
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOctahedral molybdenum cluster complexes with aromatic sulfonate ligands†
Olga A.
Efremova
*ab,
Yuri A.
Vorotnikov
c,
Konstantin A.
Brylev
cd,
Natalya A.
Vorotnikova
c,
Igor N.
Novozhilov
c,
Natalia V.
Kuratieva
cd,
Mariya V.
Edeleva
e,
David M.
Benoit
a,
Noboru
Kitamura
f,
Yuri V.
Mironov
cd,
Michael A.
Shestopalov
 *cd and
Andrew J.
Sutherland
b
*cd and
Andrew J.
Sutherland
b
aDepartment of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: o.efremova@hull.ac.uk
bAston Materials Centre, Aston University, Aston Triangle, Birmingham, B4 7ET, UK
cNikolaev Institute of Inorganic Chemistry SB RAS, 3 Acad. Lavrentiev Ave., 630090 Novosibirsk, Russian Federation
dNovosibirsk State University, 2 Pirogova Str., 630090 Novosibirsk, Russian Federation
eNovosibirsk Institute of Organic Chemistry SB RAS, 9 Acad. Lavrentiev ave., 630090 Novosibirsk, Russian Federation
fDepartment of Chemistry, Faculty of Science, Hokkaido University, 060-0810 Sapporo, Japan
First published on 30th August 2016
Abstract
This article describes the synthesis, structures and systematic study of the spectroscopic and redox properties of a series of octahedral molybdenum metal cluster complexes with aromatic sulfonate ligands (nBu4N)2[{Mo6X8}(OTs)6] and (nBu4N)2[{Mo6X8}(PhSO3)6] (where X− is Cl−, Br− or I−; OTs− is p-toluenesulfonate and PhSO3− is benzenesulfonate). All the complexes demonstrated photoluminescence in the red region and an ability to generate singlet oxygen. Notably, the highest quantum yields (>0.6) and narrowest emission bands were found for complexes with a {Mo6I8}4+ cluster core. Moreover, cyclic voltammetric studies revealed that (nBu4N)2[{Mo6X8}(OTs)6] and (nBu4N)2[{Mo6X8}(PhSO3)6] confer enhanced stability towards electrochemical oxidation relative to corresponding starting complexes (nBu4N)2[{Mo6X8}X6].
Introduction
Octahedral molybdenum cluster complexes of the general formula [{Mo6X8}L6] (where X− are inner halide ligands and L are apical organic or inorganic ligands) have been known for several decades since the structure, luminescence and redox properties of the [{Mo6Cl8}Cl6]2− anionic unit were established and studied comprehensively.1–8 However, in the last decade there has been a resurgence of interest in related octahedral molybdenum complexes. This is due to recent reports detailing the outstanding photoluminescence properties of some molybdenum octahedral cluster complexes. In general, these studies showed that upon UV-visible excitation cluster complexes based on a {Mo6X8}4+ core demonstrated emission in both the red and near-infrared regions with high quantum yields and photoluminescence lifetimes of up to several hundreds of microseconds.9–15 Moreover, these cluster complexes also demonstrated the capability to generate singlet oxygen efficiently.13–16 These photoemissive properties combined with the relative chemical robustness of the {Mo6X8}4+ cluster core make such complexes excellent candidates for photonic (e.g. materials for lasers17 and optical waveguides18), photovoltaic,19 biological and medical (e.g. imaging agents,20,21 bactericides,22 photo-dynamic therapy23,24) and environmental protection (e.g. hazardous organic waste decomposition25,26) applications.It was shown recently that complexes with a {Mo6I8}4+ cluster core ligated by residues of some strong oxoacids – either organic (fluorinated aliphatic acids9,13) or inorganic acids (nitric acid11,27) – demonstrated outstanding photophysical properties in comparison with those based on a {Mo6Cl8}4+ or {Mo6Br8}4+ core. These include not only significantly higher photoluminescence quantum yields at ambient conditions, but also comparatively narrow emission spectra profiles.
Therefore, we were keen to understand whether the same tendency in photophysical properties of {Mo6X8}4+ cluster complexes is retained in cases, when they are coordinated by sulfonate aromatic outer ligands that are also anions of strong acids. Accordingly, we report the synthesis and systematic study of the photophysical and electrochemical properties of molybdenum cluster complexes (nBu4N)2[{Mo6X8}L6], where L− = p-toluenesulfonate (OTs−) and X− = Cl− (1) Br− (2) or I− (3) or L = benzenesulfonate (PhSO3−) and X− = Cl− (4), Br− (5) or I− (6).
Experimental section
Materials
Cs2[{Mo6X8}X6],28,29 (nBu4N)2[{Mo6X8}X6],30–32 silver benzenesulfonate33 and 2,3-diphenyl-p-dioxene34 were synthesised according to previously reported procedures. All other reactants and solvents were purchased from Fisher, Alfa Aesar and Sigma-Aldrich and used as received.Instrumentation
NMR spectra of 1–6 were recorded on a Bruker Avance 300 NMR spectrometer equipped with a solution-state dual channel probe working at 300.13 MHz for 1H and 75 MHz for 13C. NMR spectra were measured in CDCl3 or in acetone-d6 in the standard way using the zg30 pulse program for 1H. Elemental analyses were obtained using a EuroVector EA3000 Elemental Analyser. FTIR spectra were recorded on a Bruker Vertex 80 as KBr disks. Absorption spectra were recorded on a PerkinElmer Lambda35 UV-vis spectrometer.Cyclic voltammetry
Cyclic voltammetry (CV) was performed using a Metrohm 797 VA Computrace instrument with a glassy carbon electrode as the working electrode and a saturated silver/silver chloride (Ag/AgCl) in 3.5 M KCl as a reference electrode, against which the half-wave potential of the ferrocene/ferrocenium (Fc/Fc+) redox couple (E1/2, Fc/Fc+) was found to be 0.598 V. The potentials were related to the standard platinum electrode. A 0.15 M solution of tetra-n-butylammonium perchlorate (nBu4NClO4) in acetonitrile was used as the electrolyte. Solutions of the samples in the electrolyte (1–2 mM) were degassed by purging with argon prior to recording the CV measurements. Compounds 1–6 as well as starting compounds (nBu4N)2[{Mo6X8}X6] (X− = Cl−, Br−, I−) were investigated voltammetrically within the potential window from –2 V to 2.3 V at 25 °C. The formal half-wave potentials (E1/2) were calculated as the midpoint between the anodic and cathodic peak potentials of the first oxidation of the cluster complex.Photoluminescence measurements
For photoluminescence measurements, powdered samples of the complexes were placed between two non-fluorescent glass plates. The absorbance of acetonitrile solutions was set at <0.1 at 355 nm. The solutions were poured into quartz cuvettes. To deaerate, the solutions were purged with an Ar-gas stream for 30 min and then the cuvettes were sealed. Measurements were carried out at 298 K. The samples were excited by 355 nm laser pulses (6 ns duration, LOTIS TII, LS-2137/3). Corrected emission spectra were recorded on a red-light-sensitive multichannel photodetector (Hamamatsu Photonics, PMA-11). For emission decay measurements, the emission was analysed using a streakscope system (Hamamatsu Photonics, C4334 and C5094). The emission quantum yields were determined using an Absolute Photo-Luminescence Quantum Yield Measurement System (Hamamatsu Photonics, C9920-03), which comprised a xenon excitation light source (the excitation wavelength was set at 400 nm), an integrating sphere, and a red-sensitive multichannel photodetector (Hamamatsu Photonics, PMA-12).Synthesis
1: Yellow. Yield: 130 mg (86%). Anal. calcd for C74H114Cl8Mo6N2O18S6: C 37.5, H 4.8, N 1.2, S 8.1; found: C 37.5, H 4.8, N 1.2, S 8.4. UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 350 (sh, 3.2 × 103). FTIR (KBr, cm−1): νas(SO2) – 1295s, 1283s; νs(SO2) – 1159s; ν(SO) – 958. 1H NMR (acetone-d6) δ (ppm): 7.67 [12H, d, 6 × 2Hortho], 7.29 [12H, d, 6 × 2Hmeta], 3.43 [16H, t, 2 × 4CH2], 2.39 [18H, s, 6 × CH3], 1.81 [16H, quin, 2 × 4CH2], 1.43 [16H, sex, 2 × 4CH2], 0.97 [24H, t, 2 × 4CH3].
