
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Ligand coordination modulates reductive elimination from aluminium(III)†
Stephanie J.
Urwin
,
David M.
Rogers
,
Gary S.
Nichol
and
Michael J.
Cowley
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Edinburgh, EH9 3FJ, UK. E-mail: michael.cowley@ed.ac.uk
First published on 4th August 2016
Abstract
Oxidative addition of inert bonds at low-valent main-group centres is becoming a major class of reactivity for these species. The reverse reaction, reductive elimination, is possible in some cases but far rarer. Here, we present a mechanistic study of reductive elimination from Al(III) centres and unravel ligand effects in this process. Experimentally determined activation and thermodynamic parameters for the reductive elimination of Cp*H from Cp*2AlH are reported, and this reaction is found to be inhibited by the addition of Lewis bases. We find that C–H oxidative addition at Al(I) centres proceeds by initial protonation at the low-valent centre.
Reductive elimination is a key reaction in organometallic chemistry, and is frequently both the product-forming and rate-determining step in important stoichiometric and catalytic transformations.1 The facility with which transition metal systems can undergo reversible oxidative addition and reductive elimination reactions is central to their widespread applications in catalysis. In this context, the analogy between the reactivity of transition metals and low-valent main-group compounds2 has concentrated effort on expanding their capability towards oxidative addition and reductive elimination reactivity.
The mechanisms of oxidative addition and reductive elimination at main group centres are diverse. Low valent group 14 carbene and alkyne analogues cleave dihydrogen through a concerted mechanism that involves simultaneous electron donation and acceptance to and from dihydrogen and the group 14 centre.3–9 Stannylenes activate the N–H bond of ammonia in an apparently similar process, yet in this reaction a coordination/deprotonation mechanism involving two equivalents of NH3 seems to be operative.6,10 Activation of ammonia, as well as other protic compounds, by constrained geometry phosphorus(III) species probably follows a similar pathway.11–15 Treatment of disilanes with Lewis bases can induce a formal reductive elimination, resulting in SiCl4 and base-coordinated SiCl2 fragments.16,17 Meanwhile, reductive elimination of H2 from arylstannanes, RSnH3, is also promoted by the addition of bases; in this case, the base does not coordinate the tin centre but instead initially deprotonates the tin hydride.18 Although a stepwise reaction, this formally heterolytic (ionic) reductive elimination of dihydrogen is reminiscent of the concerted heterolytic dihydrogen activation achieved by frustrated Lewis pairs.19
In transition metal chemistry, robust guiding principles exist that enable chemists to predict and select for oxidative addition/reductive elimination reactivity. In order to understand if the development of such principles for main-group systems is possible, mechanistic studies of a range of main-group oxidative additions and reductive eliminations are required.
Aluminium(I) compounds have been shown to readily activate H–C, H–P, H–N, H–Si and H–B bonds through oxidative addition,20 though the mechanism of these reactions is not well-understood. Recently, Fischer reported the striking reductive elimination of Cp*H from Cp*2AlH, 1 to yield the tetramer (Cp*Al)42 (Scheme 1).21 In this communication, we report the effect of coordinated ligands on reductive elimination from Cp*2AlH to form Cp*Al and Cp*H, and demonstrate that increasing coordination number and electron density at the Al(III) centre inhibits reductive elimination. Through a detailed mechanistic study of the reductive elimination of Cp*H from 1, we also reveal the important role of the Cp* ligands in enabling this transformation.
