gabidullin, A. T. Gubaidullin, M. Gruner, W. D. Habicher, A. R. Burilov and M. A. Pudovik, Tetrahedron Lett., 2014, 55, 7209 CrossRef.
- B. S. Creaven, D. F. Donlon and J. McGinley, Coord. Chem. Rev., 2009, 253, 893 CrossRef CAS.
- M. Kadirov, I. Nizameev and L. Zakharova, J. Phys. Chem. C, 2012, 116, 11326 Search PubMed; I. R. Nizameev, M. K. Kadirov, V. A. Semyonov, L. Ya. Zakharova, T. I. Ismaev, R. A. Safiullin, I. Kh. Rizvanov and V. M. Babaev, Dalton Trans., 2016, 45, 11035 RSC.
- C. C. L. McCrory, A. Devadoss, X. Ottenwaelder, R. D. Lowe, T. D. P. Stack and C. E. D. Chidsey, J. Am. Chem. Soc., 2011, 133, 3696 CrossRef CAS PubMed.
- M. Kadirov, A. Bosnjakovic and S. Schlick, J. Phys. Chem. B, 2005, 109, 7664 CrossRef CAS PubMed; M. Danilczuk, A. Bosnjakovic, M. Kadirov and S. Schlick, J. Power Sources, 2007, 172, 78 CrossRef; A. Bosnjakovic, M. Kadirov and S. Schlick, Res. Chem. Intermed., 2007, 33, 677 CrossRef.
- T. Pashirova, E. Zhil'tsova, R. Kashapov, S. Lukashenko, A. Litvinov, M. Kadirov, L. Zakharova and A. Konovalov, Russ. Chem. Bull., 2011, 59, 1745 CrossRef.
- L. Zakharova, Y. Kudryashova, N. Selivanova, M. Voronin, A. Ibragimova, S. Solovieva, A. Gubaidullin, A. Litvinov, I. Nizameev and M. Kadirov, et al. , J. Membr. Sci., 2010, 364, 90 CrossRef CAS.
- M. Kadirov, Yu. Budnikova, T. Gryaznova, O. Sinyashin, M. Bubnov, A. Krashilina and V. Cherkasov, Russ. Chem. Bull., 2007, 56, 104 CrossRef CAS; M. Kadirov, E. Tretyakov, Y. Budnikova, M. Valitov, K. Holin, T. Gryaznova, V. Ovcharenko and O. Sinyashin, J. Electroanal. Chem., 2008, 624, 69 CrossRef; Y. Budnikova, T. Gryaznova, M. Kadirov, E. Tret'yakov, K. Kholin, V. Ovcharenko, R. Sagdeev and O. Sinyashin, Russ. J. Phys. Chem. A, 2009, 83, 1976 CrossRef; M. Kadirov, E. Tret'yakov, Y. Budnikova, K. Kholin, M. Valitov, V. Vavilova, V. Ovcharenko, R. Sagdeev and O. Sinyashin, Russ. J. Phys. Chem. A, 2009, 83, 2163 CrossRef; M. Kadirov, Y. Budnikova, K. Kholin, M. Valitov, S. Krasnov, T. Gryaznova and O. Sinyashin, Russ. Chem. Bull., 2010, 59, 466 CrossRef; J. Ciszewski, D. Mikhaylov, K. Holin, M. Kadirov, Y. Budnikova, O. Sinyashin and D. Vicic, Inorg. Chem., 2011, 50, 8630 CrossRef PubMed; R. Galimullina, M. Valitov, Y. Spiridonova, E. Musina, S. Krasnov, M. Kadirov, A. Karasik, Y. Budnikova and O. Sinyashin, Russ. J. Phys. Chem. A, 2011, 85, 2214 CrossRef; M. Kadirov, K. Kholin, E. Tselishcheva, V. Burilov and A. Mustafina, Russ. Chem. Bull., 2013, 62, 1327 CrossRef; K. Kholin, M. Valitov, V. Burilov, E. Tselischeva, S. Strekalova, A. Mustafina, Yu. Budnikova and M. Kadirov, Electrochim. Acta, 2015, 182, 212 CrossRef.
- Y. Wang, D. Lu, H. Yan and R. Thomas, J. Phys. Chem. B, 1997, 101, 3953 CrossRef CAS.
- M. Kadirov, A. Litvinov, I. Nizameev and L. Zakharova, J. Phys. Chem. C, 2014, 118, 197854 Search PubMed.
- M. Stöcker, E. Tangstad, N. Aas and T. Myrstad, Catal. Lett., 2000, 69, 223 CrossRef.
- V. V. Khrizanforova, I. R. Knyazeva, V. I. Matveeva Sokolova, I. R. Nizameev, T. V. Gryaznova, M. K. Kadirov, A. R. Burilov, O. G. Sinyashin and Y. H. Budnikova, Electrocatalysis, 2015, 6, 357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: AFM images of the nickel complexes of thiophosphorylated calix[4]resorcinol show the ligand in the cone or chair conformations, polarization and power density curves of the fuel cell with electrodes composed of the immobilized catalysts on carbon black and carbon black-supported Pt. See DOI: 10.1039/c6dt02881k |
| This journal is © The Royal Society of Chemistry 2016 |