2: Yellow. Yield: 112 mg (90%) Anal. calcd for C74H114Br8Mo6N2O18S6: C 32.6, H 4.2, N 1.0, S 7.1; found: C 32.6, H 4.2, N 1.0, S 6.8. UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 320 (5.0 × 103), 363 (sh, 3.4 × 103). FTIR (KBr, cm−1): νas(SO2) – 1293s, 1277s; νs(SO2) – 1158s; ν(SO) – 964. 1H NMR (acetone-d6) δ: 7.64 [12H, d, 6 × 2Hortho], 7.28 [12H, d, 6 × 2Hmeta], 3.44 [16H, t, 2 × 4CH2], 2.39 [18H, s, 6 × CH3], 1.81 [16H, quin, 2 × 4CH2], 1.43 [16H, sex, 2 × 4CH2], 0.97 [24H, t, 2 × 4CH3].
3: Orange. Yield: 95 mg (87%) Anal. calcd for C74H114I8Mo6N2O18S6: C 28.6, H 3.7, N 0.9, S 6.2; found: C 28.7, H 3.6, N 0.9, S 6.0. UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 347 (5.2 × 103), 394 (4.3 × 103). FTIR (KBr, cm−1): νas(SO2) – 1269s; νs(SO2) – 1157s; ν(SO) – 982. 1H NMR (acetone-d6) δ: 7.59 [12H, d, 6 × 2Hortho], 7.27 [12H, d, 6 × 2Hmeta], 3.43 [16H, t, 2 × 4CH2], 2.39 [18H, s, 6CH3], 1.81 [16H, quin, 2 × 4CH2], 1.43 [16H, sex, 2 × 4CH2], 0.97 [24H, t, 2 × 4CH3].
4: Yellow. Yield: 101 mg (67%). Anal. calcd for C68H102Cl8Mo6N2O18S6: C 35.7, H 4.5, N 1.2, S 8.4; found: C 35.7, H 4.5, N 1.3, S 8.1. UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 348 (sh, 3.5 × 103). FTIR (KBr, cm−1): νas(SO2) – 1298s, 1285s; νs(SO2) – 1161s; ν(SO) – 957. 1H NMR (acetone-d6) δ: 7.85–7.72 [12H, m, 6 × 2Hortho], 7.54–7.41 [18H, m, 6 × (2Hmeta + Hpara)], 3.43 [16H, t, 2 × 4CH2], 1.80 [16H, quin, 2 × 4CH2], 1.42 [16H, sex, 2 × 4CH2], 0.96 [24H, t, 2 × 4CH3].
5: Yellow. Yield: 90 mg (72%) Anal. calcd for C68H102Br8Mo6N2O18S6: C 30.9, H 3.9, N 1.1, S 7.3; found: C 30.6, H 3.7, N 1.0, S 7.6 UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 317 (5.2 × 103), 367 (3.6 × 103). FTIR (KBr, cm−1): νas(SO2) – 1282s; νs(SO2) – 1162s; ν(SO) – 965. 1H NMR (acetone-d6) δ: 7.80–7.71 [12H, m, 6 × 2Hortho], 7.55–7.41 [18H, m, 6 × (2Hmeta + Hpara)], 3.43 [16H, t, 2 × 4CH2], 1.80 [16H, quin, 2 × 4CH2], 1.43 [16H, sex, 2 × 4CH2], 0.98 [24H, t, 2 × 4CH3].
6: Orange. Yield: 94 mg (87%). Anal. calcd for C68H102I8Mo6N2O18S6: C 27.1, H 3.4, N 0.9, S 6.4; found: C 27.4, H 3.3, N 0.9, S 6.2. UV-vis (CH2Cl2): λmax, nm (ε, M−1 cm−1) = 347 (5.8 × 103), 391 (4.7 × 103). FTIR (KBr, cm−1): νas(SO2) – 1272s; νs(SO2) – 1156s; ν(SO) – 985. 1H NMR (acetone-d6) δ: 7.75–7.67 [12H, m, 6 × 2Hortho], 7.52–7.43 [18H, m, 6 × (2Hmeta + Hpara)], 3.43 [16H, t, 2 × 4CH2], 1.81 [16H, quin, 2 × 4CH2], 1.43 [16H, sex, 2 × 4CH2], 0.97 [24H, t, 2 × 4CH3].