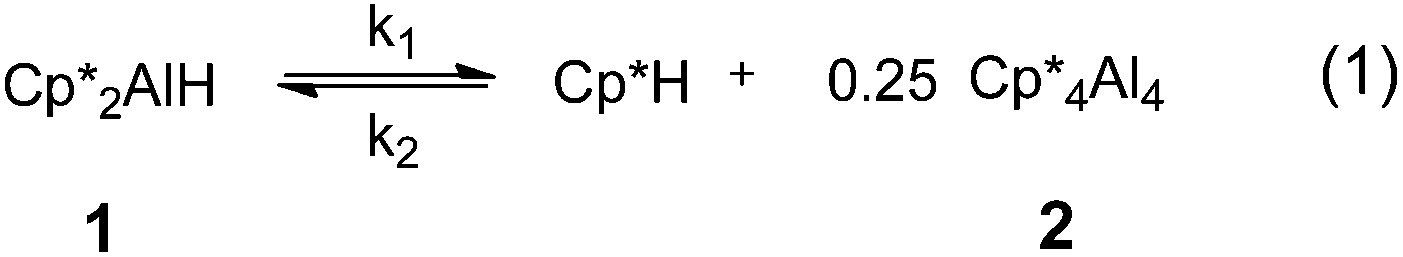 | ||
| Scheme 1 Reversible reductive elimination of Cp*H from Cp*2AlH, forming Cp*4Al42. | ||
With the diverse effects of Lewis bases on reductive elimination from silicon and tin centres, we were interested in how Lewis bases would interact with the reductive elimination chemistry of Cp*AlH, 1. Treatment of Cp*2AlH with N-heterocyclic carbenes (3a, 1,3,4,5-tetramethylimidazol-2-ylidene; 3b, 1,3-diisopropyl-4,5-dimethylimidazol-2-ylidene) or dimethyl aminopyridine (DMAP) results in the formation of 4-coordinate aluminium adducts 4a–c in high yields (Scheme 2).‡ No reaction was observed between 1 and the bulky NHC IPr (IPr = C{N(2,6-iPr2C6H3)CH}2),22 probably due to steric factors.
 | ||
| Scheme 2 Synthesis of base coordinated adducts of Cp*2AlH. | ||
The coordination of the NHC ligands 3a or 3b to Cp*2AlH 1 was readily apparent in the 1H NMR spectra of 4a and 4b. A dative Al–C interaction is confirmed by new signals observed for the now inequivalent methyl or isopropyl C–H groups of the NHC ligands (4aδ = 1.29 and 1.15 ppm; 4bδ = 6.08 and 3.76 ppm), which also display the expected downfield shifts observed for coordinated NHC ligands.23 The typical upfield shift of NHC donor carbon resonances upon coordination could not be confirmed because these signals were not observable for 4a or 4b, likely because of line broadening due to quadrupolar 27Al. The chemical shift of the Cp* methyl groups is only slightly perturbed by coordination of the NHC ligands (4aδ = 1.98 ppm; 4bδ = 2.06 ppm; 1δ = 1.91 ppm) and remains a lone singlet, indicating rapid sigmatropic shifts of the cyclopentadienyl substituents.24,25
Coordination of the DMAP ligand in the adduct 4c is confirmed by the observation of two upfield-shifted signals (δ = 7.52 3JH–H = 6.0 Hz; δ = 5.59 3JH–H = 7.0 Hz) for the aromatic protons of the DMAP ligand.
