DOI:
10.1039/C6DT02596J
(Paper)
Dalton Trans., 2016,
45, 12933-12950
Dinuclear metal(II)-acetato complexes based on bicompartmental 4-chlorophenolate: syntheses, structures, magnetic properties, DNA interactions and phosphodiester hydrolysis†
Received
1st July 2016
, Accepted 21st July 2016
First published on 21st July 2016
Abstract
A series of dinuclear metal(II)-acetato complexes: [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1), [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2), [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3), [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4), [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) and [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6), where LClO− = 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenolate, were synthesized. The complexes were structurally characterized by spectroscopic techniques and single crystal X-ray crystallography. Six-coordinate geometries with doubly bridged acetato ligands were found in Ni(II), Zn(II) and Mn(II) complexes 1, 2, 5 and 6, whereas with Cu(II) complexes a five-coordinate species was obtained with 4, and mixed five- and six-coordinate geometries with a doubly bridged dimetal core were observed in 3. The magnetic properties of complexes 1–4 and 6 were studied at variable temperatures and revealed weak to very weak antiferromagnetic interactions in 1, 2, 4 and 6 (J = −0.55 to −9.4 cm−1) and ferromagnetic coupling in 3 (J = 15.4 cm−1). These results are consistent with DFT calculations performed at the B3LYP/def2-TZVP(-f) level of theory. Under physiological conditions, the interaction of the dinculear complexes 1–5 with supercoiled plasmid ds-DNA did not show any pronounced nuclease activity, but Ni(II) complexes 1 and 2 revealed a strong ability to unwind the supercoiled conformation of ds-DNA. The mechanistic studies performed on the interaction of the Ni(II) complexes with DNA demonstrated the important impact of the nickel(II) ion in the unwinding process. In combination with the DNA study, the phosphatase activity of complexes 1, 3, and 5 was examined by the phosphodiester hydrolysis of bis(2,4-dinitrophenol)phosphate (BDNPP) in the pH range of 5.5–10.5 at 25 °C. The Michaelis–Menten kinetics performed at pH 7 and 10.7 showed that catalytic efficiencies kcat/KM (kcat = catalytic rate constant, KM = substrate binding constant) decrease in the order: Ni(II), 1 > Zn(II), 5 > Cu(II), 3. A similar trend was also observed with the turnover numbers at pH = 7. The results are discussed in relation to the coordination geometry and nature of the metal center as well as the steric environment imposed by the compartmental phenoxido ligand.
Introduction
In the last decade, a large number of bi-compartmental phenolate ligands with symmetrical and asymmetrical pendant chelating arms, including pyridyl and substituted pyridyl groups, attached to the 2- and 6-positions of the phenol ring were synthesized and structurally characterized. These ligands due to their ability to bind two identical or different 3d metal ions which are simultaneously bridged through the deprotonated phenolic group resulted in the formation of homo- and hetero-dinuclear metal complexes.1–6 In some cases, the presence of hydroxide and acetate ions may lead to further bridging and hence to doubly or triply bridged dinuclear metal complexes where the two metal centers are in close proximity in the range of 2.9–4.0 Å.7–21 Such coordination environments around the central metal ions together with the possible existence of the “coordinatively unsaturated” metal ion(s) and center with a “weakly bound” ligand(s) made these complexes attractive targets to mimic the active sites in the biological systems in order to elucidate the mechanism and the structural parameters of metalloproteins, e.g. hemocyanin,12 metallo-β-lactamases (MβL),13 catecholase oxidases,8,14,15 Mn catalases,16,17 and the phosphodiester hydrolysis of biomolecules such as purple acid phosphatases (PAPs), phosphoesterases and DNA nucleases.10,15,18–25 In addition to the possible use of these compounds in modeling the biological systems, they provide a wide range of ferro-/antiferro-magnetic couplings between the two paramagnetic metallic centers (3d5–9), bridged via the phenoxido group and through other ligands, which allows to probe the electronic structure of these compounds and compare them with the natural systems.1,9,11,23
Recently, dinuclear Zn(II), Cu(II), Co(II) and Fe(II) based phenolate systems have been extensively used as “artificial nucleases” to study the catalytic hydrolysis of phosphodiester compounds where the close proximity and the geometrical nature around the two metal ions as well possible “cooperativity” may enhance the P–O bond rupture.15,18–24 The P–O bonds in the phosphodiester linkages of DNA and RNA strands exhibit remarkable stability towards hydrolysis which is one of the most essential requirements for the survival and maintenance of life.26 Under physiological conditions, the half-life t1/2 for the hydrolysis of DNA was estimated to be ∼130![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years.25 This unusual stability of the P–O bonds towards hydrolysis in DNA was overcome in nature by the development of a number of hydrolytic metalloenzymes that efficiently catalyze the hydrolysis of P–O bonds of the DNA phosphate backbone.26 These metalloenzymes contain metal ions in their active sites. Therefore, modeling of these hydrolytic enzymes is a fundamental step in designing “artificial nucleases” capable of competing with natural ones.27–31
000 years.25 This unusual stability of the P–O bonds towards hydrolysis in DNA was overcome in nature by the development of a number of hydrolytic metalloenzymes that efficiently catalyze the hydrolysis of P–O bonds of the DNA phosphate backbone.26 These metalloenzymes contain metal ions in their active sites. Therefore, modeling of these hydrolytic enzymes is a fundamental step in designing “artificial nucleases” capable of competing with natural ones.27–31
Two mechanistic pathways have been found for DNA cleavage by small metal complexes. The first mechanism involves oxidative cleavage that occurs via reactive oxygen species (ROS: reactive singlet oxygen, 1O2; superoxide, O2−; hydroxyl radical, OH˙) and this requires addition of external agents such as light, and oxidative and/or reductive species to initiate the cleavage.31–36 Also, this mechanism generates fragments that damage the ribose sugar and/or nucleic bases of DNA and hence hamper their use in vivo.37 The second mechanism is hydrolytic cleavage that requires activation of a nucleophile (OH−) in proximity to the phosphoester moiety,30,32,38,39 and this mechanism does not suffer from the drawbacks of the oxidative cleavage mechanism because the generated DNA products can be enzymatically relegated.40–46 However, this raises a question concerning the validity of substrate compounds generally used to promote the hydrolysis of simple phosphodiester “model systems” since the operating mechanism is not necessarily the same and the reactivity might therefore differ significantly.
In addition to the cleavage of the supercoiled circular dsDNA (SC-form or form I) to the relaxed open circular form of DNA (OC-form or form II) and/or the linear DNA (L-form or form III) by small metal(II) complexes,33–39 it is possible that some of these molecules, especially those derived from Ni(II), Cu(II) and Zn(II) can bind DNA in a similar fashion to intercalators and induce single- or double-strand breaks leading to “unwinding” (i.e. relaxing) of SC-DNA to create the OC- or L-form of DNA.47–51 This binding alters the DNA winding which lengthens and stiffens. These structural changes may cause interference in the recognition and function of DNA-binding proteins.51–53
Therefore, we believe that the bicompartmental ligands 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-substituted-phenol (LR-OH) are suitable candidates to synthesize relevant dinuclear metal(II) complexes. The close proximity of the bridged metal ions may allow their cooperativity and hence enhances the capability of the complexes in promoting the cleavage of P–O bonds in phosphodiester compounds and DNA. Therefore, herein, we report the synthesis and structural characterization of six new dinuclear compounds with additional acetate bridges, [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1), [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2), [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3), [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4), [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) and [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) together with their magnetic properties (1–5). The bridged μ-acetato compounds 1, 3 and 5 were also selected to study the hydrolysis of bis(2,4-dinitrophenyl)phosphate (BDNPP) and the DNA cleavage/unwinding. The reactivity and the mechanistic pathways of these complexes are evaluated and compared to other related systems.
Results and discussion
Synthesis of the complexes
The reaction of a methanolic solution containing 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH)1a,b (Chart 1) and two equivalents of M(OAc)2·nH2O (M = Ni, n = 4; M = Cu, n = 1; M = Zn, n = 2) in the presence of NaClO4 or NH4PF6 afforded the dinuclear complexes [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1), [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2), [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3), [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4) and [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) in moderate yields (∼60%) for complexes 1–4 and a high yield (90%) for the Zn(II) complex, 5. [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) was obtained in 40% yield by the reaction of Mn(ClO4)2·6H2O and LClOH in MeOH, followed by the addition of sodium acetate. The complexes were soluble in MeOH, CH3CN and acetone. However, upon dissolution of the blue complexes 1, 2 and 4 in CH3CN there was a change in the color to green. Single crystals suitable for X-ray structure determination were obtained either directly from the methanolic solutions (3–5) or by further recrystallization of the complexes from MeOH (1, 6), whereas X-ray quality single crystals for complex 2 were obtained from recrystallization of the complex from acetone. The isolated complexes were characterized by elemental microanalyses, molar conductivity, IR and UV-VIS spectroscopy and single crystal X-ray crystallography.
 |
| | Chart 1 Structural formula of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol. | |
The molar conductivities of the synthesized complexes, ΛM were measured in CH3CN and the values are summarized in Table 1. The values (ΛM = 131–167 Ω−1 cm2 mol−1) reveal the 1:1 electrolytic behavior for complexes 1, 2, 4, 5 and 6. The color change associated with the dissolution of complexes 1 and 2 in CH3CN is attributed to bond rupture of the bridging acetato ligand(s) and the formation of the six-coordinate monocationic acetonitrile species [Ni2(μ-LClO)(μ-OAc)(OAc)(CH3CN)]+ or [Ni2(μ-LClO)(OAc)2(CH3CN)2]+ without the release of the OAc− ion. In contrast, the value of ΛM = 280 Ω−1 cm2 mol−1 observed for [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3) is consistent with an 1:2 electrolyte behavior, attributed to the dissociation of the weakly bound coordinated perchlorate ligand to produce [Cu2(μ-LClO)(μ2-OAc)]2+ + 2 ClO4−. A similar conductivity trend was reported for [Cu2(μ-LClO)(μ-pz)(ClO4)](ClO4), where pz = pyrazole anion.1b Four of the synthesized complexes revealed doubly bridged acetate, [M2(μ-LClO)(μ2-OAc)2]+ (M = Ni(II) in complexes 1 and 2; M = Zn (II) in 5; M = Mn(II) in 6). With the Cu(II) complexes two different coordination modes were observed for the acetate anion: a single bridging acetate in [Cu2(μ-LClO)(μ2-OAc)(ClO4)]+ (3) and two mono-dentate acetates in [Cu2(μ-LClO)(OAc)2]+ (4), depending on the nature of the counter ion (see X-Ray crystal structure analysis section).
Table 1 UV-Vis spectroscopic and molar conductivity data for 1–6 in CH3CN
| Complex |
λ
max (εmax, M−1 cm−1) |
Λ
M (Ω−1 cm2 mol−1) |
| [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1) |
490 (sh), 645 (11.5), ∼960 (25.6, b) |
131 |
| [Ni2(μ-LClO)(μ2-OAc)2](ClO6)·CH3COCH3 (2) |
490 (sh), 644 (7.2), ∼958 (22, b) |
152 |
| [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4)·2H2O (3) |
426 (465), ∼800 (202, b) |
290 |
| [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4) |
404 (713), ∼670 (155, b) |
131 |
| [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) |
303 (2610) |
132 |
| [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) |
316 (3550), ∼500 (sh) |
167 |
Spectroscopic characterization of the complexes
The IR spectra of the complexes display some common features. Complexes 1, 4 and 6 show a broad absorption band over the frequency range 3430–3450 cm−1 due to the ν(O–H) stretching vibration of the lattice water. The perchlorate complexes 2, 3 and 6 display the ν(Cl–O) band as a broad strong absorption over the region 1120–1090 cm−1. The split of this band into two or three bands in complexes 2 and 3, respectively is most likely due to the reduction of the ClO4− symmetry from Td to C3v or C2v symmetries as a result of the coordination of ClO4− to the metal center as in complex 3 and/or to the involvement of the counter ClO4− ion in H-bonding with the ligand or its presence in a distorted location. The corresponding hexafluorophosphate complexes 1, 4 and 5 show a strong absorption band around 840 cm−1 due to νas(P–F). The complexes also display a series of strong to weak intensity bands over the 1610–1440 cm−1 region which are attributed to the pyridyl and C–O of the acetato group moieties.40
The UV-Vis spectral data of complexes 1–6, recorded in CH3CN, are summarized in Table 1. The Ni(II) complexes 1 and 2 display similar spectra with three absorption maxima located at 960, 655 and 490 nm which indicates the same coordination environment around the central Ni(II) ions. This spectral pattern is consistent with an octahedral geometry around the 3d8 Ni(II) ion and results from the electronic d–d transitions 3T2g(F) ← 3A2g(F), 3T1g(F) ← 3A2g(F) and 3T1g(P) ← 3A2g(F), respectively.54 The spectrum of the copper(II) complex [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4) reveals the presence of two maxima at 404 and 670 nm, assigned to the dxz, dyz → dx2−y2 and dxy → dx2−y2 transitions in a distorted square pyramidal (SP) environment around the central Cu2+ ions.55 The corresponding complex [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3) displays a similar spectral pattern with two maxima at 426 and a broad band around 800 nm. This is attributed to the presence of a distorted trigonal bipyramidal geometry (TBP) around the central Cu2+ ions, [Cu2(μ-LClO)(μ2-OAc)]2+ (the weakly coordinated ClO4− ligand in complex 3 is dissociated in CH3CN as indicated by the conductivity measurements).46a,55,56 The electronic spectrum of [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) exhibits a very intense band at 316 nm with a shoulder around 500 nm (Table 1). The former band results from an M → L CT transition in a distorted octahedral environment.54 The Zn(II) complex 5 displays a very strong maximum at 303 nm which is assigned to an L → M CT transition.54a
The 1H and 13C NMR spectra of [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) were recorded in deuterated dimethylsulfoxide (DMSO-d6) (Fig. S1 and S2,† respectively). The spectra did not confirm the equivalency of the two binding sites as indicated by the appearance of sixteen aromatic carbon and the corresponding nine proton resonances. This behavior indicates that the two pyridyl groups within each site are not identical. Moreover, five resonances were observed for the methylene protons of –N–CH2–py and –N–CH2–ph where each proton showed two closely related resonances over the chemical shift region δ = 3.69–4.41 ppm indicating that these protons were not coupled equally. The structure of the complex cation and its atom numbering scheme is illustrated in Chart S1 (ESI†).
