
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMultifunctional Eu-doped NaGd(MoO4)2 nanoparticles functionalized with poly(L-lysine) for optical and MRI imaging†
Mariano
Laguna
a,
Nuria O.
Nuñez
*a,
Verónica
Rodríguez
a,
Eugenio
Cantelar
b,
Grazyna
Stepien
c,
María Luisa
García
d,
Jesús M.
de la Fuente
e and
Manuel
Ocaña
a
aInstituto de Ciencia de Materiales de Sevilla, CSIC, Américo Vespucio 49, 41092, Isla de la Cartuja, Sevilla, Spain. E-mail: nurianu@icmse.csic.es
bDpto. Física de Materiales, Universidad Autónoma de Madrid, Facultad de Ciencias, Francisco Tomás y Valiente 7, 28049 – Madrid, Spain
cInstituto de Nanociencia de Aragón, University of Zaragoza. Edificio I+D, C/Mariano Esquillor s/n, 50018, Zaragoza, Spain
dAndalusian Centre for Nanomedicine and Biotechnology, BIONAND Parque Tecnológico de Andalucía, Málaga, Spain
eInstituto de Ciencia de Materiales de Aragón, CSIC/University of Zaragoza, Edificio I+D, C/Mariano Esquillor s/n, 50018, Zaragoza, Spain
First published on 23rd September 2016
Abstract
A method for the synthesis of non-aggregated and highly uniform Eu3+ doped NaGd(MoO4)2 nanoparticles is reported for the first time. The obtained particles present tetragonal structure, ellipsoidal shape and their size can be varied by adjusting the experimental synthesis parameters. These nanoparticles, which were coated with citrate anions and functionalised with PLL, have also been developed in order to improve their colloidal stability in physiological medium (2-(N-morpholino)ethanesulfonic acid, MES). A study of the luminescent dynamics of the samples as a function of the Eu doping level has been conducted in order to find the optimum nanophosphors, whose magnetic relaxivity and cell viability have also been evaluated for the first time for this system, in order to assess their suitability as multifunctional probes for optical (in vitro) and magnetic bioimaging applications.
1. Introduction
Luminescent materials based on rare-earth (RE) compounds, consisting of a crystalline host doped with a small amount of lanthanide (Ln) cations, have recently attracted much attention because of their important applications in several fields of science and technology. Of particular interest is their use in many biotechnological applications including biosensing and biological imaging,1,2 to mention a few. Such interest mainly arises from the lower toxicity and higher chemical and optical stability of these phosphors when compared with other luminescent materials (quantum dots, organic dye-based systems, etc.).The luminescent characteristics of this kind of material are determined by the nature of the doping cations. Thus, down conversion (emissions at lower energy than that of excitation) or up-conversion (emissions at higher energy than that of excitation) phosphors can be obtained by a suitable selection of the emitting cations. In the former case, the most appropriate Ln doping cations emitting in the visible region for bio-applications are Eu3+, whose most intense emissions lie in the red region, where the auto-fluorescence of tissues is minimized in this wavelength range.3 It is also important to mention that the presence of Gd3+ cations in these phosphors is also of high interest due to the large magnetic moment and nanosecond scale electronic relaxation time of these cations, which confers to such materials an additional functionality as contrast agents for magnetic resonance imaging (MRI).4
For most biotechnological applications, the size and shape of these multifunctional materials are very important parameters to be controlled since such characteristics affect some physical properties, including luminescence and magnetic relaxivity5 and, consequently, their performance for optical and magnetic imaging. Particle size has also a strong influence on other important processes involved in their in vivo applications such as the circulation behaviour of nanoparticles in the body, their biodistribution and excretion pathway.6 In addition, such nanoparticles must be obviously non-toxic and present high colloidal stability in physiological media. To meet the latter criteria, the modification of the nanoparticle surface with ligands having different functional groups (carboxylate, amino, imino, etc.) is usually required, which helps to achieve a high colloidal stability of the nanoparticles by increasing the surface charge or by steric hindrance7 and also provides anchors for the further bioconjugation of the nanoparticles with molecules of biological interest.8 Several organic polymers have been tested for the functionalization of nanoparticles including poly(L-lysine) (PLL), a positively charged amino acid polymer having both the amine (–NH2) and carboxylic acid (–COOH) groups. This polymer offers several advantages, such as good biocompatibility, relatively good solubility in water and plentiful active amino groups,9 which makes it very interesting for the development of carriers for gene delivery.10 It has also been shown that materials with high levels of positive charge are capable of conferring antimicrobial properties because these cationic species are able to disrupt bacterial membranes.11,12
A large variety of RE-based compounds have been considered as a host matrix, including oxides,13 phosphates,14 vanadates,15 and fluorides.16 Molybdate compounds have also been shown to be very advantageous hosts due to their ability to absorb energy in the UV region and further transfer such energy to some Ln3+ cations, which considerably increases the intensity of their emissions.17 However, to the best of our knowledge, very little attention has been paid to these systems, including those systems based on NaGd(MoO4)2, which are of special interest owing to their additional magnetic imaging capability associated with the Gd ions. The main reason might be the scarce number of methods for the synthesis of nanoparticles of RE molybdate materials available in the literature. In fact, to our knowledge, the synthesis of uniform NaGd(MoO4)2 nanoparticles, either undoped or doped with Ln cations, has not been yet achieved.
Herein, we report for the first time in the literature a method for the synthesis of non-aggregated and highly uniform Eu3+ doped NaGd(MoO4)2 nanoparticles with a tetragonal structure, which are surface-modified with citrate anions. This procedure, based on a homogeneous precipitation process in a polyol-based medium, has been shown to be quite suitable to obtain nanoparticles of several RE-based compounds18–20 due to the ability of these alcohols to limit particle growth. The obtained particles present ellipsoidal shape and their size can be varied by adjusting the experimental synthesis parameters. A procedure for the further functionalization of these nanophosphors with PLL has also been developed in order to improve their colloidal stability in physiological medium (2-(N-morpholino)ethanesulfonic acid, MES). A study of the luminescent dynamics of the samples as a function of the Eu doping level has been conducted in order to find the optimum nanophosphors, whose magnetic relaxivity and cell viability have also been evaluated for the first time for this system, in order to assess their suitability as multifunctional probes for optical and magnetic bioimaging applications.