Crystal structure determination
Single-crystal X-ray diffraction data were collected at 150 K on a Bruker Nonius X8 Apex 4 K CCD diffractometer fitted with graphite monochromatised MoKα radiation (λ = 0.71073 Å). All crystallographic information is summarised in Table 1S.† Absorption corrections were made empirically using the SADABS program.35 The structures were solved by the direct method and further refined by the full-matrix least-squares method using the SHELXTL program package.35 All non-hydrogen atoms were refined anisotropically. The positions of the hydrogen atoms of the tetra-n-butylammonium cation, organic ligands and solvent molecules, apart from water molecules, were calculated corresponding to their geometrical conditions and refined using the riding model. The positions of hydrogen atoms for water molecules weren't localised. In the crystal structures of 2–4 and 6 the disorder of sulfonate groups was caused by the rotation of terminal groups around the C–S, S–O or Mo–O bonds.CCDC 1425980–1425985 contain the supplementary crystallographic data for this paper.
Singlet oxygen generation
The ability of 1–6 to generate singlet oxygen was investigated semi-quantitatively as follows: 0.6 mL of an acetone-d6 solution containing 0.0012 mmol of a cluster complex analyte and 0.12 mmol of the singlet oxygen trap molecule 2,3-diphenyl-p-dioxene36,37 was placed in a conventional NMR tube and purged with oxygen gas for 5 min. The tube was sealed and then irradiated using filtered light (λ ≥ 400 nm) from a DRSh-500 mercury lamp. 1H NMR (200 MHz) spectra were recorded on a Bruker Avance 200 NMR spectrometer after 0, 1, 3 and 5 h of irradiation. The conversion of the trap molecule was calculated as a ratio of the aliphatic hydrogen peak integral of the oxidation product of 2,3-diphenyl-p-dioxene, namely ethylene glycol dibenzoate, to the sum of the integrals of the aliphatic hydrogen peaks of 2,3-diphenyl-p-dioxene and the oxidation product, ethylene glycol dibenzoate (see Fig. 1S and 2S†).Results and discussion
Synthesis and characterization of (nBu4N)2[{Mo6X8}L6] (L = OTs−, X− = Cl− (1), Br− (2) or I− (3); L− = PhSO3−, X− = Cl− (4), Br− (5) or I− (6))
To obtain (nBu4N)2[{Mo6X8}L6] we used a well-established approach, in which tetra-n-butylammonium salts of molybdenum halide anions [{Mo6X8}X6]2− (X− = Cl−, Br− or I−) were allowed to react with a silver salt of the corresponding sulfonic acid – either p-toluenesulfonic acid (HOTs) or benzenesulfonic acid (PhSO3H). In these reactions – driven to completion by the formation of poorly-soluble and thermodynamically favourable silver halides – only the terminal halides were substituted by anionic ligands.9–15,38–42 In all cases the reactions proceeded smoothly and resulted in the formation of (nBu4N)2[{Mo6X8}(OTs)6] (X− = Cl− (1), Br− (2), I− (3)) and (nBu4N)2[{Mo6X8}(PhSO3)6] (X− = Cl− (4), Br− (5), I− (6)), respectively. The purity of compounds 1–6 was confirmed by both elemental analysis and NMR spectroscopy.As expected, due to the symmetry of the cluster anions, only one set of signals, associated with the organic ligands, was observed in the 1H NMR, with the ratio of signals from nBu4N+ and the organic ligands being 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 (Fig. 3S†). Moreover, in each case the good agreement of the elemental analysis data with the theoretically calculated formula also supports the proposed compositions of the compounds synthesised.
:3 (Fig. 3S†). Moreover, in each case the good agreement of the elemental analysis data with the theoretically calculated formula also supports the proposed compositions of the compounds synthesised.
It should be mentioned that the above reaction, although conducted in a different solvent (CH2Cl2 instead of acetone), has been used previously to synthesise 1.39,41 While Johnston et al. did not succeed in the structural characterization of the complex, Sokolov et al.41 obtained crystals of solvates 1·2CH2Cl2 (space group Pbca) and 1·2CH3CN (space group P21/c) by slow diffusion of diethyl ether vapour into either a CH2Cl2 or an acetonitrile solution of 1 respectively.
We crystallised 1–6 by slow diffusion of diethyl ether vapour into acetone solutions to generate crystals suitable for characterisation by single crystal XRD. The crystallographic data obtained are summarised in Table 1S.† In our case, all six complexes crystallised in the triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) crystal system. It is clear that the choice of solvent resulted in crystals of 1 attaining a different space group from those published previously.41 Moreover, in our work, crystals of compound 1 did not contain any solvate molecules. Indeed, only compounds 2 and 3 crystallised as solvates: with acetone, diethyl ether and water (2) or acetone and water (3).
) crystal system. It is clear that the choice of solvent resulted in crystals of 1 attaining a different space group from those published previously.41 Moreover, in our work, crystals of compound 1 did not contain any solvate molecules. Indeed, only compounds 2 and 3 crystallised as solvates: with acetone, diethyl ether and water (2) or acetone and water (3).
Although compounds 1–3 and 4–6 are cluster complexes of very similar compositions, only crystals of pairs of compounds 2 and 3, and 4 and 5 have similar crystal structures. In the independent part of the crystal structures of 2–5 there are two halves of the cluster anions [{Mo6X8}L6]2− and two nBu4N+ cations, while compounds 1 and 6 have only a half of the corresponding cluster anion and a nBu4N+ cation in the independent part. In all cases the centre of the [{Mo6X8}L6]2− cluster anion coincides with the inversion centre.
Similar to other octahedral molybdenum cluster complexes, the Mo6 octahedra in compounds 1–6 are coordinated by eight μ3-type halide ligands and, additionally, each molybdenum is coordinated by a terminal ligand – p-toluenesulfonate or benzenesulfonate ion. The organic ligands are coordinated in a monodentate mode via an oxygen atom of the SO3 group, as shown in Fig. 1. The important average bond lengths and angles in the crystal structures of compounds 1–6 are summarised in Table 2S.† The average bond lengths Mo–O are between 2.100–2.165 Å and they tend to increase with increasing size of the cluster core. Analysis of the Cambridge Structural Database (CSD)43 data on the typical Mo–OSO2 bond values show that the values found for 1–6 are towards the shorter range of these bond lengths. This observation indicates significant covalent contribution in the bonding. The Mo–O–S bond angles also tend to increase (i.e. deviate more significantly from the ideal tetrahedral bond angle) with increasing size of the cluster core, with the highest values, 140.6° and 142.1°, found in structures 3 and 6, respectively. These values are slightly higher than the most typical bond angles between sulfonate groups and molybdenum atoms (128–138° according to CSD).