Crystal structures of the complexes (1–6)
The perspective views of complexes 1–6 together with partial atom numbering schemes are given in Fig. 1. The common features of the six complexes are dinuclear complex cations, bridged by a deprotonated O1 oxygen atom of a central 4-chloro-phenolate moiety of LClO− (Table 2). Each M(II) center within a dinuclear complex cation is further coordinated to three N donor atoms of one bis-pyridylaminomethyl-group. The coordination number 6 with a distorted octahedral geometry is completed in compounds 1, 2, 5 and 6 by oxygen atoms of two μ(O,O′)-bridging acetate groups. The Ni–O/N, Zn–O/N and Mn–O/N bond distances are in the range of 2.0097(16)–2.1297(14), 2.005(3)–2.228(3) and 2.0903(11)–2.3319(12) Å, respectively. In the crystal structure of 3, the two Cu(II) centers are further linked by one μ(O,O′)-bridging acetate group. The axial site of the distorted square pyramid around the Cu1 center is occupied by O1 (τ-value = 0.20).57 The coordination number six around the Cu2 center is completed by the O5 atom of a perchlorate anion [2.781(2) Å]. In the case of 4 distorted SP geometry around each Cu(II) center is completed by an O atom of the terminal acetate group. Both axial sites are occupied by the bridging phenoxido oxygen atom O1 (τ-values are 0.16 and 0.17, respectively). The dinuclear complex cations co-crystallize with PF6−/ClO4− counter anions and solvent molecules (CH3COCH3 for 2, water for 1 and 4). Selected bond parameters and possible hydrogen bonds are presented in Tables S2–S8 (see ESI†) for complexes 1–6, respectively.
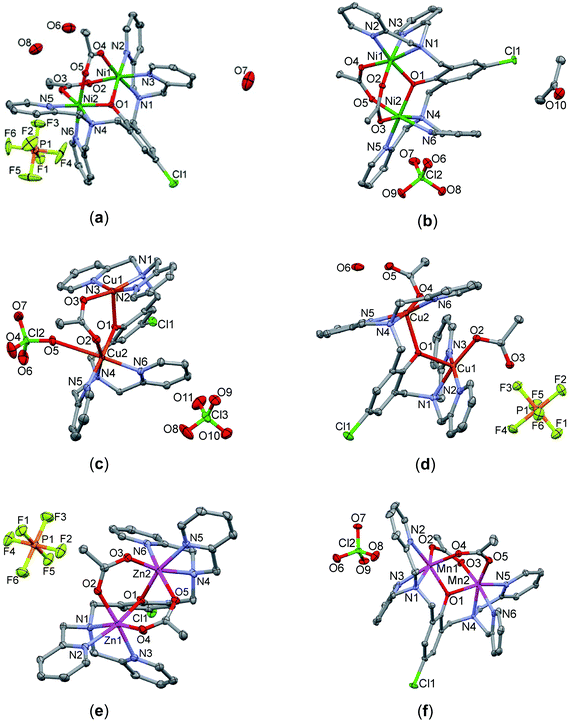 |
| | Fig. 1 Perspective views of complexes 1–6 with partial atom numbering schemes: (a) [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1), (b) [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2), (c) [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3), (d) [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4), (e) [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) and (f) [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6). | |
Table 2 Geometric parameters of the M−O−M bridges in the dinuclear units of 1–6
| Compound |
M1⋯M2 (Å) |
M1−O1−M2 (°) |
M1−O1 (Å) |
M2−O1 (Å) |
| [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1) |
3.4131(5) |
115.70(7) |
2.0097(16) |
2.0216(15) |
| [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2) |
3.4346(3) |
116.43(6) |
2.0209(12) |
2.0198(12) |
| [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3) |
3.5711(6) |
118.83(9) |
2.2089(19) |
1.9362(19) |
| [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4) |
3.9388(3) |
128.13(7) |
2.1972(14) |
2.1826(14) |
| [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) |
3.3935(8) |
113.71(11) |
2.040(2) |
2.013(2) |
| [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) |
3.4515(3) |
109.11(4) |
2.0903(11) |
2.1216(10) |
Magnetic properties
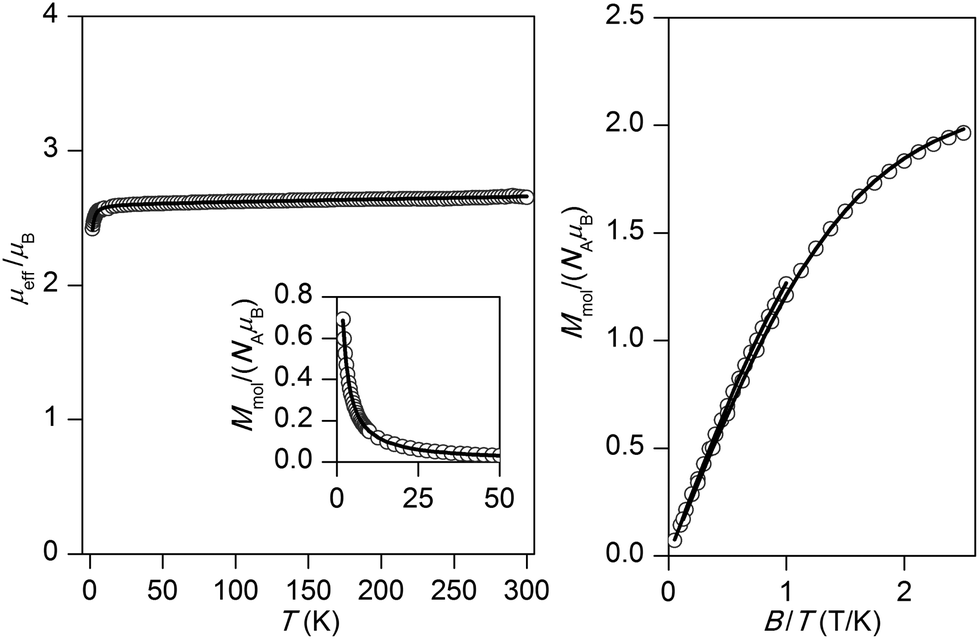
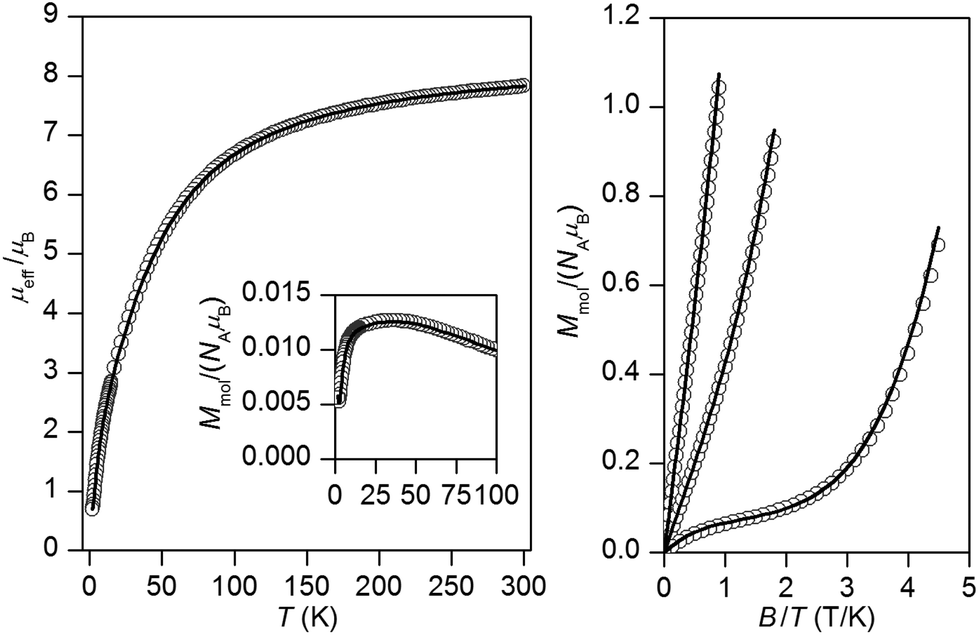
The experimental magnetic data of complexes 1–4 and 6 are shown in Fig. 2–6, respectively. The room temperature values of the effective magnetic moments of the Ni(II) compounds (μeff/μB = 4.41 for 1 and 4.45 for 2) can be compared with the expected spin-only magnetic moment μeff = 4.00μB of a system with two uncoupled spins S1 = S2 = 1 (g = 2.0). The slightly higher experimental values are due to the angular momentum contribution to the ground state from excited states. The decrease of μeff/μB on cooling is observed for both Ni(II) compounds, suggesting the presence of antiferromagnetic exchange, however in compound 1, the decrease of μeff/μB is more pronounced with a maximum at Tmax = 5.4 K in the Mmolvs. T curve, which demonstrates a stronger magnetic interaction in 1. The magnetic behavior of the Cu(II) compounds is different. In the case of 3, μeff/μB increases upon cooling as a result of ferromagnetic exchange (Fig. 4), while the μeff/μB of 4 is almost constant over the whole temperature range (Fig. 5). Finally, the magnetic behavior of the Mn(II) compound 6 manifests strong antiferromagnetic exchange by declination of μeff/μB from the theoretically expected value of 8.37 (g = 2.0) for two uncoupled spins S1 = S2 = 5/2 even at room temperature (μeff/μB = 7.83 for 6) and existence of a maximum located at Tmax = 35.0 K in the Mmolvs. T curve (Fig. 6). Since all the investigated compounds are dinuclear, the analysis of the magnetic data is based on the spin Hamiltonian for dinuclear systems of the form:58| |  | (1) |
where the isotropic exchange (J), zero-field splitting (D) and Zeeman term (g) are incorporated. The parameter a defines orientation of the magnetic field vector, Ba = B(sinθcosϕ, sinθsinϕ, cosθ). In the case of Mn(II) and Cu(II) compounds, the magnetic anisotropy was not considered, so the molar magnetization was calculated as| |  | (2) |
where Z is the partition function. However, a non-zero D-parameter was necessary in the analysis of Ni(II) compounds, and an integral average of molar magnetization was calculated in order to properly simulate the powder sample signal as| |  | (3) |
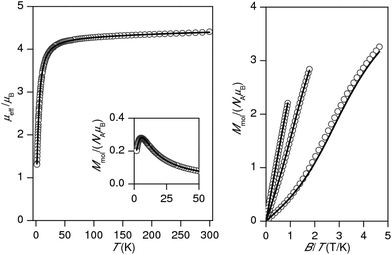 |
| | Fig. 2 Magnetic data for 1. Left: Temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: Isothermal magnetizations measured at T = 2, 5 and 10 K. Open circles: experimental data, solid lines: calculated data using eqn (1), with J = −3.70 cm−1, D = +6.38 cm−1, g = 2.16, χTIP = 4.84 × 10−9 m3 mol−1. | |
 |
| | Fig. 3 Magnetic data for 2. Left: Temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: Isothermal magnetizations measured at T = 2, 5 and 10 K. Open circles: experimental data, solid lines: calculated data using eqn (1), with J = −1.01 cm−1, D = −4.96 cm−1, g = 2.17, χTIP = 4.80 × 10−9 m3 mol−1. | |
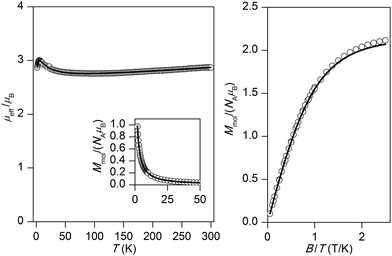 |
| | Fig. 4 Magnetic data for 3. Left: Temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: Isothermal magnetizations measured at T = 2 and 5 K. Open circles: experimental data, solid lines: calculated data using eqn (1), with J = +15.4 cm−1, g = 2.13, χTIP = 6.80 × 10−9 m3 mol−1. | |
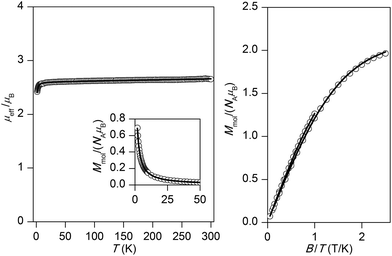 |
| | Fig. 5 Magnetic data for 4. Left: Temperature dependence of the effective magnetic moment and molar magnetization measured at B = 1 T. Right: Isothermal magnetizations measured at T = 2 and 5 K. Open circles: experimental data, solid lines: calculated data using eqn (1), with J = −0.55 cm−1, g = 2.12, χTIP = 1.58 × 10−9 m3 mol−1. | |
 |
| | Fig. 6 Magnetic data for 6. Left: Temperature dependence of the effective magnetic moment and molar magnetization measured at B = 0.1 T. Right: Isothermal magnetizations measured at T = 2, 5 and 10 K. Open circles: experimental data, solid lines: calculated data using eqn (1) with J = −9.40 cm−1, g = 2.01, xPI = 0.67%. | |
For all compounds under investigation, both temperature and field dependent magnetization data were fitted simultaneously. As a result, the fitted magnetic data which are depicted by solid lines in Fig. 2–6 showed good agreement with the experimental data. In Ni(II) compound 1, the data was fitted with J = −3.70 cm−1, D = +6.38 cm−1, g = 2.16, χTIP = 4.84 × 10−9 m3 mol−1, where χTIP is a parameter accounting for the temperature-independent paramagnetism. We have also tried to fit experimental data with a negative D-parameter but without success. In Ni(II) complex 2, it was possible to make good fits for both negative (J = −1.01 cm−1, D = −4.96 cm−1, g = 2.17, χTIP = 4.80 × 10−9 m3 mol−1 – Fig. 3) and positive (J = −1.20 cm−1, D = +2.80 cm−1, g = 2.17, χTIP = 4.77 × 10−9 m3 mol−1, Fig. S3†) D-parameters. Evidently, the antiferromagnetic exchange in 1 is stronger than that observed in 2, which can be attributed to small differences in the coordination environments around the central Ni(II) ions (Table 2) as a result of different solvents of crystallization in the two complexes. The Cu(II) ions with S = 1/2 do not have predisposition for zero-field splitting, therefore only isotropic exchange was taken into account. This treatment resulted in J = +15.4 cm−1, g = 2.13, χTIP = 6.80 × 10−9 m3 mol−1 for 3 and J = −0.55 cm−1, g = 2.12, χTIP = 1.58 × 10−9 m3 mol−1 for 4, thus confirming the significant ferromagnetic exchange in 3 and almost negligible antiferromagnetic exchange in 4. The magnetic data for the dinuclear Mn(II) complex 6 was treated with presumption that D ≈ 0 cm−1, because a large antiferromagnetic exchange results in an S = 0 ground state, which does not bear any information about D, and excited states with S > 0 are too high in energy to be used for the identification of magnetic anisotropy. As a result, the following spin Hamiltonian parameters were obtained for 6: J = −9.40 cm−1, g = 2.01, and χPI = 0.67%, where χPI is the molar fraction of the mononuclear paramagnetic impurity.