2. Experimental details
2.1. Materials
Gadolinium(III) nitrate hexahydrate (GdNO3·6H2O, Aldrich, 99.9%) and europium(III) nitrate pentahydrate (Eu(NO3)3·5H2O, Aldrich, 99.9%) were selected as Ln precursors and sodium molybdate (Na2MoO4, Aldrich, ≥98%) was used as the molybdate source. Sodium citrate (Na3C6O7H5·2H2O, Aldrich, 99.5%) was also needed as a complexing agent whereas ethylene glycol (EG, Aldrich, ≥99.5%)–water (Milli-Q) mixtures were used as solvents. For the functionalization of the nanoparticles with poly(L-lysine) (PLL), a solution 0.1% w/v in water from Aldrich was used. Sodium hydroxide (NaOH, Aldrich, BioXtra, ≥98%) was used to adjust pH.2.2. Synthesis of NaGd(MoO4)2 nanoparticles
The synthesis of NaGd(MoO4)2 nanoparticles was carried out under different experimental conditions, which are detailed in Table 1. The experimental procedure was as follows. The required amounts of Gd(NO3)3 and sodium citrate were dissolved in ethylene glycol (2.5 cm−3) under magnetic stirring while heating the vial at ∼80 °C to facilitate the dissolution process. The resulting solution was left to cool down to room temperature. In a separate vial, the desired amount of Na2MoO4 was dissolved in a certain volume of Milli-Q water (from 1 to 1.5 cm3) and then ethylene glycol was added to this aqueous solution until completing 2.5 cm3 of mixture. The latter was then added to the solution containing the Ln–citrate complex, under magnetic stirring, and the mixture was quickly introduced into an oven preheated at the desired temperature, in which it was finally aged for 20 h in tightly closed test tubes. After aging, the resulting dispersions were cooled down to room temperature, centrifuged to remove the supernatants, the precipitates were washed twice with ethanol and finally dispersed in Milli-Q water. For some analyses, the powders were dried at room temperature.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O mixtures as solvent. The standard deviations (SD) are shown in parentheses
:H2O mixtures as solvent. The standard deviations (SD) are shown in parentheses
| [Gd(NO3)3] mol dm−3 | [Na2MoO4] mol dm−3 | MoO42−/Gd3+ (mol ratio) | EG:H2O v/v (ml) |
T (°C) | Citrate/Gd3+ (mol ratio) | Particle shape | Particle size/TEM (nm) |
|---|---|---|---|---|---|---|---|
| 0.02 | 0.1 | 5 | 4:1 |
120 | — | Aggregated spheroids | — |
| 0.02 | 0.1 | 5 | 4:1 |
120 | 1 | Irregular | — |
| 0.02 | 0.1 | 5 | 4:1 |
120 | 2 | Ellipsoidal | 120(10) × 60(6) |
| 0.02 | 0.1 | 5 | 4:1 |
120 | 5 | No precipitation | — |
| 0.01 | 0.05 | 5 | 4:1 |
120 | 2 | No precipitation | — |
| 0.04 | 0.2 | 5 | 4:1 |
120 | 2 | Aggregated ovoids | — |
| 0.02 | 0.1 | 5 | 4:1 |
80 | 2 | No precipitation | — |
| 0.02 | 0.1 | 5 | 4:1 |
150 | 2 | Irregular | — |
| 0.02 | 0.1 | 5 | 5:0 |
120 | 2 | No precipitation | — |
| 0.02 | 0.1 | 5 | 3.5:1.5 |
120 | 2 | Ellipsoidal | 265(12) × 140(8) |
| 0.05 | 0.1 | 2 | 4:1 |
120 | 2 | Ellipsoidal | 70(5) × 35(6) |
| 0.02 | 0.04 | 2 | 4:1 |
120 | 2 | No precipitation | — |
2.3. Synthesis of Eu-doped NaGd(MoO4)2 nanoparticles
For the synthesis of Eu-doped NaGd(MoO4)2 nanoparticles, we proceeded as described above for the case of the undoped system but incorporating the desired amount of Eu to the starting Gd(NO3)3 solution. The Eu/(Eu + Gd) molar ratio was varied from 2 to 15% in order to investigate the effect of this doping level on the luminescent properties of the precipitated nanoparticles.For cytotoxicity assays, the pristine Eu-doped NaGd(MoO4)2 nanoparticles were previously sterilized by filtration through a 0.2 μm Millipore filter.
2.4. Functionalization with poly(L-lysine)
For functionalization, poly(L-lysine) solutions (0.1 mg mL−3) were prepared with thepH adjusted to 10 by the addition of NaOH. A weighted amount of the Eu-doped NaGd(MoO4)2 nanoparticles previously synthesized was added to such solutions so that their final concentration was 0.2 mg mL−1. The resulting dispersions, with a total volume of 10 mL, were maintained under magnetic stirring for 1 h. After this treatment, the so functionalized nanoparticles were washed several times with distilled water by centrifugation and finally redispersed in Milli-Q water.For the in vitro toxicity studies, the synthesized nanophosphors were first autoclaved at 120 °C per bar and then functionalized under sterile conditions in a laminar flow hood according to the protocol described above. The autoclavation process did not have influence on nanoparticle morphology nor stability (data not shown).
