
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure–antiproliferative activity studies on L-proline- and homoproline-4-N-pyrrolidine-3-thiosemicarbazone hybrids and their nickel(II), palladium(II) and copper(II) complexes†
Aliona
Dobrova‡
a,
Sonja
Platzer‡
a,
Felix
Bacher
a,
Miljan N. M.
Milunovic
a,
Anatolie
Dobrov
a,
Gabriella
Spengler
b,
Éva A.
Enyedy
c,
Ghenadie
Novitchi
d and
Vladimir B.
Arion
*a
aUniversity of Vienna, Institute of Inorganic Chemistry, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: vladimir.arion@univie.ac.at
bDepartment of Medical Microbiology and Immunobiology, University of Szeged, Dóm tér 10, H-6720 Szeged, Hungary
cDepartment of Inorganic and Analytical Chemistry, University of Szeged, Dóm tér 7, H-6720 Szeged, Hungary
dLaboratoire National des Champs Magnetiques Intenses-CNRS, 25 Avenue des Martyrs, 38042 Grenoble Cedex 9, France
First published on 26th July 2016
Abstract
Two water-soluble thiosemicarbazone-proline (H2L1) and thiosemicarbazone-homoproline hybrids (H2L2) were synthesised. By reaction of H2L1 with NiCl2·6H2O, PdCl2 and CuCl2·2H2O in ethanol, the series of square-planar complexes [Ni(H2L1)Cl]Cl·1.3H2O (1·1.3H2O), [Pd(H2L1)Cl]Cl·H2O (2·H2O) and [Cu(H2L1)Cl]Cl·0.7H2O (3·0.7H2O) was prepared, and starting from H2L2 and CuCl2·2H2O in methanol, the complex [Cu(H2L2)Cl2]·H2O (4·H2O) was obtained. The compounds have been characterised by elemental analysis, spectroscopic methods (IR, UV-vis and NMR spectroscopy), ESI mass spectrometry and single crystal X-ray crystallography (H2L1, 1, 2 and 4). As a solid, 1 is diamagnetic, while it is paramagnetic in methanolic solution. The effective magnetic moment of 3.26 B.M. at room temperature indicates the change in coordination geometry from square-planar to octahedral upon dissolution. The in vitro anticancer potency of ligand precursors H2L1 and H2L2 and metal complexes 1–4 was studied in three human cancer cell lines (A549, CH1 and SW480) and in noncancerous murine embryonal fibroblasts (NIH/3T3), and the mechanism of cell death was also assayed by flow cytometry. Clear-cut structure–activity relationships have been established. The metal ions exert marked effects in a divergent manner: copper(II) increases, whereas nickel(II) and palladium(II) decrease the cytotoxicity of the hybrids. The antiproliferative activity of H2L1 and metal complexes 1–3 decreases in all three tumour cell lines in the following rank order: 3 > H2L1 > 1 > 2. The role of square-planar geometry in the underlying mechanism of cytotoxicity of the metal complexes studied seems to be negligible, while structural modifications at the terminal amino group of thiosemicarbazide and proline moieties are significant for enhancing the antiproliferative activity of both hybrids and copper(II) complexes.
Introduction
α-(N)-Heterocyclic thiosemicarbazones (TSCs) are a class of highly potent inhibitors of the ribonucleotide reductase (RNR) enzyme.1,2 RNR catalyses the reduction of all four ribonucleotides to their corresponding deoxyribonucleotides and thereby provides the precursors needed for both synthesis and repair of DNA. Currently, the most studied therapeutic compound among the thiosemicarbazones is Triapine (3-aminopyridine-2-carboxaldehyde thiosemicarbazone). It is by a factor of 1000 a more potent inhibitor of hR2 RNR activity than hydroxyurea, a clinically used RNR inhibitor.3 Triapine has entered several phase II clinical trials as a chemotherapeutic agent.4 However, this investigational drug showed severe side effects, while only a small response was observed.5–8 Another promising drug, di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC), has just entered clinical trials,9 but RNR is not or only in part responsible for its biological activity.Many thiosemicarbazones are excellent chelators for transition metal ions, such as iron(II), copper(II) and zinc(II), typically coordinating via X, N, and S atoms, where X is the donor atom of the additional functional group of the aldehyde or ketone used for their preparation.10 The coordination of these TSCs can result in metal complexes with a higher cytotoxicity than that of the ligand precursors. In addition, the enhanced need for the essential metal ions of cancer cells vs. healthy cells makes the development of new chelators as anticancer agents an important task.11,12 Copper(II) complexes are promising anticancer agents,13 often exhibiting very high antiproliferative activity in vitro. In particular, copper(II) complexes with acyl diazine thiosemicarbazones bearing an N4-azabicyclo[3.2.2]nonane group showed cytotoxic activity against colon adenocarcinoma HT-29 cells with IC50 values ranging from 4 nM to 1.51 μM and human acute lymphoblastic leukemia CCRF-CEM cells with IC50 values ranging from 5 nM to 1.16 μM.14
Recently a series of copper(II)-TSC complexes have been tested for their topoisomerase IIα inhibition activity with the conclusion that these complexes are more potent topoisomerase IIα inhibitors than the corresponding ligand precursors.15 One such copper(II) complex was [CuLCl], where HL = 2-pyridinealdehyde 4-N-ethylthiosemicarbazone. The main prerequisite for their topoisomerase IIα inhibition properties was suggested to be the square-planar coordination geometry of copper(II) in these complexes.
Despite their high cytotoxicity in vitro, a low water solubility, high lipophilicity and high in vivo toxicity are the main disadvantages of TSC compounds as anticancer agents,16 so one of the current challenges is the design of new active TSCs and their metal complexes with enhanced aqueous solubility17 and a priori oriented towards a cancer specific target.18 We have shown that 2-hydroxybenzaldehyde thiosemicarbazone (STSC) can be coupled to L- or D-proline (Pro) leading to hybrids with enhanced aqueous solubility.19 Although these new systems show only moderate cytotoxic potency with IC50 values of 62 and 75 μM, respectively, in ovarian carcinoma CH1 cells and >100 μM in colon carcinoma SW480 cells, the coordination of these ligands to copper(II) resulted in a 13- and 5-fold increase in cytotoxicity in CH1 cells, based on a comparison of IC50 values, while in SW480 cells the enhancement of the antiproliferative activity was even higher. In both tested cell lines, L-Pro-STSC as well as its copper(II) complex showed slightly stronger antiproliferative activity than the compounds with a D-Pro moiety, yielding IC50 values of 4.6 and 5.5 μM for [Cu(L-Pro-STSC)Cl]Cl in CH1 and SW480 cells, respectively.19 Moreover, the preliminary data from the screening of the topoisomerase IIα inhibiting activity indicate that the square-planar copper(II) complexes prepared by us are indeed potential inhibitors of the topoisomerase IIα enzyme in contrast to the corresponding metal-free conjugates, which are devoid of such activity.
The quite recently reported results19 prompted us to prepare new 2-hydroxybenzaldehyde thiosemicarbazones substituted at the terminal nitrogen atom to enhance the lipophilicity and coupled to two related α- and β-amino acid homologs, namely proline and homoproline. We anticipated that these new conjugates will form square-planar metal complexes enabling the elucidation of (i) the role of the square-planar coordination geometry of the metal complexes in the underlying mechanism of antiproliferative activity, (ii) the effect of metal coordination and (iii) substitution at the terminal nitrogen atom of the thiosemicarbazide moiety, and (iv) the effect of the homologisation of proline to β3-homoproline on antiproliferative activity in cancer cell lines. Herein we report on the synthesis of two organic conjugates, namely 5-methyl-2-hydroxybenzaldehyde 4-N-pyrrolidine-3-thiosemicarbazone coupled to L-proline and β3-homoproline, respectively, and four new complexes with nickel(II), palladium(II) and copper(II) (Chart 1), their characterisation by elemental analysis, spectroscopic methods, and X-ray crystallography, and their antiproliferative activity in three human cancer cell lines (A549, CH1 and SW480) and noncancerous murine fibroblasts (NIH/3T3). Clear-cut structure–activity relationships (SARs) were established and discussed.