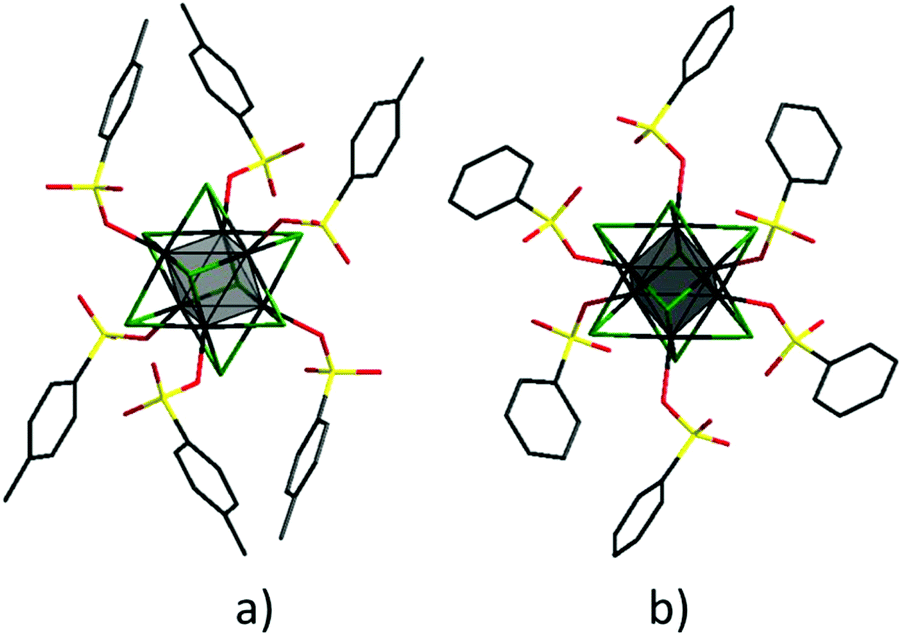 | ||
| Fig. 1 The structure of the cluster anions [{Mo6X8}(OTs)6]2− (a) and [{Mo6X8}(PhSO3)6]2− (b), where X− = Cl−, Br− or I−: Mo – black; X – green; S – yellow; O – red; C – charcoal; hydrogen atoms are omitted for clarity. | ||
The FTIR spectra of compounds 1–6 are very similar, as bands originate mostly from the organic ligands and nBu4N+ cations. The strong vibrations in the sulfonate group (Fig. 2) provide a simple way of monitoring the coordination of sulfonate ligands to the metal centres. Namely, the FTIR spectra of 1–6 further confirm the mono-dentate coordination mode of sulfonate groups. In comparison with the ν(SO2) bands observed in the FTIR spectra of p-toluenesulfonic acid and benzenesulfonic acid ν(SO2) bands observed in spectra of the obtained molybdenum cluster complexes are downshifted.44,45 Moreover, minor changes are noticeable when comparing FTIR spectra of complexes with the same ligands, but different halide ligands in the cluster core (Fig. 2). Specifically, in the series Cl–Br–I there is a noticeable upshift of ν(S–O) bands especially in case of compounds 3 and 6. This shift might indicate the change of the force constants (i.e. bonding order) for the S–O bonds, which can be additional evidence of some increase of the ionicity of the bonding between sulfonate groups and the molybdenum atoms. The DFT calculations undertaken for the crystal structures of 1–6 did not however confirm significant changes in the ionic nature of the bonding between the SO3− groups and the Mo cluster core (see ESI for details of DFT calculations, Table 3S†).
 | ||
| Fig. 2 The fingerprint regions in the FTIR spectra of 1–6. | ||
Redox properties
Oxidation and reduction of compounds 1–6 were investigated by cyclic voltammetry in acetonitrile and the results were compared with those obtained for the (nBu4N)2[{Mo6X8}X6] (X− = Cl−, Br− and I−) starting materials. Fig. 4S† shows the cyclic voltammograms of compounds 1–6, while those of the starting materials are shown in Fig. 5S.†Table 1 summarises the most relevant data pertaining to the redox properties of the cluster complexes.| Compound | [Mo6X8L6]2− = [Mo6X8L6]− + e | [Mo6X8L6]2− + e | E onsetox, V | E onsetred, V | Electrochemical energy gap, eVa | Optical energy gap, s eV | ||
|---|---|---|---|---|---|---|---|---|
| E 1/2, V | ΔEp, mV | I pa/Ipc | E redp, V | |||||
| a The electrochemical energy gap was estimated as a difference between Eonsetox and Eonsetred. b Recalculated vs. saturated Ag/AgCl in acetonitrile. c The first oxidation peak current potential. | ||||||||
| (nBu4N)2[{Mo6Cl8}Cl6] | 1.78 (1.7813)b | 105 | 1.00 | −1.40 | 1.70 | −1.21 | 2.9 | 2.6 |
| (nBu4N)2[{Mo6Br8}Br6] | 1.55 (1.5713)b | 100 | 1.04 | −1.35 | 1.51 | −1.26 | 2.8 | 2.4 (2.452) |
| (nBu4N)2[{Mo6I8}I6] | 1.02c (1.1213)b | — | — | −1.25 | 0.70 | −1.04 | 1.7 | 2.1 |
| 1 | 1.81 | 110 | 0.85 | −1.09 | 1.72 | −0.89 | 2.6 | 2.7 |
| 2 | 1.88 | 111 | 0.82 | −1.14 | 1.77 | −0.87 | 2.6 | 2.6 |
| 3 | 1.54 | 94 | 0.86 | −1.13 | 1.44 | −1.00 | 2.4 | 2.3 |
| 4 | — | — | — | −1.15 | 2.7 | |||
| 5 | 1.93 | 97 | 0.85 | −1.13 | 1.81 | −0.81 | 2.6 | 2.4 |
| 6 | 1.54 | 170 | 0.89 | −1.10 | 1.42 | −0.81 | 2.2 | 2.2 |
Oxidation of the starting complexes (nBu4N)2[{Mo6X8}X6] (X− = Cl−, Br− and I−) has been reported previously by Kirakci et al.13 Our CV measurements confirmed the earlier data that [{Mo6Cl8}Cl6]2− and [{Mo6Br8}Br6]2− undergo one-electron reversible oxidation in the positive potential region, while [{Mo6I8}I6]2− undergoes irreversible oxidation, with the oxidation potential lower than that for the lighter cluster anions (Table 1).13
CV measurements of compounds 1–3, 5 and 6 showed quasi-reversible oxidation in the positive region at somewhat higher potentials than those observed for the corresponding (nBu4N)2[{Mo6X8}X6] complexes (Table 1). For compound 4 no oxidation peak was recorded up to a potential of 2 V. In summary it appears that substitution of the apical halide ligands by sulfonated aromatic ligands – benzenesulfonate and p-toluenesulfonate – increases the stability of the corresponding cluster complexes towards oxidation. Likewise, in analogous fashion to the starting complexes, oxidation of the complexes with heavier (iodide) cluster cores takes place at significantly lower potential, which signifies the noteworthy input of iodide ligand orbitals to the HOMO.