DFT calculations
Our recent studies on a series of dinuclear doubly bridged Cu(II) complexes of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) and various bridging ligands (OH−, O2P(OC6H5)2−, C3H3N2−) showed that the DFT calculations of the isotropic exchange parameters J based on the B3LYP hybrid functional are helpful in understanding the efficiency of magnetic super-exchange pathways in these complexes.1b Therefore, the exchange mechanisms for the complexes under investigation were studied using the same functional.1b The calculations were performed on the molecular cations [Ni2(μ-LClO)(μ2-OAc)2]+1 and 2, [Cu2(μ2-LClO)(μ-OAc)(ClO4)]+3, [Cu2(μ-LClO)(OAc)2]+4 and [Mn2(μ-LClO)(μ2-OAc)2]+6 by the aid of the well-established ORCA 3.0 computational package using the def2-TZVP(-f) basis set. The J-values were evaluated from the energy difference Δ, between the high spin (HS) and broken-symmetry (BS) statesby both Ruiz's approach59| | | JRuiz = 2Δ/[(S1 + S2)(S1 + S2 + 1)] | (5) |
and Yamaguchi's approach60| | | JYam = 2Δ/[〈S2〉HS − 〈S2〉BS] | (6) |
where the following spin Hamiltonian for the dinuclear system was used| | 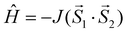 | (7) |
The results of the DFT calculations are summarized in Table 3 and the calculated spin densities for the selected compounds are depicted in Fig. 7. In all the cases, the DFT calculations resulted in J values (JRuiz/JYam) close to parameters extracted from the experimental magnetic data (Jmag), except for the copper(II) complexes 3 (Cu(II) ions adopt different geometries, square pyramidal for Cu(1) and octahedral for Cu(2)), and 4 (the two Cu(II) ions adopt the square pyramidal geometries), where the calculations slightly overestimated the antiferromagnetic exchange (Table 3). The non-orthogonal magnetic orbitals of the broken symmetry solution with the highest overlap Sαβ for the three compounds 1, 3 and 6 are shown in Fig. 8. In the case of the singly bridged-phenoxido Cu(II) complex 4 where SP geometries are adopted, the unpaired electrons reside in the dx2−y2 orbitals, where these magnetic orbitals are well isolated and hence the magnetic interaction is almost negligible. However, the magnetic orbitals in compound 3 are close enough and at the same time they are orthogonal, which explains the observed ferromagnetic interaction in this complex (Fig. 8). The nickel(II) complexes are octahedral with the unpaired electrons in the dz2 and dx2−y2 orbitals, and to a large extent, the spin density is delocalized on all donor atoms. The super-exchange pathway should be efficient through the phenoxido and acetato ligands, but the largest overlap was observed through the phenoxido-bridge (Fig. 8). In Mn(II) complex 6, where all d-orbitals contain unpaired electrons, a smaller extent of spin delocalization is observed. Surprisingly, the magnetic orbitals with the highest overlap are located outside the metal–donor bonds (Fig. 8).
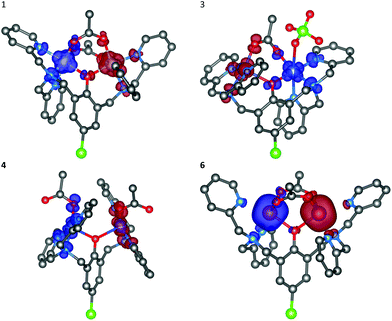 |
| | Fig. 7 Calculated isodensity surfaces of the broken symmetry spin states for molecular fragments [Ni2(μ-LClO)(μ2-OAc)2]+ of 1, [Cu2(μ-LClO)(μ2-OAc)(ClO4)]+ of 3, [Cu2(μ-LClO)(OAc)2]+ of 4 and [Mn2(μ-LClO)(μ2-OAc)2]+ of 6 using B3LYP/def2-TZVP(-f). Positive and negative spin densities are represented by dark blue, and dark red surfaces, respectively. The isodensity surfaces are plotted with the cut-off values of 0.005ea0−3. Hydrogen atoms are omitted for clarity. | |
 |
| | Fig. 8 The non-orthogonal magnetic orbitals with the highest overlap Sαβ of the broken-symmetry spin state visualized for [Ni2(μ-LClO)(μ2-OAc)2]+ of 1, [Cu2(μ-LClO)(μ2-OAc)(ClO4)]+ of 3 and [Mn2(μ-LClO)(μ2-OAc)2]+ of 6. Hydrogen atoms are omitted for clarity. | |
Table 3 DFT-calculated net Mulliken spin densities (ρ), expected values <S2>, overlap Sαβ between the corresponding orbitals and isotropic exchange parameters (J) from high-spin (HS) and broken symmetry (BS) states, compared with the experimental exchange parameters, of the dinuclear complexes [Ni2(μ-LClO)(μ2-OAc)2]+1 and 2, [Cu2(μ-LClO)(μ2-OAc)(ClO4)]+3, [Cu2(μ-LClO)(OAc)2]+4 and [Mn2(μ2-LClO)(μ2-OAc)2]+6 using the B3LYP functional and experimental structural parameters
| |
1
|
2
|
3
|
4
|
6
|
|
ρ
HS(M1)/ρHS(M2) |
1.65/1.65 |
1.65/1.65 |
0.61/0.64 |
0.62/0.62 |
4.85/4.85 |
|
ρ
BS(M1)/ρBS(M2) |
−1.64/1.64 |
−1.65/1.65 |
−0.61/0.64 |
−0.62/0.62 |
−4.85/4.85 |
| <SHS2>/<SBS2> |
6.01/2.01 |
6.01/2.01 |
2.01/1.00 |
2.01/1.01 |
30.01/5.00 |
|
S
αβ
|
0.04841 |
0.04388 |
0.02459 |
0.00468 |
0.07766 |
| 0.03511 |
0.03374 |
|
|
0.04656 |
|
|
|
|
|
0.02303 |
|
|
|
|
|
0.01745 |
|
|
|
|
|
0.00813 |
|
Δ/cm−1 |
−16.514 |
−7.701 |
+12.066 |
−0.279 |
−195.733 |
|
J
Ruiz/JYam (cm−1) |
−5.50/−8.25 |
−2.57/−3.85 |
+12.1/+24.1 |
−0.279/−0.558 |
−13.0/−15.7 |
|
J
mag/cm−1 |
−3.70 |
−1.01/−1.20 |
+15.4 |
−0.55 |
−9.40 |
| ∠(M–OPh–M)/° |
115.70 |
116.43 |
118.83 |
128.14 |
109.11 |
|
d(M–M)/10−10 m |
3.413 |
3.435 |
3.571 |
3.939 |
3.452 |
Interactions of metal complexes with DNA
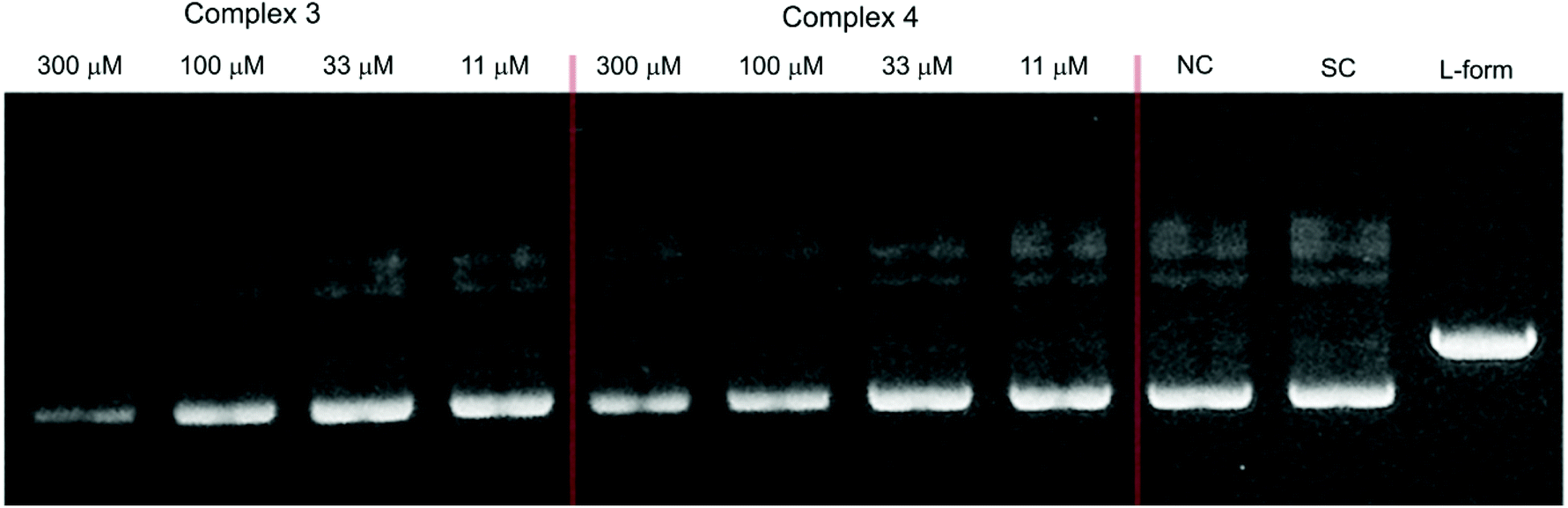
Some copper(II), zinc(II), nickel(II) and cobalt(II) complexes have been reported to serve as efficient artificial nucleases for DNA cleavage.25,33–36,39,47,56 In general, it is well established that the supercoiled circular form of plasmid ds-DNA (SC-form) is cleaved to the relaxed open circular form of DNA (OC-form), which upon further cleavage results in the formation of the linear DNA (L-form). In this work, the interaction of the dinuclear complexes 1–5 with DNA has been investigated under the physiological conditions. The interaction of the copper(II) (3 and 4) and zinc(II) (5) complexes with DNA did not show any nuclease activity as clearly illustrated in Fig. 9 and 10, respectively. Also, it emerges that the concentration of the SC-form of plasmid DNA was decreased without showing any sign of increase neither of the OC-form nor the linear L-form. On the other hand, the observed effect is not augmented by longer incubation of plasmid DNA with these complexes. As a model molecule, copper(II) complex 3 was examined at a longer incubation time and at different complex concentrations. The gel electrophoretic band pattern remained the same even if the interaction time was prolonged up to 22 hours (Fig. S4†). The observed effects might be explained by a direct competition of these complexes with the ethidium bromide (EtBr) which was used in the gel electrophoresis; no damaging/cleavage of DNA was noticed neither after a long reaction time nor at high complex concentrations. Replacing the intercalated EtBr from its binding sites on DNA by zinc(II) or copper(II) complexes could lead to the reduction of EtBr fluorescence on the gel as it has been seen from the decrease of intensities of the individual bands.