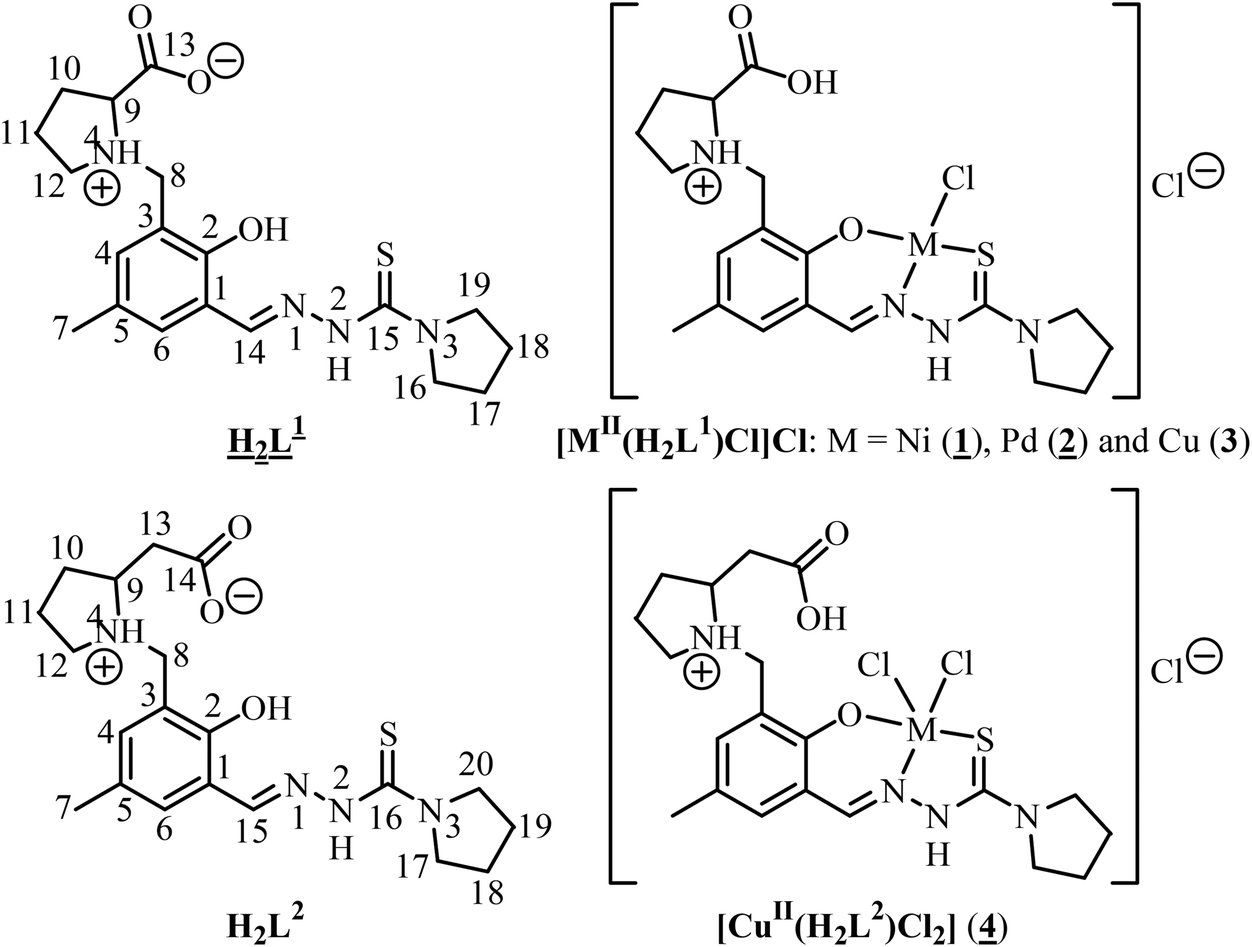 | ||
| Chart 1 Line drawings of L-proline and β3-homoproline thiosemicarbazone conjugates H2L1 and H2L2, as well as their nickel(II), palladium(II) and copper(II) complexes. We use for consistency the same abbreviations H2L1 and H2L2 for the overall charge-neutral ligand precursors and ligands, although the charge distributions in these species are clearly different. The underlined formula and numbers indicate compounds studied by X-ray crystallography. | ||
Experimental section
Chemicals
All reagents were used as purchased from commercial suppliers. L-Proline was from Alfa Aesar. 4-Pyrrolidine-3-thiosemicarbazide20 was obtained in 60% yield by refluxing methyl hydrazinecarbodithioate21 and pyrrolidine in water for 6 h. 3-Chloromethyl-2-hydroxy-5-methylbenzaldehyde was synthesised by following a modified published procedure.22Z-L-Proline23 and β3-homoproline24 were synthesised according to previously reported protocols. All utilised solvents were of HPLC grade and used without further purification.Synthesis of ligand precursors
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of ethyl acetate/hexane as the eluent (Rf = 0.14). The yellow oil was dried in vacuo. Yield: 1.18 g, 80.0%. 1H NMR (500.10 MHz, DMSO-d6): δ 10.24 (s, 1H, HC
:1 mixture of ethyl acetate/hexane as the eluent (Rf = 0.14). The yellow oil was dried in vacuo. Yield: 1.18 g, 80.0%. 1H NMR (500.10 MHz, DMSO-d6): δ 10.24 (s, 1H, HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 7.36 (d, J = 2.0 Hz, 1H, Ar–H), 7.25 (d, J = 2.0 Hz, 1H, Ar–H), 4.16 (d, J = 14.1 Hz, 1H, Ar–CH2–N), 3.60 (s, 3H, OCH3), 3.54 (d, J = 14.2 Hz, 1H, Ar–CH2–N), 2.98–2.91 (m, 1H, Pyrr), 2.91–2.85 (m, 1H, Pyrr), 2.81–2.74 (m, 1H, Pyrr), 2.45 (dd, J = 7.6 and 8.1 Hz, 2H, CH2COOMe), 2.33–2.27 (m, 1H, Pyrr), 2.24 (s, 3H, CH3), 2.08–2.00 (m, 1H, Pyrr), 1.76–1.67 (m, 3H, Pyrr), 1.61–1.53 (m, 1H, Pyrr) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ 191.78, 172.18, 159.25, 136.54, 128.19, 127.61, 125.57, 122.37, 61.01, 54.81, 53.44, 51.80, 38.39, 30.53, 22.38, 20.35 ppm. ESI-MS in MeOH (positive): m/z 291.6 ([M + H]+). ESI-MS in MeOH (negative): m/z 289.3 ([M − H]−).
350), 300sh (14000), 288 (16265), 249sh (10910), 227 (19380). 1H NMR (500.10 MHz, DMSO-d6): δ 11.24 (s, 1H, NH), 8.47 (s, 1H, HCN), 7.22 (s, 1H, Ar), 7.19 (s, 1H, Ar), 4.17 (d, J = 13.5 Hz, 1H, CH2), 3.99 (d, J = 13.5 Hz, 1H, CH2), 3.66 (s, 4H, proline or pyrrolidine), 3.56–3.52 (m, 2H, proline), 3.30–3.23 (m, 2H, proline), 2.85–2.77 (m, 1H, proline), 2.26 (s, 3H, CH3), 2.21–2.12 (m, 1H, proline), 2.01–1.90 (m, 4H, pyrrolidine), 1.90–1.82 (m, 1H, proline), 1.75–1.65 (m, 1H, proline) ppm. ESI-MS in MeOH (positive): m/z 391 ([H2L1 + H]+). Single crystals of X-ray diffraction quality were obtained by slow diffusion of diethyl ether into an ethanolic solution of H2L1.
327), 300 (15771), 288 (20648), 227 (22884). For the atom labeling scheme and assignment of NMR resonances see Chart 1.
O), 7.36 (d, J = 2.0 Hz, 1H, Ar–H), 7.25 (d, J = 2.0 Hz, 1H, Ar–H), 4.16 (d, J = 14.1 Hz, 1H, Ar–CH2–N), 3.60 (s, 3H, OCH3), 3.54 (d, J = 14.2 Hz, 1H, Ar–CH2–N), 2.98–2.91 (m, 1H, Pyrr), 2.91–2.85 (m, 1H, Pyrr), 2.81–2.74 (m, 1H, Pyrr), 2.45 (dd, J = 7.6 and 8.1 Hz, 2H, CH2COOMe), 2.33–2.27 (m, 1H, Pyrr), 2.24 (s, 3H, CH3), 2.08–2.00 (m, 1H, Pyrr), 1.76–1.67 (m, 3H, Pyrr), 1.61–1.53 (m, 1H, Pyrr) ppm. 13C{1H} NMR (125.76 MHz, DMSO-d6): δ 191.78, 172.18, 159.25, 136.54, 128.19, 127.61, 125.57, 122.37, 61.01, 54.81, 53.44, 51.80, 38.39, 30.53, 22.38, 20.35 ppm. ESI-MS in MeOH (positive): m/z 291.6 ([M + H]+). ESI-MS in MeOH (negative): m/z 289.3 ([M − H]−).
350), 300sh (14000), 288 (16265), 249sh (10910), 227 (19380). 1H NMR (500.10 MHz, DMSO-d6): δ 11.24 (s, 1H, NH), 8.47 (s, 1H, HCN), 7.22 (s, 1H, Ar), 7.19 (s, 1H, Ar), 4.17 (d, J = 13.5 Hz, 1H, CH2), 3.99 (d, J = 13.5 Hz, 1H, CH2), 3.66 (s, 4H, proline or pyrrolidine), 3.56–3.52 (m, 2H, proline), 3.30–3.23 (m, 2H, proline), 2.85–2.77 (m, 1H, proline), 2.26 (s, 3H, CH3), 2.21–2.12 (m, 1H, proline), 2.01–1.90 (m, 4H, pyrrolidine), 1.90–1.82 (m, 1H, proline), 1.75–1.65 (m, 1H, proline) ppm. ESI-MS in MeOH (positive): m/z 391 ([H2L1 + H]+). Single crystals of X-ray diffraction quality were obtained by slow diffusion of diethyl ether into an ethanolic solution of H2L1.
327), 300 (15771), 288 (20648), 227 (22884). For the atom labeling scheme and assignment of NMR resonances see Chart 1.
Synthesis of metal complexes
100), 293 (38900), 259 (62300), 236 (69600). ESI-MS in methanol (positive): m/z 447.26 [Ni(HL1)]+. Calcd for C19H25NiN4O3S m/z 447.10. X-ray diffraction quality single crystals were grown by slow diffusion of diethyl ether into the solution of the complex in methanol. Magnetic susceptibility measurements indicate its diamagnetism in the solid state.