Similarly to earlier reported data on oxidation of octahedral molybdenum13,46 and rhenium47–49 cluster complexes, the difference between anodic and cathodic potential peaks (ΔEp) was higher than the “ideal” value of 59 mV in the Nernst equation for a one electron process. The increase of ΔEp typically occurs because of uncompensated solution resistance and/or non-linear diffusion. Indeed, the anodic and cathodic peak currents, Ip(a) and Ip(c), respectively, were proportional to the square root of the scan rate, which agrees with Randles–Sevcik theory for processes controlled by diffusion. Moreover, the values of Ip(a)/Ip(c) for 1–3, 5 and 6 indicate that some adsorption of the reactant could take place.50
In the negative potential region, both the starting compounds (nBu4N)2[{Mo6X8}X6] (X− = Cl−, Br− and I−) and compounds 1–6 underwent irreversible reduction. Interestingly, the reduction of all compounds took place in a very narrow range of voltages ∼−1.1 V (Table 1, Fig. 4S and 5S†). Such similarity in reduction potentials suggests that the LUMO orbital in the studied molybdenum complexes does not have significant input from the halide (inner) ligand orbitals. The irreversible nature of reduction is likely to be associated with the destruction of the cluster complexes, as it has been suggested previously for [{Mo6Cl8}Cl6]2−.51
It is known that cyclic voltammetry can be used to estimate the energy gap between the HOMO and the LUMO (also referred to as the electrochemical energy gap) as the difference between onsets of oxidation and reduction peaks. The values of the electrochemical band gap for the starting materials and the studied compounds decrease in the order Cl > Br > I for given apical ligands (Table 1).
Absorption properties
The UV-vis absorption spectra of all the cluster complexes were recorded in dichloromethane and are shown in Fig. 3. Comparison of the absorption spectra of (nBu4N)2[{Mo6X8}X6]13 with those of 1–6 shows that the substitution of apical halide ligands by sulfonate ones causes a blue shift of the absorption bands, especially in the case of the cluster complexes based on {Mo6I8}4+ core. These observations further support the observation from electrochemical data about the significant input from the apical ligands in the HOMO. Taking into account the suggested nature of the HOMO and the LUMO, the lowest energy electronic transitions arise from ligand to metal charge transfer (LMCT). This is directly analogous to previously reported suggestions, based on theoretical calculations, for [{Mo6X8}(NCS)6]2− (X− = Cl−, Br−, I−)53 and isoelectronic rhenium clusters54–56 with 24 valence electron counts (VEC).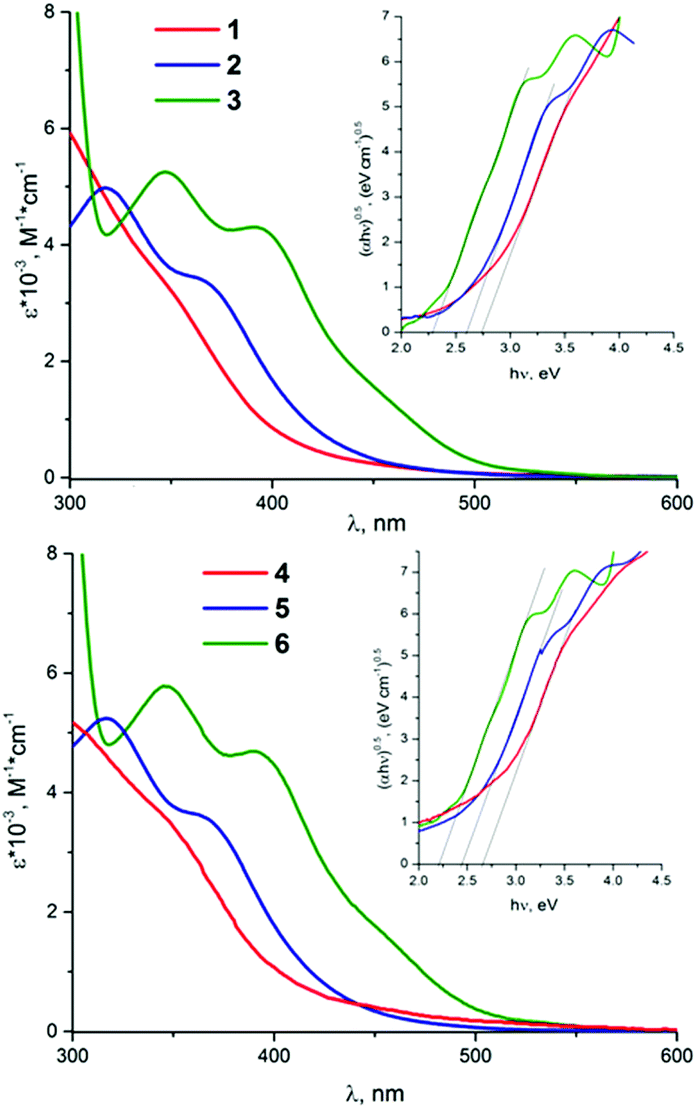 | ||
| Fig. 3 The UV-vis spectra of 1–3 (top) and 4–6 (bottom) in dichloromethane. The inserts are the Tauc plots used to estimate excited state energy. | ||
UV-vis spectra of 1–6 show that the maxima of the absorptions as well as the values of the extinction coefficients do not depend on whether the apical ligand is benzenesulfonate or p-toluenesulfonate. A pronounced red shift of absorption band maxima is observed in the series Cl–Br–I. Similar bathochromic shifts of absorption maxima have been reported previously for [{Mo6X8}X6]2−, [{Mo6X8}(NCS)6]2− (X = Cl−, Br−, I−) and [{Mo6X8}(CF3COO)6]2−.13,57,58 These shifts signify that the energy gap between the HOMO and the LUMO in cluster complexes [{Mo6X8}L6]2− decreases in the order Cl > Br > I for given apical ligands. This order was further confirmed, when the energy gaps (also known as the optical energy gap) were estimated via Tauc plot (see inserts in Fig. 3 as well as Fig. 6S†). These optical energy gaps, obviously, refer to spin-allowed (i.e. singlet state to singlet state) transitions. The above values for the electrochemical band gap for the starting materials and the studied compounds are comparable with the optical energy band gaps found from absorption spectra (Table 1). Moreover the same trend in terms of the widths of the energy gaps is also observed, i.e. the optical energy gap decreases in the order Cl > Br > I for given apical ligands.