X-ray diffraction of single crystals of 4a–c confirm our NMR spectroscopic assignments. All compounds possess the expected tetrahedral aluminium centre, with both of the Cp* substituents η1 coordinated (Fig. 1). The long C–Al distances for the alkene ring carbons of the Cp* substituents in 4a–c preclude any Al–C bonding interactions. This differs from the reported structure of 1, where the two Cp* rings are η2 and η3 coordinated.21 Clearly, the coordination of strong σ-donor to the aluminium centre of 1 is favoured over the weaker donation of electron density from the π-system of the Cp* ligands. Compound 4a is isostructural with its gallium analogue,26 and the NHC bond distances in 4a and 4b are directly comparable to the very few reported NHC adducts of aluminium.27,28
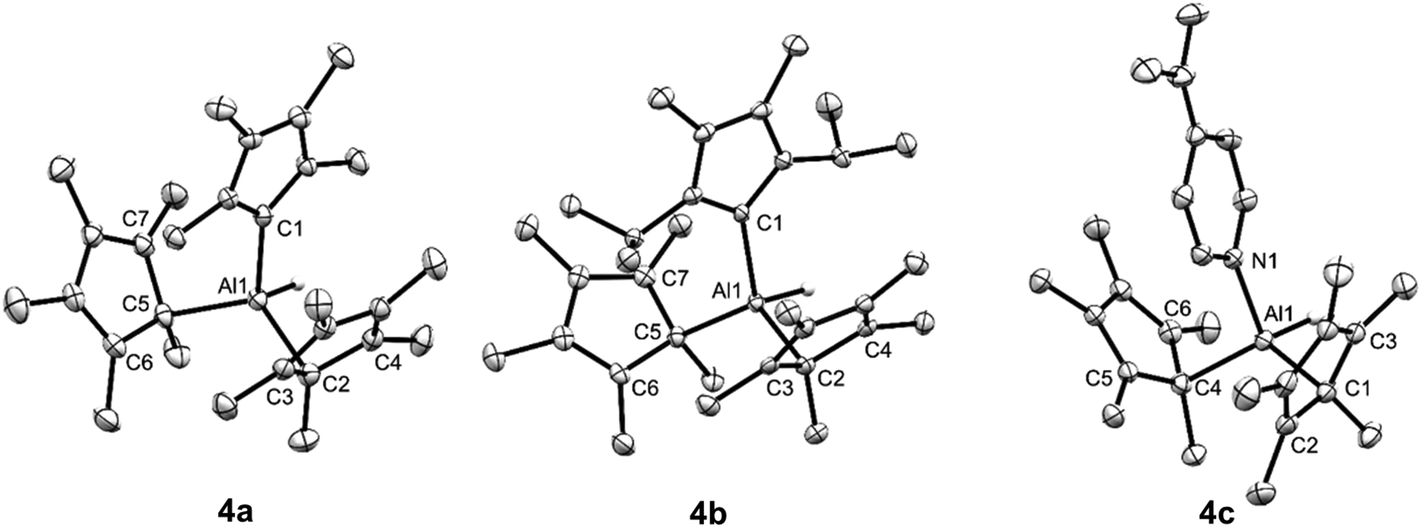 | ||
| Fig. 1 X-Ray crystal structures of NHC coordinated adducts of 1. Thermal ellipsoids at 50% probability and hydrogen atoms (except Al–H) omitted for clarity. Selected bond distances (Å). 4a: Al1–C1 2.0571(15), Al1–C2 2.0857(16), Al1–C3 = 2.65437(8), Al1–C4 = 2.79902(7), Al1–C5 2.0901(15), Al1–C6 3.03754(10), Al1–C7 2.70808(8); 4b Al1–C1 2.069(2), Al1–C2 2.082(2), Al1–C3 2.7948(3), Al1–C4 2.92435(18), Al1–C5 2.072(2), Al1–C6 3.1378(2), Al1–C7 2.8882(3); 4c Al1–N1 1.943(2), Al1–C1 2.081(3), Al1–C2 2.70138(8), Al1–C3 2.92094(8), Al1–C4 2.067(3), Al1–C5 2.81996(7), Al1–C6 2.66689(18). | ||
In contrast to the group 14 systems mentioned previously, the interaction of Lewis bases with the aluminium hydride 1 does not result in reductive elimination reactivity. Even after heating the NHC adducts 4a or 4b at 100 °C for several days, no elimination of Cp*H was observed.29 However, heating solutions of the DMAP adduct 4c at 80 °C resulted in reductive elimination of Cp*H and formation of tetramer 2 as the only aluminium-containing product, along with uncoordinated DMAP. The rate of Cp*H elimination from 4c is significantly slower than that from Cp*2AlH 1 (for example, after 100 minutes at 353 K, 31.3% of 4c was converted to the tetramer 2 whilst 90.7% of 1 had been converted).