340), 320 (31575), 244 (60220). 1H NMR (500.10 MHz, DMSO-d6): δ 9.99 (br, 1H, N2H), 8.31 (s, 1H, C14H), 7.27 (d, J = 2.0 Hz, 1H, C6H), 7.22 (d, J = 2.0 Hz, 1H, C4H), 4.55–4.47 (m, 1H, C9H), 4.42 (dd, J = 13.0, 5.0 Hz, 1H, C8Hb), 4.31 (dd, J = 13.0, 5.0 Hz, 1H, C8Ha), 3.64–3.55 (m, 1H, C12Hb), 3.53–3.47 (m, 2H, C16H), 3.43–3.34 (m, 1H, C12Ha), 2.50–2.41 (m, 1H, C10Hb), 2.21 (s, 3H, C7H3), 2.11–1.98 (m, 2H, C10Ha + C11Hb), 1.94–1.86 (m, 1H, C11Ha) ppm. 13C NMR (126 MHz,) δ 171.20, 170.35, 158.62, 146.79, 137.09, 135.24, 122.92, 120.03, 118.87, 66.01 (C9H), 62.69, 55.95 (C8H), 55.14 (C12H), 51.24 (C16H), 34.63, 28.42 (C10H), 26.03 (C17H), 22.86 (C11H), 20.14 (C7), 14.29 ppm. ESI-MS in methanol (negative): m/z 530.93 [M − HCl − H]−. Calcd for C19H24ClN4O3PdS m/z 531.03. Single crystals were grown from dimethyl sulfoxide at room temperature.
347), 333sh (10050), 311sh (21397), 299 (29236), 267 (18430), 260sh (18410). ESI-MS in methanol (positive): m/z 452 [Cu(HL1)]+.
457), 327 (14377), 297 (35476), 263 (24855).
Crystallographic structure determination
X-ray diffraction measurements of H2L1 and 1 were performed on a Bruker D8 Venture, while those of 2 and 4 were performed on Bruker X8 APEXII CCD and STOE diffractometers, respectively, both equipped with an Oxford Cryosystem cooler device. The single crystals of H2L1, 1, 2 and 4 were positioned at 35, 35, 40 and 40 mm from the detector, and 1401, 1071, 636 and 2136 frames were measured, each for 20, 10, 20 and 38 s over 0.4, 0.5, 1 and 1° scan width, respectively. The data were processed using SAINT software.25 Crystal data, data collection parameters, and structure refinement details are given in Table 1. The structures were solved by direct methods and refined by full-matrix least-squares techniques. All non-hydrogen atoms were refined with anisotropic displacement parameters. H atoms were inserted in calculated positions and refined with a riding model. The pyrrolidine and proline rings in one of the two crystallographically independent molecules of 1 were found to be disordered over two positions with an s.o.f. of 0.65:0.35 and 0.75:0.25, respectively, without using any restraints implemented in SHELXL2014. SADI and EADP restraints were applied to resolve the static disorder observed in the co-crystallised solvent molecules in H2L1. The single crystal of 4 was weakly diffracting and the resolution of the collected X-ray data was estimated to be 1.02 Å. Nevertheless, the structure could be solved and the electron density of the molecule is well-defined, allowing for the determination of the atomic connectivity and refinement with anisotropic temperature factors for all non-hydrogen atoms. The following software programs and computer were used: structure solution, SHELXS-97; refinement, SHELXL-97;26 molecular diagrams, ORTEP-3;27 computer, Intel CoreDuo. CCDC 1471829–1471831 and 1492553.
| Compound | H2L1 | 1 | 2 | 4 |
|---|---|---|---|---|
| a R 1 = ∑‖Fo| − |Fc‖/∑|Fo|. b wR2 = {∑[w(Fo2 − Fc2)2]/∑[w(Fo2)2]}1/2. c GOF = {∑[w(Fo2 − Fc2)2]/(n − p)}1/2, where n is the number of reflections and p is the total number of parameters refined. | ||||
| Empirical formula | C20.25H31N4O4.25S | C19H26Cl2N4NiO3S | C19H26Cl2N4O3PdS | C20H28Cl2CuN4O3S |
| F w | 430.55 | 520.11 | 567.80 | 538.96 |
| Space group | C2 | P21 | P21 | C2/c |
| a [Å] | 10.5018(4) | 6.9292(2) | 6.9628(5) | 20.3880(16) |
| b [Å] | 24.7063(9) | 18.4742(8) | 18.5602(13) | 14.1122(7) |
| c [Å] | 16.9193(7) | 17.4103(7) | 17.4498(12) | 15.9901(12) |
| β [°] | 90.967(2) | 101.171(2) | 101.011(2) | 100.503(6) |
| V [Å3] | 4389.3(3) | 2186.49(14) | 2213.5(3) | 4523.6(5) |
| Z | 8 | 4 | 4 | 8 |
| λ [Å] | 0.71073 | 0.71073 | 0.71073 | 1.54186 |
| ρ calcd [g cm−3] | 1.303 | 1.580 | 1.704 | 1.583 |
| Crystal size [mm] | 0.20 × 0.13 × 0.12 | 0.32 × 0.20 × 0.20 | 0.15 × 0.05 × 0.05 | 0.08 × 0.04 × 0.02 |
| T [K] | 100(2) | 130(2) | 100(2) | 100(2) |
| μ [mm−1] | 0.182 | 1.257 | 1.203 | 4.658 |
|
R
1a |
0.0543 | 0.0342 | 0.0418 | 0.0555 |
| wR2b |
0.1492 | 0.0827 | 0.0899 | 0.1307 |
| Flack parameter | 0.01(2) | 0.001(8) | −0.04(2) | |
| GOFc | 1.029 | 1.024 | 1.023 | 0.942 |
One-dimensional 1H and 13C NMR and two-dimensional 1H–1H COSY and 1H–13C HMBC NMR spectra were recorded on two Bruker Avance III spectrometers at 500.32 or 500.10 (1H) and 125.82 or 125.76 (13C) MHz, respectively, at room temperature, and using standard pulse programs. 1H and 13C shifts are quoted relative to the solvent residual signals. Electrospray ionisation mass spectrometry (ESI-MS) was carried out with a Bruker Esquire 3000 instrument and the samples were dissolved in methanol. Elemental analyses were performed at the Microanalytical Laboratory of the University of Vienna with a Perkin Elmer 2400 CHN Elemental Analyzer (Perkin Elmer, Waltham, MA). IR spectra were recorded on a Bruker Vertex 70 Fourier transform IR spectrometer by using the ATR technique. UV-vis absorption spectra were recorded on a JASCO V770 spectrophotometer, while CD spectra were recorded on a JASCO J1500 spectrometer. The spectra were recorded at room temperature in a cuvette with 10 mm path length.
Antiproliferative activity
CH1 cells (human ovarian carcinoma) were a generous gift from Lloyd R. Kelland, CRC Centre for Cancer Therapeutics, Institute of Cancer Research, Sutton, UK. SW480 (human adenocarcinoma of the colon) and A549 (human nonsmall cell lung cancer) cells were kindly provided by Brigitte Marian (Institute of Cancer Research, Department of Medicine I, Medical University of Vienna, Austria). NIH/3T3 (ATCC CRL-1658) murine embryonic fibroblasts were cultured according to the ATCC (American Type Culture Collection) protocol. All cell culture media and reagents were purchased from Sigma-Aldrich Austria and plastic ware from Starlab Germany. CH1, SW480, and A549 cells were grown in 75 cm2 culture flasks as adherent monolayer cultures in Minimum Essential Medium (MEM) supplemented with 10% heat-inactivated fetal calf serum, 1 mM sodium pyruvate, 4 mM L-glutamine and 1% non-essential amino acids (from 100× ready-to-use stock). NIH/3T3 cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% of bovine calf serum. Cultures were maintained at 37 °C under a humidified atmosphere containing 95% air and 5% CO2.Cytotoxic effects of the test compounds were determined by means of a colorimetric microculture assay [MTT assay; MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide]. Cisplatin (Teva) was used as a positive control. Cells were harvested from culture flasks by trypsinization and seeded by using a pipetting system (Biotek Precision XS Microplate Sample Processor) at densities of 3 × 103 (A549), 1 × 103 (CH1) and 2 × 103 (SW480) in 100 μL per well aliquots in 96-well microculture plates. For 24 h, cells were allowed to settle and resume proliferation. Test compounds were then dissolved in DMSO, diluted in complete culture medium and added to the plates where the final DMSO content did not exceed 0.5%. After 96 h of drug exposure, the medium was replaced with 100 μL per well of a 1:7 MTT/RPMI 1640 mixture (MTT solution, 5 mg mL−1 of MTT reagent in phosphate-buffered saline; RPMI 1640 medium, supplemented with 10% heat-inactivated fetal bovine serum and 4 mM L-glutamine), and plates were incubated at 37 °C for a further 4 h. Subsequently, the solution was removed from all wells, and the formazan crystals formed by viable cells were dissolved in 150 μL of DMSO per well. Optical densities at 550 nm were measured with a microplate reader (Biotek ELx808) by using a reference wavelength of 690 nm to correct for unspecific absorption. The quantity of viable cells was expressed relative to untreated controls, and 50% inhibitory concentrations (IC50) were calculated from concentration–effect curves by interpolation. Evaluation is based on the means from three independent experiments. A slightly modified procedure was used to test the antiproliferative effect in NIH/3T3 murine embryonal fibroblast cells, which is provided in the ESI.†
Mechanisms of cell death: assay for apoptosis induction