Photoluminescence properties
The normalised photoluminescence spectra of samples 1–6 in the solid state and in deaerated acetonitrile solutions are shown in Fig. 4. The photophysical characteristics, namely emission maximum wavelengths (λem), quantum yields (Φem) and lifetimes (τem) with the corresponding amplitudes (A), of all samples measured in the solid state as well as in air-equilibrated and deaerated acetonitrile solutions are summarised in Table 2. | ||
| Fig. 4 Normalised emission spectra of 1–6 in the solid state (top) and in deaerated acetonitrile solutions (bottom). | ||
 ; Φem is photoluminescence quantum yield
; Φem is photoluminescence quantum yield
| (nBu4N)2[{Mo6X8}(OTs)6] | (nBu4N)2[{Mo6X8}(C6H5SO3)6] | ||||||
|---|---|---|---|---|---|---|---|
| λ em, nm (fwhm, cm−1) | τ em, μs (A) | Φ em | λ em, nm (fwhm, cm−1) | τ em, μs | Φ em | ||
| X = Cl | Powdered sample | 723 (∼4000) | τ 1 = 228 (0.7) | 0.34 | 719 (∼4200) | τ 1 = 127 (0.3) | 0.13 |
| τ 2 = 174 (0.3) | τ 2 = 75 (0.7) | ||||||
| Aerated solution | 735 (∼3850) | τ 1 = 5.1 (0.3) | <0.01 | 723 (∼4050) | τ 1 = 3.9 (0.4) | <0.01 | |
| τ 2 = 1.2 (0.7) | τ 2 = 0.5 (0.6) | ||||||
| Deaerated CH3CN solution | 735 (∼3850) | τ 1 = 9.8 (0.2) | <0.01 | 723 (∼4050) | τ 1 = 9.2 (0.1) | <0.01 | |
| τ 2 = 3.0 (0.8) | τ 2 = 2.6 (0.9) | ||||||
| X = Br | Powdered sample | 708 (∼4100) | τ 1 = 185 (0.7) | 0.29 | 718 (∼4350) | τ 1 = 124 (0.3) | 0.13 |
| τ 2 = 86 (0.3) | τ 2 = 52 (0.7) | ||||||
| Aerated CH3CN solution | 717 (∼4150) | 15 | <0.01 | 750 (∼4150) | 13 | <0.01 | |
| Deaerated CH3CN solution | 717 (∼4150) | 243 | 0.26 | 750 (∼4150) | 179 | 0.17 | |
| X = I | Powdered sample | 662 (∼2300) | τ 1 = 135 (0.6) | 0.44 | 657 (∼2200) | τ 1 = 183 (0.6) | 0.62 |
| τ 2 = 56 (0.4) | τ 2 = 57 (0.4) | ||||||
| Aerated CH3CN solution | 667 (∼2400) | 5.0 | 0.01 | 665 (∼2400) | 5.2 | 0.01 | |
| Deaerated CH3CN solution | 667 (∼2400) | 305 | 0.65 | 665 (∼2400) | 263 | 0.60 | |
As Table 2 shows, the photophysical characteristics of the complexes are strongly dependent on the halide ligand in the cluster core and only slightly on the composition of the apical ligands.
Generally, all of the complexes, in both the solid state and in acetonitrile solution, exhibited red photoluminescence with broad spectral emission profiles ranging from ∼550 to longer than 900 nm. These are typical photoluminescence emission profiles for compounds based on {Mo6X8}4+ (X− = Cl−, Br− or I−) cluster cores.9–15,38 Notably, the emission profiles of iodide complexes 3 and 6 are significantly narrower and blue shifted compared with the emission profiles of the chloride and bromide-containing clusters. In combination absorption and photoluminescence spectra of 1–6 show that the emission in the case of {Mo6I8}4+ occurs mostly from the triplet state that is closer to the excited singlet state than that in the case of {Mo6Cl8}4+ and {Mo6Br8}4+. This intriguing feature of [{Mo6I8}L6]2− cluster complexes, where L is a residue oxygenated acid has been noticed earlier by us and other groups.9–11,13,38
The solid state luminescence properties of complexes 1–6 are highly encouraging being characterised by quantum yields, from 0.13 to 0.62. The highest quantum yields were found for complexes 3 and 6, i.e. those comprising the {Mo6I8}4+ cluster core. The luminescence lifetimes of all the solid samples were fitted by double exponential decays that are usually explained by an efficient excitation energy migration and subsequent energy trap/emission in the crystalline phase.
The emission spectra for each compound, in both aerated and deaerated solutions, have almost identical profiles. For solutions of the bromide and iodide complexes (2, 3, 5 and 6) the deaerated solutions are characterised by significantly higher quantum yields and longer lifetimes than those observed for the corresponding aerated systems (Table 2). This difference may be explained readily by the fact that the long-lived luminescence of hexanuclear cluster complexes is well known to be quenched efficiently by oxygen.9–11,38
Chloride complexes 1 and 4 behaved differently from their bromide and iodide analogues. Even in a deaerated acetonitrile solution these complexes demonstrated very weak and relatively short-lived luminescence despite the relatively high τem and Φem values found for them in the solid state (Table 2). Similar low levels of luminescence in solution with a double exponential emission decay and relatively small lifetimes have also been reported previously for {Mo6Cl8}4+-based clusters coordinated by fluorinated carboxylate apical ligands.9,13
In contrast, as in our work, the emissions of dissolved related bromide, and especially, iodide clusters were characterised by very high emission quantum yields with decay profiles fitted well by single exponential functions and τem values of hundreds of microseconds.9,13 Since oxygen quenches the luminescent emissions of cluster complexes 1–6 effectively (Table 4S†), we were keen to explore the potential of these materials in applications associated with the use of singlet oxygen generated in situ. Therefore, we compared the ability of each of the cluster complexes 1–6 to generate singlet oxygen using a semi-quantitative test based on oxidation of the singlet oxygen trap molecule, 2,3-diphenyl-p-dioxene.