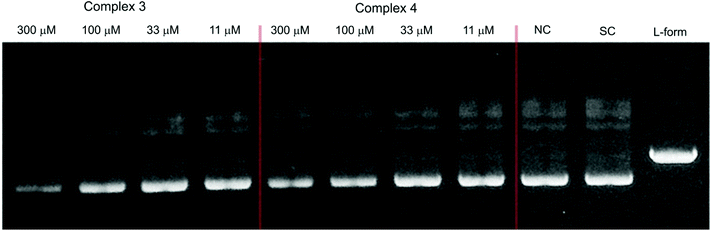 |
| | Fig. 9 Agarose gel (in TBE buffer with pH 8.4 at room temperature) electrophoresis patterns for the effect of concentrations of Cu(II) complexes 3 (left) and 4 (middle) (11–300 μM) on the native pUC19 plasmid DNA [the calculated concentration of base pairs (bp) = 23 μM]. NC is the negative control, SC is the supercoiled plasmid DNA, and L-form represents the sample of native plasmid linearized by HindIII endonuclease. | |
 |
| | Fig. 10 Agarose gel (in TBE buffer with pH 8.4 at room temperature) electrophoresis patterns for the effect of concentrations of Zn(II) complex 5 (left) (11, 33, 100 and 300 μM) on the native pUC19 plasmid DNA [the calculated concentration of base pairs (bp) = 23 μM]. NC is the negative control, SC is the supercoiled plasmid DNA, and L-form represents the sample of native plasmid linearized by HindIII endonuclease. | |
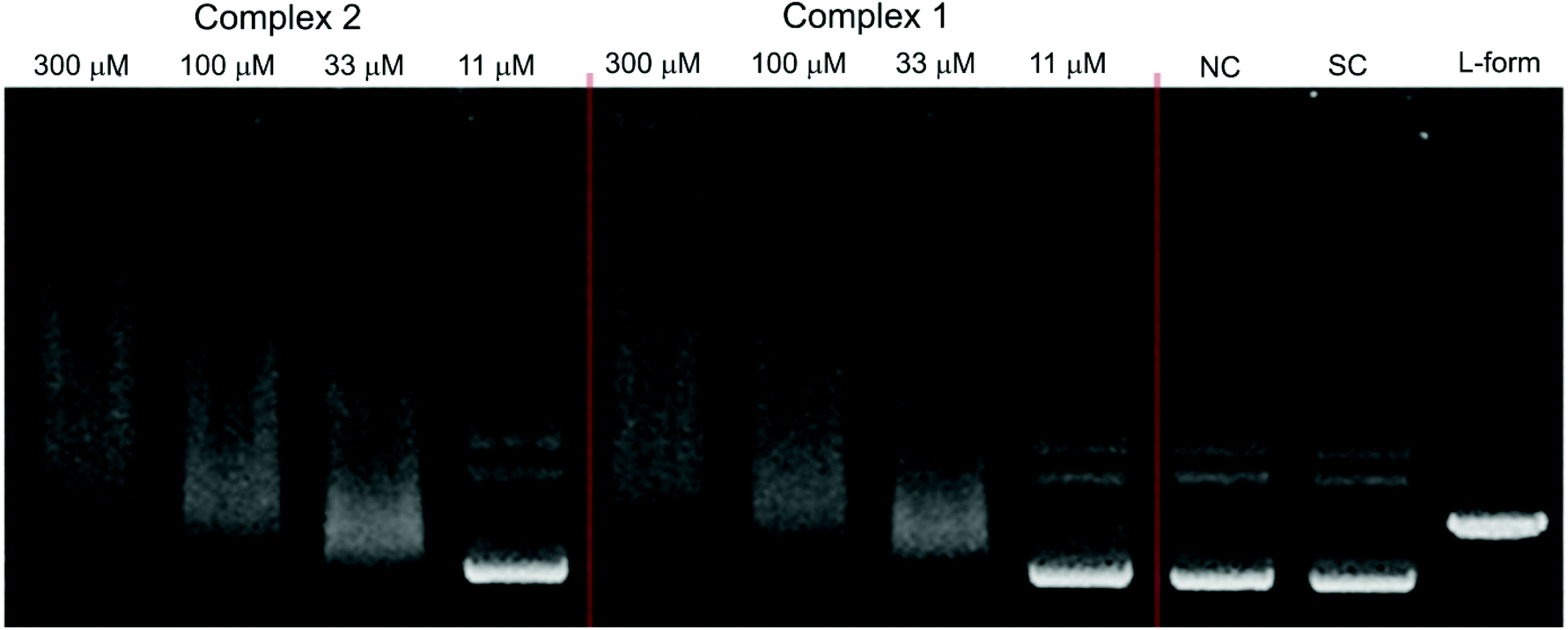
The catalytic activity of DNA was also investigated in the presence of Ni(II) complexes 1 and 2 in a similar fashion to that described for complexes 3–5. The results which are depicted by the gel electrophoresis in Fig. 11 showed that no nuclease activity was observed with the two complexes. However, unlike the copper(II) (3 and 4) and zinc(II) (5) complexes, the nickel(II) complexes [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1) and [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2) revealed a strong ability to unwind the supercoiled plasmid dsDNA and behave as intercalators. Unwinded and partially unwinded plasmid conformations were observed in the plasmid-DNA gel electrophoresis as a “smear” (Fig. 11). A similar trend was recently reported by A. Terenzi et al. in the interaction of the Ni(II)-salphen complex (salphen = the anion of N,N-bis-salicylidene-1,2-phenylenediamine) with native DNA.48 In addition, the unwinding activity by the two nickel(II) complexes 1 and 2 was associated with a “smear” of DNA molecules which is slower than that detected for the OC-form. Perhaps, this could be caused either by the neutralization of the negative charge of DNA or due to cross-linking of the DNA molecules or a combination of both effects which led to an increase in their molecular weights and hence changes in the conformations. The dependence of the total amounts of the plasmid SC-form as a function of the concentration of complexes 1–5 is shown in Fig. 12.
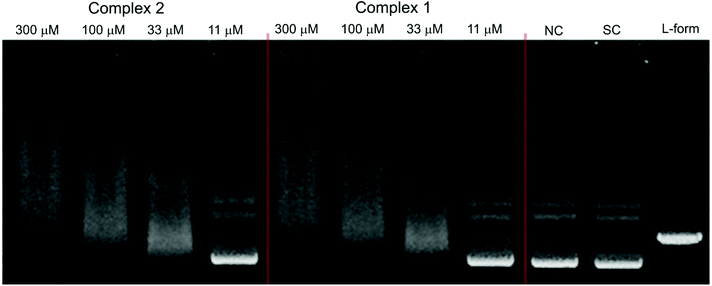 |
| | Fig. 11 Agarose gel (in TBE buffer with pH 8.4 at room temperature) electrophoresis patterns of the interaction of Ni(II) complexes 2 (left) and 1 (middle) at different concentrations (11–300 μM) with the native pUC19 plasmid [the calculated concentration of base pairs (bp) = 23 μM], where NC is the negative control, SC is the supercoiled plasmid DNA, and L-form represents the sample of native plasmid linearized by HindIII endonuclease. | |
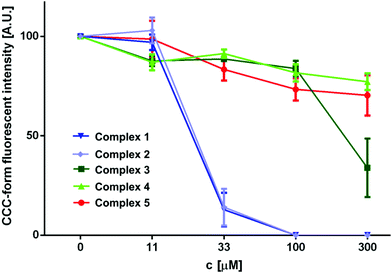 |
| | Fig. 12 The graphical interpretation of the concentration-dependent decline in the portion of the supercoiled plasmid DNA (also referred to as SC-form) in the samples showing the effective interaction of Ni(II) complexes with plasmid DNA leading to the formation of interconnected multiplexes or decrease of the surface charge of the polynucleotide backbone (or a combination of both). Other complexes showed the ability to quench the fluorescence of the ethidium bromide–plasmid DNA complex, which is evident from the linear dependence of the decreasing fluorescence of the SC-form with the increasing concentration of the applied transition metal complex. | |
In order to understand the mechanistic pathway responsible for the attenuation of the supercoiled plasmid DNA observed in the fluorescence electrophoretograms for the Ni(II) complexes 1 and 2, the interaction of the Ni(II) complexes was performed in the presence of oxidative scavengers (DMSO and KI), the metal competitor MgSO4, and the metal chelator EDTA (the complex/inhibitor molar ratio is 1:1).38,39,46 Therefore, a series of experiments for the DNA–Ni(II) interaction were conducted at two different Ni(II) concentrations (33 and 300 μM) in the presence of these inhibitors (DMSO, KI, MgSO4 and EDTA) and the results are illustrated in Fig. 13 and 14. Inspection of these figures demonstrates that MgSO4, DMSO and KI had no effect for the complexes on the DNA structure. However, in the case of complex 2, EDTA was able to attenuate the unwinding of supercoiled plasmid DNA (Fig. 14 and 15) at a low complex concentration (33 μM). These results are quantified in Fig. 15 which indicates that the nickel(II) ion is important for the plasmid DNA unwinding, where the bond between complexes 1 and 2 with DNA is stronger than that of Ni(II)–EDTA.
 |
| | Fig. 13 The electrophoretogram showing the effect of the addition of hydroxyl scavenger DMSO and increased ionic strength on the interactions of Ni(II) complexes 2 and 1 with plasmid DNA. NC is the negative control with the appropriate concentration of an additional substance. The indicated concentration levels represent both the concentrations of applied complexes, as well as the concentration of added substances (molar ratio 1:1). | |
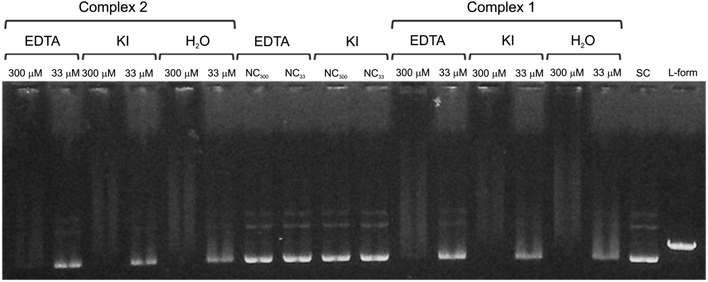 |
| | Fig. 14 The electrophoretogram showing the effect of addition of different inhibitors (and H2O as a positive control) on the interactions of the Ni(II) complexes 2 and 1 with plasmid DNA. NC is the negative control with the appropriate concentration of an additional substance. The indicated concentration levels represent both the concentrations of applied complexes, as well as the concentration of added substances (molar ratio 1:1). | |
 |
| | Fig. 15 The diagram shows the effect of different inhibitors on the decline in the portion of the supercoiled plasmid DNA (also referred to as SC-form) in the samples showing the effective interaction of the Ni(II) complexes 1 and 2 with plasmid DNA (at the 33 μM concentration level, leading to the formation of interconnected multiplexes). The importance of transition metal function in the formation of the mentioned superstructures was confirmed as the portion of the SC-form of plasmid DNA in the group with added EDTA at the 33 μM level which differs significantly from the positive control (p < 0.05). | |
Phosphodiester hydrolysis
The phosphatase reactivity of complexes 1, 3, and 5 was probed with a well-established spectroscopic assay using the activated phosphodiester BDNPP as a model substrate.21,61–63 Cleavage of the P–O bond was followed at 25 °C by monitoring the hydrolysis product 2,4-dinitrophenolate by its strong absorption at 400 nm (ε = 12000 M−1 cm−1).63 All measurements were carried out in 1:1 acetonitrile–buffer mixtures. The pH dependence of the activity was studied by varying the pH of the multicomponent buffer (pH 5.5–pH 10.5) in the assay; the plots obtained for complexes 1 and 5 are presented in Fig. 16. Complex 3 did not show a significant increase of the BDNPP hydrolysis rate. The data were fitted to eqn (8), which was based on a model for a diprotic system with two active species:64| |  | (8) |
Here, v0 is the initial rate and vmax is the maximum reaction rate that is reached under given conditions. The factor γ is related to the relative activity of the two active species in equilibrium (EnS and En−1S); a value of γ less than unity corresponds to a more active EnS adduct and a value higher than one considers the deprotonated adduct En−1S as more active.64,65 The resulting pKa and γ values are listed in Table 4.