The assay was carried out using an Annexin V-FITC Apoptosis Detection Kit (Cat. No. PF 032) from Calbiochem according to the manufacturer's instructions. The concentration of the A549 cell suspension was adjusted to approximately 0.5 × 106 cells per mL in MEM and the cell suspension was distributed in 1 mL aliquots into a 24-well plate, and then incubated overnight at 37 °C and 5% CO2. The next day, the medium was removed and replaced by 1 mL MEM containing the compounds except the control samples. A549 cells were incubated in the presence of the compounds at 20 μM in the 24-well plate at 37 °C for 3 h, and 12H-benzo[α]phenothiazine (M627)28 and cisplatin (Teva) at 20 μM were used as positive controls. After the incubation period the samples were washed with PBS and fresh MEM medium was added to the samples. The cells were incubated overnight at 37 °C and 5% CO2. The next day, 200 μL 0.25% Trypsin (Trypsin-Versen) was added to the samples until cells appeared detached followed by the addition of 400 μL of MEM supplemented with 10% bovine serum. The cells were collected in Eppendorf tubes and centrifuged at 2000g for 2 min. The harvested cells were resuspended in fresh serum free MEM culture medium. After this step, the apoptosis assay was carried out according to the rapid protocol of the kit. The fluorescence was analysed immediately using a ParTec CyFlow flow cytometer (Partec GmbH, Münster, Germany).Results and discussion
Synthesis and characterization of proligands and metal complexes
The chiral 2-hydroxy-3-methyl-(S)-pyrrolidine-2-carboxylate-5-methylbenzaldehyde 4-N-pyrrolidine-3-thiosemicarbazone was prepared in three steps as described recently for the unsubstituted at the terminal nitrogen atom of the thiosemicarbazide moiety derivative.19 First, 3-chloromethyl-2-hydroxy-5-methylbenzaldehyde21 was allowed to react with L-proline methyl ester hydrochloride in the presence of triethylamine with the formation of the desired conjugate. Condensation reaction of this latter compound with 4-N-pyrrolidine-3-thiosemicarbazide16 followed by hydrolysis of the methyl ester group afforded the corresponding TSC coupled via a methylene group to L-Pro moiety (H2L1). The formation of desired species has been confirmed by 1H and 13C NMR measurements, as well as by ESI mass spectrometry. The mass spectrum recorded in the positive ion mode showed peaks at m/z 391 due to [M + H]+ ions. Aqueous solution of H2L1 at neutral pH is optically active. The racemate H2L2 was obtained similarly by using the (R/S)-β3-homoproline methyl ester instead of the L-proline methyl ester hydrochloride. The latter was prepared via a four-step procedure by exploring the Arndt–Eistert reaction as a common method of homologisation of an α-amino acid into β-amino acid (e.g., proline to β3-homoproline) as shown in Scheme S1.†29 Briefly, in the first step the secondary amine in L-proline was protected by reaction with benzyl chloroformate in aqueous KOH solution. Then the N-protected L-proline was converted into the acyl halide by treating with excess oxalylchloride in dichloromethane followed by methylation with (trimethylsilyl)diazomethane and formation of the α-diazotylketone. This latter conversion, known as Wolff rearrangement, is the key step involving the insertion of a CH2-group.29 The α-diazotylketone was further rearranged in dry methanol in the presence of silver benzoate and triethylamine. Finally the amino group was deprotected with H2 in the presence of 10% PdC in dry methanol. The synthesis can be performed on a large scale with 51% overall yield of β3-homoproline methyl ester. The reaction of β3-homoproline methyl ester with 3-chloromethyl-2-hydroxy-5-methylbenzaldehyde in THF/CH2Cl2 afforded [1-(3-formyl-2-hydroxy-5-methylbenzyl)-(R/S)-pyrrolidin-2-yl]acetic acid methyl ester, which was purified by column chromatography to give an orange oil in 80% yield. Condensation of this product with 4-N-pyrrolidine-3-thiosemicarbazide accompanied by hydrolysis of the ester group resulted in H2L2 in 43% yield. The formation of the desired ligand precursor was confirmed by ESI mass spectrometry, which showed the presence of a peak with m/z 405, attributed to [M + H]+ ions, as well as by 1H and 13C NMR spectroscopy.By reaction of H2L1 with NiCl2·6H2O in ethanol at room temperature the red complex [Ni(H2L1)Cl]Cl·1.3H2O was synthesised in 82% yield. Similarly, starting from PdCl2 and CuCl2·2H2O the complexes [Pd(H2L1)Cl]Cl·H2O and [Cu(H2L1)Cl]Cl·0.7H2O were isolated in 47 and 59% yields, respectively. ESI mass spectra of 1 and 3 recorded in the positive ion mode exhibit a strong peak at m/z 447.26 and m/z 452, which can be assigned to [Ni(HL1)]+ and [Pd(HL1)]+, respectively, while the negative ion mass spectrum of 2 contains a strong signal at m/z 530.93 due to [Pd(L1)Cl]−. By reaction of a methanolic solution of CuCl2·2H2O with H2L2 the complex [Cu(H2L2)Cl2]·H2O was obtained in 55% yield. ESI mass spectra, recorded in the positive ion mode, showed the presence of a strong peak at m/z 466.31 attributed to [CuII(HL2)]+, while in the negative ion mode, peaks with m/z 464.10 and 500.05 due to [CuII(L2-H)]− and [CuII(L2)Cl]−, respectively, were observed, in agreement with the X-ray diffraction structure (vide infra).
X-ray crystallography
Fig. 1 shows the structure of the chiral ligand precursor H2L1 with selected bond distances and angles quoted in the legend. The compound crystallizes in the monoclinic space group C2 with two crystallographically independent molecules in the asymmetric unit. The conformation adopted by H2L1 is very close to that of the ligand in complexes 1 and 2 (vide infra). The prolinic moiety is in the zwitterionic form, which makes the atom N4a in addition to C14a chiral. A strong intramolecular hydrogen bonding O1a–H⋯N1a with an O1a⋯N1a of 2.595(4) Å and an O1a–H⋯N1a of 143.4° is evident in the structure of H2L1. The atoms N2a and N4a act as proton donors to O3ai(−x + 1, y, −z + 1) and O2bi(−x + 1, y, −z + 1), respectively, forming hydrogen bonds N2a–H⋯O3ai and N4a–H⋯O2bi with an N2a⋯O3ai of 2.736(5) Å, an N2a–H⋯O3ai of 155.5° and an N4a⋯O2bi of 2.857(5) Å, an N2a–H⋯O3ai of 136.7°.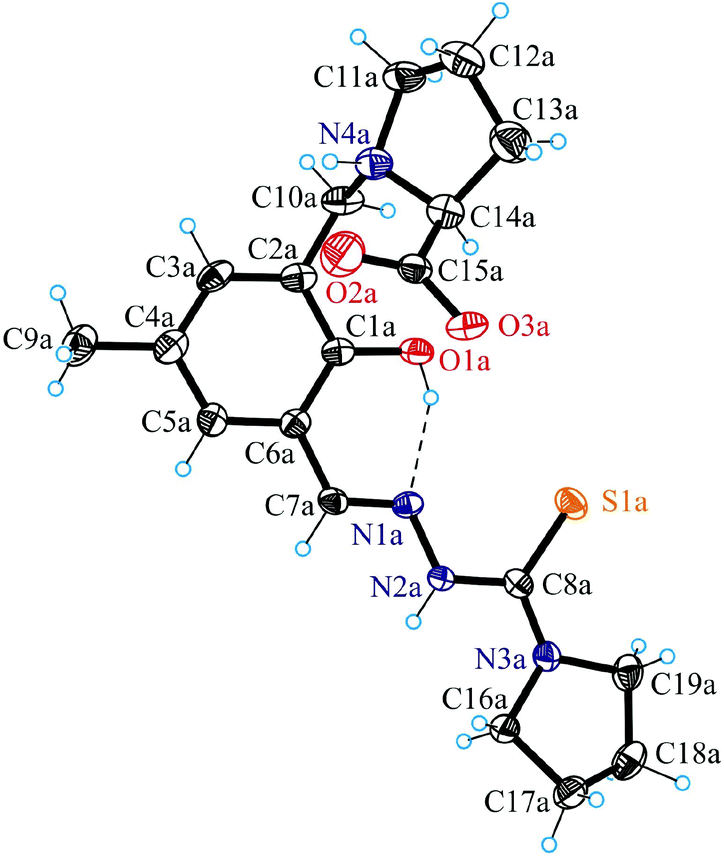 | ||
| Fig. 1 ORTEP view of H2L1 with thermal displacement parameters drawn at the 50% probability level. Selected bond distances (Å) and torsion angles (°): C1a–O1a 1.352(4), N1a–N2a 1.367(3), N2a–C8a 1.368(4), C8a–S1a 1.694(3), C8a–N3a 1.335(4); C1a–C2a–C10a–N4a −91.6(3); C10a–N4a–C14a–C15a −100.8(3). | ||
The results of X-ray diffraction studies of 1, 2 and 4 are shown in Fig. 2 and 3. Compounds 1 and 2 are isostructural and crystallised in the noncentrosymmetric monoclinic space group P21 as pure enantiomers, as also confirmed by the Flack parameters quoted in Table 1, with two chiral atoms of the same configuration (vide infra). Complex 4 crystallised in the centrosymmetric monoclinic space group C2/c. The asymmetric unit in 1 and 2 consists of two crystallographically independent cations [M(H2L1)Cl]+ (M = Ni, Pd) and two chloride counterions, where H2L1 is the charge-neutral proline–thiosemicarbazone hybrid, while that of 4 consists of one complex, [Cu(H2L2)Cl2]. The ligands H2L1 in 1 and 2 and H2L2 in 4 act as a tridentate binding to the metal(II) via the phenolate oxygen atom O1, the nitrogen atom N1 and the thione sulfur S1 as shown in Fig. 2 and 3 for one of the two crystallographically independent cations [Ni(H2L1)Cl]+, [Pd(H2L1)Cl]+ and the complex [Cu(H2L2)Cl2]. The ligands are deprotonated at O1, but protonated at N4, so that the overall protonation level does not change. Protonation at N4 makes this atom chiral (S configuration) in 1 and 2. The second chiral centre C14 has the same S configuration. According to the literature, the nitrogen atom of the L-prolinate ligand or moiety upon coordination to metal ions or protonation in most cases adopts the same configuration as the asymmetric carbon atom,30 although cases where the two atoms adopt opposite configurations are also known.31 The coordination geometry of nickel(II) and palladium(II) is square-planar. The fourth coordination place is occupied by a chlorido ligand.