Accordingly, 1H NMR was used to trace oxidation of 2,3-diphenyl-p-dioxene in the presence of the cluster complexes. Specifically, 1:100 molar mixtures of each cluster complex:trap were irradiated for periods of 1, 3 and 5 h. The results obtained unambiguously confirmed the successful generation of singlet oxygen by all six complexes 1–6 (Fig. 5, 1S and 2S, Table 4S†). The rate of oxidation of the 2,3-diphenyl-p-dioxene was dependent on the halide ligand present in the cluster core and decreased in the order I > Br > Cl for given apical ligands. This order of singlet oxygen generation efficiencies correlates well with the photoluminescence quantum yields recorded in deaerated acetonitrile solutions of 1–6.
 | ||
| Fig. 5 The conversion of 2,3-diphenyl-p-dioxene into ethylene glycol dibenzoate in the presence of 1–6 after photoirradiation with λ ≥ 400 nm. | ||
Conclusions
In this work we synthesised and characterised, in detail, octahedral molybdenum cluster complexes, where the cores {Mo6X8}4+ (X− = Cl−, Br− or I−) are coordinated by aromatic sulfonates – either p-toluenesulfonate or benzenesulfonate. Our study clearly confirms the strong effect that the inner halide ligands exert on the photophysical and redox properties of the obtained cluster complexes. Specifically UV-vis and CV studies showed that the energy gap between ground state and the first singlet (exited) state decreases in the order Cl > Br > I, while the gap between emissive (triplet) state and the ground state increases in the same order. Furthermore, our data also suggest that the HOMO orbital has a significant input from halide ligands, especially in the case of iodide cluster complexes, while the LUMO is predominantly Mo6 derived in nature.All of the cluster complexes displayed strong red luminescence in the solid state, while only those with heavier cluster cores (i.e. Br− and I− inner ligands) showed outstanding photoluminescence in deaerated acetonitrile solutions. Notably, cluster complexes with {Mo6I8}4+ cores showed the highest quantum yields and the narrowest and most strongly blue-shifted emission spectra. Moreover, [{Mo6I8}(OTs)6]2− and [{Mo6I8}(PhSO3)6]2− also showed the highest singlet oxygen generation efficiency in comparison with the chloride and bromide analogues. This study thus further suggests that synthesis of highly photoactive compounds and materials may be generally achieved by coordinating of octahedral cluster {Mo6I8}4+ by residues of strong oxoacids. In order to facilitate the rational design of materials with bespoke photophysical properties, detailed study should be undertaken to uncover whether there is any dependence between the acidy of an oxoacid and the photoluminescence parameters of the corresponding octahedral molybdenum cluster complexes.
Acknowledgements
This work was supported by a Marie-Curie Intra-European Fellowship (No. 327440), by the Russian Foundation for Basic Research (Grant No. 14-03-92612), the Royal Society (IE140281) and by the Grant of the President of the Russian Federation (MK 4054.2015.3). K. A. Brylev thanks the Japan Society for the Promotion of Science (JSPS) for a post-doctoral fellowship for foreign researchers.References
- J. C. Sheldon, Nature, 1959, 184, 1210–1213 CrossRef CAS.
- J. C. Sheldon, J. Chem. Soc., 1960, 1007–1014 RSC.
- J. C. Sheldon, Chem. Ind., 1961, 323 CAS.
- J. C. Sheldon, J. Chem. Soc., 1963, 4183–4186 RSC.
- A. W. Maverick and H. B. Gray, J. Am. Chem. Soc., 1981, 103, 1298–1300 CrossRef CAS.
- A. W. Maverick, J. S. Najdzionek, D. MacKenzie, D. G. Nocera and H. B. Gray, J. Am. Chem. Soc., 1983, 105, 1878–1882 CrossRef CAS.
- D. G. Nocera and H. B. Gray, J. Am. Chem. Soc., 1984, 106, 824–825 CrossRef CAS.
- Y. Saito, H. K. Tanaka, Y. Sasaki and T. Azum, J. Phys. Chem., 1985, 89, 4413–4415 CrossRef CAS.
- M. N. Sokolov, M. A. Mihailov, E. V. Peresypkina, K. A. Brylev, N. Kitamura and V. P. Fedin, Dalton Trans., 2011, 40, 6375–6377 RSC.
- M. N. Sokolov, M. A. Mikhailov, K. A. Brylev, A. V. Virovets, C. Vicent, N. B. Kompankov, N. Kitamura and V. P. Fedin, Inorg. Chem., 2013, 52, 12477–12481 CrossRef CAS PubMed.
- O. A. Efremova, M. A. Shestopalov, N. A. Chirtsova, A. I. Smolentsev, Y. V. Mironov, N. Kitamura, K. A. Brylev and A. J. Sutherland, Dalton Trans., 2014, 43, 6021–6025 RSC.
- K. Kirakci, K. Fejfarova, M. Kucerakova and K. Lang, Eur. J. Inorg. Chem., 2014, 2331–2336 CrossRef CAS.
- K. Kirakci, P. Kubat, J. Langmaier, T. Polivka, M. Fuciman, K. Fejfarova and K. Lang, Dalton Trans., 2013, 42, 7224–7232 RSC.
- K. Kirakci, P. Kubat, M. Dusek, K. Fejfarova, V. Sicha, J. Mosinger and K. Lang, Eur. J. Inorg. Chem., 2012, 3107–3111 CrossRef CAS.
- K. Kirakci, V. Sicha, P. Holub and K. Lang, Inorg. Chem., 2014, 53, 13012–13018 CrossRef CAS PubMed.
- J. A. Jackson, M. D. Newsham, C. Worsham and D. G. Nocera, Chem. Mater., 1996, 8, 558–564 CrossRef CAS.
- S. Chenais and S. Forget, Polym. Int., 2011, 61, 390–406 CrossRef.
- H. Liu, J. B. Edel, L. M. Bellan and H. G. Craighead, Small, 2006, 2, 495–499 CrossRef CAS PubMed.
- B. C. Rowan, L. R. Wilson and B. S. Richards, IEEE J. Sel. Top. Quantum Electron., 2008, 14, 1312–1322 CrossRef CAS.
- C. Vonesch, F. Aguet, J.-L. Vonesch and M. Unser, IEEE Signal Process. Mag., 2006, 20–31 CrossRef.
- J. F. Lovell, T. W. B. Liu, J. Chen and G. Zheng, Chem. Rev., 2010, 110, 2839–2857 CrossRef CAS PubMed.