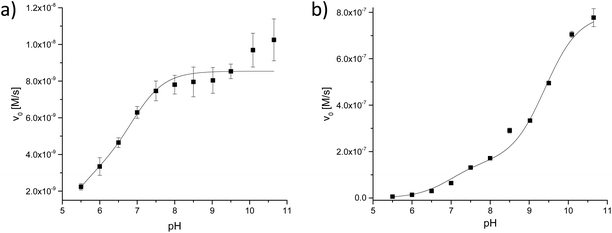 |
| | Fig. 16 pH dependence of the BDNPP (2.5 mM) hydrolysis activity by complexes (0.02 mM) (a) 5 and (b) 1 (the pH values refer to the aqueous component). | |
Table 4 Kinetic data: kcat in (10−3 s−1), KM in (mM) and kcat/KM in (s−1 M−1) of the BDNPP hydrolysis (all kinetic studies were done in a mixture of acetonitrile:aqueous buffer = 1:1)a
| Complex |
pKa(I) |
pKa(II) |
γ
|
pHb |
k
cat (10−3 × s−1) |
K
M (mM) |
k
cat/KM (10−3 × s−1 mM−1) |
|
The pH values refer to the aqueous component; the pH of a 1:1 mixture of buffer and acetonitrile is the same within the error as in an aqueous solution of the buffer.66
pH of aqueous buffer solution used for substrate dependent assays (Michaelis–Menten measurements).
|
|
5
|
5.30 ± 0.60 |
6.90 ± 0.31 |
2.52 ± 0.80 |
7 |
0.48 ± 0.05 |
2.55 ± 0.66 |
0.19 ± 0.28 |
|
|
|
|
|
10.5 |
8.24 ± 2.35 |
54.28 ± 16.90 |
0.15 ± 0.42 |
|
1
|
6.97 ± 0.07 |
9.41 ± 0.22 |
5.03 ± 0.74 |
7 |
2.80 ± 0.13 |
0.21 ± 0.11 |
13.33 ± 0.56 |
|
|
|
|
|
10.5 |
64.71 ± 0.81 |
2.81 ± 0.08 |
23.03 ± 0.03 |
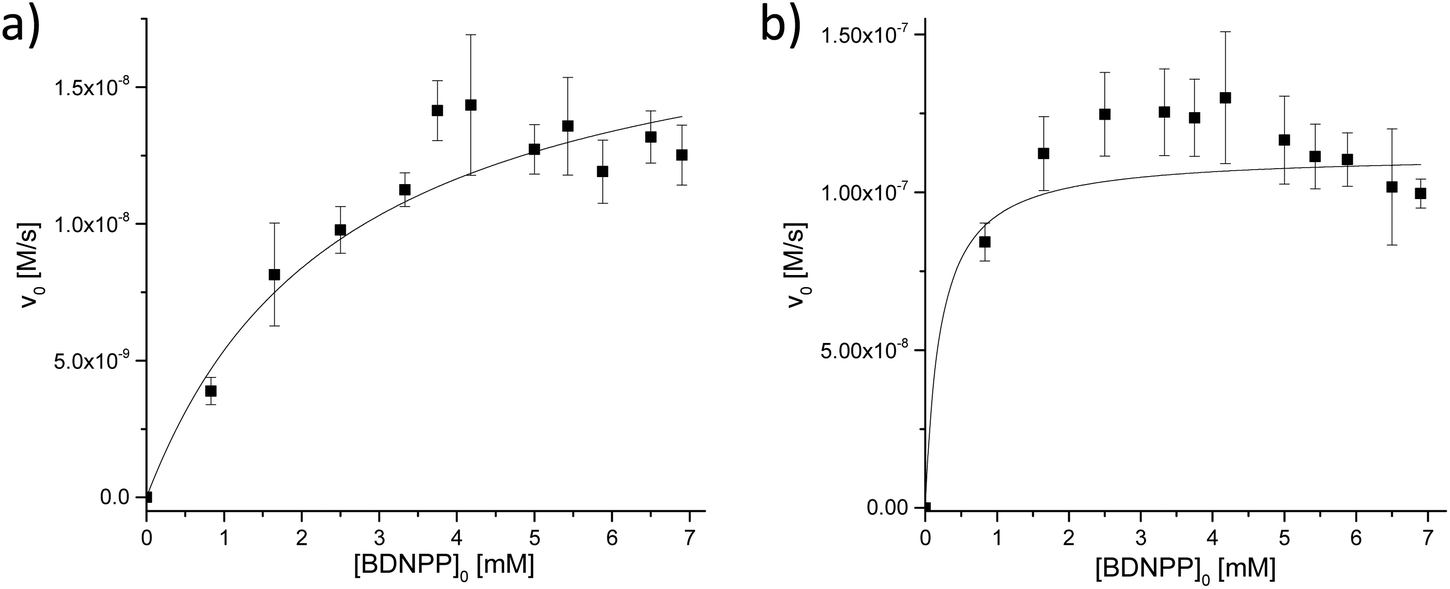
The initial rate vs. pH profiles for complexes 1 and 5 showed different shapes. The curve shape of the nickel(II) complex 1 indicates that three different complex species are present in the pH range of importance. An active species is generated in a first deprotonation step (pKa = 6.97); the second deprotonation step (pKa = 9.41) increases its activity further. This behavior is different from that for the zinc(II) complex 5, which shows only a slight increase in reactivity over the pH range from 9 to 10.
The dependence of the BDNPP hydrolysis rate of complexes 1 and 5 on the substrate concentration was determined at pH 7 and pH 10.5 (Fig. 17 and 18). Fitting the resulting data to the Michaelis–Menten equation provides values for the parameters kcat and KM, which are listed in Table 4 (the catalytic efficiencies kcat/KM have also been determined but the corresponding standard deviations do not allow a meaningful interpretation in all cases). Comparison of the two complexes allows an assessment of the impact of the metal center of the catalyst. The zinc(II) complex 5 shows lower hydrolysis rates compared to the nickel(II) complex (1) at pH 7 and pH 10.5. Moreover, the substrate affinity of 1 is also higher than that for 5 for both pH values investigated, resulting in higher catalytic efficiencies. Therefore, the catalytic activities of the complexes are found to decrease in the order: Ni(II) > Zn(II) > Cu(II). This order contrasts the published results for the phosphomonoester hydrolysis activities of dinuclear copper(II), zinc(II) and nickel(II) complexes of a macrocyclic ligand.67
 |
| | Fig. 17 Substrate concentration dependence of the BDNPP hydrolysis activity by complexes (0.04 mM; pH = 7) (a) 5 and (b) 1. | |
 |
| | Fig. 18 Substrate concentration dependence of the BDNPP hydrolysis activity by complexes (0.04 mM; pH = 10.5) (a) 5 and (b) 1. | |
To account for the two observed reactive species, we can propose that in aqueous acetonitrile and in the presence of BDNPP, one of the acetato groups in the doubly bridged diacetato complex [M2(μ-LClO)(μ2-OAc)2]+ is displaced by BDNPP and two water molecules; one coordinated to each metal ion: [M2(μ-LClO)(OAc)(H2O)2(BDNPP)]+ (Scheme 1). In the neutral pH region (pKa1 = 6.97 ± 0.07 and 5.3 ± 0.60 for complexes 1 and 5, respectively), a hydroxo intermediate [M2(μ-LClO)(OAc)(OH)(H2O)(BDNPP)] is considered to be the predominant species in solution, where the coordinated cis-hydroxo group attacks in an intramolecular nucleophilic reaction the phosphorous atom of the mono-coordinated BDNPP, leading to the hydrolysis of BDNPP and the formation of the product with a bridged-mono-phosphatoester (path A in Scheme 1). A similar dinuclear complex with a bridging μ2-phenylphosphate was isolated and structurally characterized with Cu(II).1b In basic medium (pKa2 = 9.41 ± 0.22 and 6.90 ± 0.31 for 1 and 5, respectively), the second predominant hydroxido species in which the hydroxide and BDNPP are coordinated to the same metal center undergoes a similar hydrolytic process, resulting in the generation of 2,4-dinitrophenolate and the four-membered chelated monophosphatoester (path B).
 |
| | Scheme 1 A tentative mechanism for the hydrolysis of BDNPP by the dinuclear metal(II) complexes (M = Ni(II) or Zn(II)) around the neutral pH region in 1:1 acetonitrile–buffer mixtures. | |
The turn over numbers (TONs) of complexes 1, 3, and 5 were also investigated. The studies were conducted at pH 7 at room temperature and the samples were taken from the assays at various intervals during the studies, diluted with solvent, and their UV-Vis spectra were recorded to determine the amount of phosphoester hydrolysis. The increase in the absorbance at 400 nm, associated with the hydrolysis product 2,4-dinitrophenolate, was monitored over time and TON values were calculated after eight days. The resulting data are given in Table 5.
Table 5 TON of the BDNPP hydrolysis at pH 7 with complexes 1, 3 and 5
| Complex |
TON after 5 days |
TON after 8 days |
|
3
|
1 ± 1 |
— |
|
1
|
66 ± 1 |
79 ± 1 |
|
5
|
14 ± 1 |
16 ± 1 |
In accordance with the kinetic studies, the nickel(II) complex shows the highest TON and the TONs follow the order: Cu(II) < Zn(II) < Ni(II). This behavior is in contrast to two principles: (a) the exchange rates of metal-coordinated water molecules increase in the order Ni(II) < Zn(II) < Cu(II), and (b) the acidity of the coordinated water molecules mainly depends on the electronegativity of the metal ions, as the metal ions in consideration have the same charge. The electronegativities follow the order: Zn < Cu ∼ Ni.67 For these reasons, we believe that the coordination geometry of the metal ions has a significant influence on the reactivity of the complexes. An interesting observation therefore is that the two Cu(II) ions in 3 are five- and six-coordinate, respectively, whereas both Ni(II) ions in 1 and both Zn(II) ions in 5 are six-coordinate.
Experimental
Materials and physical measurements
Bis(2-pyridylmethyl)amine (DPA) was purchased from TCI-America. All other chemicals were commercially available and used without further purification. 2,6-Bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenol (LCl-OH) was synthesized and characterized as recently described.1a Infrared spectra were recorded on a JASCO FTIR-480 plus spectrometer as KBr pellets. Electronic spectra were recorded using an Agilent 8453 HP diode array UV-Vis spectrophotometer. 1H and 13C NMR spectra for the zinc(II) complex were obtained at room temperature on a Varian 400 NMR spectrometer operating at 400 MHz (1H) and 100 MHz (13C). 1H and 13C NMR chemical shifts (δ) are reported in ppm and were referenced internally to residual solvent resonances (DMSO-d6: δH = 2.49, δC = 39.4 ppm). The conductivity measurements were performed using a Mettler Toledo Seven Easy conductivity meter, calibrated by the aid of a 1413 μS cm−1 conductivity standard. Elemental analyses were carried out by the Atlantic Microlaboratory, Norcross, Georgia, USA.
Caution: Salts of perchlorate and their metal complexes are potentially explosive and should be handled with great care and in small quantities.
Syntheses of the complexes
[Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1).
To a mixture of Ni(OAc)2·4H2O (0.100 g, 0.40 mmol) and 2,6-bis[bis(2-pyridylmethyl)amino]-4-chlorophenol (0.112 g, 0.20 mmol) dissolved in MeOH (25 mL), NH4PF6 (0.130 g, 0.80 mmol) was added. The resulting solution was heated on a steam-bath for 10 min, filtered while hot through Celite and then allowed to stand at room temperature. After ca. 3 h, the light blue single crystals which separated were collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 120 mg, 61%). Characterization for 1: Calcd for C36H42ClF6Ni2N6O8P (MM = 984.52 g mol−1): C, 43.92; H, 4.30; N, 8.54%. Found: C, 44.23; H, 4.30; N, 8.57%. Selected FTIR bands (ν, cm−1): 3432 (m, b) ν(O–H), 1604 (vs) ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C); 1422 (m), ν(CN); 845 (vs) νas(P–F). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 490 (sh), 645 (11.5), ∼960 (25.6, b). Molar conductivity in CH3CN, ΛM = 131 Ω−1 cm2 mol−1.