 | ||
| Fig. 2 ORTEP view of one of the two crystallographically independent complex cations in the asymmetric unit of [Ni(H2L1)Cl]Cl (left) and [Pd(H2L1)Cl]Cl (right) with atom labeling schemes; thermal ellipsoids were drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°) [Ni(H2L1)Cl]Cl: Ni1a–Cl1a 2.184(3), Ni1a–O1a 1.858(7), Ni1a–N1a 1.848(10), Ni1a–S1a 2.147(3), C1a–O1a 1.300(14), N1a–N2a 1.392(12), N2a–C8a 1.340(15), C8a–S1a 1.708(13), C8a–N3a 1.311(15); O1a–Ni1a–N1a 93.6(4), N1a–Ni1a–S1a 88.8(3). Selected bond distances (Å) and bond angles (°) [Pd(H2L1)Cl]Cl: Pd1a–Cl1a 2.3049(12), Pd1a–O1a 2.016(3), Pd1a–N1a 1.976(4), Pd1a–S1a 2.2470(11), C1a–O1a 1.320(5), N1a–N2a 1.382(5), N2a–C8a 1.350(5), C8a–S1a 1.722(5), C8a–N3a 1.318(5); O1a–Pd1a–N1a 92.39(14), N1a–Pd1a–S1a 86.53(11). | ||
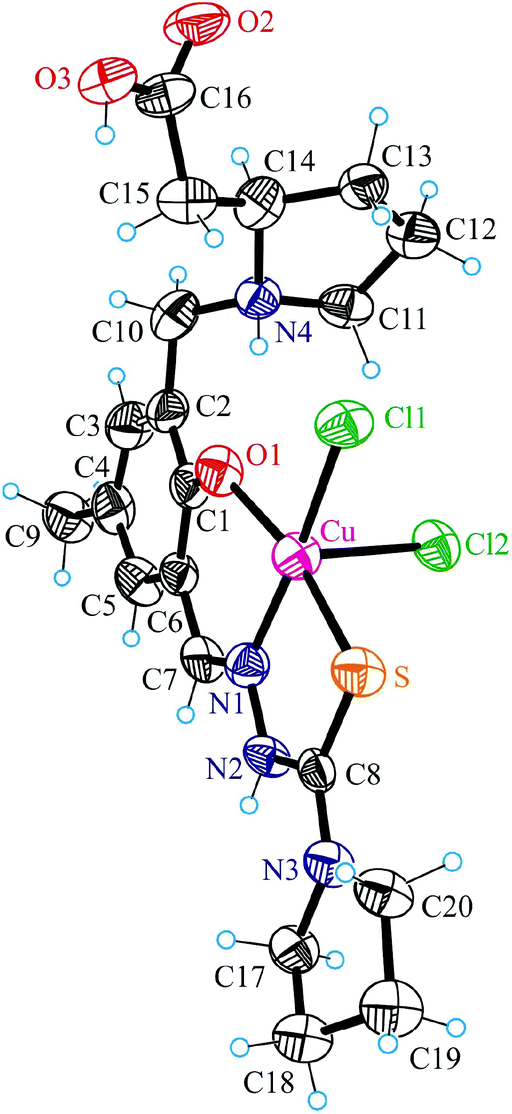 | ||
| Fig. 3 ORTEP view of 4 with atom labeling schemes; thermal ellipsoids were drawn at the 50% probability level. Selected bond distances (Å) and bond angles (°): Cu–Cl1 2.294(2), Cu–Cl2 2.732(3), Cu–N1 2.001(8), Cu–O1 1.916(6), Cu–S 2.262(3), C1–O1 1.339(11), N1–N2 1.373(10), N2–C8 1.376(11), C8–S 1.702(10), C8–N3 1.320(12); O1–Cu–N1 90.3(3), N1–Cu–S 86.1(2). | ||
The overall positive charge of the complex cation is counterbalanced by another chloride ion.
The Pd–O, Pd–N, Pd–Cl and Pd–S bond lengths in 2 are well-comparable to those found in the complex [Pd(HSal4Et)Cl]·H2O, where H2Sal4Et = 2-hydroxybenzaldehyde 4-N-ethylthiosemicarbazone, at 2.019(2), 1.965(2), 2.3078(8) and 2.2456(9) Å, respectively.32 The protonated atom N4a in cation A acts as a proton donor in bifurcated hydrogen bonding interaction to O2a and Cl2i (x − 1, y, z − 1), while N4b in cation B acts as a proton donor to O1B and O2B as shown in Fig. S1.† In addition, strong hydrogen bonding interactions N2a–H⋯Cl2a [N2a⋯Cl2a 3.157(6) Å, N2a–H⋯Cl2a 160.2°], N2b–H⋯Cl2bii (−x + 1, y − 0.5, −z + 1) [N2b⋯Cl2bii 3.162(0) Å, N2b–H⋯Cl2bii 156.5°] and O3b–H⋯Cl2aii (−x + 1, y + 0.5, −z + 1) [O3b⋯Cl2aii 2.926(0) Å, O3b–H⋯Cl2aii 170.4°] are evident in the crystal structure of 2. Closely similar hydrogen bonding interactions for 1 are quoted in Table S1.†
A feature of note is the presence of short intermolecular contacts Ni⋯S and Pd⋯S between neighbouring complex cations of ca. 3.69–3.70 and 3.59–3.60 Å, respectively, as shown in Fig. S2 and S3.† The interplanar separation between the aromatic rings of the interacting species is ca. 3.25 and 3.16 Å, in 1 and 2, respectively. Formation of short contacts of this type is not characteristic of the complex [Pd(HSal4Et)Cl]·H2O mentioned previously.
The coordination polyhedron of copper(II) in 4 is best described as a square-pyramid (τ = 0.15).33 The tridentate ligand H2L2 and one chloride occupy the basal plane, while the second chlorido ligand occupies the apical position. Comparison of the bond lengths Cu–Cl1 and Cu–Cl2 (see the legend to Fig. 3) indicates markedly weaker binding of the apical chlorido ligand to copper(II). The protonated atom N4 acts as a proton donor in intramolecular bifurcated hydrogen bonding interaction to O1 and Cl2 [N4⋯O1 2.624(10) Å, N4–H⋯O1 132.5°; N4⋯Cl1 3.684(8) Å, N4–H⋯Cl1 149.0°]. In addition, strong intermolecular hydrogen bonding interactions N2–H⋯Cl2ii (−x + 1, −y + 1, −z) [N2⋯Cl2ii 3.197(8) Å, N2–H⋯Cl2ii 156.6°] and O3–H⋯Cl2i (x, −y + 1, z + 0.5) [O3⋯Cl2i 2.925(7) Å, O3–H⋯Cl2i 167.5°] are evident in the crystal structure of 4.
NMR spectroscopy
The 1H NMR spectra of 2 and H2L1 have been analysed. The 2D homonuclear COSY 1H–1H and heteronuclear HMBC 1H–13C correlations were applied to assign the NMR signals (see Fig. S4†). The NMR spectra of H2L1 are consistent with the X-ray diffraction structure. We note here the particular splitting of protons of the methylene group which connects the aromatic and proline rings. The two protons C8Ha and C8Hb are magnetically inequivalent due to their different spatial orientation with respect to the bond C8H2–N4C9C12. The simulation of the 1H NMR spectra (Fig. 4) resulted in the geminal 2JC8Ha–C8Hb constant of 13 Hz and chemical shifts δC8Ha = 4.177 and δC8Hb = 4.005 ppm. The proton in the α position (C9HδC9H = 3.554 ppm) of the proline ring is split due to its vicinal proton–proton interaction with the two protons C10H2. The approximate dihedral angle for the conformation adopted in this case (Fig. 4, right) can be calculated by using the Karplus equation:34,35
 | ||
| Fig. 4 Experimental (500 MHz) and simulated 1H NMR spectra of 2 (left) and H2L1 (right). | ||
The conformation of the proline ring in H2L1 is consistent with previously reported data.36,37
As can be seen from Fig. 3, the C8H2 group in 2 has a different splitting pattern and chemical shift in comparison with H2L1. A similar change in chemical shift is observed for C9H. The downfield shift of proton resonances of the proline ring in 2 can be related to double protonation of the proline group (amine nitrogen and carboxylato groups) in the complex in comparison with single protonation in the zwitterionic form of the ligand precursor. The single crystal X-ray analysis shows that the proline nitrogen (N4) is protonated in both H2L1 and in 2. In 2 an additional protonation of the ligand at O3a was also proved by X-ray analysis. The protonation state of N4 is also preserved in solution (DMSO), in accord with the additional splitting of two protons of the C8H2 group. The modification in the splitting pattern of C8H2 signals can be associated with the change in spatial orientation of the C8H2 group after coordination. According to the simulation of the NMR spectra the best fit between theoretical and experimental spectra indicates the same magnitude of vicinal coupling constant between the protons C8Ha and HN4 and C8Hb and HN4 and corresponds to the dihedral angle of 40° (3JC8H–HN4 = 5.0 Hz) (Fig. 3). This value corresponds to an average conformational position upon rotation around the C8H2–N4C9C12 single bond on the time scale of NMR spectroscopy. The protonation also affects the splitting of the α proton (C9H), and all 1H NMR resonances are upfield shifted (δC8Ha = 4.419, δC8Hb = 4.307 and δC9H = 4.509 ppm). The HN4 signal appears in the 1H NMR spectra at 9.97 ppm as a broad signal (13.0 Hz) as a consequence of splitting and chemical exchange in solution. The correlations between HN4 with C8H2 and C9H were confirmed by the presence of cross peaks in COSY 1H–1H spectra (Fig. S5†). The geminal 2JHa–Hb constant is of the same magnitude of 13.0 Hz.