In order to explain our observations, we propose a mechanism involving the reversible dissociation of DMAP from the adduct 4c under the reaction conditions. Reductive elimination to form 2 can only take place from 1; the DMAP adduct 4c does not itself eliminate Cp*H (Scheme 3). The formation of (Cp*Al)4 is not observed when the NHC adducts 4a and 4b are heated because of the stronger coordination of these ligands to the aluminium centre.
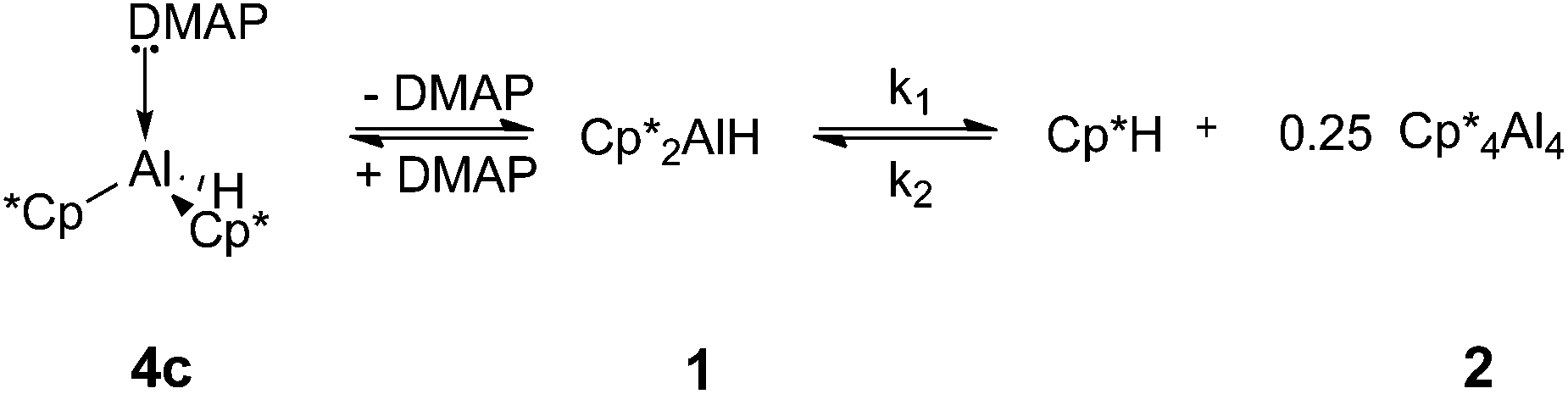 | ||
| Scheme 3 Reversible coordination of DMAP to 1. | ||
The proposed reversible coordination of DMAP to 1 at higher temperatures is supported by the observation of time-averaged chemical shifts for the DMAP aromatic CH protons. For example, when a sample of 4c in d8-toluene is heated to 363 K, broad resonances are observed in the 1H NMR spectrum at δ = 7.71 and 5.88 (at 300 K: 4cδ = 7.52, 5.59; DMAP δ = 8.44, 6.10). Monitoring the rate of reductive elimination of Cp*H from Cp*2AlH 1 and from 4c confirms that DMAP inhibits Cp*H elimination. Upon heating a solution of 1 for 150 minutes, equilibrium was reached with 95.9% conversion to 2 and Cp*H. However, at equilibrium solutions of 4c only displayed 35.9% conversion to 2.
Why does base coordination to 1 inhibit reductive elimination, when in other main-group systems reductive elimination can be promoted by the coordination of donor ligands? We sought to understand this observation by undertaking a mechanistic study of reductive elimination from 1.