C); 1422 (m), ν(CN); 845 (vs) νas(P–F). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 490 (sh), 645 (11.5), ∼960 (25.6, b). Molar conductivity in CH3CN, ΛM = 131 Ω−1 cm2 mol−1.
[Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2).
To a mixture of Ni(OAc)2·4H2O (0.100 g, 0.40 mmol) and 2,6-bis[bis(2-pyridylmethyl)amino]-4-chlorophenol (0.112 g, 0.20 mmol) dissolved in MeOH (25 mL), NaClO4 (0.100 g, 0.80 mmol) was added. The resulting solution was heated on a steam-bath for 10 min, filtered while hot through Celite and then allowed to stand at room temperature. The crude solid which separated was collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 105 mg, 56%). Recrystallization of the product from acetone afforded aqua-blue crystals suitable for X-ray structure determination. Characterization for 2: Calcd for C39H42ClNi2N6O10 (MM = 943.09 g mol−1): C, 49.67; H, 4.49; N, 8.91%. Found: C, 48.28; H, 4.32; N, 9.15%. Selected FTIR bands (ν, cm−1): 1604 (vs), 1593 (m) ν(CC); 1430 (s), 1415 (s), ν(CN); 1121 (s), 1095 (s) νas(Cl–O). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 490 (sh), 642 (7.2), ∼955 (22, b). ΛM (CH3CN) = 152 Ω−1 cm2 mol−1.
[Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3).
A procedure similar to that described for 2 was used but Cu(OAc)2·H2O (0.100 g, 0.40 mmol) was used instead of Ni(OAc)2·4H2O. After one day, the green compound which separated was collected by filtration and further recrystallized from MeOH. The green crystals were collected by filtration, washed with propan-2-ol and Et2O and then dried in air (overall yield: 120 mg, 61%). Characterization for 3: Calcd for C34H33Cl3Cu2N6O11 (MM = 935.11 g mol−1): C, 43.38; H, 3.56; N, 8.99%. Found: C, 43.52; H, 3.72; N, 8.56%. Selected FTIR bands (ν, cm−1): 1637 (vs) ν(CC); 1439 (w), ν(CN); 1120 (m), 1108 (m), 1091 (m) νas(Cl–O). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 426 (465), ∼800 (202, b). ΛM (CH3CN) = 280 Ω−1 cm2 mol−1.
[Cu2(μ-LClO)(OAc)2](PF6)·H2O (4).
A procedure similar to that described for complex 3 was used but NH4PF6 (0.100 g, 0.80 mmol) was used instead of NaClO4. The crude solid which was separated in the following day was collected by filtration. Further recrystallization of the product from MeOH afforded blue crystals which were collected by filtration, washed with propan-2-ol and Et2O and dried in air (overall yield: 120 mg, 63%). Characterization for 4: Calcd for C36H38ClCu2F6N6O6P (MM = 958.25 g mol−1): C, 45.12; H, 4.00; N, 8.77%. Found: C, 45.21; H, 4.01; N, 8.76%. Selected FTIR bands (ν, cm−1): 3432 (s) ν(O–H); 1612 (vs) ν(CC); 1587 (s), 1486 (w), 1459 (m), 1445 (s), 1398 (s) ν(CN); 842 (vs) νas(P–F). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 404 (713), ∼670 (155, b). ΛM (CH3CN) = 131 Ω−1 cm2 mol−1.
[Zn2(μ-LClO)(μ2-OAc)2](PF6) (5).
A procedure similar to that described for complex 4 was followed except that Zn(OAc)2·2H2O (0.088 g, 0.40 mmol) was used instead of Cu(OAc)2·H2O. The crude solid which was separated in the following day was collected by filtration. Recrystallization of the product from MeOH afforded colorless long needles of X-ray quality. These were collected by filtration, washed with propan-2-ol and Et2O and dried in air (overall yield: 170 mg, 90%). Characterization for 5: Calcd for C34H37Cl3Cu2N6O13 (MM = 943.93 g mol−1): C, 50.22; H, 4.46; N, 10.34%. Found: C, 49.93; H, 4.51; N, 10.28%. Selected FTIR bands (ν, cm−1): 1604 (vs), 1574 (m), 1465 (m), 1429 (s), 843 (vs). UV-Vis spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 303 (2610). ΛM (CH3CN) = 132 Ω−1 cm2 mol−1. 1H NMR (DMSO-d6, 400 MHz, δ in ppm): 2.00 (s, 3H, CH3-acetate), 2.50 (s, 2H, H13), 3.34–3.38 (m, 6H, H13a, H13b, H22a, H22b, H11a, H11b), 3.69 (d, 1H, H12a), 3.75 (d, 1H, H12b), 4.07 (d, 1H, H20a), 4.41 (d, 1H, H 20b), 6.63 (d, 1H, H7), 6.72 (s, 1H, H16), 7.17 (t, 1H, H4), 7.48 (t, 1H, H9), 7.54 (t, 1H, H8), 7.63 (d, 1H, H2), 8.03 (t, 1H, H3), 8.20 (d, 1H, H10), 8.69 (d, 1H, H5). 13C NMR (DMSO-d6, 400 MHz): 24.84 (CH3–, acetate), 56.94 (C11), 58.31 (C12), 59.37 (C13), 117.59 (C5), 121.04 (C10), 122.99 (C9), 124.20 (C3), 124.41 (C8), 125.08 (C4), 129.57 (C9), 138.16 (C2), 139.56 (C7), 145.79 (C18), 147.29 (C16), 149.60 (C19), 154.44 (C15), 154.72 (C17), 160.55 (C14), 177.80 (–CO, acetate). The atom numbering of the complex ion [Zn2(μ-LClO)(μ2-OAc)2]+ is shown in Chart 1.
Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6).
A mixture containing Mn(ClO4)2·6H2O (0.150 g, 0.40 mmol) and 2,6-bis[bis(2-pyridylmethyl)amino]-4-chlorophenol (0.112 g, 0.20 mmol) dissolved in MeOH (25 mL) was heated on a steam bath for 10 min, then NaOAc (0.108 g, 0.80 mmol) was added followed by heating for another 10 min, filtered while hot through Celite and then it was allowed to stand at room temperature. In the following day, the off white long needles which separated were collected by filtration, washed with propan-2-ol and Et2O and then dried at room temperature (overall yield: 72 mg, 40%). Anal. Calcd for 6: C36H38Cl2Mn2N6O10 (MM = 895.53 g mol−1): C, 48.28; H, 4.28; N, 9.38%. Found: C, 47.88; H, 4.16; N, 9.23%. Selected FTIR bands (ν, cm−1): 3448 (w), 1589 (vs), 1427 (s), 1320 (w), 1093 (s). UV-VIS spectrum {λmax, nm (ε, M−1 cm−1)} in CH3CN: 316 (3550), ∼500 (sh). ΛM (CH3CN) = 167 Ω−1 cm2 mol−1.
X-Ray crystal structure analysis
The X-ray single-crystal data of compounds 1–6 were collected on a Bruker-AXS APEX CCD diffractometer at 100(2) K. The crystallographic data, conditions retained for the intensity data collection and some features of the structure refinements are listed in Table S1 (see the ESI†). The intensities were collected with Mo-Kα radiation (λ = 0.71073 Å). Data processing, Lorentz-polarization and absorption corrections were performed using APEX, and the SADABS computer programs.68 The structures were solved by direct methods and refined by full-matrix least-squares methods on F2, using the SHELXTL69 program package. All non-hydrogen atoms were refined anisotropically. The hydrogen atoms were located from difference Fourier maps, assigned with isotropic displacement factors and included in the final refinement cycles by the use of HFIX (parent C atom) or DFIX (parent O atom) utility of the SHELXTL program. Molecular plots were performed with the Mercury program.70
Magnetic measurements
Magnetic data of nickel (1 and 2) and manganese (6) complexes were measured with a PPMS Dynacool VSM magnetometer (T = 1.9–300 K at B = 0.1/1 T; B = 0–9 T at T = 2, 5 and 10 K). The copper complexes (3 and 4) were measured with a MPMS XL7 SQUID magnetometer (T = 1.9–300 K at B = 1 T; B = 0–5 T at T = 2 and 5 K). The magnetic data were corrected for diamagnetic susceptibilities and the signal of the sample holder.
DFT calculations
The DFT calculations were performed with the ORCA 3.0.3 computational package.71 The hybrid B3LYP functional72 and the polarized triple-ζ quality basis set def2-TZVP(-f) proposed by Ahlrichs and co-workers was used for all atoms.73 The calculations utilized the RI approximation with the decontracted auxiliary def2-TZV/J Coulomb fitting basis sets and the chain-of-spheres (RIJCOSX) approximation to exact exchange as implemented in ORCA.74 Increased integration grids (Grid5 in ORCA convention) and tight SCF convergence criteria were used in all calculations. The spin densities were visualized with the program VESTA 3.75
DNA study
Interactions of the complexes with plasmid DNA.
To determine the nuclease activity of complexes 1–5, 300 ng (i.e. 23.1 μM of base pairs) in the 20 μL of the reaction mixture of the native supercoiled pUC19 plasmid DNA was incubated with different concentrations of the tested complexes which were dissolved in 25% (v/v) acetonitrile at 37 °C and allowed to interact for 2 h. Immediately after that, the samples were then quickly cooled to 4 °C and mixed with gel loading buffer [containing 30% (v/v) glycerol, 0.25% (w/v) bromophenol blue] and subsequently loaded on 0.8% (w/v) agarose gel in TBE buffer (containing 45 mM Tris-borate buffer and 1 mM EDTA) impregnated with 0.15 μg mL−1 of ethidium bromide (EtBr). The electrophoretogram was analyzed by the AlphaEaseFC version 4.0.0.34 software (Alpha Innotech, USA) and the relative amounts of the supercoiled circular (SC-form), single-strand nicked (OC-form) and linear (L-form) forms were evaluated. The quantification of the SC-form of plasmid DNA was corrected by a factor of 1.47.39
Effect of incubation time on the interaction of complex 3 with plasmid DNA.
To evaluate the effect of incubation time on the cleaving activity of the compounds, Cu(II) complex 3 was selected as a model compound. The compound was incubated at the concentrations of 300 and 30 μM with 300 ng of supercoiled plasmid DNA for 2, 4, 6, and 22 h. After the incubation, the samples were analyzed by gel electrophoresis as described above.
Effect of oxidative scavengers and inhibitors on the interactions of Ni(II) complexes with plasmid DNA.
The Ni(II) complexes 1 and 2 were selected to determine whether their interactions with supercoiled plasmid DNA could be modulated in the presence of different inhibitors. Therefore, the ROS scavengers DMSO, and KI,39,46a the metal competitor MgSO4 and highly efficient metal chelator EDTA were added in a molar ratio of 1:1 with 300 and 33 μM of the complexes and these were incubated with 300 ng of plasmid DNA at 37 °C for 2 h in a similar manner as described above.
Kinetics of the phosphodiester hydrolysis.
Phosphodiester hydrolysis activity was probed for complexes 1, 3 and 5. BDNPP was used as a phosphodiester model substrate in the assay. It was synthesized following a published procedure with minor modifications.76 Cleavage of BDNPP was followed spectrophotometrically by monitoring the generated product, 2,4-dinitrophenolate, by its strong absorption at 400 nm (ε = 12100 M−1 cm−1). The spectra were recorded at 25 °C with a JASCO V-570 spectrophotometer in 10 mm or 2 mm quartz cuvettes. All measurements were carried out in 1:1 acetonitrile–buffer mixtures and performed in triplicate. The aqueous buffer consisted of 2-(N-morpholino)-ethanesulfonic acid (MES) (50 mM; pH range: 5.5–6.7), 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (50 mM; pH range: 6.8–8.2), 2-(cyclohexylamino)ethanesulfonic acid (CHES) (50 mM; pH range: 8.6–10.0), 3-(cyclohexylamino)-1-propanesulfonic acid (CAPS) (50 mM; pH range: 9.7–11.1) and lithium perchlorate (250 mM) for ionic strength control. The desired pH of the buffers was adjusted by addition of aqueous sodium hydroxide solution. Subsequent treatment with Chelex® (Chelex 100 sodium form) overnight and filtration with 45 μm syringe filters ensured the absence of metal ions in the buffer solutions. BDNPP was initially prepared as a 15 mM stock solution in acetonitrile and the complex stock solutions were 1 mM in acetonitrile. The complex was allowed to equilibrate in the acetonitrile–buffer mixture for one minute prior to addition of the substrate. When the substrate was added to the reaction mixture the starting hydrolysis activity was monitored in the time between 15 and 195 seconds and analyzed by linear regression. For each experiment autohydrolysis assays were conducted by measuring the hydrolysis rate under the same conditions, but without the complex, and were subtracted from the derived data. The pH dependent assays contained the complex at 0.02 mM and BDNPP at 2.5 mM in the cuvette. The substrate concentration dependent assays were 0.04 mM in the complex. The experimental data obtained were fitted by the Origin (OriginLab) program. Studies of the TON were conducted at 10 μM in the complex and 3 mM in BDNPP. Samples were taken at various intervals during the experiment, diluted with solvent (final concentration: 0.5 nM in complex and 0.15 mM in BDNPP), and their UV-vis spectra were recorded. The increase in the absorbance at 400 nm, assigned to 2,4-dinitrophenolate, was monitored over time and TON values were calculated using the Beer–Lambert Law.