Dissolution of the red solid of 1 in methanol resulted in the formation of a green solution suggesting a change in coordination geometry of nickel(II). The 1H NMR spectrum of 1 showed broad lines, which indicated the presence of paramagnetic species in solution. We determined the effective magnetic moment of 1 in CD3OD (1.58 × 10−2 M) at 298 K by using the NMR method of Evans.38 The value amounts to 3.26 B.M., which is typical for octahedral nickel(II) complexes with d8 electronic configuration.39 The CD spectrum of a methanolic solution of 1 (Fig. 4, green trace) indicates the presence of asymmetric centres in the molecule. The CD signals are correlated to the absorption bands for the ligand precursor and show the absence of CD signals in the visible region. The absence of CD signals in the visible region of the spectrum indicates that the proline moiety with its two asymmetric centres is not bound to the nickel(II) in solution.
The electronic absorption spectrum of 1 in methanol in the visible and NIR regions shows three bands with a λmax of 865 (11560 cm−1), 770 (12987 cm−1) and 620 (16129 cm−1) nm (Fig. 5) which can be attributed to 3A2g → 3T2g(F), 3A2g → 1Eg(D) and 3A2g → 3T1g(F) transitions of Ni(II) in an octahedral environment, respectively.40 Spin-allowed transitions are known to give broad absorption bands, while spin-forbidden transitions are usually sharp.41 For nickel(II) (d8) in an octahedral environment 3A2g is the ground term and the spin allowed transition 3A2g → 3T2g(F) is equal to the value of the crystal field splitting (10 Dq).42 The value (11560 cm−1) is consistent with those reported for octahedral nickel(II) complexes.43 The third characteristic transition 3A2g → 3T1g(P) is expected in the range of 19000–27000 cm−1, which is obscured by strong intraligand absorption bands.
 | ||
| Fig. 5 CD (green trace) and UV-vis absorption spectra (blue trace) of 1 in methanol. | ||
Stability in aqueous solution
Quite recently we reported on complex formation reactions of copper(II) with L-Pro-STSC in 30% (w/w) DMSO/H2O.19 Several determined thermodynamic cumulative stability constants (logβ([CuLH]) = 21.58(3), logβ([CuL]) = 17.54(3) and logβ([CuLH−1]) = 6.97(4)) along with the computed pM (= −log[M]) value of 13.4 (pH 7.4, cCu = 1 μM; cL/cCu = 10) undoubtedly indicate significantly high stability of copper(II) complexes, which are closely related to those studied in this work (3 and 4). In addition, we monitored the behaviour of 4 in aqueous solution with 1% DMSO by UV-vis spectroscopy. The data (Fig. S6 and S7†) indicate that the complex remains intact over 24 h in solution.
Cytotoxicity and the mechanism of cell death
The antiproliferative activity of the two organic hybrids H2L1 and H2L2 and the four metal complexes 1–4 was investigated in the human cancer cell lines A549 (nonsmall cell lung carcinoma), CH1 (ovarian carcinoma) and SW480 (colon carcinoma) by means of the colorimetric MTT assay. Generally, CH1 cells are more sensitive to the compounds investigated in this work, giving up to 5 times lower IC50 values than SW480 cells, while A549 cells are less sensitive, giving up to 9 times higher IC50 values than SW480 cells. The ligand precursors H2L1 and H2L2 and complexes 1–4 show different cytotoxic potencies, with IC50 values ranging from 0.9 to 308 μM (Table 2). The following structure–activity relationships are of note: (i) impact of metal coordination as compared to the ligand precursors; (ii) negligible role of square-planar coordination geometry of metal complexes in the underlying mechanism of their cytotoxicity; (iii) the impact of substitution at the terminal nitrogen atom of the thiosemicarbazide moiety and (iv) the effect of increasing the structural flexibility of the amino acid moiety by the insertion of a methylene group between the pyrrolidine ring and the carboxylic group (proline vs. homoproline). The impact of the central metal ion identity on the activity of ligands H2L1 and H2L2 is pronounced, and notably it is divergent in the case of H2L1. Coordination of H2L1 to nickel(II) and palladium(II) weakens markedly the antiproliferative activity in all three cell lines, in line with other reported data.44 The IC50 values of the nickel(II) complex 1 indicate roughly 10- (A549), 7.5- (CH1) and 2.7-fold (SW480) reduction and those of palladium(II) complex 2 a 13- (A549), 23- (CH1) and 26-fold (SW480) drop of cytotoxicity when compared to that of H2L1. In contrast, binding of H2L1 to copper(II) results in a 9.5- (A549), 3- (CH1) and 10-fold (SW480) increase of cytotoxicity based on IC50 values. Similarly, coordination of H2L2 to copper(II) gives a 3.8- (A549), 3.4- (CH1) and 8.6-fold (SW480) increase of cytotoxicity. Since complexes 1–3 are square-planar based on the X-ray diffraction data of 1 and 2 and the X-ray crystallography data of the previously studied copper(II) complex [Cu(D-Pro-STSC)Cl]Cl,19 which is closely related to 3, we can conclude that the role of coordination geometry in the underlying mechanism of cytotoxicity seems to be marginal, if any, when we compare the IC50 values of 1–3 with those of H2L1. This conclusion remains valid if we exclude 1 from the series due to the change in coordination geometry in solution to octahedral. Substitution at the terminal nitrogen atom of the thiosemicarbazide moiety, namely pyrrolidine vs. NH2, has a marked effect on cytotoxicity as well, which can be compared with that of metal coordination. A 23- (CH1) and more than 10-fold (SW480) decrease of IC50 values is achieved via this substitution for metal-free compounds, and a 5- to 5.5-fold decrease is achieved for the same structural modification in copper(II) complexes.19 Insertion of a CH2 group between the pyrrolidine ring and the COOH group of the amino acid moiety reduces markedly the antiproliferative activity of both the ligand precursor and its copper(II) complex. Based on IC50 values a maximal 8-fold drop in cytotoxicity was observed on going from 3 to 4. The antiproliferative activity of complex 3 is slightly lower, but comparable to the clinical drug cisplatin. Compounds found to be active (H2L1, H2L2, complexes 3 and 4) in human cancer cells were tested for their antiproliferative activity in the noncancerous murine embryonal fibroblast (NIH/3T3) cell line as well. They showed no selectivity as their IC50 values for the normal cells are comparable to those obtained for the cancer cells.| IC50 value ± SD | ||||
|---|---|---|---|---|
| A549 | CH1 | SW480 | NIH/3T3a | |
| a Applied protocol described in the ESI. b Taken from ref. 44. c Taken from ref. 45. d Not determined. | ||||
| H2L1 | 22.9 ± 3.0 | 2.7 ± 0.2 | 9.8 ± 1.0 | 21.0 ± 1.0 |
| H2L2 | 72 ± 5 | 6.7 ± 0.1 | 32 ± 2 | 34.7 ± 0.2 |
| 1 | 226.2 ± 17.1 | 20.3 ± 4.5 | 26.0 ± 4.3 | n.d. |
| 2 | 307.9 ± 6.5 | 62.0 ± 3.3 | 252.1 ± 19.6 | n.d. |
| 3 | 2.4 ± 0.3 | 0.90 ± 0.08 | 0.99 ± 0.09 | 3.4 ± 0.3 |
| 4 | 19 ± 3 | 2.0 ± 0.1 | 3.7 ± 0.1 | 13.8 ± 0.8 |
| CuCl2·2H2Ob | n.d.d | 43 ± 3 | >160 | n.d. |
| Cisplatin | 1.3 ± 0.4c | 0.16 ± 0.03c | 3.5 ± 0.3c | 2.3 ± 0.4 |
The mechanism of cytotoxicity induced by H2L1, H2L2 and complexes 3 and 4 was assessed by analysis of A549 cells stained with Annexin-V and propidium iodide (PI) using flow cytometry. Compounds M627 and cisplatin were used as positive controls. Apoptosis is a fundamental mode of cell death which performs a regulatory function during normal development, in tissue homeostasis, and in some disease processes. In normal viable cells phosphatidylserine (PS) is located on the cytoplasmic surface of the cell membrane. Upon induction of apoptosis, rapid alterations in the organisation of phospholipids in most cell types occur, leading to exposure of PS on the cell surface. In this assay a fluorescein isothiocyanate (FITC) conjugate of Annexin-V (a Ca-dependent phospholipid-binding protein with a high affinity for PS) was used, allowing detection of apoptosis. Since membrane permeabilisation is observed in necrosis, necrotic cells will also bind Annexin-V-FITC. PI is used to distinguish between viable, early apoptotic and necrotic or late apoptotic cells. Necrotic cells bind Annexin-V-FITC and stain with PI, while PI is excluded from viable (FITC negative) and early apoptotic (FITC positive) cells. In the absence of phagocytosis the final stages of apoptosis involve necrotic-like disintegration of the total cell; thus cells in late apoptosis will be labeled with both FITC and PI. The fluorescence of PI (FL3) is plotted versus Annexin-V fluorescence (FL1) as shown in Fig. 6 for the positive controls and complex 4. The early apoptotic cells in percentage were compared for the tested compounds (Fig. S8†). Elevated early apoptosis was observed for one of the studied ligand precursors (H2L2) and complexes 3 and 4 as well as for cisplatin and M627 compared with the DMSO control. According to these data the studied compounds can be considered to be moderate apoptosis inducers. The percentage of late apoptosis and necrosis gated events was found to be significantly higher for the ligand precursors (H2L1: 51.9%, H2L2: 68.7% vs. complex 3: 24.7% and 4: 30.1%) at the applied concentration (20 μM).