We initially confirmed Fischer's report21 that reductive elimination of Cp*H from the hydride 1 is reversible, and determined equilibrium constants for this process. Monitoring a d8-toluene solution of 1 by 1H NMR spectroscopy reveals 100% conversion to 2 and Cp*H at 100 °C; upon cooling to 70 °C and then to 28 °C, compound 1 was cleanly regenerated and the conversion to 2 fell to 91.3 and 88.5% respectively (Fig. S9 and S11†). By measuring the concentrations of (Cp*Al)42, Cp*2AlH 1 and Cp*H we determined Keq. for the equilibrium depicted in Scheme 1 at a range of temperatures (Table S3†). We were thus able to determine ΔG°300 as +13.83 ± 0.48 kJ mol−1, indicating reductive elimination from 1 to 2 is an endothermic process, as might be expected for the reduction of AlIII to AlI.30
Having established experimental values for thermodynamic parameters of Cp*H reductive elimination, we studied the kinetics of this reaction. An important assumption we make is that the tetramerisation of Cp*Al to (Cp*Al)4, and the reverse process, proceeds with lower barriers than reductive elimination of oxidative addition of Cp*H. The tetramerisation energy for Cp*Al has been measured experimentally as 150 ± 20 kJ mol−1, and tetramer and monomer are in rapid equilibrium under our reaction conditions.31
Oxidative addition of Cp*H to Cp*Al is significantly faster than reductive elimination from 1; fitting our experimental data to the model in Scheme 1 we determined rate constants k1 and k2 at 333 K as 1.46 × 10−3 ± 0.04 × 10−3 s−1 and 35 × 10−3 ± 4 × 10−3 M−1 s−1 respectively. An Eyring plot (Fig. S13†) reveals an activation barrier of 95.48 ± 3.95 kJ mol−1 for reductive elimination (EREa) of Cp*H from 1. We could only obtain rate data for oxidative addition of Cp*H to Cp*Al at a limited range of temperatures, so are unable to accurately determine a value for the activation barrier of this reaction. However, EOAa can be estimated by subtracting ΔG°300 for reaction (1) from EREa giving a value of 81.65 ± 3.97 kJ mol−1. This value correlates well with the value we estimated from an Eyring plot with limited rate data (Fig. S14†) which was 92.80 ± 5.32 kJ mol−1. Unexpectedly, the entropy of activation for reductive elimination is close to zero, and slightly negative, at −0.167 ± 2.64 J K−1 mol−1, rather than the positive figure that could be expected for a reductive elimination reaction.
Although coordination of an external Lewis base to 1 does not promote reductive elimination of Cp*H, we questioned if one of the Cp* ligands of 1 could play this role, particularly since X-ray crystallography reveals that the two Cp* ligands of 1 adopt η2 and η3 coordination modes.21 A shift to higher hapticity of one Cp* ligand could explain the slightly negative entropy of activation for reductive elimination. An alternative explanation could be an ionic-type mechanism involving the dissociation of a Cp*− ligand to form a transient [Cp*AlH]+ species, with solvent ordering around the charged intermediates being responsible for the negative entropy of activation.32 We examined the reductive elimination of Cp*H from Cp*2AlH using DFT (Fig. 2) in order to better understand the mechanism.