2.5. Characterization
The shape of the nanoparticles was examined by transmission electron microscopy (TEM, Philips 200CM). For this, a droplet of an aqueous suspension of the samples was deposited on a copper grid coated with a transparent polymer and dried. The particle size distributions were obtained from the micrographs by counting about one hundred particles, as well as by dynamic light scattering (DLS) measurements conducted using a Malvern Zetasizer Nano-ZS90 apparatus. The same equipment was employed to obtain Z potential values. The colloidal stability of nanoparticle suspensions (nanoparticle concentration = 0.5 mg mL−3) in milliQ water at pH 6.8 and in 50 mM MES solutions at pH 6.5, was monitored by analyzing the particle size distribution (hydrodynamic diameter, dh) obtained from dynamic light scattering (DLS) measurements (Zetasizer NanoZS90, Malvern). The quantitative composition of the samples was analyzed by inductively coupled plasma atomic emission spectroscopy (ICP-AES, Horiba Jobin Yvon, Ultima 2). The crystalline structure of the prepared nanoparticles was identified by X-ray diffraction (XRD, Panalytical X'Pert Pro with an X-Celerator detector). The XRD data were collected at intervals of 0.02° (2θ) and an accumulation time of 4000 s. The infrared spectra (FTIR) of the nanoparticles diluted in KBr pellets were recorded in a Jasco FT/IR-6200 Fourier transform spectrometer. Thermogravimetric analyses (TGA) were performed in air at a heating rate of 10 °C min−1, using a Q600 TA Instrument.For an appropriate comparison of the luminescent properties of these nanophosphors containing different Eu doping levels, aqueous suspensions of the samples having the same concentration (0.5 mg cm−3) were prepared. The excitation and emission spectra of these dispersed samples were measured in a Horiba Jobin Yvon spectrofluorimeter (Fluorolog FL3-11) operating with a slit of 1.5 nm. Both spectra were corrected by the spectral response of the equipment and the intensity of the excitation source. The lifetime measurements for the Eu-doped nanophosphors were obtained for powdered samples under pulsed excitation at 532 nm by using the second harmonic of a Nd:YAG laser (Spectra Physics model DCR 2/2A 3378) with a pulse width of 10 ns and a repetition rate of 10 Hz. The fluorescence was analysed through an ARC monochromator model SpectraPro 500-i and then detected synchronously with an EMI-9558QB photomultiplier and recorded by a Tektronix TDS420 digital oscilloscope. The photograph showing the luminescence of the aqueous suspension of the Eu-doped nanophosphors was taken under illumination with ultraviolet radiation (λ = 312 nm).
1H NMR relaxation times T1 and T2 were measured at 1.5 T in a Relaxiometer (Bruker Minispec spectrometer) at different concentrations of nanoparticles in water at 298 K. Aqueous solutions of nanoparticles at concentrations ranging from 0.01 mM to 0.2 mM in Gd as determined by Plasma ICP (ICP-AES, Horiba Jobin Yvon, Ultima 2) were prepared and transferred into an 8-well chamber (Ibidi, Martinsried, Germany). T1 values were determined using a saturation-recovery spin-echo sequence (TR values from 50 ms to 10 s) and T2 values using a 64-echo Carr-Purcell-Maiboom-Gill (CPMG) sequence (TE values from 7.5 ms to 640 ms). Relaxivities (r1, r2) were obtained from the slopes of the curves 1/T1 or 2vs. the concentration of Gd(III) expressed in mM. T1 and T2 relaxivities were also measured at high field (9.4 T) on a Bruker Biospec MRI system equipped with 400 mT m−1 field gradients and a 40 mm quadrature bird-cage resonator at 298 K. Phantoms were obtained at 9.4 Tesla in a BrukerBiospec MRI.
2.6. Cytotoxicity assay
Cell viability and proliferation were analyzed by the MTT colorimetric assay21 using Vero cells (monkey kidney epithelial cells) cultured in Dulbecco's Modified Eagle's Medium (DMEM), supplemented with 10% fetal bovine serum (FBS), 5% glutamine and 5% penicillin/streptomycin. Cell cultures were incubated at 37 °C under a 5% CO2 atmosphere. For the cytotoxicity assay 1 × 104 cells were seeded in each well of 96-well plates and cultured for 24 h. After 24 h, the medium was replaced with a fresh medium containing the different types of nanoparticles in varying concentrations. After cultivation again for 24 h, the cells were thoroughly washed with DPBS and 20 μL of MTT dye solution (5 mg mL−1 in PBS) was added to each well. Following 4 h of incubation at 37 °C, formazan crystals were dissolved with 200 μL of DMSO. The absorbance of each well was determined on a microplate reader (Thermo Scientific Multiskan GO UV/Vis microplate spectrophotometer) at λ = 740 nm. The spectrophotometer was calibrated to zero absorbance using a culture medium without cells. The relative cell viability (%) related to control wells containing a cell culture medium without nanoparticles was calculated by [A]test/[A]control × 100. Each measurement was repeated at least five times to obtain the mean values and the standard deviation.3. Results and discussion
3.1. Undoped NaGd(MoO4)2 nanoparticles
It has been well documented that homogeneous precipitation is a suitable procedure for the formation of uniform particles in solution. Such a homogeneous process can be achieved by a controlled release of anions or cations, so that the kinetics of precipitation is similar throughout the solution.22 In our case, we have selected the controlled release of Gd3+ cations through the formation of a Ln3+–Cit3− complex in the initial solution, which can be thermally decomposed slowly releasing the Ln3+ cations involved in the formation of the solid phase.23 It is also known that to obtain uniform particles by these procedures, a specific kinetic of precipitation is required, which must be found through an appropriate adjustment of the experimental parameters (reagent concentrations, temperature and nature of solvent). In our case, this task was first addressed for the undoped NaGd(MoO4)2 system, for simplicity. The results of this study are summarized in Table 1.In this table, it can be observed that the formation of uniform particles only took place under a very restrictive set of conditions. In particular, uniform nanoparticles with ellipsoidal shape (Fig. 1a) and narrow size distribution (120 ± 10 × 60 ± 6 nm, Table 1) were obtained by aging at 120 °C for 20 h, a 0.02 mol dm−3 Gd(III) and 0.1 mol dm−3 Na2MoO4 (MoO42−/Gd3+ ratio = 5) solution having citrate ions (citrate/Gd3+ mol ratio = 2) in an EG:H2O mixture (volumetric ratio 4:1). The formation of the Gd3+–citrate complex is essential for the formation of uniform particles since in the absence of citrate, heterogeneous ovoids with a certain degree of aggregation were obtained (Fig. S1a in the ESI†). This was the expected result, since without the formation of the Gd–citrate complex, the controlled release of cations into solution does not take place, not achieving the conditions required for homogeneous precipitation. The amount of citrate anions added is also critical since for a large excess of citrate ions (citrate/Gd3+ mol ratio = 5), no appreciable precipitation was detected, indicating that the decomposition of the Ln–citrate complex does not occur or that the kinetics of precipitation is very low under these conditions. On the other hand, when the citrate/Gd3+ mol ratio was diminished from 2 to 1, aggregated and more heterogeneous particles were obtained (Fig. S1b†), which suggests that such an amount of citrate is insufficient to achieve a complete chelating interaction between the citrate and rare-earth ions resulting in a heterogeneous kinetic of precipitation.