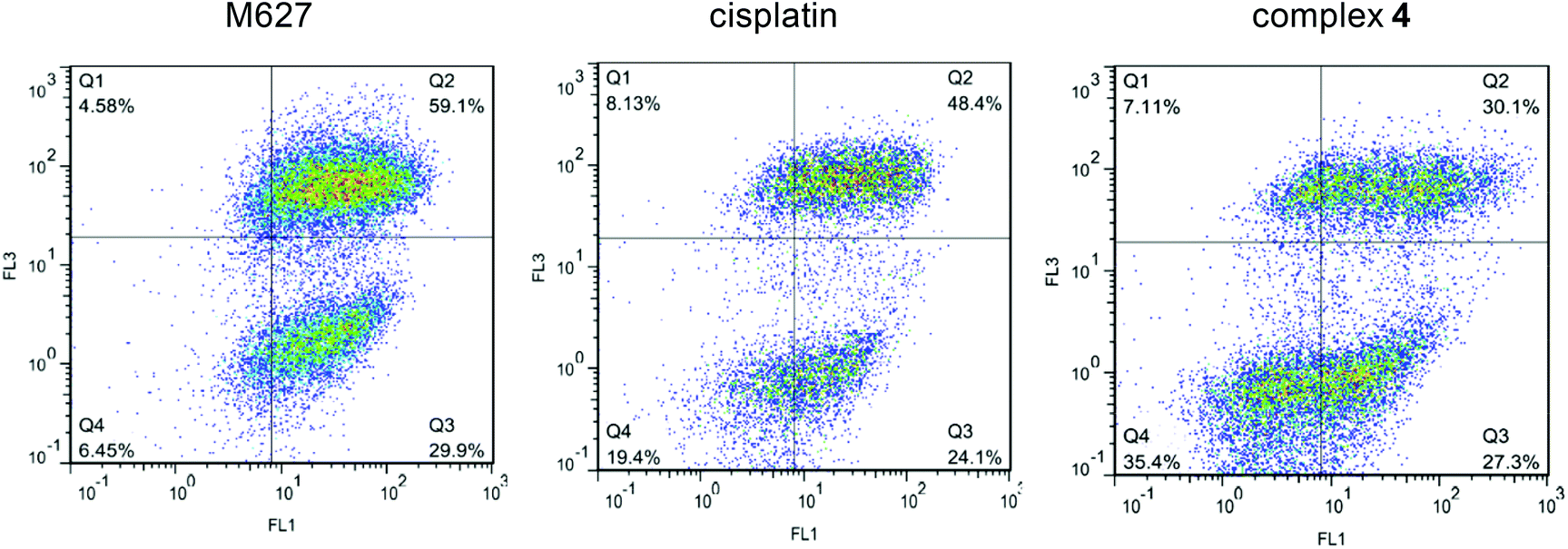 | ||
| Fig. 6 Quantification of apoptosis in cells treated with 4 and M627 and cisplatin (as positive controls) using the Annexin-V/PI double staining assay. A549 cells were treated with 20 μM of compounds. The dual parametric dot plots combining Annexin-V (FL1) and PI (FL3) fluorescence show the viable cell population in the lower left quadrant Annexin-V−/PI− (Q4), the early apoptotic cells in the lower right quadrant Annexin-V+/PI− (Q3), and the late apoptotic cells in the upper right quadrant Annexin-V+/PI+ (Q2). (Number of cells counted: 19914 (M627), 9823 (cisplatin), and 17166 (4)). | ||
Conclusions
Two homologous L-proline- and β3-homoproline-thiosemicarbazone conjugates H2L1 and H2L2 were synthesised via multistep procedures. Substitution of the terminal amino group by pyrrolidine should presumably increase the lipophilicity, while insertion of one CH2 group between the pyrrolidine moiety and the COOH group of the amino acid enhanced the structural flexibility of the potential ligand. Both ligands were found to form square-planar and square-pyramidal complexes [Ni(H2L1)Cl]Cl·1.3H2O (1·1.3H2O), [Pd(H2L1)Cl]Cl·H2O (2·H2O), [Cu(H2L1)Cl]Cl·0.7H2O (3·0.7H2O) and [Cu(H2L2)Cl2]·H2O (4·H2O), by reactions of the ligand precursors with the corresponding metal salts in ethanol or methanol. The two homologous ligand precursors and four metal complexes proved to be suitable for performing antiproliferative activity assays and the establishment of notable and clear-cut structure–cytotoxicity relationships for three human cancer cell lines (A549, CH1 and SW480). The metal ions exert marked effects in a divergent manner: copper(II) modulates the cytotoxic potency of H2L1 and H2L2 in a beneficial way, whereas coordination to nickel(II) and palladium(II) impairs the antiproliferative activity of H2L1. The cytotoxicity of H2L1 and metal complexes 1–3 decreases in all three cell lines in the following rank order: 3 > H2L1 > 1 > 2. Complex 3 exhibits the highest biological activity in all three cell lines with IC50 values of 2.4, 0.9 and 1.0 μM in A549, CH1 and SW480 cells, respectively. On the other hand, compounds H2L1 and H2L2 and complexes 3 and 4 do not display selectivity to cancerous cell lines over normal cells. The flow cytometry analysis of A549 cells doubly stained with Annexin-V/PI showed induced early apoptosis in the case of H2L2 and complexes 3 and 4. The role of the square-planar geometry in 1–3 (or more strictly in 2 and 3) in the underlying mechanism of cytotoxicity seems to be negligible, while structural modifications at the terminal amino group of thiosemicarbazide and amino acid moieties have a significant impact on the antiproliferative activity of both ligand precursors and copper(II) complexes. Substitution of the terminal NH2 group of the thiosemicarbazide moiety by a pyrrolidinyl one resulted in a 23- (in CH1 cells) and >10-fold enhancement of cytotoxicity of metal-free compounds, and a 5- to 5.5-fold increase for the corresponding copper(II) complexes. In contrast, homologisation via insertion of a CH2 group between the pyrrolidine ring and the carboxylate moiety of the amino acid reduced markedly the cytotoxicity of the ligand and its copper(II) complex. The SARs found will be explored further for the development of effective anticancer agents in the reported series of thiosemicarbazone–proline hybrids.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
We are indebted to the Austrian Science Fund (FWF) for the financial support of the project P28223-N34. We also thank Maria S. Novak and Dr Michael Jakupec for performing the antiproliferative activity assays in human cancer cell lines, Alexander Roller from the X-ray diffraction centre of the Faculty of Chemistry of the University of Vienna and Dr Jozef Kožíšek from Slovak Technical University for the collection of X-ray data, and Prof. Markus Galanski for recording the NMR spectra.References
- L. Thelander and A. Gräslund, J. Biol. Chem., 1983, 258, 4063–4066 CAS.
- Y. Yu, J. Wong, D. B. Lovejoy, D. S. Kalinowski and D. R. Richardson, Clin. Cancer Res., 2006, 12, 6876–6883 CrossRef CAS PubMed.
- R. A. Finch, M.-C. Liu, A. H. Cory, J. G. Cory and A. C. Sartorelli, Adv. Enzyme Regul., 2000, 39, 3–12 CrossRef.
- A. M. Traynor, J. W. Lee, G. K. Bayer, J. M. Tate, S. P. Thomas, M. Mazurczak, D. L. Graham, J. M. Kolesar and J. H. A. Schiller, Invest. New Drugs, 2010, 28, 91–97 CrossRef CAS PubMed.
- S. Wadler, D. Makower, C. Clairmont, K. Lambert, K. Fehn and M. Sznol, J. Clin. Oncol., 2004, 22, 1553–1563 CrossRef CAS PubMed.
- M. J. Mackenzie, D. Saltman, H. Hirte, J. Low, C. Johnson, G. Pond and M. J. Moore, Invest. New Drugs, 2007, 25, 553–558 CrossRef CAS PubMed.
- J. Kolesar, R. C. Brundage, M. Pomplun, D. Alberti, K. Holen, A. Traynor, P. Ivy and G. Wilding, Cancer Chemother. Pharmacol., 2011, 67, 393–400 CrossRef CAS PubMed.