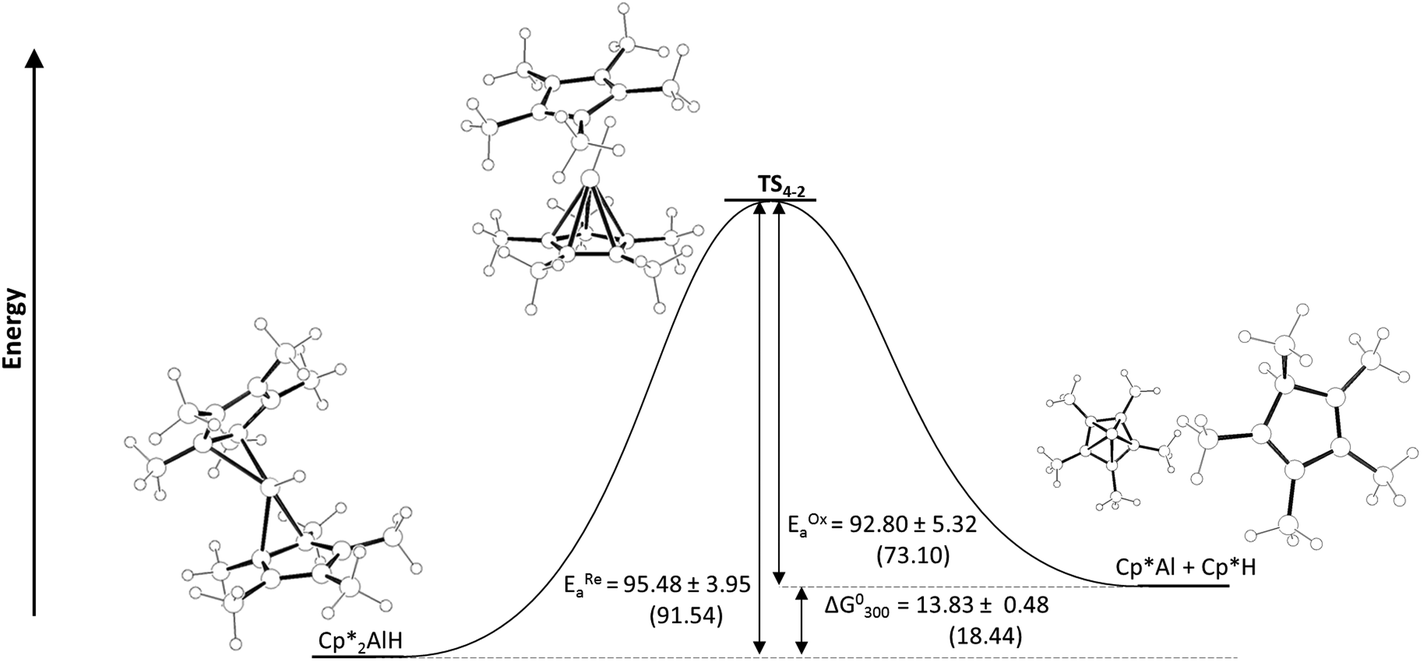 | ||
| Fig. 2 Potential energy diagram with energies (theoretical) stated in kJ mol−1. Calculated energies predicated at the BP86/def-TZVPP level of theory using the BP86/def2-SVP optimised geometries (shown). | ||
Geometry optimisations were performed for compounds 1, 2, and Cp*H and the transition state that links them (geometries were optimised at the BP86/def2-SVP level of theory, and confirmed as minima by frequency calculations (ref to ESI†)). The transition state for reductive elimination of Cp*H from 1, TS1–2 was identified by a transition state search at the BP86/def2-SVP level of theory. Energies were calculated at the BP86/def2-TZVPP level of theory. The calculated geometries of 1 and 2, are consistent with experimental observations, and predicted ΔG°300 and activation barriers for reductive elimination of Cp*H from 1 are in excellent agreement with those determined experimentally (ΔG°300 = +18.44 vs. +13.83 ± 0.48 kJ mol−1; EREa = 91.54 vs. 95.48 ± 3.95 kJ mol−1).
The geometry of TS1–2 is informative in explaining why base coordination to 1 inhibits reductive elimination of Cp*H. In TS1–2, one Cp* ligand is η5 with C–Al distances essentially identical to those in Cp*Al (average C–Al distance for η5 Cp* in TS1–2 = 2.358 Å; Cp*Al = 2.355 Å). This interaction can not take place whilst an external Lewis base is coordinated.