 | ||
| Fig. 1 TEM images of the nanoparticles obtained under the following experimental conditions: (a) MoO42−/Gd3+ mol ratio = 5 ([Gd(III)] = 0.02 M, [Na2MoO4] = 0.1 M); (b) volumetric ratio EG:H2O (3.5:1.5) and (c) MoO42−/Gd3+ mol ratio = 2 ([Gd(III)] = 0.05 M, [Na2MoO4] = 0.1 M). In all cases the temperature and the mol ratio citrate/Gd3+ remained constant as described in Table 1. | ||
Another factor that plays a decisive role in the formation of uniform ellipsoidal nanoparticles is the presence of water in the reaction medium. Thus, when using pure EG as a solvent, no appreciable precipitation was observed (Table 1). This behavior may be ascribed to the high viscosity of the solvent that slows down the diffusion processes implicated in the solid phase formation and particle growth. In agreement with this interpretation would be the increase of the particle size from 120 × 60 nm to 265 × 140 nm observed when the EG:water volumetric ratio was reduced from 4:1 to 3.5:1.5 (Table 1, Fig. 1b), which involves a decrease of viscosity.
The formation of uniform ellipsoids also occurs within a narrow range of precursor concentrations in the starting solutions. Thus, by decreasing the concentration of Gd(III) from 0.02 mol dm−3 to 0.01 mol dm−3 (keeping constant the MoO42−/Gd3+ ratio and the other synthesis parameters), precipitation was not detected indicating that the minimum concentration for nucleation was not achieved, while by increasing such a magnitude from 0.02 mol dm−3 to 0.04 mol dm−3, heterogeneous particles were obtained (Fig. S1c†). In the latter case, due to the high concentration of precursors, several nucleation events may take place, which would lead to heterogeneous systems, according to the classical model proposed by LaMer and Dinegar.24 Moreover, it was observed that with the increase of the temperature to 150 °C, heterogeneous particles were obtained (Fig. S1d†), whereas when the temperature was lowered to 80 °C no appreciable precipitation was observed (Table 1). These changes observed when varying the synthesis temperature must also be ascribed to variations in the kinetics of precipitation, which affect the nucleation and growth processes.
Finally, the effects of the MoO42−/Gd3+ mol ratio on the precipitation process were also investigated. It was found that keeping constant all the conditions required to obtain the particles shown in Fig. 1a, but reducing such a mol ratio from 5 to 2 by increasing the amount of Gd3+ from 0.02 to 0.05 mol dm−3, a significant decrease in the mean particle size from 120 × 60 nm to 70 × 35 nm was observed (Table 1), keeping the same morphology (Fig. 1c). However and surprisingly, no precipitation was detected for the same value of the MoO42−/Gd3+ mol ratio (2) when it was achieved by reducing the amount of molybdate ions from 0.1 to 0.04 mol dm−3. It should be noted that the total amount of Na+ ions present in the solution (coming from both molybdate and citrate precursors) is much lower in the latter case (0.2 mol dm−3) than in the other experiments conducted with uniform ellipsoids (≥0.32 mol dm−3). This observation seems to suggest that the precipitation of the NaGdMoO4 phase under the studied conditions also requires a large excess of Na+ ions in the initial solutions.
According to XRD, all samples consisting of uniform ellipsoids were crystalline, having a tetragonal NaGd(MoO4)2 structure (ICDD file: 25-0828), as illustrated in Fig. 2 for the smallest particles (Fig. 1c).
 | ||
| Fig. 2 X-ray diffraction patterns obtained for the nanoparticles shown in Fig. 1c. The reference pattern corresponding to the tetragonal phase of NaGd(MoO4)2 is also included. | ||
3.2. Ln-doped NaGd(MoO4)2 nanoparticles
In order to synthesize Eu-doped nanophosphors, we addressed the doping of the NaGd(MoO4)2 nanoparticles with Eu3+ cations using the conditions involved in the synthesis of the sample shown in Fig. 1c composed by the smallest particles here obtained (70 × 35 nm, Table 1), which are more appropriate for bioapplications. For doping, the total Ln concentration (0.05 mol dm−3) was kept constant, whereas the doping level was varied between 2 and 15% (Eu/Eu + Gd mol ratio) in order to optimize the luminescent properties of these nanophosphors.It was found that the addition of Eu to the starting solutions did not produce noticeable changes in the morphological characteristics of the precipitated nanoparticles whose shape and size were similar to those obtained for the undoped systems (Fig. S2†). The XRD patterns of the doped samples (Fig. S3 in the ESI†) were also similar to that of undoped NaGd(MoO4)2, indicating that they also crystallized in the tetragonal phase.
The success of the doping procedure was confirmed by ICP analyses, which showed that the experimental Eu/(Eu + Gd) ratio obtained for all doped samples was very similar to the nominal values (Table 2).
| Eu/(Eu + Gd) | |
|---|---|
| Nominal [%] | ICP [%] |
| 2 | 2.05 |
| 5 | 5.5 |
| 10 | 10.8 |
| 15 | 15.2 |
FTIR analyses gave additional information on the composition of the doped nanoellipsoids. Thus, as shown for the Eu(5%):NaGd(MoO4)2 nanoparticles, their infrared spectrum (Fig. 3) displayed bands corresponding to the absorbed water (3400 and 1630 cm−1) and to the Mo–O stretching vibration of the MoO42− anion (<1000 cm−1)25 along with some other bands (1590–1400 cm−1 region), which can be associated with the carboxylate groups coming from the adsorbed citrate ions.26 Therefore, the method of synthesis developed in this work produces uniform NaGd(MoO4)2 nanoparticles coated with citrate ions, which hereafter will be labeled as NaGd(MoO4)2@citrate. Thermogravimetric analyses performed for these nanoparticles (Fig. 4) gave an estimation on the extent of coating. Thus, a weight loss of ∼5 wt% was observed in the temperature range of 25–250 °C, due to the elimination of absorbed water, whereas an additional weight loss of ∼4.5 wt% took place at higher temperatures (250–800 °C), which can be associated with the decomposition of the adsorbed citrate ions.