Conclusions
Six dinuclear metal(II)-acetato complexes [Ni2(μ-LClO)(μ2-OAc)2](PF6)·3H2O (1), [Ni2(μ-LClO)(μ2-OAc)2](ClO4)·CH3COCH3 (2), [Cu2(μ-LClO)(μ2-OAc)(ClO4)](ClO4) (3), [Cu2(μ-LClO)(OAc)2](PF6)·H2O (4), [Zn2(μ-LClO)(μ2-OAc)2](PF6) (5) and [Mn2(LCl-O)(μ2-OAc)2](ClO4)·H2O (6) have been synthesized in order to test their efficiencies in catalyzing the P–O bonds in DNA and in promoting the hydrolysis of BDNPP. These complexes were structurally and magnetically characterized. The backbone of all complexes consists of 2,6-bis[bis(2-pyridylmethyl)aminomethyl]-4-chlorophenolate (LCl-O−) linking the two metal ions through the deprotonated phenolate group, the acetate ligands are further bridging the metal ions in complexes 1, 2, 3, 5 and 6 but are simple monodentate donors in 4. The magnetic measurements revealed antiferromagnetic coupling for complexes 1, 2, 4 and 6, and ferromagnetic coupling in 3, and these results were supported by the DFT calculations.
The hydrolysis of the phosphodiester bis(2,4-dinitrophenol)phosphate (BDNPP), used as a model substrate for the P–O bond cleavage in DNA, was examined with complexes 1, 3 and 5 over the pH range 5.5–10.5 at 25 °C. Michaelis–Menten kinetics (pH = 7 and 10.5) showed that catalytic efficiencies kcat/KM decrease in the order Ni(II), 1 > Zn(II), 5 > Cu(II), 3. Parallel to the phosphatase reactivity study, the nuclease activity of complexes 1–5 were employed for studying the supercoiled plasmid ds-DNA cleavage under the physiological conditions. Surprisingly, none of the complexes showed any sign of cleavage activity but instead only the two nickel complexes 1 and 2 revealed a strong ability to unwind the supercoiled plasmid ds-DNA. These results raise a question about the validity of a direct comparison of model metal complexes used for the hydrolysis of phosphodiesters with DNA cleavage, where two mechanistic pathways (hydrolysis and oxidative cleavage) exist. In addition to the present results, [Zn(TPA)(H2O)]2+ (TPA = tris(2-pyridylmethyl)amine) was shown to exhibit an enhanced phosphodiester hydrolysis rate77 but the corresponding catalytic cleavage reaction for DNA was insignificant.46b
Comparative studies for supercoiled ds-DNA cleavage efficiency by a number of metal complexes, when different metals exist in the same coordination environment and are bound to the same ligand, have been performed.42a,46b For example in the cleavage of DNA by the structurally characterized hexa-coordinate complexes [M(bpa)(NO3)]+, where bpa = N-(2-ethoxyethanol)-bis(2-picolyl)amine, the efficiency decreased in the order: Cu(II) > Co(II) > Zn(II) ≈ Ni(II)42a and in a comparable study using the five-coordinate [M(TPA)(H2O)]2+, the reactivity order was Co(II) > Cu(II) ≫ Zn(II).46b These two sets of data demonstrate that the observed reactivity is not attributed to a specific metal ion nor to the lability of metal ions.78 In addition, our recent work on the DNA cleavage by a series of sterically hindered TBP Co(II) complexes derived from the substituted N4-tripod TPA39 and on the dinuclear Cu(II)-bdpaTCl complexes, where bdpaTCl = 2-chloro-4,6-bis(di-2-picolylamino)-1,3,5-triazine,36 showed that the steric environment imposed by the ligands around the central metal ions has a strong influence on supressing the approach of DNA to the metal center and this may lower or even inhibit the reactivity of DNA cleavage.36,39 However, it is important to mention that this may not be the case in the hydrolysis of simple phosphodiesters as these molecules have a very small size compared to DNA. Therefore, aside from the mechanistic complications in the DNA cleavage reactions, the observed lack of reactivity of complexes 1–5 in the DNA cleavage reactions and the observed efficiency by complexes 1 and 5 in promoting the hydrolysis of BDNPP may be due to the large steric effect imposed by the coordinated organic ligands which prohibits the ds-DNA from closely approaching the metal centers while this presents no problem for BDNPP. In conclusion, careful attention must be paid to the results in which metal complexes are used to mimic the hydrolysis of simple phosphodiester compounds as “model systems” for the natural biological phosphoesters, DNA or RNA.
Acknowledgements
S. S. M. acknowledges the financial support of this research by the Department of Chemistry-University of Louisiana at Lafayette. R. H., J. H. and Z. T. gratefully thank the National Program of Sustainability I (LO1305) of the Ministry of Education, Youth and Sports of the Czech Republic for financial support. F. A. M. acknowledges the support by NAWI Graz. S. B. and P. C. gratefully acknowledge support by the German Science Foundation (DFG), the Deutscher Akademischer Austauschdienst (DAAD) and the University of Heidelberg.
References
-
(a) S. S. Massoud, M. Spell, C. Ledet, T. Junk, R. Herchel, R. C. Fischer, Z. Travnicek and F. A. Mautner, Dalton Trans., 2015, 44, 2110–2121 RSC;
(b) S. S. Massoud, T. Junk, F. R. Louka, R. Herchel, Z. Travnicek, R. C. Fischer and F. A. Mautner, RSC Adv., 2015, 5, 87139–87150 RSC.
- G. Ambrosi, M. Formica, V. Fusi, L. Giorgi and M. Micheloni, Coord. Chem. Rev., 2008, 252, 1121–1152 CrossRef CAS.
-
(a) S. Svane, F. Kryuchkov, C. J. Lennarston, C. K. McKenzie and F. Kjeldsen, Angew. Chem., Int. Ed., 2012, 51, 3216–3219 CrossRef CAS PubMed;
(b) R. K. Edgal, A. D. Bond and C. J. Mckenzie, Dalton Trans., 2009, 3833–3839 Search PubMed;
(c) R. K. Edgal, F. B. Larsen, A. D. Bond and C. J. Mckenzie, Inorg. Chim. Acta, 2005, 358, 376–382 CrossRef.
-
(a) A. Boisen, A. Hazell and C. J. Mckenzie, Chem. Commun., 2001, 2136–2137 RSC;
(b) P. Dalgaard, A. Hazell, C. J. McKenzie, B. Moubaraki and K. S. Murray, Polyhedron, 2000, 19, 1909–1015 CrossRef CAS;
(c) H. Adams, D. Bradshaw and D. E. Fenton, Inorg. Chim. Acta, 2002, 332, 195–200 CrossRef CAS.
-
(a) A. S. Borovik, M. P. Hendrich, T. R. Holman, E. Munck, V. Papaefthymiou and L. Que Junior, J. Am. Chem. Soc., 1990, 112, 6031–6038 CrossRef CAS;
(b) A. S. Borovik and L. Que Jr., J. Am. Chem. Soc., 1988, 110, 2345–2347 CrossRef CAS.
- F. R. Xavier, A. Neves, A. Casellato, R. A. Peralta, A. J. Bortoluzzi, B. Szpoganicz, P. C. Severino, H. Terenzi, Z. Tomkowicz, S. Ostrovsky, W. Haase, A. Ozarowski, J. Krzystek, J. Telser, G. Schenk and L. R. Gahan, Inorg. Chem., 2009, 48, 7905–7921 CrossRef CAS PubMed.
-
(a) F. Michel, P. Torelli, St. F. Thomas, C. Duboc, C. Philouze, S. Belle, E. Hamman, S. Saint-Aman and J. L. Pierre, Angew. Chem., Int. Ed., 2005, 44, 438–441 CrossRef CAS PubMed;
(b) R. C. Holz and M. J. Brink, Inorg. Chem., 1994, 33, 4609–4610 CrossRef CAS.
- S. Torelli, C. Belle, I. Gautier-Luneau, J. L. Pierre, E. Saint-Aman, J. M. Latour, L.-L. Pape and D. Luneau, Inorg. Chem., 2000, 39, 3526–3536 CrossRef CAS PubMed.
- M. Jarenmark, M. Haukka, S. Demeshko, F. Tuczek, L. Zuppiroli, F. Meyer and E. Nordlander, Inorg. Chem., 2011, 50, 3866–3887 CrossRef CAS PubMed.
- A. Neves, M. Lanznaster, A. J. Bortoluzzi, R. A. Peralta, A. Casellato, E. E. Castellano, P. Herrald, M. J. Riley and G. Schenk, J. Am. Chem. Soc., 2007, 129, 7486–7487 CrossRef CAS PubMed.
-
(a) S. S. Massoud, T. Junk, R. Herchel, Z. Travnicek, M. Mikuriya, R. C. Fischer and F. A. Mautner, Inorg. Chem. Commun., 2015, 60, 1–3 CrossRef CAS;
(b) S. S. Massoud, T. Junk, M. Mikuriya, N. Naka and F. A. Mautner, Inorg. Chem. Commun., 2014, 50, 48–50 CrossRef CAS.
- K. D. Karlin, Z. Tyeklár, A. Farooq, M. S. Haka, P. Ghosh, R. W. Cruse, Y. Gultneth, J. C. Hayes, P. J. Toscano and J. Zubieta, Inorg. Chem., 1992, 31, 1436–1451 CrossRef CAS.
- L. J. Daumann, J. A. Larrabee, P. Comba, G. Schenk and L. R. Gahan, Eur. J. Inorg. Chem., 2013, 3082–3089 CrossRef CAS.
- S. J. Smith, C. J. Noble, R. C. Palmer, G. R. Hanson, G. Schenk, L. R. Gahan and M. J. Riley, J. Biol. Inorg. Chem., 2008, 13, 499–510 CrossRef CAS PubMed.
- T. P. Camargo, F. F. Maia, C. Chaves, B. de Souza, A. J. Bortoluzzi, N. Castilho, N. T. Bortolotto, H. Terenzi, E. E. Castellano, W. Haase, Z. Tomkowicz, R. A. Peralta and A. Neves, J. Inorg. Biochem., 2015, 146, 77–88 CrossRef CAS PubMed.
- R. L. Lomoth, P. Huang, J. Zheng, L. Sun, L. Hammarström, B. Akermark and S. Styring, Eur. J. Inorg. Chem., 2002, 5965–2974 Search PubMed.
- Y. Gultneh, Y. T. Tesema, T. B. Yisgedu, R. J. Butcher, G. Wang and G. T. Yee, Inorg. Chem., 2006, 45, 3023–3033 CrossRef CAS PubMed.
- M. Ghiladi, C. K. Mckenzie, A. Meler, A. K. Powell, J. Ulstrup and S. Wocadlo, J. Chem. Soc., Dalton Trans., 1997, 4011–4018 RSC.
- P. V. Bernhardt, S. Bosch, P. Comba, L. R. Gahan, G. R. Hanson, V. Mereacre, C. J. Noble, A. K. Powell, G. Schenk and H. Wadepohl, Inorg. Chem., 2015, 54, 7249–7263 CrossRef CAS PubMed.
- P. Comba, L. R. Gahan, V. Mereacre, G. R. Hanson, A. K. Powell, G. Schenk and M. Zajaczkowski-Fischer, Inorg. Chem., 2012, 51, 12195–12209 CrossRef CAS PubMed.
- S. Bosch, P. Comba, L. R. Gahan and G. Schenk, Inorg. Chem., 2014, 53, 9036–9051 CrossRef CAS PubMed.
- B. Das, H. Daver, M. Pyrkosz-Bulska, E. Perscha, S. K. Barman, R. Mukherjee, E. Gumienna-Kontecka, M. Jarenmark, F. Himo and E. Nordlander, J. Inorg. Biochem., 2014, 132, 6–17 CrossRef CAS PubMed.
- L. J. Daumann, P. Comba, J. A. Larrabee, G. Schenk, R. Stranger, G. Cavigliasso and L. R. Gahan, Inorg. Chem., 2013, 52, 2029–2043 CrossRef CAS PubMed.
- D. Montagner, V. Gandin, C. Marzano and A. Erxleben, Eur. J. Inorg. Chem., 2014, 4084–4092 CrossRef CAS.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and P. H. Terenzi, Inorg. Chem., 2007, 46, 348–350 CrossRef CAS PubMed.
-
(a) G. K. Schroeder, C. Lad, P. Wyman, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4052–4255 CrossRef CAS PubMed;
(b) J. A. Cowan, Chem. Rev., 1998, 98, 1067–1088 CrossRef CAS PubMed.
-
P. Hendry and A. M. Sargeson, Progress Inorg. Chem.: Bioinorganic Chemistry, ed. S. Lippard, 1990, vol. 38, pp. 201–258 Search PubMed.
- J. A. Cowan, Nucleic Acids Mol. Biol., 2004, 14, 339–360 CAS.
- C. Liu, M. Wang, T. Zhang and H. Sun, Coord. Chem. Rev., 2004, 248, 147–168 CrossRef CAS.
- P. Molenveld, J. F. J. Engbersen and D. N. Reinhoudt, Chem. Soc. Rev., 2000, 29, 75–86 RSC.
-
(a)
M. Pitie, C. Boldron and G. Pratviel, Advances in Inorganic Chemistry, ed. R. van Eldick and J. Reedijk, 2006, vol. 58, pp. 77–130 Search PubMed;
(b)
G. Pratviel, A. Bernadou and B. Meunier, Advances in Inorganic Chemistry, ed. G. A. Sykes, Academic Press, London, 1997, vol. 45, pp. 251–312 Search PubMed.
-
(a) Y. Jin, M. A. Lewis, N. H. Gokhale, E. C. Long and J. A. Cowan, J. Am. Chem. Soc., 2007, 129, 8353–8361 CrossRef CAS PubMed;
(b) Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 127, 8408–8415 CrossRef CAS PubMed.
-
(a) L. Qian, W. R. Browne and G. Roelfes, Inorg. Chem., 2011, 50, 8318–8325 CrossRef PubMed;
(b) Q. Li, W. R. Browne and G. Roelfes, Inorg. Chem., 2010, 49, 11009–11017 CrossRef CAS PubMed.
-
(a) D.-D. Li, J.-L. Tian, W. Gu, X. Liu, H.-H. Zeng and S.-P. Yan, J. Inorg. Biochem., 2011, 105, 894–901 CrossRef CAS PubMed;
(b) D.-D. Li, F.-P. Huang, G.-J. Chen, Y.-C. Gao, J.-L. Tian, W. Gu, X. Liu and S.-P. Yan, J. Inorg. Biochem., 2010, 104, 431–441 CrossRef CAS PubMed.
-
(a) H. Prakash, A. Shodal, H. Yasui, H. Sakurai and H. S. Hirota, Inorg. Chem., 2008, 47, 5045–5047 CrossRef CAS PubMed;
(b) H. Li, X.-Y. Le, D. W. Pang, H. Deng, Z.-H. Xu and Z.-H. Lin, J. Inorg. Biochem., 2005, 99, 2240–2247 CrossRef CAS PubMed.
- S. S. Massoud, F. R. Louka, W. Xu, R. Perkins, R. Vicente, J. H. Albering and F. A. Mautner, Eur. J. Inorg. Chem., 2011, 3469–3479 CrossRef CAS.
- O. I. Aruoma, B. Halliwell and M. Dizdaroglu, J. Biol. Chem., 1989, 264, 13024–13028 CAS.
- Q. Jiang, N. Xiao, P. Shi, Y. Zhu and Z. Guo, Coord. Chem. Rev., 2007, 251, 1951–1972 CrossRef CAS.
- S. S. Massoud, R. S. Perkins, F. R. Louka, W. Xu, A. Le Roux, Q. Dutercq, R. C. Fischer, F. F. A. Mautner, M. Handa, Y. Hiraoka, G. L. Kreft, T. Bortolotto and H. Terenzi, Dalton Trans., 2014, 43, 10086–10103 RSC.
- K. E. Erkkila, D. T. Odom and J. K. Barton, Chem. Rev., 1999, 99, 2777–2796 CrossRef CAS PubMed.
- M. S. Deshpande, A. A. Kumbhar and A. S. Kumbhar, Inorg. Chem., 2007, 46, 5450–5452 CrossRef CAS PubMed.
-
(a) S. I. Kirlin, C. M. Happel, S. Hrubanova, T. Weyhermüller, C. Klein and N. M. Notle, Dalton Trans., 2004, 1201–1207 RSC;
(b) T. Itoh, H. Hisada, T. Sumiya, T. Hosono, Y. Usui and Y. Fujii, Chem. Commun., 1997, 677–678 RSC.
- J. T. Wang, Q. Xia, X.-H. Zheng, H.-Y. Chen, H. Chao, Z.-W. Mao and L.-N. Ji, Dalton Trans., 2010, 39, 2128–2136 RSC.
- S. Dhar, P. A. N. Reddy and A. R. Chakravarty, Dalton Trans., 2004, 697–698 RSC.
- J. He, J. Sun, Z. W. Mao, L. N. Ji and H. Z. Sun, J. Inorg. Biochem., 2009, 103, 851–856 CrossRef CAS PubMed.
-
(a) S. S. Massoud, R. S. Perkins, K. D. Knierim, S. P. Comiskey, K. H. Otero, C. L. Michel, W. M. Juneau, J. H. Albering, F. A. Mautner and W. Xu, Inorg. Chim. Acta, 2013, 399, 177–184 CrossRef CAS;
(b) W. Xu, F. A. Craft, P. R. Fontenot, B. Marion, K. D. Knierim, J. H. Albering, F. A. Mautner and S. S. Massoud, Inorg. Chim. Acta, 2011, 373, 159–166 CrossRef CAS.
- G. Barone, A. Terenzi, A. Lauria, A. M. Almerico, J. M. Leal, N. Busto and B. García, Coord. Chem. Rev., 2013, 257, 2848–2862 CrossRef CAS.
- A. Terenzi, C. Ducani, L. Male, G. Barone and M. J. Hannon, Dalton Trans., 2013, 42, 11220–11226 RSC.
-
(a) A. J. Clarke, N. Yamamoto, P. Jensen and T. W. Hambley, Dalton Trans., 2009, 10787–10798 RSC;
(b) A. Hille, I. Ott, A. Kitanovic, I. Kitanovic, H. Alborzinia, E. Lederer, S. Wolfl, N. Metzler-Nolte, S. Schafer, W. S. Sheldrick, C. Bischof, U. Schatzschneider and R. Gust, J. Biol. Inorg. Chem., 2009, 14, 711–725 CrossRef CAS PubMed.
- A. Arola-Arnal, J. Benet-Buchholz, S. Neidle and R. Vilar, Inorg. Chem., 2008, 47, 11910–11919 CrossRef CAS PubMed.
- M. J. Hannon, Chem. Soc. Rev., 2007, 36, 280–295 RSC.
- R. Martinez and L. Chacon-Garcia, Curr. Med. Chem., 2005, 12, 127–151 CrossRef CAS PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565–4579 RSC.
-
(a)
G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorg. Chem, Pearson, Boston, USA, 5th edn, 2014, pp. 412–431 Search PubMed;
(b)
C. E. Housecroft and A. G. Sharpe, Inorg. Chem, Pearson, Harlow, England, 4th edn, 2012, pp. 687–697 Search PubMed.
-
B. J. Hathaway, in Comprehensive Coordination Chemistry, ed. G. Wilkinson, R. D. Gillard and J. A. McCleverty, Pergamon Press, Oxford, England, 1987, vol. 5, p. 533 Search PubMed.
-
(a) S. S. Massoud, F. R. Louka, R. N. David, M. J. Dartez, Q. L. Nguyn, N. J. Labry, R. C. Fischer and F. A. Mautner, Polyhedron, 2015, 90, 258–265 CrossRef CAS;
(b) S. S. Massoud, F. R. Louka, Y. K. Obaid, R. Vicente, J. Ribas, R. C. Fischer and F. A. Mautner, Dalton Trans., 2013, 42, 3968–3978 RSC;
(c) S. S. Massoud, L. Le Quan, K. Gatterer, J. H. Albering, R. C. Fischer and F. A. Mautner, Polyhedron, 2012, 31, 601–606 CrossRef CAS;
(d) F. A. Mautner, R. Vicente and S. S. Massoud, Polyhedron, 2006, 25, 1673–1680 CrossRef CAS;
(e) U. Mukhopadhyay, I. Bernal, S. S. Massoud and F. A. Mautner, Inorg. Chim. Acta, 2004, 357, 3673–3682 CrossRef CAS.
- A. W. Addison, T. N. Rao, J. Reedijk, J. V. Rijin and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
R. Boča, Theoretical Foundations of Molecular Magnetism, Elsevier, Amsterdam, 1999 Search PubMed.
-
(a) E. Ruiz, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 1999, 20, 1391–1400 CrossRef CAS;
(b) E. Ruiz, A. Rodríguez-Fortea, J. Cano, S. Alvarez and P. Alemany, J. Comput. Chem., 2003, 24, 982–989 CrossRef CAS PubMed.
-
(a)
K. Yamaguchi, Y. Takahara and T. Fueno, in Applied Quantum Chemistry, ed. V. H. Smith, Dordrecht, Reidel, Dordrecht, 1986, pp. 155–184 Search PubMed;
(b) T. Soda, Y. Kitagawa, T. Onishi, Y. Takano, Y. Shigeta, H. Nagao, Y. Yoshioka and K. Yamaguchi, Chem. Phys. Lett., 2000, 319, 223–230 CrossRef CAS.
- L. R. Gahan, S. J. Smith, A. Neves and G. Schenk, Eur. J. Inorg. Chem., 2009, 2745–2758 CrossRef CAS.
- D. Desbouis, I. P. Troitsky, M. J. Belousoff, L. Spiccia and B. Graham, Coord. Chem. Rev., 2012, 256, 897–937 CrossRef CAS.
- R. Peralta, A. J. Bortoluzzi, B. de Souza, R. Jovito, F. R. Xavier, R. A. A. Couto, A. Casellato, F. Nome, A. Dick, L. R. Gahan, G. Schenk, G. R. Hanson, F. C. S. de Paula, E. C. Pereira-Maia, S. Machado, P. C. Severino, C. Pich, F. L. Fischer, H. Terenzi, E. E. Castellano, A. Neves and M. J. Riley, Inorg. Chem., 2010, 49, 11421–11438 CrossRef CAS PubMed.
-
I. H. Segel, Enzyme Kinetics - Behavior and Analysis of Rapid Equilibrium and Steady-State Enzyme Systems, Wiley-VCH, New York, 1975 Search PubMed.
- A. Kantacha, R. Buchholz, S. J. Smith, G. Schenk and L. R. Gahan, J. Biol. Inorg. Chem., 2011, 16, 25–32 CrossRef CAS PubMed.
-
(a) L. J. Daumann, L. Marty, G. Schenk and L. R. Gahan, Dalton Trans., 2013, 42, 9574–9584 RSC;
(b) L. J. Daumann, L. R. Gahan, P. Comba and G. Schenk, Inorg. Chem., 2012, 51, 7669–7681 CrossRef CAS PubMed.
- S. Anbu and M. Kandaswamy, Inorg. Chim. Acta, 2012, 385, 45–52 CrossRef CAS.
-
(a)
Bruker, SAINT v. 7.23, Bruker AXS Inc., Madison, Wisconsin, USA, 2005 Search PubMed;
(b)
G. M. Sheldrick, SADABS v. 2, University of Goettingen, Germany, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, T. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
-
(a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS;
(c) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS;
(d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
-
(a) A. Schafer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef;
(b) A. Schafer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef;
(c) F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
-
(a) F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS;
(b) R. Izsak and F. Neese, J. Chem. Phys., 2011, 135, 144–150 CrossRef PubMed.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2011, 44, 1272–1276 CrossRef CAS.
- C. A. Bunton and S. J. Farber, J. Org. Chem., 1969, 34, 767–772 CrossRef CAS.
- G. Feng, J. C. Mareque-Rivas, R. T. M. de Rosales and N. H. Williams, J. Am. Chem. Soc., 2005, 127, 13470–13471 CrossRef CAS PubMed.
- H. Krüger, Chem. Soc. Rev., 1982, 11, 227–255 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data and processing parameters, and selected bond parameters for compounds 1–6 are summarized in Tables S1 and S2–S7, respectively. Possible hydrogen bonds for compounds 1 and 4 are given in S8, respectively. The 1H and 13C NMR spectra for complex 5 are illustrated in Fig. S1 and S2, respectively and the atom numbering scheme is shown in Chart S1. The magnetic data of complex 2, and the incubation data time for complex 3 with DNA, are shown in Fig. S3 and S4, respectively. CCDC 1444958–1444963 for 1–6. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02596j |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence c,
Jan
Hošek
c,
Zdeněk
Trávníček
*c,
Roland C.
Fischer
d and
Franz A.
Mautner
*e
c,
Jan
Hošek
c,
Zdeněk
Trávníček
*c,
Roland C.
Fischer
d and
Franz A.
Mautner
*e