Although the geometry around the departing Cp*(H) ring is planar in TS1–2, there is a clear interaction between a Cp* ring carbon and the Al–H functionality, with a C–H distance (1.461 Å) almost suggestive of a deprotonation of a Cp*AlH+ species by Cp*−. The calculated Al–H bond distance increases dramatically from 1 to TS1–2 (1.579 to 1.837 Å). Consistent with this, when NPA charges on the Al–H were compared, a substantial depletion of negative charge at the hydride was observed when moving from 1 to TS1–2 (from −0.373 to −0.049). Notably, TS1–2 is very similar to that very recently calculated by Cao and Zhang for the oxidative addition of Cp*H to Roesky's NacNacAlI compound (NacNac = HC[CMeN(2,6-iPr2-C6H3)]2).33
We conclude that Cp*Al, like NacNacAlI, activates acidic C–H bonds via an initial proton transfer from C–H to the aluminium(I) centre. Ligand effects are important: ΔG°298 for oxidative addition of Cp*H to NacNacAlI (calculated by Cao and Zhang to be −100.9 to −108.0 kJ mol−1) is significantly higher than that for Cp*Al (ΔG°300 measured by us to be −13.83 ± 0.48 kJ mol−1). Thus, it seems that Cp* can stabilise AlI more effectively than the NacNac ligand; the aromatisation of the η5 Cp* ligand in 2 almost certainly offsets the thermodynamically unfavourable transformation from AlIII to AlI. In the same way, the aromatisation of the Cp* ligand in TS1–2 lowers the barrier to reductive elimination of Cp*H (which we estimate at 80–90 kJ mol−1) compared to the calculated value for NacNacAlI (167–188 kJ mol−1), rendering the oxidative addition of Cp*H to Cp*Al reversible, when that to NacNacAlI is not. As might be expected, the coordination of strong σ-donors to the aluminium centre of 1 inhibits reductive elimination. This effect is twofold in origin. Firstly, the presence of a strong electron donor substantially stabilises the high(er) oxidation state aluminium centre secondly, coordination inhibits the aromatisation of the Cp* ligands required to enable reductive elimination. The combined effects of the π-donating Cp* ligands and the coordination of strong σ-donors in modulating the AlIII/AlI process is similar to the recently reported effect of strong σ-donors in oxidative addition to germylenes.34 Such ligands not only enable oxidative addition reactivity by narrowing the HOMO/LUMO gap in the low-valent species, but also favour the low oxidation state species by providing increased electron density.
Continued study of reaction mechanisms of (reversible) oxidative addition and reductive elimination in low-valent main-group systems will be essential in developing effective principles for ligand design.
The authors would like to thank the European Commission, the EPSRC and the University of Edinburgh for financial support. This work was supported by a career integration grant, funded by the FP7 Marie Curie Actions of the European Commission (PCIG14-GA-2013-631483). MJC would like to thank Prof. Polly Arnold for helpful discussions.
Notes and references
- R. H. Crabtree, The Organometallic Chemistry of the Transition Metals, John Wiley & Sons Ltd, 6th edn, 2014 Search PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- G. H. Spikes, J. C. Fettinger and P. P. Power, J. Am. Chem. Soc., 2005, 127, 12232–12233 CrossRef CAS PubMed.
- G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439–442 CrossRef CAS PubMed.
- Y. Peng, M. Brynda, B. D. Ellis, J. C. Fettinger, E. Rivard and P. P. Power, Chem. Commun., 2008, 6042–6044 RSC.
- Y. Peng, J. Guo, B. D. Ellis, Z. Zhu, J. C. Fettinger, S. Nagase and P. P. Power, J. Am. Chem. Soc., 2009, 131, 16272–16282 CrossRef CAS PubMed.
- J. Li, C. Schenk, C. Goedecke, G. Frenking and C. Jones, J. Am. Chem. Soc., 2011, 133, 18622–18625 CrossRef CAS PubMed.
- L. Zhao, F. Huang, G. Lu, Z. Wang and P. V. R. Schleyer, J. Am. Chem. Soc., 2012, 134, 8856–8868 CrossRef CAS PubMed.