 | ||
| Fig. 3 Infrared spectra obtained for the Eu doped-NaGd(MoO4)2 nanoparticles before and after functionalization with PLL. The infrared spectrum of sodium citrate and PLL are also included for comparison. | ||
 | ||
| Fig. 4 TGA curves obtained for the NaGd(MoO4)2 nanoparticles before and after functionalization with PLL. | ||
The photoluminescent properties of Eu:NaGd(MoO4)2@citrate nanoparticles are displayed in Fig. 5. The excitation spectrum recorded for the nanoparticles with a Eu content of 5%, monitored at the most intense Eu3+ emission band (614 nm), is presented in Fig. 5a. As is observed, this spectrum displayed a strong broad band whose maximum intensity lies beyond the detection limit of our spectrofluorimeter (∼250 nm). This feature has been reported to correspond to an energy transfer (ET) process from the excited levels of the MoO42− group to the Eu3+ cations.21 The bands resulting from the direct excitation of Eu3+ (the most intense being that appearing at 393 nm, which is due to the f–f electronic transition characteristics of the Eu3+ ions),27 were also found (inset in Fig. 5a), although they were much weaker.
 | ||
| Fig. 5 (A) Excitation spectrum (λem = 614 nm) of the citrate coated-Eu(5%):NaGd(MoO4)2 nanoparticles. Inset in (A) magnification of the 350–480 nm region for a better visualization of the electronic transitions corresponding to the 4f6 configuration of Eu3+ ions. (B) Emission spectra recorded for the same nanoparticles using different excitation wavelengths. Inset in (B) photograph of the nanoparticles in water suspension taken under UV illumination at λ = 312 nm. (C) Emission spectra (λexc = 250 nm) of the Eu-doped NaGd(MoO4)2@citrate nanoparticles with a different Eu content; and (D) temporal evolution of the 5D0 → 7F2 luminescence (614 nm) for the NaGd(MoO4)2@citrate nanoparticles with increasing amounts of Eu3+ (excitation at 532 nm). | ||
Upon excitation within the ET band (λex = 250 nm), the emission band characteristics of the electronic transitions from the 5D1 to 7F1 (535 nm) and from the 5D0 to the 7FJ (J = 1, 2, 3, and 4) levels of Eu3+ (λem > 550 nm) were detected (Fig. 5b). It is well known that the relative intensities of 5D0–7F2 (614 nm) and 5D0–7F1 (593 nm) transitions strongly depend on the local symmetry of the Eu3+ ions, since the latter transition is magnetic-dipole allowed and insensitive to the local environment of the Eu(III) cations, whereas the 5D0–7F2 electric-dipole transition becomes the strongest one when the Eu local symmetry is lowered.28 In our case, the emission originating from the 5D0–7F2 transition is clearly dominant, indicating that the Eu3+ occupies a crystallographic site in the host lattice with no inversion center.18 The much higher intensity of the 5D0–7F2 transition when compared with the one of the other bands gives rise to a visible red emission with high color purity, as is illustrated in the inset of Fig. 5b.
In Fig. 5b, it can also be observed that the emission intensity observed after excitation through the ET band was much higher than that corresponding to the direct excitation of Eu3+ cations (393 nm), indicating that the former excitation path is more favorable.
To study the effects of the Eu content on the luminescence of the Eu–NaGd(MoO4)2@citrate samples, they were excited through the energy transfer band (λex = 250 nm). In Fig. 5c, it can be observed that the increase of the Eu doping level from 2% to 5% gave rise to an important increase of the intensity of the emission bands as a consequence of the increase in the number of emission centers. However, such intensity decreased significantly when the Eu content was increased to 10% suggesting the presence of a concentration quenching effect.
In order to gain more information on the luminescence efficiency of these samples, lifetime measurements were also carried out. The decay curves obtained for the 5D0 → 7F2 transition (614 nm), after pulsed excitation at λex = 532 nm, as a function of the Eu concentration are shown in Fig. 5d. Under these conditions, the Eu3+ ions are excited to the 5D1 multiplet, and then they decay partially non-radiatively to the 5D0 level, from where the emissions to the lower 5FJ ground state multiplets are observed.29 In all cases, the decays are not single exponentials, but can be fitted by using a bi-exponential temporal dependence according to eqn (1):
| I(t) = I01exp(−t/τ1) + I02exp(−t/τ2) | (1) |
| Eu/(Eu + Gd) nominal [%] | I 01 | τ 1 (ms) | I 02 | τ 2 (ms) | <τ> (ms) |
|---|---|---|---|---|---|
| 2 | 0.35 | 0.33 | 0.65 | 0.80 | 0.70 |
| 5 | 0.40 | 0.48 | 0.60 | 0.93 | 0.79 |
| 10 | 0.30 | 0.24 | 0.70 | 0.66 | 0.59 |
| 15 | 0.29 | 0.17 | 0.71 | 0.52 | 0.47 |
Considering the non-exponential character of the decay curves, they are usually33 characterized by an average decay time, defined as  , which in the case of a bi-exponential behaviour becomes:
, which in the case of a bi-exponential behaviour becomes:
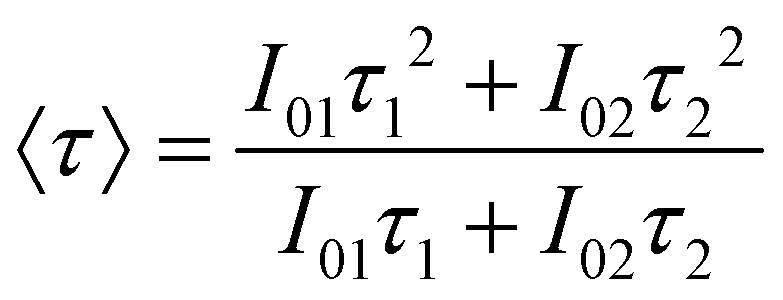 | (2) |
As is observed, these values are dependent on the Eu concentration, the sample with an Eu content of 5% showing the higher average lifetime value (τ = 0.79 ± 0.04 ms), which is in agreement with the optimum doping level suggested by the emission spectra. For Eu concentrations above 5%, the average lifetime values experienced a reduction (Table 3), indicating that at these doping levels concentration quenching becomes operative by the increase of the amount of luminescent centers. As is shown in Fig. S5,† the concentration dependence of the lifetime can be fitted considering a lifetime reduction from the low concentration value (τ0) due to a quenching term, Wq, proportional to the square of Eu concentration:  , a characteristic dependence also found in other materials.34,35 It must be mentioned that the lifetime values obtained for our optimum sample is much higher than those reported in the literature for tetragonal Eu:NaGd(MoO4)2 phosphors (≤0.47 ms),23,36,37 which might suggest the higher luminescence efficiency of our nanophosphors.