- F. Vatansever, W. C. M. A. de Melo, P. Avci, D. Vecchio, M. Sadasivam, A. Gupta, R. Chandran, M. Karimi, N. A. Parizotto, R. Yin, G. P. Tegos and M. R. Hamblin, Microbiol. Rev., 2013, 37, 955–989 CAS.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- P. Agostinis, K. Berg, K. A. Cengel, H. Foster, A. W. Girotti, S. O. Gollnick, S. M. Hahn, M. R. Hamblin, A. Juzeniene, D. Kessel, M. Korbelik, J. Moan, P. Mroz, D. Nowis, J. Piette, B. C. Wilson and J. Golab, CA-Cancer J. Clin., 2011, 61, 250–281 CrossRef PubMed.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- A. Barras, S. Cordier and R. Boukherroub, Appl. Catal., B, 2012, 123–124, 1–8 CrossRef CAS.
- A. O. Solovieva, Y. A. Vorotnikov, K. E. Trifonova, O. A. Efremova, A. A. Krasilnikova, K. A. Brylev, E. V. Vorontsova, P. A. Avrorov, L. V. Shestopalova, A. F. Poveshchenko, Y. V. Mironov and M. A. Shestopalov, J. Mater. Chem. B, 2016, 4, 4839–4846 RSC.
- A. Flemström, T. K. Hirsch, L. Eriksson and S. Lidin, Solid State Sci., 2004, 6, 509–517 CrossRef.
- K. Kirakci, S. Cordier and C. Perrin, Z. Anorg. Allg. Chem., 2005, 631, 411–416 CrossRef CAS.
- W. Preetz and K. Harder, J. Alloys Compd., 1992, 183, 413–429 CrossRef CAS.
- P. Bruckner, W. Preetz and M. Punjer, Z. Anorg. Allg. Chem., 1997, 623, 8–17 CrossRef.
- K. Kirakci, S. Cordier, T. Roisnel, S. Golhen and C. Perrin, Z. Kristallogr. - New Cryst. Struct., 2005, 220, 116–118 CAS.
- A. P. Cote and G. K. H. Shimizu, Inorg. Chem., 2004, 43, 6663–6676 CrossRef CAS PubMed.
- R. K. Summerbell and D. R. Berger, J. Am. Chem. Soc., 1959, 81, 633–639 CrossRef CAS.
- Bruker, APEX2 (Version 1.08), SAINT (Version 7.03), SADABS (Version 2.11), SHELXTL (Version 6.12), Bruker AXS Inc., Madison, WI, USA, 2004 Search PubMed.
- L. Gao, M. A. Peay and T. G. Gray, Chem. Mater., 2010, 22, 6240–6245 CrossRef CAS.
- M. A. Shestopalov, K. E. Zubareva, O. P. Khripko, Y. I. Khripko, A. O. Solovieva, N. V. Kuratieva, Y. V. Mironov, N. Kitamura, V. E. Fedorov and K. A. Brylev, Inorg. Chem., 2014, 53, 9006–9013 CrossRef CAS PubMed.
- M. A. Mikhailov, K. A. Brylev, A. V. Virovets, M. R. Gallyamov, I. N. Novozhilov and M. N. Sokolov, New J. Chem., 2016, 40, 1162–1168 RSC.
- D. H. Johnston, D. C. Gaswick, M. C. Lonergan, C. L. Stern and D. F. Shriver, Inorg. Chem., 1992, 31, 1869–1873 CrossRef CAS.
- P. Braack, M. K. Simsek and W. Preetz, Z. Anorg. Allg. Chem., 1998, 624, 375–380 CrossRef CAS.
- M. N. Sokolov, M. A. Mihailov, P. A. Abramov and V. P. Fedin, J. Struct. Chem., 2012, 53, 197–201 CrossRef CAS.
- M. N. Sokolov, M. A. Mikhailov, A. V. Virovets, K. A. Brylev, R. A. Bredikhin, A. M. Maksimov, V. E. Platonov and V. P. Fedin, Russ. Chem. Bull., 2013, 62, 1764–1767 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., 2016, 72, 171–179 CrossRef CAS PubMed.
- S. Detoni and D. Hadzi, Spectrochim. Acta, 1956, 11, 601–608 CrossRef.
- S. M. Holmes, S. G. Mckinley and G. S. Girolami, Inorg. Synth., 2002, 33, 91–93 CAS.
- K. Kirakci, S. Cordier, O. Hernandez, T. Roisnel, F. Paul and C. Perrin, J. Solid State Chem., 2005, 178, 3117–3129 CrossRef CAS.
- T. Yoshimura, K. Umakoshi, Y. Sasaki, S. Ishizaka, H. B. Kim and N. Kitamura, Inorg. Chem., 2000, 39, 1765–1772 CrossRef CAS PubMed.
- Y. Molard, A. Ledneva, M. Amela-Cortes, V. Circu, N. G. Naumov, C. Meriadec, F. Artzner and S. Cordier, Chem. Mater., 2011, 23, 5122–5130 CrossRef CAS.
- Z. P. Zheng, T. G. Gray and R. H. Holm, Inorg. Chem., 1999, 38, 4888–4895 CrossRef CAS PubMed.
- R. H. Wopschall and I. Shain, Anal. Chem., 1967, 39, 1514–1527 CrossRef CAS.
- P. A. Barnard, I.-W. Sun and C. L. Hussey, Inorg. Chem., 1990, 29, 3670–3674 CrossRef CAS.
- P. Kumar, S. Kumar, S. Cordier, S. Paofai, R. Boukherroub and S. L. Jain, RSC Adv., 2014, 4, 10420–10423 RSC.
- R. Ramirez-Tagle and R. Arratia-Perez, Chem. Phys. Lett., 2008, 460, 438–441 CrossRef CAS.
- R. Arratia-Perez and L. Hernandez-Acevedo, J. Chem. Phys., 1999, 111, 168–172 CrossRef CAS.
- R. Arratia-Perez and L. Hernandez-Acevedo, J. Chem. Phys., 1999, 110, 2529–2532 CrossRef CAS.
- L. Hernandez-Acevedo and R. Arratia-Perez, J. Chil. Chem. Soc., 2003, 48, 125–128 CAS.
- R. Rramirez-Tagle and R. Arratia-Perez, Chem. Phys. Lett., 2008, 460, 438–441 CrossRef.
- R. Rramirez-Tagle and R. Arratia-Perez, Chem. Phys. Lett., 2008, 455, 38–41 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: DFT calculations data, single crystal data; singlet oxygen-generation data; examples of NMR spectra; cyclic voltammograms; the Tauc plot of the absorption spectra of (nBu4N)2[{Mo6X8}X6]; average values of the important bond lengths and angles. CCDC 1425980–1425985. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02863b |
| This journal is © The Royal Society of Chemistry 2016 |