- (a) J. E. Karp, F. J. Giles, I. Gojo, L. Morris, J. Greer, B. Johnson, M. Thein, M. Sznol and J. Low, Leuk. Res., 2008, 32, 71–77 CrossRef CAS PubMed; (b) J. F. Zeidner, J. E. Karp, A. L. Blackford, B. D. Smith, I. Gojo, S. D. Gore, M. J. Levis, H. E. Garraway, J. M. Greer, S. P. Ivy, K. W. Pratz and M. A. McDevitt, Haematologica, 2014, 99, 672–678 CrossRef CAS PubMed.
- (a) https://clinicaltrials.gov/ct2/show/NCT02688101?term=NCT02688101&rank=1 ; (b) P. J. Jansson, D. S. Kalinowski, D. J. Lane, Z. Kovacevic, N. A. Seebacher, L. Fouani, S. Sahni, A. M. Merlot and D. R. Richardson, Pharmacol. Res., 2015, 100, 255–260 CrossRef CAS PubMed; (c) A. E. Stacy, D. Palanimuthu, P. V. Berhhardt, D. S. Kalinowski, P. J. Jansson and D. R. Richardson, J. Med. Chem., 2016, 59, 4965–4984 CrossRef CAS PubMed.
- J. Garcia-Tojal, R. Gil-Garcia, P. Gomez-Saiz and M. Ugalde, Curr. Inorg. Chem., 2011, 1, 189–210 CrossRef CAS.
- Y. Yu, D. S. Kalinowski, Z. Kovacevic, A. R. Siafakas, P. J. Jansson, C. Stefani, D. B. Lovejoy, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2009, 52, 5271–5294 CrossRef CAS PubMed.
- D. S. Kalinowski and D. R. Richardson, Pharmacol. Rev., 2005, 57, 547–583 CrossRef CAS PubMed.
- F. Tisato, C. Marzano, M. Porchia, M. Pellei and C. Santini, Med. Res. Rev., 2010, 30, 708–749 CAS.
- J. Easmon, G. Puerstinger, G. Heinisch, T. Roth, H. H. Fiebig, W. Holzer, W. Jaeger, M. Jenny and J. Hofmann, J. Med. Chem., 2001, 44, 2164–2171 CrossRef CAS PubMed.
- B. M. Zeglis, V. Divilov and J. S. Lewis, J. Med. Chem., 2011, 54, 2391–2398 CrossRef CAS PubMed.
- M.-C. Liu, T.-S. Lin and A. C. Sartorelli, J. Med. Chem., 1992, 35, 3612–3611 Search PubMed.
- J. Li, C.-S. Niu, X. Li, T. W. Doyle and S.-H. Chen, Patent U.S, US5767134A19980616, 1998 Search PubMed.
- (a) G. Sava, G. Jaouen, E. A. Hillard and A. Bergamo, Dalton Trans., 2012, 41, 8226–8234 RSC; (b) W. N. Hait, Cancer Res., 2009, 69, 1263–1267 CrossRef CAS PubMed; (c) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- M. N. M. Milunovic, E. A. Enyedy, N. V. Nagy, T. Kiss, R. Trondl, M. A. Jakupec, B. K. Keppler, R. Krachler, G. Novitchi and V. B. Arion, Inorg. Chem., 2012, 51, 9309–9321 CrossRef CAS PubMed.
- S. Maylonas and A. Mamalis, J. Heterocycl. Chem., 2005, 42, 1273–1281 CrossRef.
- D. L. Klayman, J. F. Bartosevich, T. S. Griffin, C. J. Mason and J. P. Scovill, J. Med. Chem., 1979, 22, 855–862 CrossRef CAS PubMed.
- (a) Q. Wang, C. Wilson, J. A. Blake, R. S. Collinson, A. P. Tasker and M. Schröder, Tetrahedron Lett., 2006, 47, 8983–8987 CrossRef CAS; (b) M. Huisman, I. A. Koval, P. Gamez and J. Reedijk, Inorg. Chim. Acta, 2006, 359, 1786–1794 CrossRef CAS.
- E. J. Corey, S. Shibata and R. K. Bakshi, J. Org. Chem., 1988, 53, 2861–2863 CrossRef CAS.
- J.-M. Cassal, A. Fürst and W. Meier, Helv. Chim. Acta, 1976, 59, 1917–1924 CrossRef CAS.
- SAINT-Plus, version 7.06a and APEX2, Bruker-Nonius AXS Inc., Madison, WI, 2004 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 46, 112–122 CrossRef PubMed.
- G. K. Johnson, Report ORNL-5138, OAK Ridge National Laboratory, Oak Ridge, TN, 1976 Search PubMed.
- I. Mucsi, A. Varga, M. Kawase, N. Motohashi and J. Molnar, Anticancer Res., 2002, 22, 2833–2836 CAS.
- F. Arndt and B. Eistert, Ber. Dtsch. Chem. Ges., 1935, 68, 200–208 CrossRef.
- (a) K. Sunkel, W. Hoffmuller and W. Beck, Z. Naturforsch., 1998, 53b, 1365–1368 Search PubMed; (b) T. Poth, H. Paulus, H. Elias, C. Dücker-Benfer and R. van Eldik, Eur. J. Inorg. Chem., 2001, 1361–1369 CrossRef CAS; (c) R. I. Yousef, M. Bette, G. N. Kaluderović, R. Paschke, C. Yiran, D. Steinborn and H. Schmidt, Polyhedron, 2011, 30, 1990–1996 CrossRef CAS; (d) D. Carmona, F. J. Lahoz, R. Atencio, L. A. Oro, M. P. Lamata, F. Viguri, E. S. José, C. Vega, J. Reyes, F. Joó and Á. Kathó, Chem. – Eur. J., 1999, 5, 1544–1564 CrossRef CAS.
- (a) D. Carmona, M. P. Lamata, F. Viguri, I. Dobrinovich, F. L. Lahoz and L. A. Oro, Adv. Synth. Catal., 2002, 344, 499–502 CrossRef CAS; (b) F. Bacher, É. A. Enyedy, N. V. Nagy, A. Rockenbauer, G. M. Bognár, R. Trondl, M. S. Novak, E. Klapproth, T. Kiss and V. B. Arion, Inorg. Chem., 2013, 52, 8895–8908 CrossRef CAS PubMed.
- D. Kovala-Demertzi, P. N. Yadav, M. A. Demertzis, J. P. Jasiski, F. J. Andreadaki and I. D. Kostas, Tetrahedron Lett., 2004, 45, 2923–2926 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. Karplus, J. Am. Chem. Soc., 1963, 85, 2870–2871 CrossRef CAS.
- M. J. Minch, Concepts Magn. Reson., 1994, 6, 41–56 CrossRef CAS.
- M. Cai, Y. Huang, J. Liu and R. Krishnamoorthi, J. Biomol. NMR, 1995, 6, 123–128 CrossRef CAS PubMed.
- R. J. Abraham, B. D. Hudson and W. A. Thomas, Magn. Reson. Chem., 1986, 24, 812–815 CrossRef CAS.
- (a) D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC; (b) S. K. Sur, J. Magn. Reson., 1989, 82, 169–173 CAS.
- (a) O. Kahn, Molecular Magnetism, VCH Publishers, Inc., New York, Weinheim, Cambridge, 1993, 393 p Search PubMed; (b) R. Carlin, Magnetochemistry, Springer Verlag, 1986, 320 p Search PubMed.
- E. González, A. Rodrigue-Witchel and C. Reber, Coord. Chem. Rev., 2007, 251, 351–363 CrossRef.
- (a) M. A. Robinson, J. D. Curry and D. H. Busch, Inorg. Chem., 1963, 2, 1178–1181 CrossRef CAS; (b) A. Dobrov, V. B. Arion, S. Shova, A. Roller, E. Rentschler and B. K. Keppler, Eur. J. Inorg. Chem., 2008, 4140–4145 CrossRef CAS.
- Y. Tanabe and S. Sugano, J. Phys. Soc. Jpn., 1954, 9, 753–766 CrossRef CAS.
- A. B. P. Lever, Inorganic Electronic Spectroscopy, Elsevier Pub. Co., 1968, 420 p Search PubMed.
- (a) P. Chellan, N. Shunmoogam-Gounden, D. T. Hendricks, J. Gut, P. J. Rosenthal, C. Lategan, P. J. Smith, K. Chibale and G. S. Smith, Eur. J. Inorg. Chem., 2010, 3520–3528 CrossRef CAS; (b) M. F. Primik, G. Mühlgassner, M. A. Jakupec, O. Zava, P. J. Dyson, V. B. Arion and B. K. Keppler, Inorg. Chem., 2010, 49, 302–311 CrossRef CAS PubMed.
- V. Pichler, S. M. Valiahdi, M. A. Jakupec, V. B. Arion, M. Galanski and B. K. Keppler, Dalton Trans., 2011, 40, 8187–8192 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis pathway of H2L2 (Scheme S1), ORTEP view of another crystallographically independent cation of 2 showing intermolecular hydrogen bonding interactions (Fig. S1), fragments of the crystal structure of 1 and 2 revealing intermolecular Pd⋯S contacts between two crystallographically independent cations (Fig. S2 and S3), COSY 1H–1H NMR spectra of 2 (Fig. S4 and S5), UV-vis spectra of H2L2 and 4 (Fig. S6 and S7), hydrogen bonding interactions in 1 (Table S1) and selected matrix elements for simulation of 1H NMR spectra of H2L1 and 2 (Table S2); a description of the assay for antiproliferative effects used to test the compounds in NIH/3T3 fibroblasts cells; gated events (%) in the apoptosis assay obtained for the tested compounds (Fig. S8). CCDC 1471829–1471831 and 1492553. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02784a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |