
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDinuclear clathrochelate complexes with pendent cyano groups as metalloligands†
Mathieu
Marmier
a,
Giacomo
Cecot
a,
Anna V.
Vologzhanina
b,
José L.
Bila
a,
Ivica
Zivkovic
c,
Henrik M.
Ronnow
c,
Balint
Nafradi
c,
Euro
Solari
a,
Philip
Pattison
cd,
Rosario
Scopelliti
a and
Kay
Severin
 *a
*a
aInstitut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch; Fax: (+41) 21-693-9305
bNesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 119991 Moscow, Russia
cInstitute of Physics, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland
dSwiss-Norwegian Beam Lines at ESRF, 6 rue Jules Horowitz, 38043 Grenoble Cedex, France
First published on 12th September 2016
Abstract
Dinuclear clathrochelate complexes with two, three, four, or five cyano groups in the ligand periphery were prepared following two distinct synthetic strategies: (a) zinc(II)- or cobalt(II)-templated polycondensation reactions of CN-functionalized arylboronic acids and phenoldioximes, or (b) postsynthetic cross-coupling reactions of polybrominated zinc(II) clathrochelates with 4-cyanophenylboronic acid. The new clathrochelate complexes were used as metalloligands for the construction of heterometallic Zn2+/Ag+ and Co2+/Ag+ coordination polymers (CPs), which were characterized by single crystal X-ray diffraction and FT-IR. A one-dimensional CP was observed for ditopic clathrochelates, whereas two- and three-dimensional CPs were generated from tetra- and pentatopic metalloligands. The three-dimensional network is unique as it displays an unprecedented network topology with the point symbol (8·102)(82·104)(82·10)(83·103). Furthermore, it is a self-catenated net with an extremely high topological density.
Introduction
Silver(I) ions tend to form labile complexes with a flexible coordination number and geometry.1 These features are attractive for the preparation of coordination polymers (CPs)2 because the reversible and malleable coordination chemistry facilitates crystallization, and thus characterization. A sizable fraction of the silver(I) CPs reported to date is based on polycyano ligands. Various ligands have been used in this context, including compounds with two,3 three,4 four,5 and more6 cyano groups. In addition to purely organic ligands, the utilization of metalloligands with cyano groups in the ligand periphery has been explored. Some selected examples are shown in Fig. 1.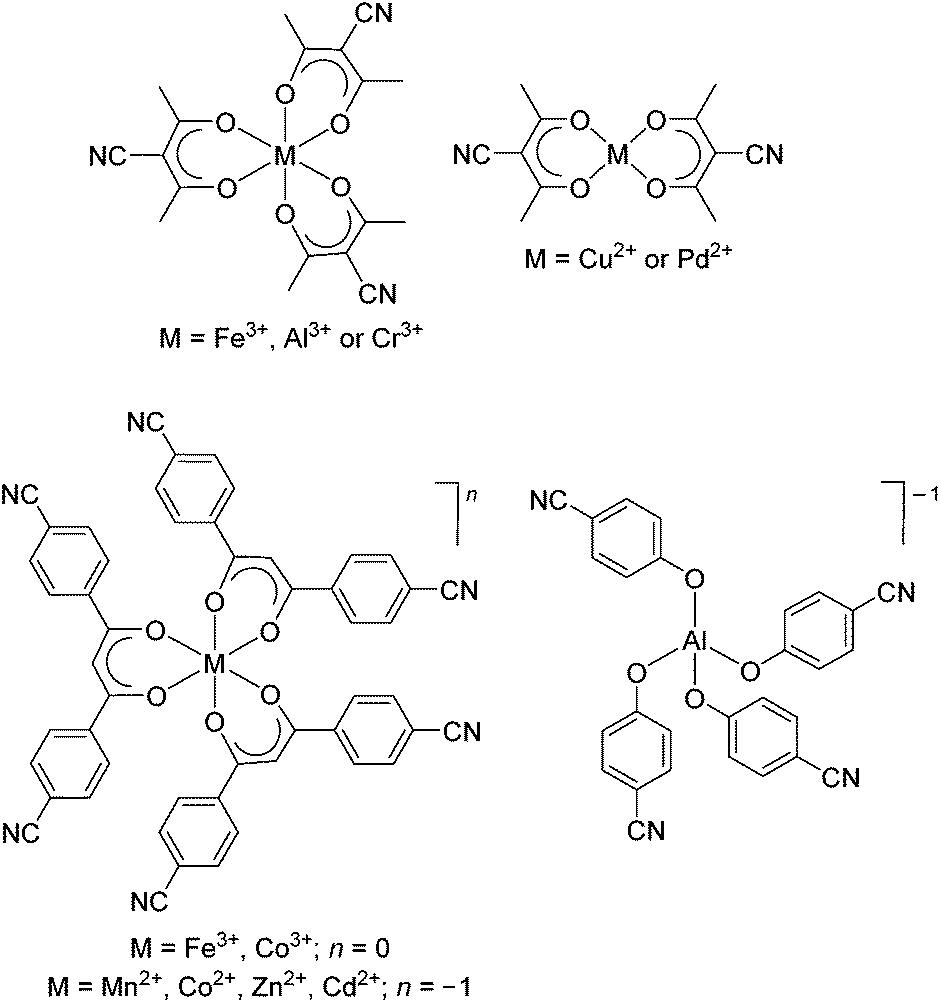 | ||
| Fig. 1 Examples of neutral and anionic metalloligands with pendent cyanogroups. The complexes were used to prepare heterometallic coordination polymers with silver(I) salts.7,8 | ||
The groups of Englert, Hosseini, Burrows and Mahon used homo- and heteroleptic metalloligands containing 3-cyanoacetylacetonato ligands for the construction of silver(I) CPs.7 Tritopic metalloligands were obtained with iron(III),7h aluminum(III),7c and chromium(III),7g whereas ditopic metalloligands were formed with copper(II)7h and palladium(II).7b Carlucci and coworkers have used β-diketonate ligands with two benzonitrile groups to make hexadentate metalloligands. The complexes display an overall charge of zero or minus one, depending on the central metal ion. Very similar three-dimensional CPs were obtained with these metalloligands, despite the difference in overall charge. Notably, it was possible to perform anion exchange in single crystal to single crystal processes. Schulz and coworkers have prepared silver(I) CPs with a diamond-like topology using the tetrahedral [Al(OC6H4CN)4]−1 anion.8 Due to network interpenetration, large channels or pores were not observed.
We have recently started to explore the potential of dinuclear clathrochelate complexes as metalloligands.9,10 These complexes can be synthesized in metal-templated one-pot reactions from easily accessible starting materials. Their characteristics make them interesting building blocks for structural supramolecular chemistry: they are rigid, relatively large, luminescent (for M = Zn), and anionic. Furthermore, the metalloligands display an unusual trigonal bipyramidal geometry. So far, efforts have focused on clathrochelates with pendent pyridyl9 or carboxylic acid groups.10 Below, we show that clathrochelates can also be decorated with cyano groups. Following two distinct synthetic strategies, we were able to prepare clathrochelates containing two, three, four, or five cyano groups which are oriented in a divergent fashion. The utility of these new metalloligands is demonstrated by the synthesis of one-, two-, and three-dimensional coordination polymers, in which the clathrochelate ligands are linked by Ag+ ions.
Results and discussion
Direct synthesis of CN-functionalized clathrochelates
Dinuclear clathrochelate complexes with boronate ester capping groups are generally prepared by polycondensation of a boronic acid with a phenoldioxime in the presence of a divalent metal ion (Mn2+, Zn2+, or Co2+).9–12 In order to introduce cyano groups in apical position, we have performed a similar condensation reaction using commercially available 4-cyanophenylboronic acid in combination with 2,6-diformyl-4-tert-butylphenol dioxime (L1). As metal salts, we have employed either [Co(H2O)6](NO3)2 or Zn(OTf)2. The reactions proceeded in a clean fashion, and the desired clathrochelates 1 and 2 could be isolated in high yield following addition of tetraethylammonium hydroxide (Scheme 1). Both complexes were analyzed by Fourier-transform infrared spectroscopy (FT-IR) and high-resolution mass spectrometry. The diamagnetic zinc complex 1 was also analyzed by NMR spectroscopy (DMSO-d6). Only one set of signals for the protons of the phenolatodioximato ligand were observed, corroborating the expected pseudo-C3 symmetry of the complex. The FT-IR spectra showed a characteristic band at 2220 cm−1, which can be assigned to the absorption frequency of the cyano groups. In line with earlier observations,9,10 complex 1 was found to be luminescent with an emission maximum at 445 nm (DMF, λex = 335 nm, Fig. S15†).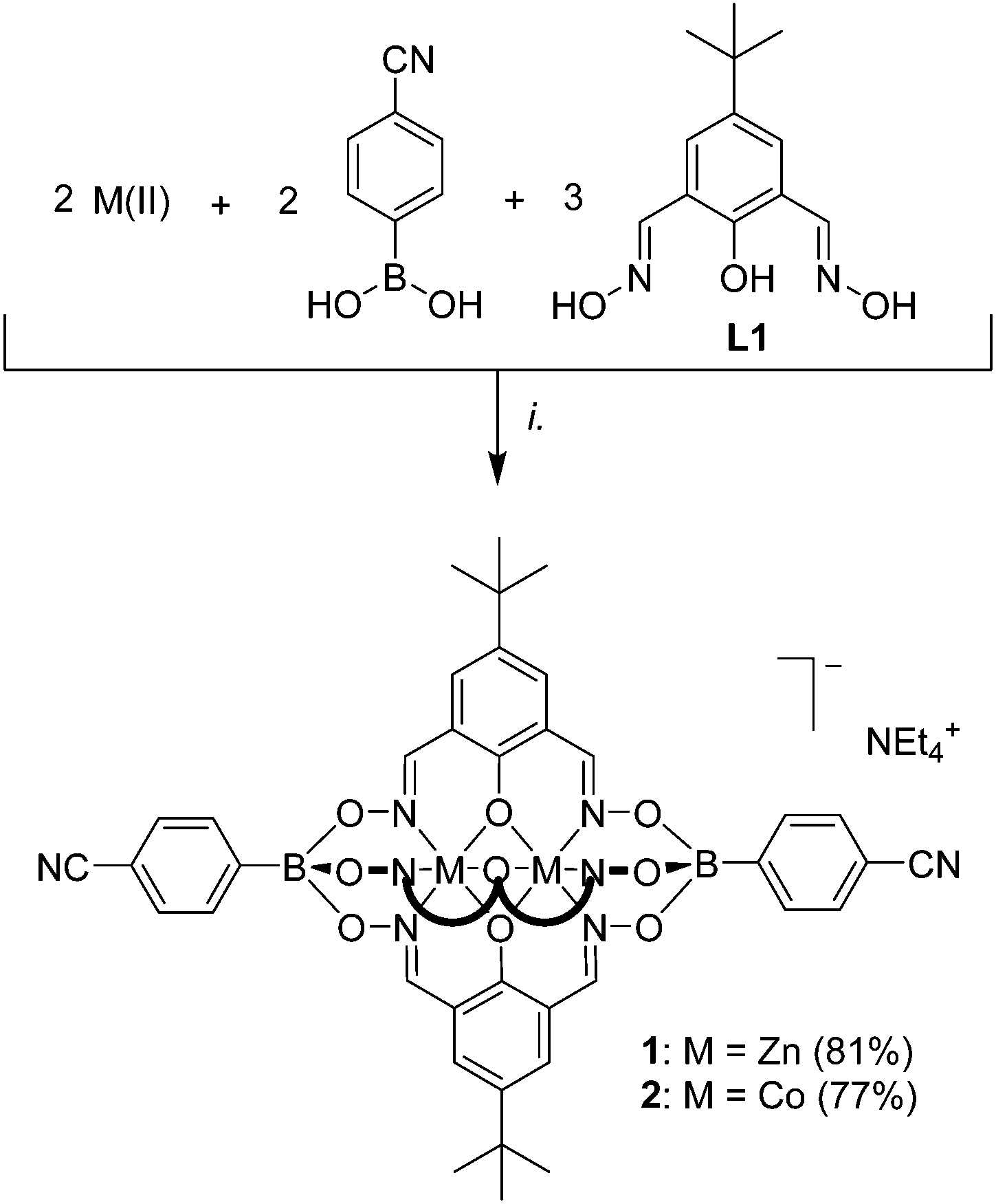 | ||
| Scheme 1 Synthesis of the ditopic clathrochelate complexes 1 and 2. Reagents and conditions: (i.): [Co(H2O)6](NO3)2 or Zn(OTf)2, EtOH/MeOH, 70 °C, then NEt4OH. | ||
Previously, we had investigated the magnetism of dinuclear Co clathrochelates with pendent carboxylic acid groups using the Evans method. The data suggested that the Co(II) centers have a high spin configuration. For complex 2, we have now performed a more detailed analysis using a superconducting quantum interference device (SQUID) magnetometer. A plot of the magnetic susceptibility vs. temperature reveals an increase of susceptibility as temperature decreases and a broad maximum around 87 K below which a significant decrease of the susceptibility occurs (Fig. 2).
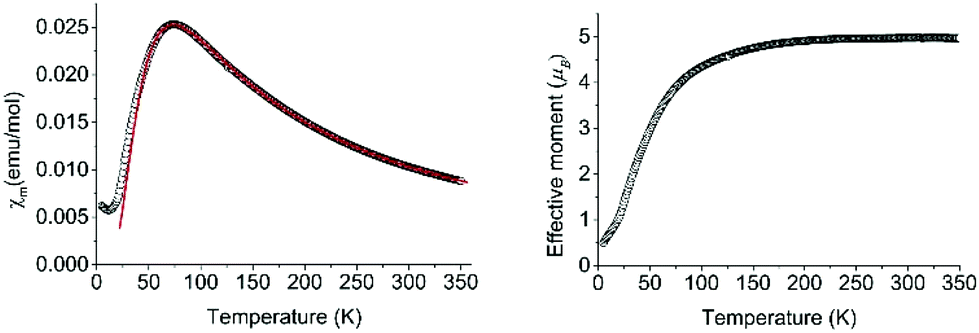 | ||
| Fig. 2 The magnetic susceptibility and the effective magnetic moment of complex 2 as a function of temperature as determined by SQUID measurements. | ||
Given that the pair of cobalt ions are well separated from other magnetic ions, the inter-cluster interaction can be neglected and the Hamiltonian of the system can be written simply as H = −2J·S1·S2, where S1 and S2 are spins of two cobalt ions that form a pair and J is the intra-cluster exchange interaction. The room temperature value of the effective moment per Co ion reaches 5.0μB, close to the theoretically maximum value for a high-spin cobalt(II) state with L = 3 orbital momentum included. The data has been analyzed using MagSaki software13 which takes into account the magnetic exchange coupling J, spin–orbit coupling λ, an orbital reduction factor κ and the axial-splitting parameter Δ. Very good agreement with measured data has been achieved with J = −20(3) cm−1, λ = −110(10) cm−1, κ = 1(0.1) and Δ = 286(30) (see red line in Fig. 2, left). These parameters correspond to significant g-factor anisotropy, gx = 4.9(0.3) and gz = 3.0(0.3), which is in agreement with preliminary ESR measurements. These values are similar to those reported for other di-nuclear Co-based compounds with much larger intra-cluster magnetic interaction.14
In order to prepare clathrochelates with cyano groups in lateral position, we have prepared the phenoldioxime ligand L2. The synthesis of this ligand was accomplished by Pd-catalyzed cross-coupling of 4-cyanophenylboronic acid and 2,6-diformyl-4-bromophenol in presence of SPhos and Pd2(dba)3,15 followed by treatment of the crude dialdehyde with hydroxylamine hydrochloride (Scheme 2).
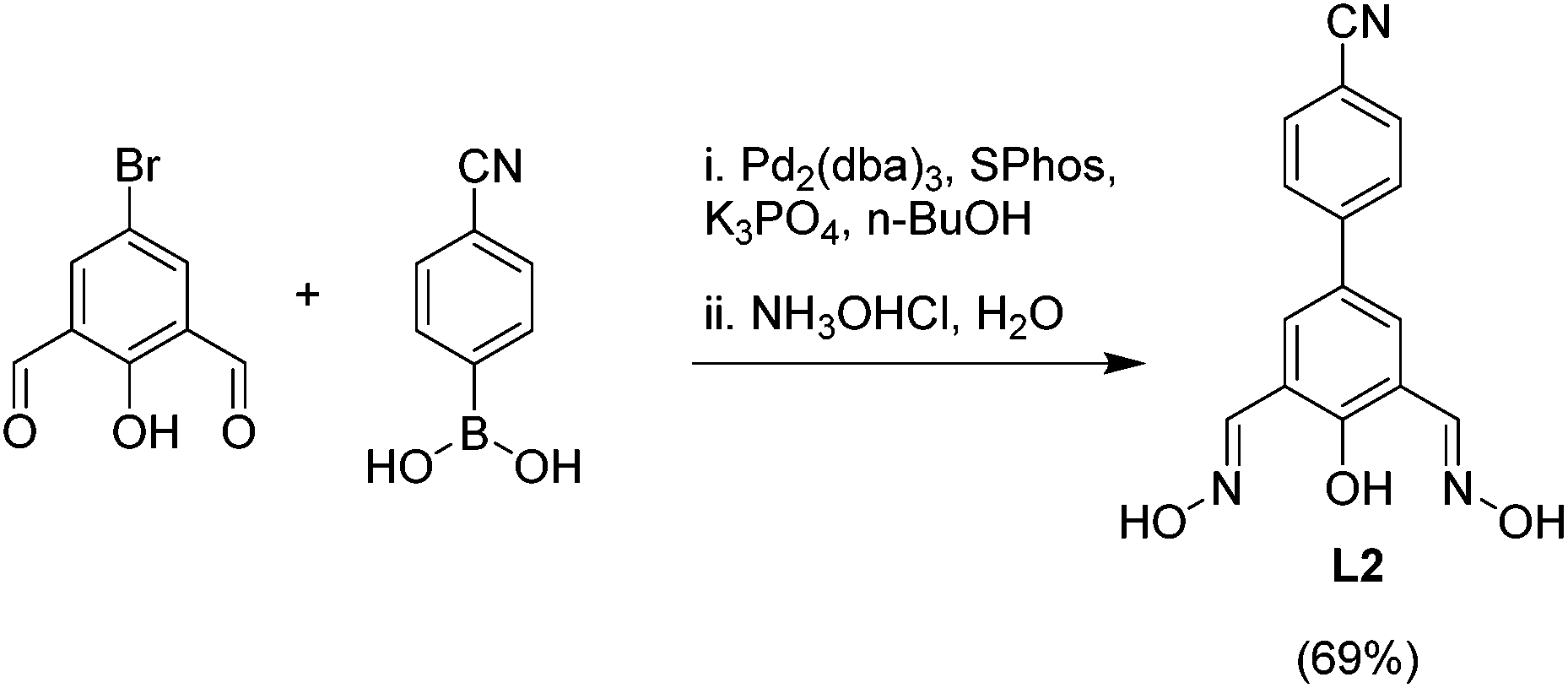 | ||
| Scheme 2 Synthesis of the dioxime ligand L2. | ||
The tri- and pentatopic clathrochelates 3–5 were then obtained by using L2 in combination of either 4-bromophenylboronic acid or 4-cyanophenylboronic following the standard protocol (Scheme 3). To the best of our knowledge, complexes 4 and 5 represent first examples of polycyano ligands with trigonal bipyramidal geometry. Zn-based clathrochelates 3 and 4 are luminescent, with emission maxima at 450 nm (DMF, λex = 335 nm, Fig. S15†). Their emissions are thus slightly red-shifted compared to what was observed for 1. The analytical data for 3–5 are in line with the expected structures. The magnetic properties of 5 were expected to be similar to what was found for 2 and additional SQUID measurements were not performed.
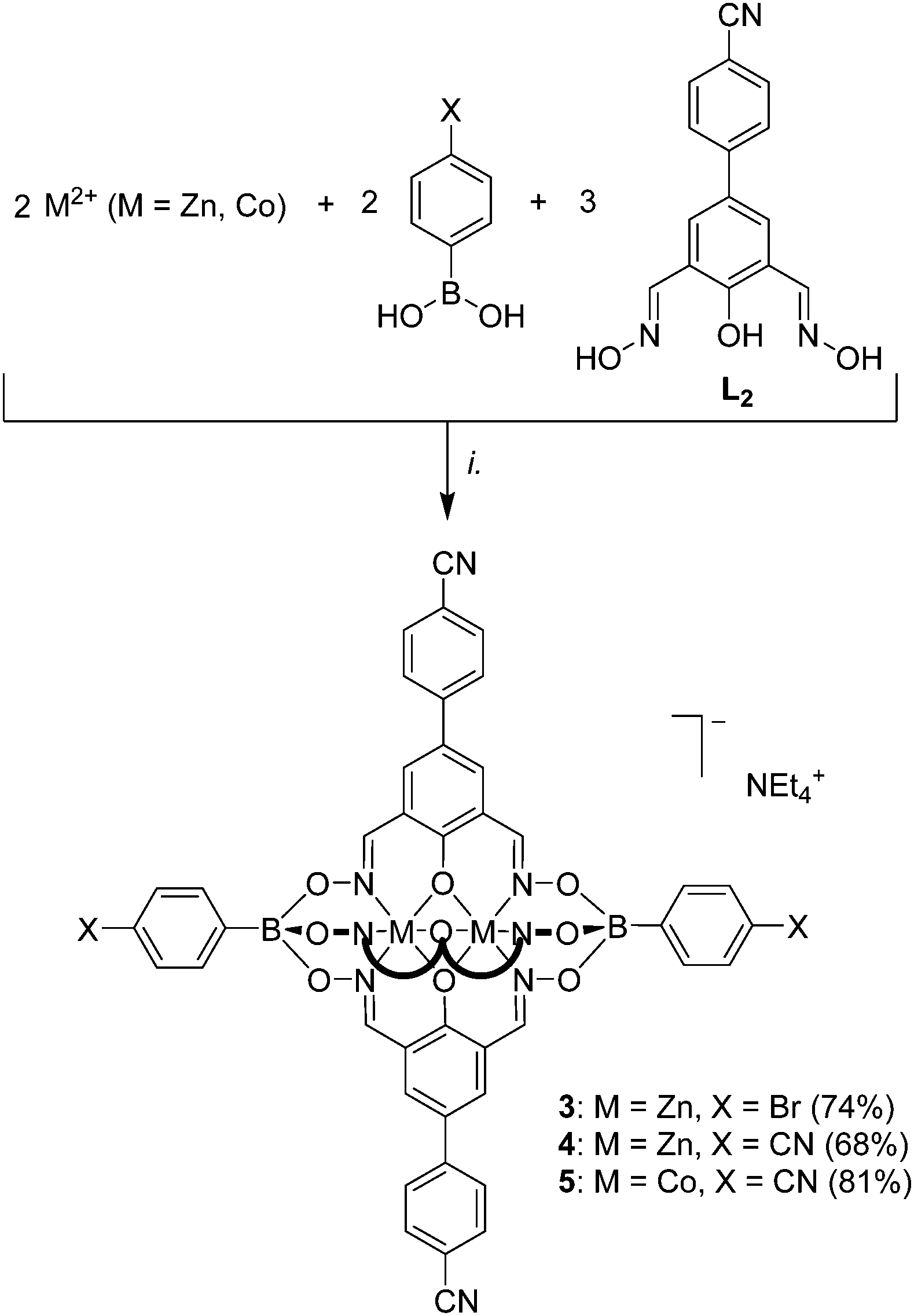 | ||
| Scheme 3 Synthesis of the tri- and pentatopic clathrochelate complexes 3–5. Reagents and conditions: (i.): 4-bromo- or 4-cyanophenylboronic acid, dioxime ligand L2, Zn(OTf)2 or [Co(H2O)6](NO3)2, MeOH/EtOH, 70 °C, then NEt4OH. | ||
Synthesis of CN-functionalized clathrochelates by postsynthetic cross-coupling reactions
Recently, we have shown that brominated clathrochelate complexes of Zn2+ and Co2+ are sufficiently stable to be used as substrates in Pd-catalyzed cross-coupling reactions, allowing the preparation of elongated polypyridyl ligands.9a We anticipated that a similar strategy could be used to prepare clathrochelate complexes with pendent benzonitrile groups. Indeed, the two-fold coupling of the previously described clathrochelate 6 featuring 3-bromophenylboronate ester caps9a with 4-cyanophenylboronic acid gave the desired complex 7 in 91% yield (Scheme 4). With regard to potential applications, it is worth noting that ditopic cyanoliogands with a bent shape have been used extensively for the preparation of silver(I) CPs.3a–g Compared to the ligands used previously, clathrochelate 7 stands out because of its size and its negative charge. Attempts to prepare a linear dicyano ligand by cross-coupling of a clathrochelate with terminal 4-bromophenylboronate ester groups were unfortunately not successful. Even though we were able to detect the desired product by mass spectrometry, we were not able to achieve a complete reaction, and separation of the side products was found to be difficult. This result indicates that coupling reactions in para position to the boronate ester function are more problematic.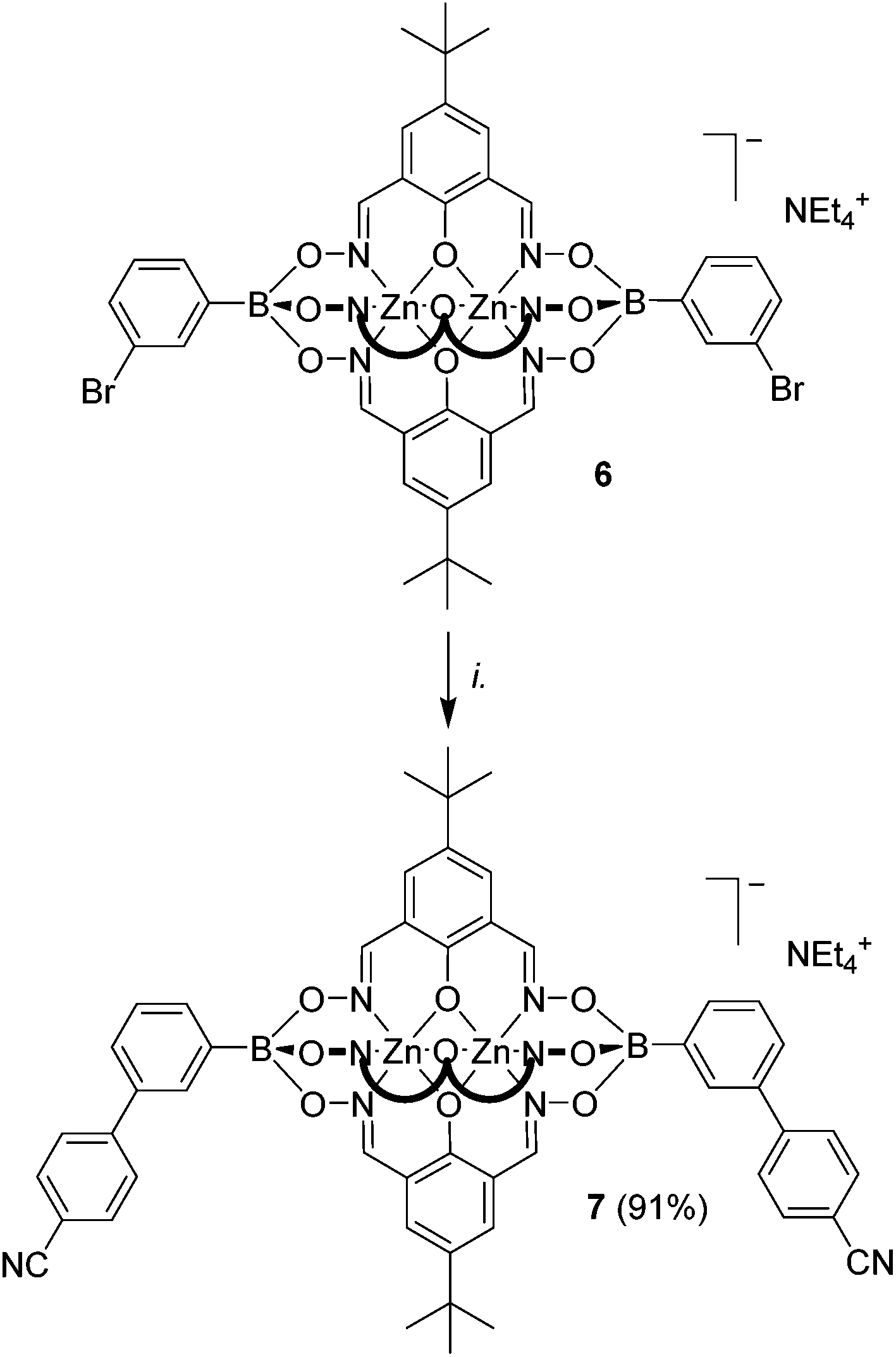 | ||
Scheme 4 Synthesis of the extended ditopic clathrochelate complex 7. Reagents and conditions: (i.): 6 (1 eq.), 4-cyanophenylboronic acid (8 eq.), K3PO4 (4 eq.), Pd2(dba)3 (5 mol%), SPhos (10 mol%), n-BuOH/toluene (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 90 °C, 14 h. :1), 90 °C, 14 h. | ||
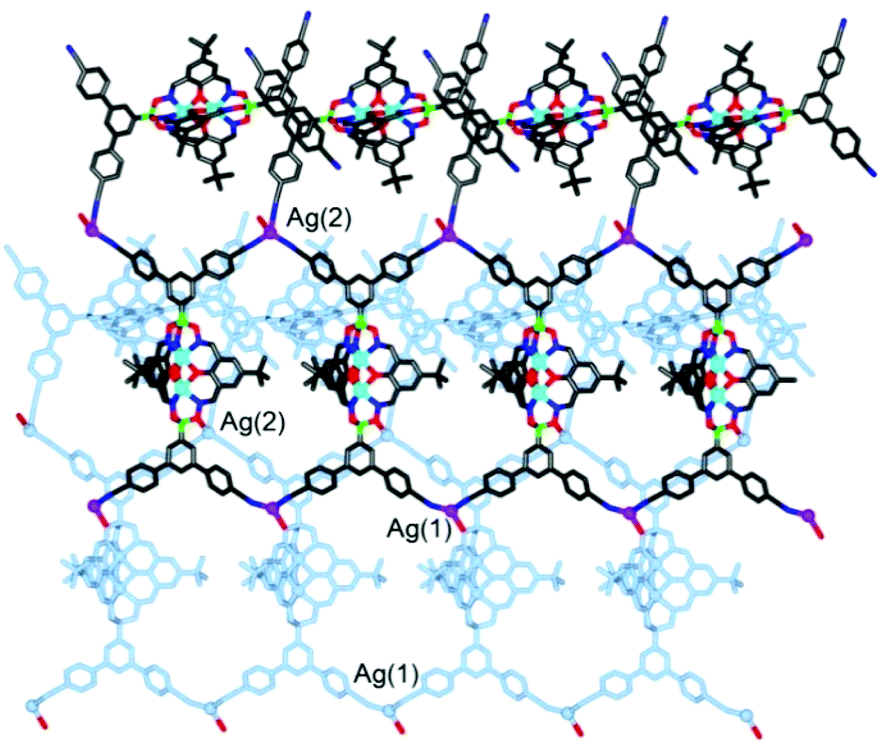
To further test the scope of the cross-coupling procedure, we prepared the tetrabrominated clathrochelate complex 8 using 3,5-dibromophenylboronic acid and dioxime L1. The subsequent four-fold coupling with 4-cyanophenylboronic acid in the presence of Pd2(dba)3 and SPhos was remarkably clean, providing the tetratopic complex 9 in 89% yield (Scheme 5).
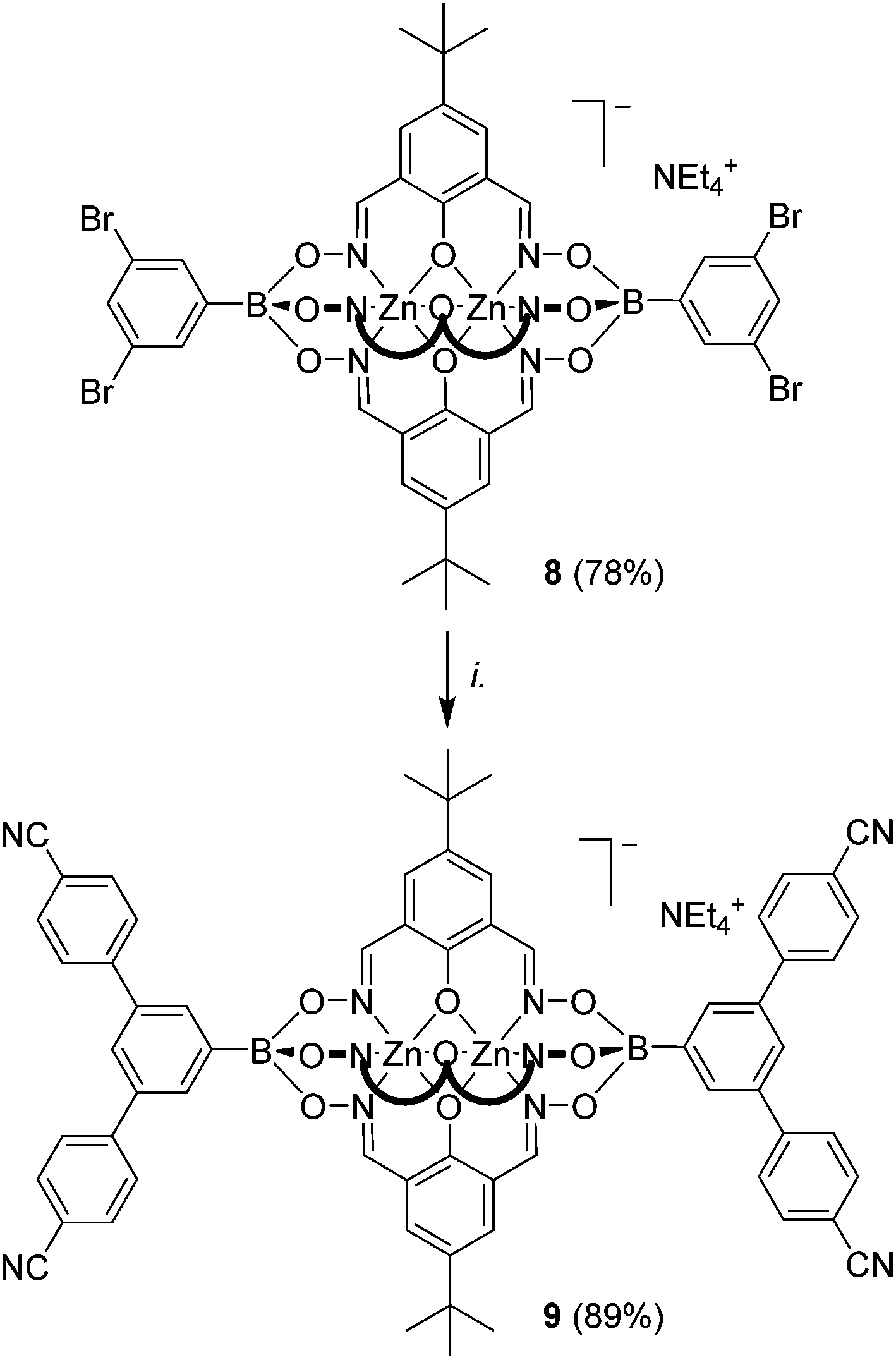 | ||
| Scheme 5 Synthesis of the tetratopic clathrochelate complex 9. Reagents and conditions: (i.): 8 (1 eq.), 4-cyanophenylboronic acid (20 eq.), K3PO4 (10 eq.), Pd2(dba)3 (5 mol%), SPhos (10 mol%), n-BuOH/toluene (1:1), 90 °C, 14 h. | ||
The elongated clathrochelates 7 and 9 are soluble in polar organic solvents such as DMSO, nitromethane and DMF, and the analytical data match the proposed structures. Both complexes are luminescent, with an emission maximum at 445 nm (DMF, λex = 335 nm, Fig. S16†).
Coordination polymers with silver(I)
After having established efficient protocols for the synthesis of CN-functionalized clathrochelate complexes, we started to explore their utilization as metalloligands in CPs. As outlined in the introduction, polycyano ligands are particularly suited for the preparation of silver(I) CPs, and we thus focused on reactions with Ag+ salts. For some clathrochelate complexes, we were able to obtain crystalline CPs upon reaction with silver salts, and the results are detailed below.Single crystals of the CPs 10 and 11 were obtained by layering a solution of AgNO3 in MeOH on top of a solution of 1 or 2 in nitromethane (Scheme 6). Crystallographic analyses revealed that both CPs are isostructural compounds having stoichiometry [Ag(1)(CH3OH)](CH3OH)1.5(CH3NO2) for 10 and [Ag(2)(CH3OH)](CH3OH)1.25(CH3NO2)1.5 for 11. The terminal cyano groups coordinate to the Ag+ ions in a slightly bent fashion (N–Ag–N = 154° and 155°) forming an infinite chain. The Ag+ ions display a trigonal pyramidal geometry, with one of the coordination sites being occupied by a methanol ligand. The fourth coordination site is occupied by the oxygen atom of an adjacent boronate ester group. It should be noted that coordination of a metal ion to the oxygen atom of clathrochelate complexes has not been observed before. As a result of this cross-linking, we observe an unusual double chain architecture (Fig. 3).3d A summary of the bond distances of the connecting silver complexes is given in Table 1.
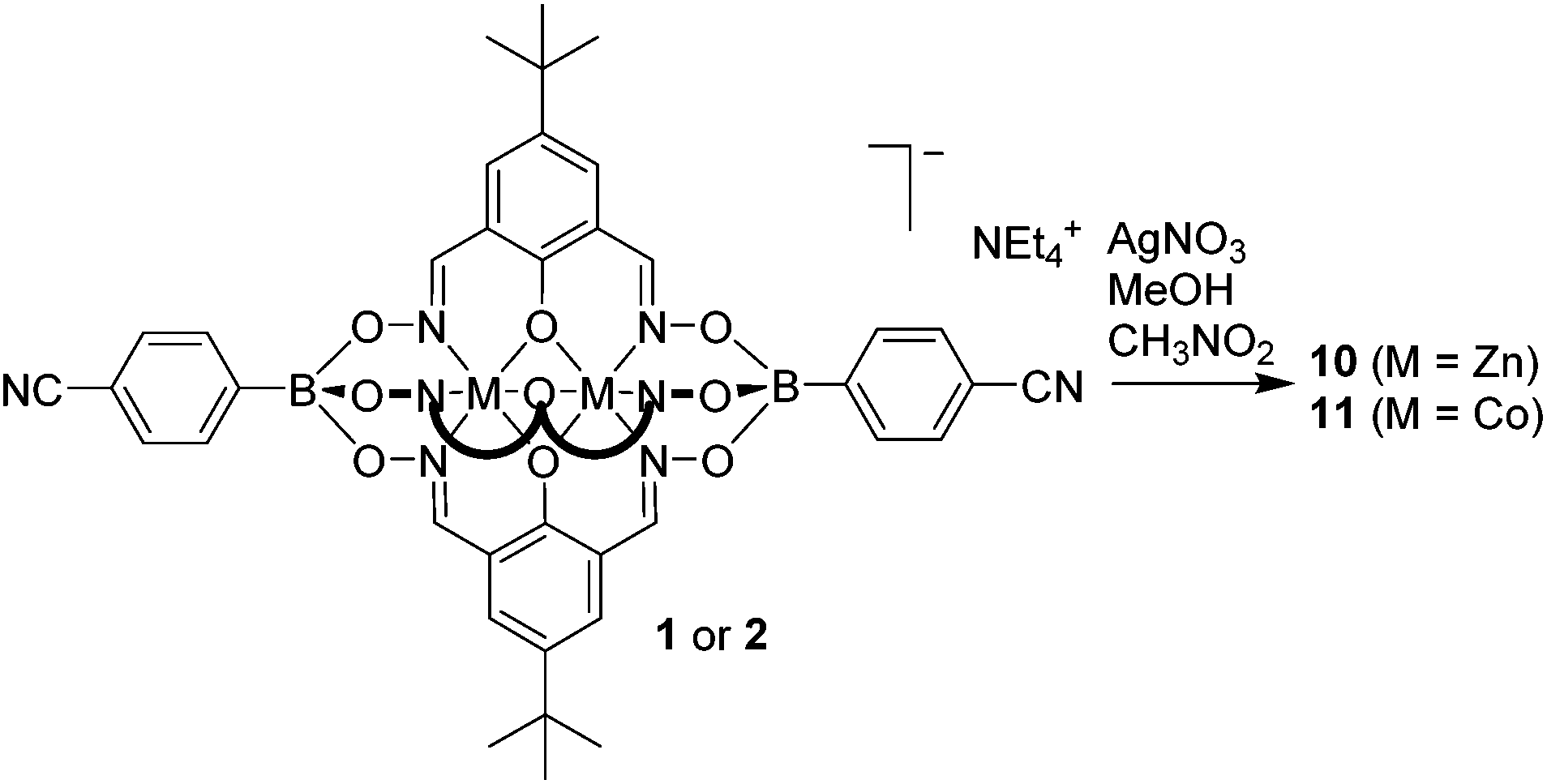 | ||
| Scheme 6 Synthesis of the heterometallic coordination polymers 10 and 11. | ||
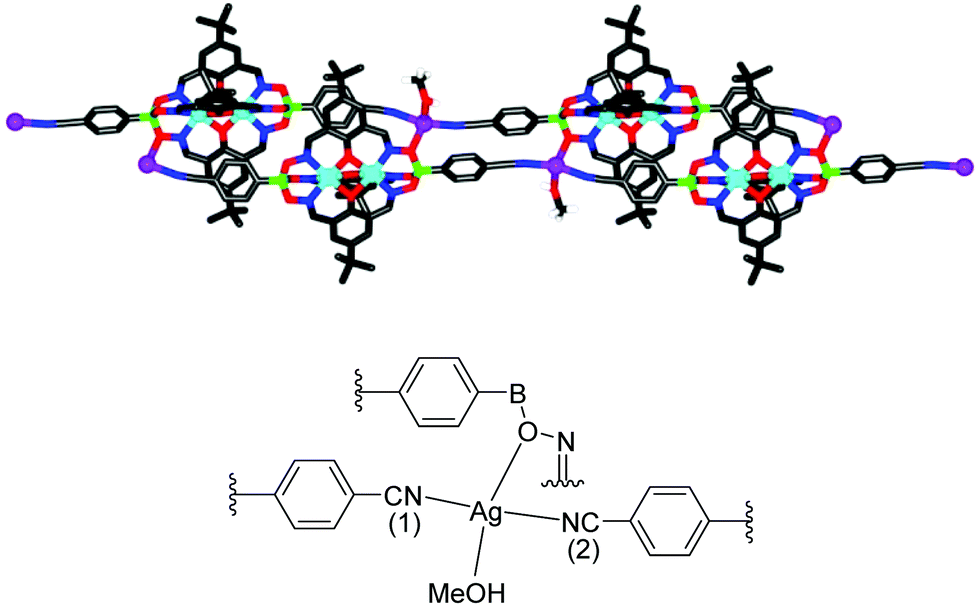 | ||
| Fig. 3 Graphical representation of the double chain structure of coordination polymer 10 (top) and the first coordination sphere of the Ag+ ions (bottom). Most hydrogen atoms and unbound solvent molecules have been omitted for clarity. Color coding: C: gray; hydrogen: white; O: red; N: blue; Ag: purple; B: green; Zn: cyan. | ||
| Complex | Ag–N(1) | Ag–N(2) | Ag–OMeOH | Ag–OOxime |
|---|---|---|---|---|
| 10 | 2.158(6) Å | 2.132(6) Å | 2.627(4) Å | 2.586(4) Å |
| 11 | 2.171(3) Å | 2.155(3) Å | 2.526(5) Å | 2.577(3) Å |
The structures of 10 and 11 do not contain nitrate anions from the silver salt because the charge compensation is achieved by the metalloligand itself. It should be noted that for previously reported silver(I) CPs, the anion derived from the silver salt was often found to influence the final structure.3d,4a,d,5b,g,16
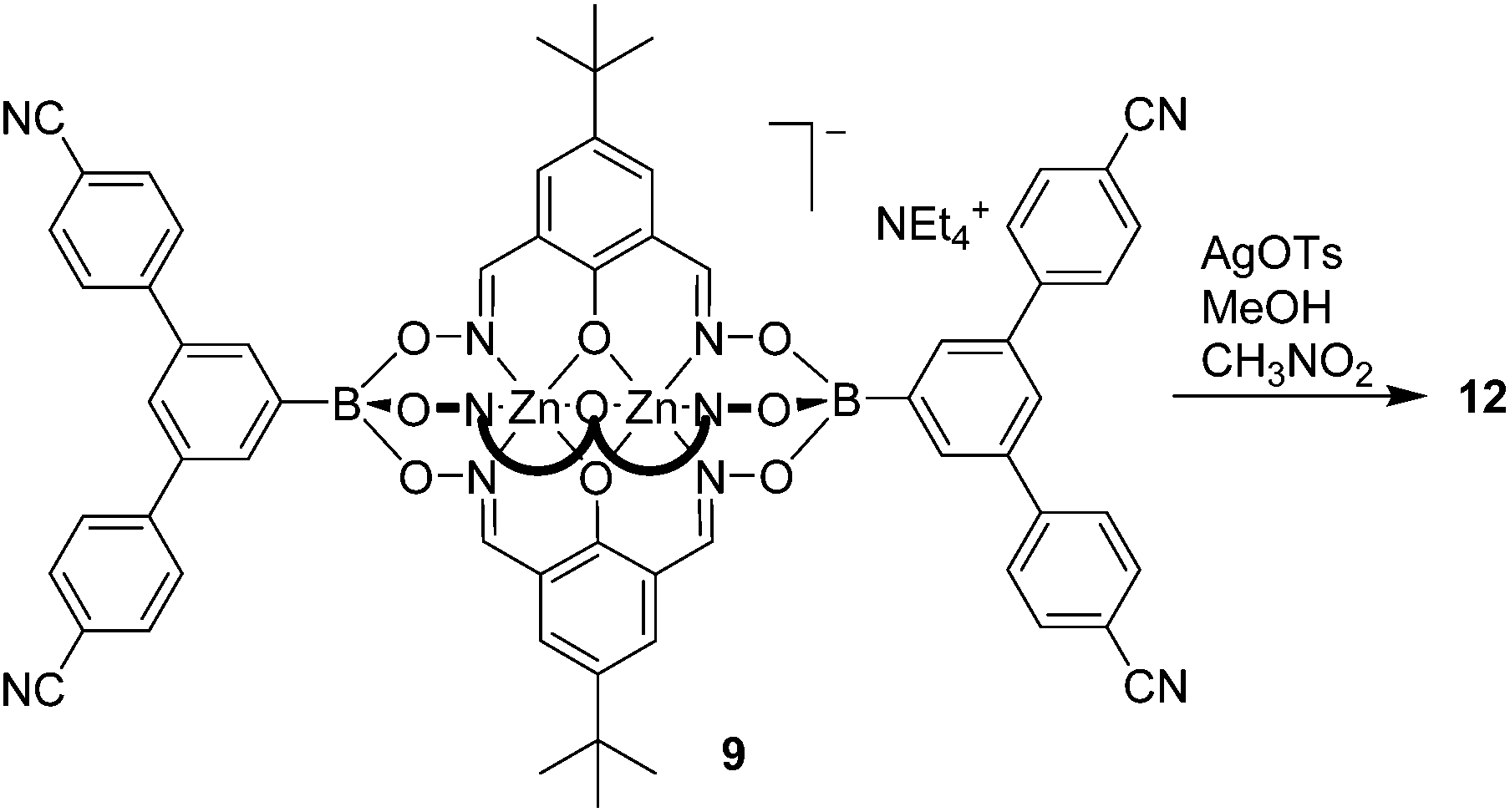
We were also successful in preparing a CP using the tetratopic metalloligand 9. Layering a solution of AgOTs in MeOH on top of a solution of 9 in nitromethane led to the formation of transparent, plate-like crystals of coordination polymer 12 (Scheme 7).
 | ||
| Scheme 7 Synthesis of the heterometallic coordination polymer 12. | ||
Single crystal X-ray analysis of 12 revealed the formation of a two-dimensional network structure with the stoichiometry [Ag2(9)2](CH3OH)1.5(CH3NO2)3. A graphical representation of the structure is depicted in Fig. 4. One can observe two distinct clathrochelate metalloligands in the structure. The first one is coordinated via all of its cyano groups to silver ions Ag(1) and Ag(2), thereby forming an infinite double chain. The second clathrochelate is coordinated via only one of the four cyano groups to silver ions Ag(2). The latter display a trigonal pyramidal geometry (Ag(2)–Nav. = 2.281 Å), with the fourth coordination site being occupied by the oxygen atom of a boronate ester group from an adjacent clathrochelate (Ag(2)–O = 2.587(6) Å). The Ag(1) ions display approximate trigonal planar geometry, with coordination of Ag(1) to two cyano groups (Ag(1)–Nav. = 2.173 Å) and one oxygen atom of a boronate ester (Ag(1)–O = 2.390(5) Å). The Ag–O–B linkages connect the double chains, resulting in an overall two-dimensional 3,6-coordinated network structure, where silver atoms act as three-connected nodes and clathrochelates as six-connected nodes (through four cyano and two boronate groups). The underlying net of this CP has the kgd topology (kagome dual, given in terms of the RCSR notation;17 see ESI, Fig. S19†) which is the most widespread topology for 3,6-coordinated two-dimensional CPs.18 As it was observed for CP 10 and 11, the charge compensation is achieved by the metalloligands, and tosylate anions from the silver salt are not found in the structure.
 | ||
| Fig. 4 Part of the structure of coordination polymer 12. The shaded pale blue part of the structure is connected via Ag–O bonds. Hydrogen atoms and solvent molecules have been omitted for clarity. Color coding: C: gray; O: red; N: blue; Ag: purple; B: green; Zn: cyan. | ||
Silver(I) CPs based on pentacyano ligands with a trigonal bipyramidal geometry are – to the best of our knowledge – not known. We were thus particularly interested to obtain CPs with the pentatopic metalloligands 4 and 5. After several attempts, we were able to crystallize CP 13 using metalloligand 5 in combination with AgClO4 in the presence of MeOH, DMF and 1,2-dichlorobenzene as solvents (Scheme 8). Single crystal X-ray analysis of 13 showed that a three-dimensional network with the stoichiometry [Ag3(5)2(OMe)](C6H4Cl2)(MeOH)(sol.)n had formed (Fig. 5). The asymmetric unit of the crystal contains three silver ions and two clathrochelate complexes 5. Although the ratio of Ag to 5 is equal to 3:2, the overall network is neutral due to presence of disordered methoxy groups which are coordinated to Ag(2) ions. Besides, the Ag(2) ions act as linkers between two cobalt-containing metalloligands (Ag(2)–Nav. = 2.119 Å). The Ag(1) and Ag(3) ions, on the other hand, are coordinated in a trigonal planar fashion to three cyano groups, with Ag(1)–N distances varying from 2.11(2) to 2.30(2) Å. Both metalloligands act as four-connected nodes through both of the apical and two of the three lateral cyano groups (one cyano group is ‘free’). The first crystallographically independent metalloligand coordinates to one Ag(2) and three Ag(1) ions, and the second one to one Ag(2) and three Ag(3) ions. As a result, the Ag(2) ions links two interpenetrating CPs of the stoichiometry [Ag(5)] and the utp topology (widespread among 3-coordinated CP nets)19 to form a three-dimensional net.
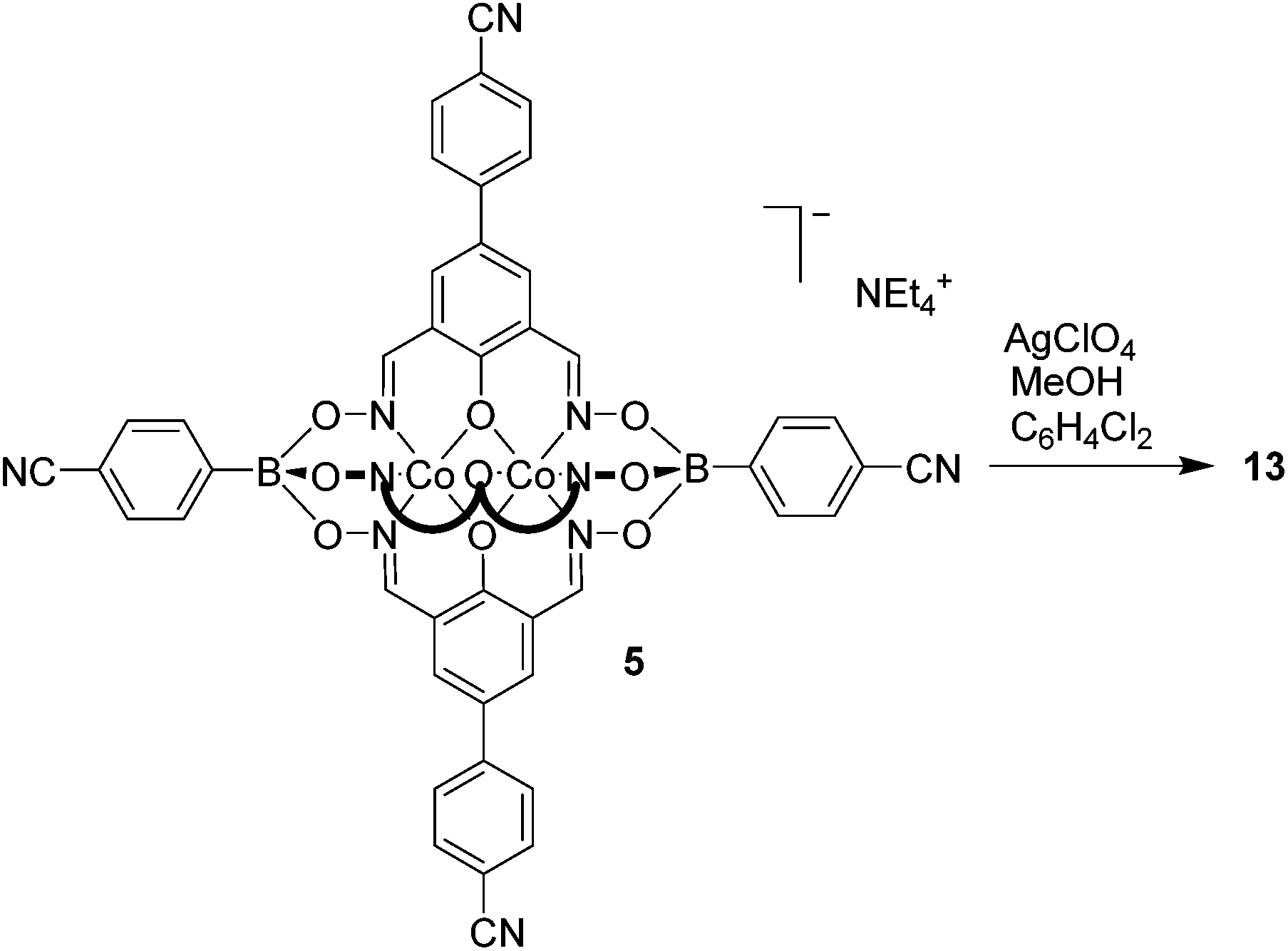 | ||
| Scheme 8 Synthesis of coordination polymer 13 using pentatopic clathrochelate 5. | ||
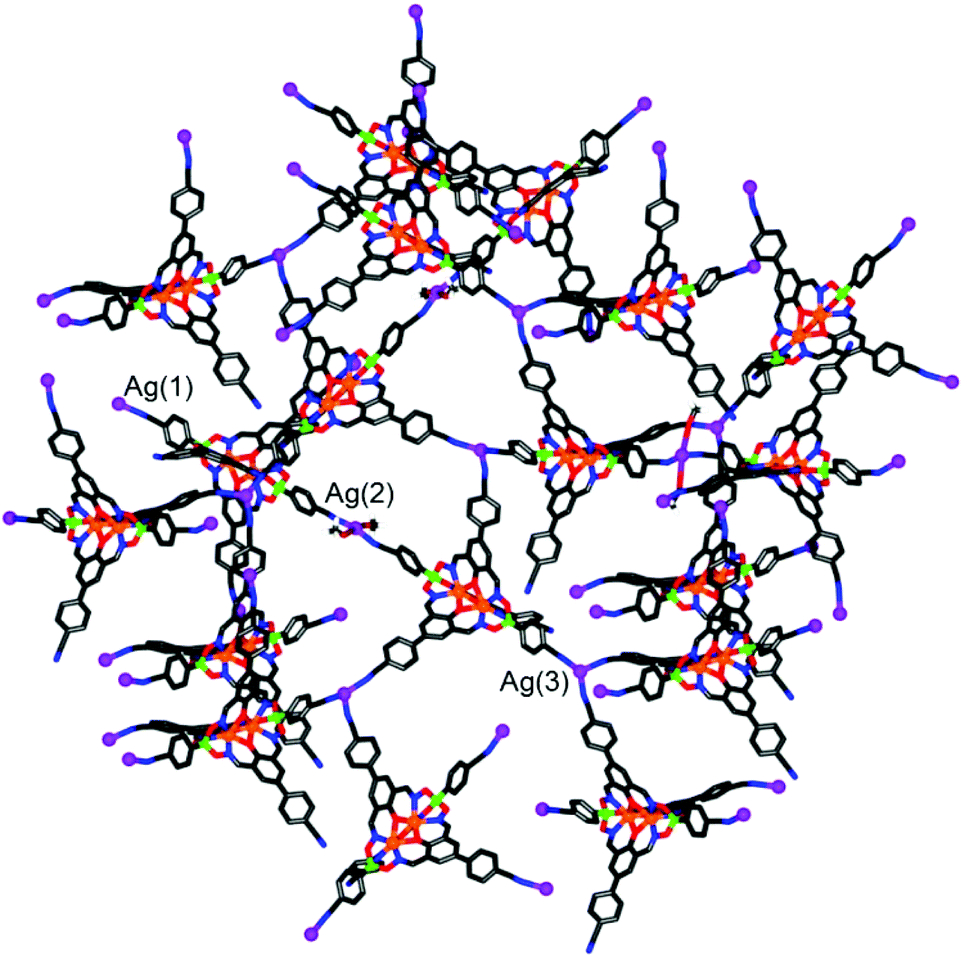 | ||
| Fig. 5 Part of the structure of the three-dimensional coordination polymer 13. Most hydrogen atoms and unbound solvent molecules have been omitted for clarity. Color coding: C: gray; H: white; O: red; N: blue; Ag: purple; B: green; Co: orange. | ||
A topological analysis of the resulting architecture by means of the ToposPro package20 revealed that the net has a point symbol (8·102)(82·104)(82·10)(83·103). To the best of our knowledge, such a network topology has not been observed in crystals before. The structure was deposited to the Topos TTD collection under the abbreviation epf1.
An intriguing structural feature of 13 is that all eight- and ten-membered circuits of the nodes are crossed by other rings, resulting in self-catenation (Fig. S20 and S21†). Self-catenated structures are single networks with regions in which chains from the same net pass through the smallest topological circuits in a fashion similar to that of interpenetrated systems. The observation of self-catenation in 13 is unusual because most of reported self-catenated nets are constructed from flexible ligands.21 The catenation in 13 is very dense from a topological point of view: the eight-membered rings are crossed by 42 other rings and the ten-membered rings are crossed by at least 57 other rings. The topological density of the net can be given as TD10 = 3127, which is the number of nodes within the first 10 coordination spheres of a given node. According to the RCSR database,17 this value represents the highest topological density among all known 3,3,4,4-coordinated nets, and is almost as high as TD10 = 3245 for a recently published 3,4-coordinated mhq-z net.22 Despite the high topological density, CP 13 displays a very large solvent-accessible volume of 53% according to calculations with Mercury.23 These voids are occupied by highly disordered solvent molecules, some of which could not be located from difference Fourier maps. The attempted desolvation of CP 13 resulted in rapid degradation of the material.
Conclusions
We have shown that dinuclear clathrochelate complexes with a trigonal bipyramidal shape can be decorated with cyano groups in apical and/or lateral position by using cyano-functionalized starting materials for the clathrochelate synthesis. Alternatively, we have been able to prepare elongated clathrochelates with pendent benzonitrile groups by post-synthetic modification of brominated clathrochelates in Pd-catalyzed cross-coupling reactions. The functionalized clathrochelates can be used as metalloligands for the preparation of heterometallic Co2+/Ag+ and Zn2+/Ag+ coordination polymers, as evidenced by the synthesis of one-, two-, and three-dimensional CPs in reactions with silver salts. Due to the negative charge of the metalloligands, the CPs are devoid of anions derived from the silver salt. A structure-directing role of the anion, as observed for many silver(I) CPs, is thereby avoided. The three-dimensional CP 13 is of special interest because it displays an unprecedented network topology and a very high topological density.Potential applications of the CN-functionalized clathrochelate complexes described above are not restricted to the formation of polymeric networks. Polycyanoligands have also been employed for the constructions of molecularly defined nanostructures using coordination-based self-assembly,24 and our new metalloligands should be useful building blocks in this context as well.
Experimental section
Materials and general procedures
Clathrochelate 6 and 2,6-diformyl-4-tert-butylphenol dioxime ligand (L1) were synthetized according to literature.10,111H and 13C NMR spectra were obtained on a Bruker Avance III (1H: 400 MHz, 13C: 100 MHz). 1H and 13C chemical shifts are reported in parts per million δ (ppm) referenced to the internal solvent. All spectra were recorded at RT. Electrospray-ionisation MS data were acquired on a Q-Tof Ultima mass spectrometer (Waters) operated in the negative ionization mode and data were processed using the MassLynx 4.1 software. APPI-FT-ICR experiments were performed on a hybrid linear ion trap Fourier transform ion cyclotron resonance mass spectrometer (LTQ FT-ICR MS, Thermo Scientific, Bremen, Germany) equipped with a 10 T superconducting magnet (Oxford Instruments Nanoscience, Abingdon, UK). Data analysis was carried out using XCalibur software (Thermo Scientific, Bremen, Germany). IR spectra were recorded on a Perkin Elmer Spectrum One Golden Gate FTIR spectrometer. Emission spectra were recorded with a Varian Cary Eclipse spectrometer. Magnetization and magnetic susceptibility measurements were performed with a Quantum Design MPMS-XL 5 T superconducting quantum interference device (SQUID) magnetometer. The powder samples were contained in plastic capsules which were incorporated into two plastic straws as sample holder. The measurements were collected as a function of the applied field of 1 T and temperatures ranging from 5 to 350 K with Zero Field Cooled method (ZFC).:1 mixture of MeOH and nitromethane (2 ml). Transparent yellowish needles of 10 (7.4 mg) were obtained within four weeks. IR: 2960, 2865, 2245, 2165, 1615, 1550, 1445, 1395, 1365, 1330, 1280, 1220, 1200, 1080, 1035, 1000, 930, 875, 840, 815, 785, 775, 690, 635, 560, 540, 530 cm−1.
:1 mixture of MeOH and nitromethane (2 ml). Orange needles of 11 (8.1 mg) were obtained within five weeks. IR: 2955, 2860, 2250, 2225, 2165, 1610, 1555, 1450, 1395, 1365, 1330, 1265, 1210, 1090, 1020, 970, 930, 840, 825, 785, 765, 695, 655, 635, 610, 570, 560 cm−1.
:1 mixture of MeOH and nitromethane (2 ml). Transparent pale-yellow plates of 12 (4.1 mg) were obtained within eight weeks. IR: 2955, 2865, 2235, 1680, 1610, 1555, 1505, 1450, 1395, 1365, 1330, 1285, 1240, 1225, 1185, 1145, 1070, 1035, 995, 930, 890, 835, 785, 725, 680, 640, 605, 590, 570, 555 cm−1.
:1 mixture of MeOH and 1,2-dichlorobenzene (2 ml). Orange plates of 13 (9.8 mg) were obtained within one week. IR: 3390, 2225, 1600, 1560, 1545, 1445, 1390, 1340, 1305, 1205, 1180, 1080, 1035, 1020, 980, 935, 910, 830, 800, 775, 750, 690, 655, 610, 550, 535 cm−1.
Acknowledgements
The work was supported by the Swiss National Science Foundation and by the Ecole Polytechnique Fédérale de Lausanne (EPFL). We are grateful to the Swiss-Norwegian Beamline Consortium for providing access to synchrotron radiation.References
- (a) T. C. W. Mak and X.-L. Zhao, Encyclopedia of Inorganic Chemistry, 2006, John Wiley & Sons Search PubMed; (b) B. S. Fox, M. K. Beyer and V. E. Bondybey, J. Am. Chem. Soc., 2002, 124, 13613 CrossRef CAS PubMed.
- (a) C.-Y. Su, C.-L. Chen, J.-Y. Zhang and B.-S. Kang, in Design and Construction of Coordination Polymers, ed. M.-C. Hong and L. Chen, 2009, John Wiley & Sons, p. 111 Search PubMed; (b) C.-L. Chen, B.-S. Kang and C.-Y. Su, Aust. J. Chem., 2006, 59, 3 CrossRef CAS.
- For examples see: (a) X.-M. Guo, L. Zhao, H.-Y. Zou, Y.-N. Yan, Y.-J. Qi and Q. Wang, Inorg. Chem. Commun., 2015, 54, 57 CrossRef CAS; (b) S. P. Anthony, L. Wang, S. Varughese and S. M. Draper, CrystEngComm, 2013, 15, 6602 RSC; (c) C. D. MacKinnon, S. L. M. Parent, R. C. Mawhinney, A. Assound and C. M. Robertson, CrystEngComm, 2009, 11, 160 RSC; (d) Y.-B. Dong, H.-X. Xu, J.-P. Ma and R.-Q. Huang, Inorg. Chem., 2006, 45, 3325 CrossRef CAS PubMed; (e) Y.-B. Dong, H.-Y. Wang, J.-P. Ma, D.-Z. Shen and R.-Q. Huang, Inorg. Chem., 2005, 44, 4679 CrossRef CAS PubMed; (f) S. P. Anthony and T. P. Radhakrishnan, Chem. Commun., 2004, 1058 RSC; (g) Y.-B. Dong, P. Wang and R.-Q. Huang, Inorg. Chem., 2004, 43, 4727 CrossRef CAS PubMed; (h) J. Zhang, M. Nieuwenhuyzen, J. P. H. Charmant and S. L. James, Chem. Commun., 2004, 2808 RSC; (i) Z. Xu, Y.-H. Kiang, S. Lee, E. B. Lobkovsky and N. Emmott, J. Am. Chem. Soc., 2000, 122, 8376 CrossRef CAS; (j) K. A. Hirsch, S. R. Wilson and J. S. Moore, Chem. – Eur. J., 1997, 3, 765 CrossRef CAS; (k) D. Venkataraman, S. Lee, J. S. Moore, P. Zhang, K. A. Hirsch, G. B. Gardner, A. C. Covey and C. L. Prentice, Chem. Mater., 1996, 8, 2030 CrossRef CAS.
- For examples see: (a) J. Ni, K.-J. Wei, Y. Liu, X.-C. Huang and D. Li, Cryst. Growth Des., 2010, 10, 3964 CrossRef CAS; (b) Y.-Q. Sun, C. Yang, Z. Xu, M. Zeller and A. D. Hunter, Cryst. Growth Des., 2009, 9, 1663 CrossRef CAS; (c) S. Banfi, L. Carlucci, E. Caruso, G. Ciani and D. M. Prosperpio, Cryst. Growth Des., 2004, 4, 29 CrossRef CAS; (d) F. C. Pigge, M. D. Burgard and N. P. Rath, Cryst. Growth Des., 2003, 3, 331 CrossRef CAS; (e) Y.-H. Kiang, G. B. Gardner, S. Lee, Z. Xu and E. B. Lobkovsky, J. Am. Chem. Soc., 1999, 121, 8204 CrossRef CAS; (f) W. Choe, Y.-H. Kiang, Z. Xu and S. Lee, Chem. Mater., 1999, 11, 1776 CrossRef CAS; (g) G. B. Gardner, Y.-H. Kiang, S. Lee, A. Asgaonkar and D. Venkataraman, J. Am. Chem. Soc., 1996, 118, 6946 CrossRef CAS; (h) D. Venkataraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600 CrossRef CAS; (i) G. B. Gardner, D. Venkataraman, J. S. Moore and S. Lee, Nature, 1994, 374, 792 CrossRef.
- For examples see: (a) Z. Zhang, H. Zhao, M. M. Matsushita, K. Awaga and K. R. Dunbar, J. Mater. Chem. C, 2014, 2, 399 RSC; (b) B. Blankschein, A. Schulz, A. Villinger and R. Wustrack, ChemPlusChem, 2014, 79, 973 CrossRef CAS; (c) C. S. Hawes, S. R. Batten and D. R. Turner, CrystEngComm, 2014, 16, 3737 RSC; (d) A. V. Mossine, P. Thavornyutikarn and J. Atwood, CrystEngComm, 2013, 15, 1673 RSC; (e) K. Akimoto, Y. Kondo, K. Endo, M. Yamada, Y. Aoyama and F. Hamada, Tetrahedron Lett., 2008, 49, 7361 CrossRef CAS; (f) Y.-B. Dong, Y.-Y. Jiang, J.-P. Ma, F.-L. Liu, B. Tang, R.-Q. Huang and S. R. Batten, J. Am. Chem. Soc., 2007, 129, 4520 CrossRef CAS PubMed; (g) K. S. Min and M. P. Suh, J. Am. Chem. Soc., 2000, 122, 6834 CrossRef CAS; (h) C. Kleina, E. Graf, M. W. Hosseini, A. De Cian and J. Fischer, Chem. Commun., 2000, 239–240 RSC; (i) I. Ino, J. Chu, M. Munakata, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and Y. Kitamori, Inorg. Chem., 2000, 39, 4273 CrossRef CAS PubMed; (j) G. Mislin, E. Graf, M. W. Hosseini, A. De Cian, N. Kyritsakas and J. Fischer, Chem. Commun., 1998, 2545 RSC; (k) F.-Q. Liu and T. D. Tilley, Inorg. Chem., 1997, 36, 5090 CrossRef CAS.
- For examples see: (a) C. A. Hollis, S. R. Batten and C. J. Sumby, Cryst. Growth Des., 2013, 13, 2350 CrossRef CAS; (b) C. A. Hollis, L. R. Hanton, J. C. Morris and C. J. Sumby, Cryst. Growth Des., 2009, 9, 2911 CrossRef CAS.
- (a) A. Wang, C. Merkens and U. Englert, CrystEngComm, 2015, 17, 4293 RSC; (b) Q. Guo, C. Merkens, R. Si and U. Emglert, CrystEngComm, 2015, 17, 4383 RSC; (c) C. Merkens, N. Becker, K. Lamberts and U. Englert, Dalton Trans., 2012, 41, 8594 RSC; (d) C. Merkens and U. Englert, Dalton Trans., 2012, 41, 4664 RSC; (e) B. Kilduff, D. Pogozhev, S. A. Baudron and M. W. Hosseini, Inorg. Chem., 2010, 49, 11231 CrossRef CAS PubMed; (f) D. Pogozhev, S. A. Baudron and M. W. Hosseini, Inorg. Chem., 2010, 49, 331 CrossRef CAS PubMed; (g) M. Kondracka and U. Emglert, Inorg. Chem., 2008, 47, 10246 CrossRef CAS PubMed; (h) A. D. Burrows, K. Cassar, M. F. Mahon and J. E. Warren, Dalton Trans., 2007, 2499–2509 RSC.
- M. Karsch, H. Lund, A. Schulz, A. Villinger and K. Voss, Eur. J. Inorg. Chem., 2012, 5542–5553 CrossRef CAS.
- (a) M. Marmier, G. Cecot, B. F. E. Curchod, P. Pattison, E. Solari, R. Scopelliti and K. Severin, Dalton Trans., 2016, 45, 8422 RSC; (b) M. Pascu, M. Marmier, C. Schouwey, R. Scopelliti, J. J. Holstein, G. Bricogne and K. Severin, Chem. – Eur. J., 2014, 20, 5592 CrossRef CAS PubMed.
- M. Marmier, M. D. Wise, J. J. Holstein, P. Pattison, K. Schenk, E. Solari, R. Scopelliti and K. Severin, Inorg. Chem., 2016, 55, 4006 CrossRef CAS PubMed.
- S. Khanra, T. Weyhermüller, E. Bill and P. Chaudhuri, Inorg. Chem., 2006, 45, 5911 CrossRef CAS PubMed.
- J. Zhang, Z. Ma, H. Zhao, C. Ge, Y. Wang and X. Zhang, Inorg. Chem. Commun., 2016, 65, 63 CrossRef CAS.
- H. J. Sakiyama, Chem. Software, 2001, 7, 171 CrossRef CAS.
- L. J. Daumann, P. Comba, J. A. Larrabee, G. Schenk, R. Stranger, G. Cavigliasso and L. R. Gahan, Inorg. Chem., 2013, 52, 2029 CrossRef CAS PubMed.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- Selected examples: (a) K.-J. Wei, J. Ni, J. Gao, Y. Liu and Q.-L. Liu, Eur. J. Inorg. Chem., 2007, 3868 CrossRef CAS; (b) A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117 CrossRef CAS.
- (a) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782 CrossRef PubMed; (b) http://rcsr.anu.edu.au/ .
- T. G. Mitina and V. A. Blatov, Cryst. Growth Des., 2013, 13, 1655 CAS.
- E. V. Alexandrov, V. A. Blatov, A. V. Kochetkov and D. M. Proserpio, CrystEngComm, 2011, 13, 3947 RSC.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576 CAS.
- For a self-penetrated net based on a rigid metalloligand see: L. Carlucci, G. Ciani, D. M. Proserpio and F. Porta, Angew. Chem., Int. Ed., 2003, 115, 331 CrossRef.
- H. Ma, D. Sun, L. Zhang, R. Wang, V. A. Blatov, J. Guo and S. Dun, Inorg. Chem., 2013, 52, 10732 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- For examples see: (a) B. Nohra, R. Réau and C. Lescop, Eur. J. Inorg. Chem., 2014, 1788 CrossRef CAS; (b) A. I. A. Perez, T. Biet, S. Graule, T. Agou, C. Lescop, N. R. Branda, J. Crassous and R. Réau, Chem. – Eur. J., 2011, 17, 1337 CrossRef PubMed; (c) Y. Yao, W. Shen, B. Nohra, C. Lescop and R. Réau, Chem. – Eur. J., 2010, 16, 7143 CrossRef CAS PubMed; (d) B. Nohra, S. Graule, C. Lescop and R. Réau, J. Am. Chem. Soc., 2006, 128, 3520 CrossRef CAS PubMed; (e) F. Fochi, P. Jacopozzi, E. Wegelius, K. Rissanen, P. Cozzini, E. Marastoni, E. Fisicaro, P. Manini, R. Fokkens and E. Dalcanale, J. Am. Chem. Soc., 2001, 123, 7539 CrossRef CAS PubMed; (f) P. Jacopozzi and E. Dalcanale, Angew. Chem., Int. Ed., 1997, 36, 613 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1481126–1481129. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02758j |
| This journal is © The Royal Society of Chemistry 2016 |