- T. J. Hadlington, M. Hermann, J. Li, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2013, 52, 10199–10203 CrossRef CAS PubMed.
- A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- A. J. Arduengo, C. A. Stewart, F. Davidson, D. A. Dixon, J. Y. Becker, B. S. A. Culley and M. B. Mizen, J. Am. Chem. Soc., 1987, 109, 627–647 CrossRef CAS.
- S. M. McCarthy, Y. Lin, D. Devarajan, J. W. Chang, H. P. Yennawar, R. M. Rioux, D. H. Ess and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 4640–4650 CrossRef CAS PubMed.
- T. P. Robinson, D. M. De Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- A. Pal and K. Vanka, Inorg. Chem., 2016, 55, 558–565 CrossRef CAS PubMed.
- G. Zeng, S. Maeda, T. Taketsugu and S. Sakaki, Angew. Chem., Int. Ed., 2014, 53, 4633–4637 CrossRef CAS PubMed.
- D. Kummer and H. Koester, Angew. Chem., Int. Ed. Engl., 1969, 8, 878–879 CrossRef CAS.
- F. Meyer-Wegner, A. Nadj, M. Bolte, N. Auner, M. Wagner, M. C. Holthausen and H.-W. Lerner, Chemistry, 2011, 17, 4715–4719 CrossRef CAS PubMed.
- C. P. Sindlinger, A. Stasch, H. F. Bettinger and L. Wesemann, Chem. Sci., 2015, 6, 4737–4751 RSC.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- T. Chu, I. Korobkov and G. I. Nikonov, J. Am. Chem. Soc., 2014, 136, 9195–9202 CrossRef CAS PubMed.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. Fischer, Chem. Commun., 2013, 49, 2858–2860 RSC.
- L. Jafarpour, E. D. Stevens and S. P. Nolan, J. Organomet. Chem., 2000, 606, 49–54 CrossRef CAS.
- X. Li, J. Su and G. H. Robinson, Chem. Commun., 1996, 2683–2684 RSC.
- P. Jutzi, Adv. Organomet. Chem., 1986, 217–295 CrossRef CAS.
- P. Jutzi, Chem. Rev., 1986, 86, 983–996 CrossRef CAS.
- J. D. Gorden, C. L. B. Macdonald and A. H. Cowley, J. Organomet. Chem., 2002, 643–644, 487–489 CrossRef CAS.
- J. M. Pietryga, J. D. Gorden, C. L. B. Macdonald, A. Voigt, R. J. Wiacek and A. H. Cowley, J. Am. Chem. Soc., 2001, 123, 7713–7714 CrossRef CAS PubMed.
- M. Wu, M. A. M. Gill, L. Yunpeng, L. Falivene, L. Yongxin, R. Ganguly, L. Cavalloc and F. García, Dalton Trans., 2015, 44, 15166 RSC.
- When we attempted treating (Cp*Al)4 with NHCs to directly synthesise base-coordinated aluminium(I) species Cp*Al·NHC, we also observed no reaction.
- This differs from the findings of Fischer in ref. 21, who reports (on the basis of DFT calculations) that this reaction is slightly exergonic.
- J. Gauss, U. Schneider, R. Ahlrichs, C. Dohmeier and H. Schnockel, J. Am. Chem. Soc., 1993, 115, 2402–2408 CrossRef CAS.
- P. K. Byers, A. J. Canty, M. Crespo, R. J. Puddephatt and J. D. Scott, Organometallics, 1988, 7, 1363–1367 CrossRef CAS.
- X. Zhang and Z. Cao, Dalton Trans., 2016, 45, 10355–10365 RSC.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2016, 22, 11685 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1489247–1489249. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02698b |
| ‡ For full experimental, spectroscopic and computational details, please refer to the ESI.† |
| This journal is © The Royal Society of Chemistry 2016 |