, a characteristic dependence also found in other materials.34,35 It must be mentioned that the lifetime values obtained for our optimum sample is much higher than those reported in the literature for tetragonal Eu:NaGd(MoO4)2 phosphors (≤0.47 ms),23,36,37 which might suggest the higher luminescence efficiency of our nanophosphors.
For further studies, we selected the sample with the strongest emission, which was that with a europium doping level of 5%.
The longitudinal (r1) and transversal (r2) relaxivity values for the Eu(5%):NaGd(MoO4)2@citrate nanoparticles were measured to evaluate their potential application as an MRI contrast agent. It is well known that the transverse relaxivity (r2) to longitudinal relaxivity (r1) ratio governs the ability of a contrast agent to be used as negative (MRI images become darker on increasing the nanoparticle concentration) or positive (MRI images become brighter on increasing the nanoparticle concentration) agents, the latter being preferred since they permit diminishing the possible interferences with other pathogenic or biological signals.38 Thus, a low r2/r1 ratio (<5) is considered to be ideal for positive MRI contrast agents.39 As is observed in Fig. 6, the r1 value obtained for the Eu(5%):NaGd(MoO4)2@citrate sample at 1.5 T was very high (8.96 mM−1 s−1), whereas r2 was 5.03 mM−1 s−1, resulting in an r2/r1 ratio of 1.78, which is among the lowest values ever reported for Gd containing nanoparticles5,16,40 making our system appropriate as a positive MRI contrast agent.
 | ||
| Fig. 6 Proton relaxivities (r1 and r2) of the citrate coated-Eu(5%):NaGd(MoO4)2 nanoparticles (top) and for the same nanoparticles functionalized with PLL (bottom), both measured at 1.5 T. | ||
Biocompatibility studies were performed by evaluating the cell viability using Vero cells through the MTT assay.21 The metabolic activity and proliferation of fibroblasts were measured after 24 hour culture and the results are shown in Fig. 7. In this figure, it can be seen that the Eu(5%):NaGd(MoO4)2@citrate nanoparticles showed negligible toxicity effects with viability percentages as high as >90% for concentrations up to 0.2 mg mL−1.
 | ||
| Fig. 7 Cytotoxicity profiles for Vero cells of the citrate coated-Eu(5%):NaGd(MoO4)2 nanoparticles (top) and for those further functionalized with PLL (bottom), determined by MTT assay. Percentage of viability of cells was expressed relative to control cells (n = 5). Results are represented as mean ± standard deviations. | ||
The colloidal stability of these nanoparticles was also evaluated to assess their suitability for biotechnological applications. It was observed that the hydrodynamic diameter (dh) obtained from the DLS measurements conducted on water suspensions (pH = 6.8) was only slightly higher (77 nm) than the mean particle size obtained from the TEM pictures (70 × 35 nm), which indicates that the nanoparticles can be well dispersed in water (Fig. 8). However, in MES physiological buffer (50 mM, pH = 6.5), the value of the hydrodynamic diameter (Fig. 8) was clearly much higher (477 nm), which clearly indicates the presence of an aggregation process, suggesting that the citrate coating is insufficient to achieve colloidal stability in such a medium.
 | ||
| Fig. 8 Hydrodynamic diameter (dh) obtained from the DLS measurements in Milli-Q water at pH = 6.8 and in biological buffer media (MES at pH = 6.5) for the citrate coated-nanoparticles (top) and for the PLL-functionalized nanoparticles (bottom). | ||
3.3. PLL-functionalization of Eu:NaGd(MoO4)2@citrate nanoparticles
To improve the colloidal stability of our Eu:NaGd(MoO4)2@citrate nanoparticles in physiological buffer, they were further functionalized with PLL even under sterile conditions. Owing to the presence of positive charges in PLL coming from the amino groups, it is expected that the molecular chains bind the particle surface by means of attractive electrostatic forces between such groups and the carboxylate groups (negatively charged) coming from the citrate anions present on the surface of the pristine nanoparticles.41Information on the PLL functionalization process was obtained from the zeta potential measurements conducted for aqueous suspensions at pH = 6.8. Thus, the zeta potential of the NaGd(MoO4)2@citrate nanoparticles was −39 mV, whereas the value of this magnitude after the treatment with PLL was +29.6 mV. This surface charge reversal clearly suggests the success of such a functionalization process.
The presence of PLL on the surface of the nanoparticles was also substantiated by FTIR spectroscopy. Thus, in addition to the vibrational features due to the non-functionalized nanoparticles, the FTIR spectrum obtained for the PLL-functionalized nanoparticles (Fig. 3) displayed some extra signals that confirm the presence of PLL on the surface of the nanoparticles. In particular, the amide I band due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group stretching mode and the amide II band resulting from C–N stretching and N–H bending modes were observed at 1647 cm−1 and 1542 cm−1, respectively.8,9
O group stretching mode and the amide II band resulting from C–N stretching and N–H bending modes were observed at 1647 cm−1 and 1542 cm−1, respectively.8,9
Thermogravimetric analyses were performed to estimate the amount of PLL species attached to the nanoparticle surface. The TGA curve obtained for the PLL-functionalized nanoparticles is shown in Fig. 4. As is observed, the total mass loss was higher (∼30.5 wt%) than that observed for the pristine citrate coated nanoparticles (∼9.5 wt%). In both cases two steps were detected, which occurred at the same temperature range. The first one (25–250 °C), due to the water release, was slightly higher (∼6.5 wt%) for the PLL coated nanoparticles than that observed before PLL functionalization (∼5 wt%). The second loss took place at 250–800 °C and must be ascribed to the decomposition of the adsorbed organic species, PLL and citrate anions. Such a loss was also much higher after (∼24 wt%) than before (4.5%) PLL coating. Therefore, it can be considered that the amount of PLL species on the surface of the nanoparticles should correspond to an ∼19.5 wt%.
The functionalization with PLL had a positive effect on the colloidal stability of the here synthesized nanophosphors. Thus, the mean hydrodynamic diameter obtained for the Eu:NaGd(MoO4)2@citrate@PLL nanoparticles from the DLS measurements in water (Fig. 8) was only slightly higher (160 nm) than that obtained for the citrate-coated NaGd(MoO4)2 system (77 nm), which can be explained by the existence of PLL on the nanoparticle surface, although the presence of a weak aggregation process cannot be discarded. The colloidal stability of this sample in MES buffer was also evaluated. It was found that the average hydrodynamic diameter of the PLL-functionalized nanoparticles remained unaltered (155 nm) when compared with the value obtained for freshly prepared dispersions in Milli-Q water at pH = 6.8 (160 nm), indicating the absence of significant aggregation also in MES buffer, and therefore, a high colloidal stability of the PLL-functionalized nanoparticles in this medium. Therefore, only after PLL functionalization, the Eu:NaGd(MoO4)2@citrate nanoparticles meet the criterion of colloidal stability required for their use in biotechnological applications.
It is important to mention that the functionalization with PLL did not affect in a significant manner the photoluminescence properties of the nanophosphors. Thus, the intensity of the emission bands (λex = 250 nm) of the PLL-functionalized sample was only slightly lower than that observed before functionalization (Fig. S6†), which indicates that the presence of PLL molecules on the surface of the nanoparticles already coated with citrate ions, did not increase the Eu3+ luminescence quenching due to the interaction of these ions with the organic species. Therefore, these PLL-modified Eu:NaGd(MoO4)2@citrate nanoparticles maintain a red luminescence and may be used as optical biolabels for “in vitro” applications.
Biocompatibility studies of these nanophosphors were also evaluated finding a slight increase of toxicity after PLL functionalization (Fig. 7), which might be due to the positive surface charge of the PLL-functionalized nanoparticles, as previously noticed for various types of nanoparticles.42,43 Nevertheless, viability percentages of around 80% were still obtained for concentrations up to 0.025 mg ml−1 (Fig. 7), which are suitable for their use as MRI contrast agents.
Finally, it was also found that the r1 value decreased (from 5.03 to 2.12 mM−1 s−1) after PLL functionalization, whereas the r2 value remained almost unchanged (8.7 mM−1 s−1) (Fig. 6) which gives an r2/r1 ratio of 4.10, indicating that the PLL-functionalized nanoparticles are more suitable as a negative MRI contrast agent. It should be noted that external organic surface coating has been previously shown to have an important effect on the magnetic relaxivities of particulate MRI contrast agents, since it would impact the access of water molecules to the superficial Gd3+ ions.44 Nevertheless, in our case the r2/r1 ratio was sufficiently low for the nanoparticles to be suitable as a positive MRI contrast agent.
The magnetic relaxivities obtained for the nanoparticles at a high magnetic field (9.4 T) were different (r1 = 1.35 mM−1 s−1 and r2 = 96.42 mM−1 s−1) to those measured at a low magnetic field (1.5 T), giving an r2/r1 ratio of 71.42. This very high value indicates that these nanoparticles will behave as a T2 rather than T1 contrast agent when using preclinical magnetic fields, as illustrated in Fig. 9 and 10.
 | ||
| Fig. 9 T 1 and T2 relaxivities of PLL-functionalized nanoparticles measured at 9.4 T. | ||
 | ||
| Fig. 10 T 1-Weighted (a) and T2-weighted (b) MR images of phantoms containing solutions of PLL-functionalized nanoparticles at concentrations ranging from 0.01 mM (top, left) to 0.2 mM (right, down). | ||
4. Conclusions
Uniform NaGd(MoO4)2 nanoparticles have been synthesized for the first time through a homogeneous precipitation method at a relatively low temperature (120 °C) using a citrate–Gd complex as a Gd precursor and ethylene glycol/water mixtures as solvent. This procedure yields nanoparticles with an ellipsoidal morphology and a tetragonal structure, which resulted in coating with citrate anions during the synthesis process. These nanoparticles could be doped with Eu cations without altering their morphology or crystalline structure. The Eu doped samples presented a red luminescence when excited through an energy transfer band from the MoO42− group to the Eu3+ centers. The maximum luminescence efficiency was achieved for a 5% doping level. These nanophosphors showed no significant toxicity for cells up to a concentration of 0.2 mg mL−1 and also presented appropriate magnetic relaxivity values (low r2/r1 ratio) for their use as a positive MRI contrast agent at a low magnetic field (1.5 T). The colloidal stability of such nanoparticles in a physiological (MES) medium was achieved after their functionalization with poly(L-lysine). It was found that the so functionalized nanoparticles maintained a visible red luminescence and cell viability at concentrations up to 0.025 mg mL−1. Finally, magnetic relaxivity measurements revealed a significant increase of the r2/r1 ratio after functionalization so that the poly(L-lysine)-functionalized nanoparticles were more suitable as a negative contrast agent for MRI, even at high magnetic fields similar to those used in preclinical tests. Therefore, the here reported multifunctional nanoparticles meet the main requirements to be good candidates for their use in optical (in vitro) and magnetic imaging applications.Acknowledgements
This work has been supported by the Spanish Ministry of Economy and Competitiveness (MINECO) (MAT2014-54852-R), CSIC (PIE 201460E005) and ERC-Starting Grant 239931-NANOPUZZLE, SAF2014-54763-C2-2-R, and Research Funding from DGA (Fondo Social Europeo).References
- F. Wang, Y. Zhang, X. Fan and M. Wang, Nanotechnology, 2006, 17, 1527 CrossRef CAS.
- D. Y. Kong, Z. L. Wang, C. K. Lin, Z. W. Quan, Y. Y. Li, C. X. Li and J. Lin, Nanotechnology, 2007, 18, 075601 CrossRef CAS PubMed.
- R. Naccache, F. Vetrone, V. Mahalingam, L. A. Cuccia and J. A. Capobianco, Chem. Mater., 2009, 21, 717 CrossRef CAS.
- F. Eyanics, P. R. Diamente, F. C. J. M. van Veggel, G. J. Stanisz and R. S. Prosser, Chem. Mater., 2006, 18, 2499 CrossRef.
- N. J. J. Johnson, W. Oakden, G. J. Stanisz, R. S. Prosser and F. C. J. M. van Veggel, Chem. Mater., 2011, 23, 3714 CrossRef CAS.
- X. Duan and Y. Li, Small, 2013, 9, 1521 CrossRef CAS PubMed.
- S. Jiang, K. Y. Win, S. Liu, C. P. Teng, Y. Zheng and M. Y. Han, Nanoscale, 2013, 5, 3127 RSC.
- C. Fang and M. Zhang, J. Mater. Chem., 2009, 19, 6258 RSC.
- C. Shan, H. Yang, D. Han, Q. Zhang and A. Ivaska, Langmuir, 2009, 25, 12030 CrossRef CAS PubMed.
- M. A. Wolfert, P. R. Dash, O. Nazaroya, D. Oupicky, L. W. Seymour, S. Smart, J. Strohalm and K. Ulbrich, Bioconjugate Chem., 1999, 10, 993 CrossRef CAS PubMed.
- S. R. Bhattarai, E. Muthuswamy, A. Wani, M. Brichacek, A. L. Castaneda, S. L. Brock and D. Outpicky, Pharm. Res., 2010, 27, 2556 CrossRef CAS PubMed.
- M. Kar, P. S. Vijayakumar, B. L. V. Prasad and S. S. Gupta, Langmuir, 2010, 26, 5772 CrossRef CAS PubMed.
- G. Tian, Z. Gu, X. Liu, L. Zhou, W. Yin, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, J. Phys. Chem. C, 2011, 115, 23790 CAS.
- N. O. Nuñez, S. Rodriguez-Liviano and M. Ocaña, J. Colloid Interface Sci., 2010, 349, 484 CrossRef PubMed.
- N. O. Nuñez, S. Rivera, D. Alcántara, J. M. de la Fuente, J. García-Sevillano and M. Ocaña, Dalton Trans., 2013, 42, 10725 RSC.
- S. Rodriguez-Liviano, N. O. Nuñez, S. Rivera-Fernández, J. M. de la Fuenta and M. Ocaña, Langmuir, 2013, 29, 3411 CrossRef CAS PubMed.
- Y. Huang, L. Zhou, L. Yang and Z. Tang, Opt. Mater., 2011, 33, 777 CrossRef CAS.
- N. O. Nuñez and M. Ocaña, Nanotechnology, 2007, 18, 455606 CrossRef.
- L. Poul, S. Ammar, N. Jouini and F. Fievet, J. Sol-Gel Sci. Technol., 2003, 26, 261 CrossRef CAS.
- C. Feldmann, Adv. Funct. Mater., 2003, 13, 101 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55 CrossRef CAS PubMed.
- E. Matijević, Chem. Mater., 1993, 5, 412 CrossRef.
- H. Wang and L. Wang, Inorg. Chem., 2013, 52, 2439 CrossRef CAS PubMed.
- V. K. LaMer and R. H. Dinegar, J. Am. Chem. Soc., 1950, 72, 4847 CrossRef CAS.
- G. S. Rama Raju, E. Pavitra, G. P. Nagaraju, R. Kandimalla and B. F. El-Rayes, Cryst. Growth Des., 2013, 13(9), 4051 Search PubMed.
- N. O. Nuñez, P. Zambrano, J. García-Sevillano, E. Cantelar, S. Rivera-Fernández, J. M. de la Fuente and M. Ocaña, Eur. J. Inorg. Chem., 2015, 27, 4546 CrossRef.
- R. Yan and Y. Li, Adv. Funct. Mater., 2005, 15, 763 CrossRef CAS.
- Y. Liu, D. Tu, H. Zhu, E. Ma and X. Chen, Nanoscale, 2013, 5, 1369 RSC.
- S. Rodriguez-Liviano, F. J. Aparicio, T. C. Rojas, A. B. Hungría, L. E. Chinchilla and M. Ocaña, Cryst. Growth Des., 2012, 12, 635 CAS.
- N. O. Nuñez, J. Sabek, J. García-Sevillano, E. Cantelar, A. Escudero and M. Ocaña, Eur. J. Inorg. Chem., 2013, 8, 1301 CrossRef.
- J. W. Stouwdam, M. Raudsepp and F. C. J. M. van Veggel, Langmuir, 2005, 21(15), 7003 CrossRef CAS PubMed.
- A. Beeby, I. M. Clarkson, R. S. Dickins, S. Faulkner, D. Parker, L. Royle, A. S. de Sousa, J. A. G. Williams and M. Woods, J. Chem. Soc., Perkin Trans. 2, 1999, 493 RSC.
- J. Liao, D. Zhou, H. You, H. Wen, Q. Zhou and B. Yang, Optik, 2013, 124, 1362 CrossRef CAS.
- M. Weber, Phys. Rev. B: Solid State, 1971, 4, 2932 CrossRef.
- M. Quintanilla, E. Cantelar, J. A. Sanz-García, G. Lifante, G. A. Torchia and F. Cussó, J. Lumin., 2008, 128, 927 CrossRef CAS.
- J. Liao, H. Huang, H. You, X. Qiu, Y. Li, B. Qiu and H. R. Wen, Mater. Res. Bull., 2010, 45, 1145 CrossRef CAS.
- Y. Jiang, Y. Liu, G. Liu, X. Dong, J. Wang, W. Yu and Q. Dong, Opt. Mater., 2014, 36, 1865 CrossRef CAS.
- H. Hifumi, S. Yamaoka, A. Tanimoto, D. Citterio and K. Suzuki, J. Am. Chem. Soc., 2006, 128(47), 15090 CrossRef CAS PubMed.
- M. Borges, S. Yu, A. Laromaine, A. Roig, S. Suárez-García, J. Lorenzo, D. Ruiz-Molina and F. Novio, RSC Adv., 2015, 5, 86779 RSC.
- F. Hu and Y. S. Zhao, Nanoscale, 2012, 4, 6235 RSC.
- M. Babic, D. Horák, M. Trchová, P. Jendelová, K. Glogarová, P. Lesný, V. Heryek, M. Hájek and E. Syková, Bioconjugate Chem., 2008, 19(3), 740 CrossRef CAS PubMed.
- S. Bhattacharjee, I. Rietjens, M. P. Singh, T. M. Atkins, T. K. Purkait, Z. Xu, S. Regli, A. Shukaliak, R. J. Clark, B. S. Mitchell, G. M. Alink, A. Marcelis, M. Fink, J. Veinot, S. M. Kauzlarich and H. Zuilhof, Nanoscale, 2013, 5(11), 4870 RSC.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897 CrossRef CAS PubMed.
- M. Cho, R. Sethi, J. S. A. Narayanan, S. S. Lee, D. N. Benoit, N. Taheri, P. Decuzzi and V. L. Colvin, Nanoscale, 2014, 6(22), 13637 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt02663j |
| This journal is © The Royal Society of Chemistry 2016 |