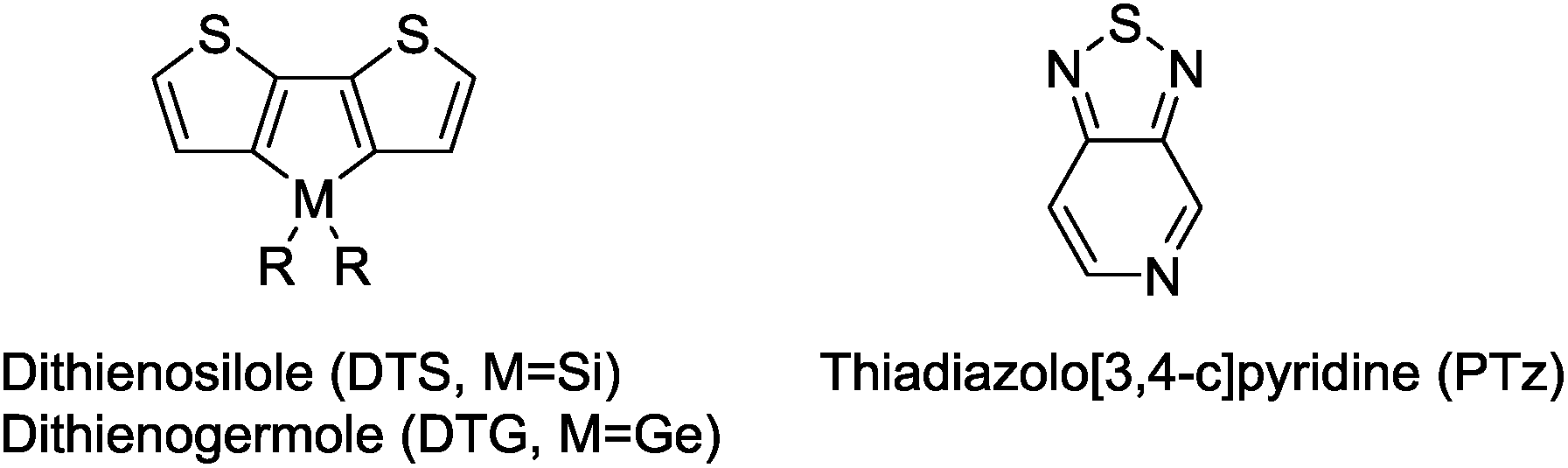DOI:
10.1039/C6DT02469F
(Paper)
Dalton Trans., 2016,
45, 13817-13826
Synthesis of organic photosensitizers containing dithienogermole and thiadiazolo[3,4-c]pyridine units for dye-sensitized solar cells†
Received
20th June 2016
, Accepted 27th July 2016
First published on 28th July 2016
Abstract
Dithienogermole (DTG) is a germanium-bridged bithiophene system that has been applied as a building unit of conjugated materials for organic electronic devices, including organic photovoltaics and organic light emitting diodes. However, DTG has not been used as a component of sensitizers for dye-sensitized solar cells (DSSCs). In this work, we have synthesized three D–π-A–π-A type sensitizers containing DTG and thiadiazolo[3,4-c]pyridine (PTz). We expected that combining DTG and a strong acceptor PTz would give rise to a strong absorption in the visible region. In addition, we introduced bulky 2-ethylhexyl groups on the germanium atom to prevent dye aggregation on TiO2 films. Three DTG-containing dyes with different anchor units were synthesized and their optical/electrochemical properties were investigated. The DTG-containing dyes exhibited broad and strong absorption bands around 600 nm on TiO2. We fabricated DSSCs based on the DTG-containing dyes. The onsets of incident photon to current conversion efficiency (IPCE) spectra reached 900 nm and a maximal power conversion efficiency of 2.76% was achieved.
Introduction
Dye-sensitized solar cells (DSSCs) have been extensively studied as a photovoltaic system with potential applications in low-cost, flexible, and lightweight modules, since O'Regan and Grätzel reported high-performance DSSCs for the first time.1 In this system, an organic dye adsorbed on a TiO2 electrode absorbs sunlight to generate photocurrent. Thus, the design of the dye molecule is the key to improving cell performance. Currently, Ru-complex dyes, such as N3, N719, and black dye, are used to fabricate high-performance cells that exhibit over 10% power conversion efficiency (PCE).2 Recently, Ru-free D (donor)–π-A (acceptor) type organic dyes have drawn attention as new low-cost photosensitizers. The D–A interaction in the π-conjugation system minimizes the HOMO–LUMO gap to extend dye absorption to the visible-NIR region, improving cell performance.3
Modified D–π-A systems have been examined to further improve the sensitizing properties of the dyes. For example, D–A–π-A and D–π-A–π-A type dyes that contain an additional acceptor linked by a π-spacer have been developed. It has been demonstrated that the introduction of a central acceptor unit, such as benzothiadiazole, quinoxaline, or diketopyrrolopyrrole, yields advantages such as photoenergy conversion in the NIR region, high electron injection efficiency, and improved stability of the sensitizers towards photoirradiation.4 For example, as high as 9.04% and 8.50% PCEs were achieved in cells fabricated with benzothiadiazole- and quinoxaline-containing D–A–π-A dyes, respectively.5 PCEs of 8.21% and 7.43% were achieved in cells made of benzothiadiazole (BT)- and diketopyrrolopyrrole-introduced D–π-A–π-A dyes.6 Recently, thiadiazolo[3,4-c]pyridine (PTz, Chart 1) was used as the central acceptor of D–π-A–π-A dyes.7 PTz is a stronger electron-withdrawing unit than common acceptors, such as BT and quinoxaline, because its extremely low-lying LUMO8 gives rise to a strong absorption in the visible region.
 |
| | Chart 1 Structures of dithienosilole, dithienogermole, and thiadiazolo[3,4-c]pyridine. | |
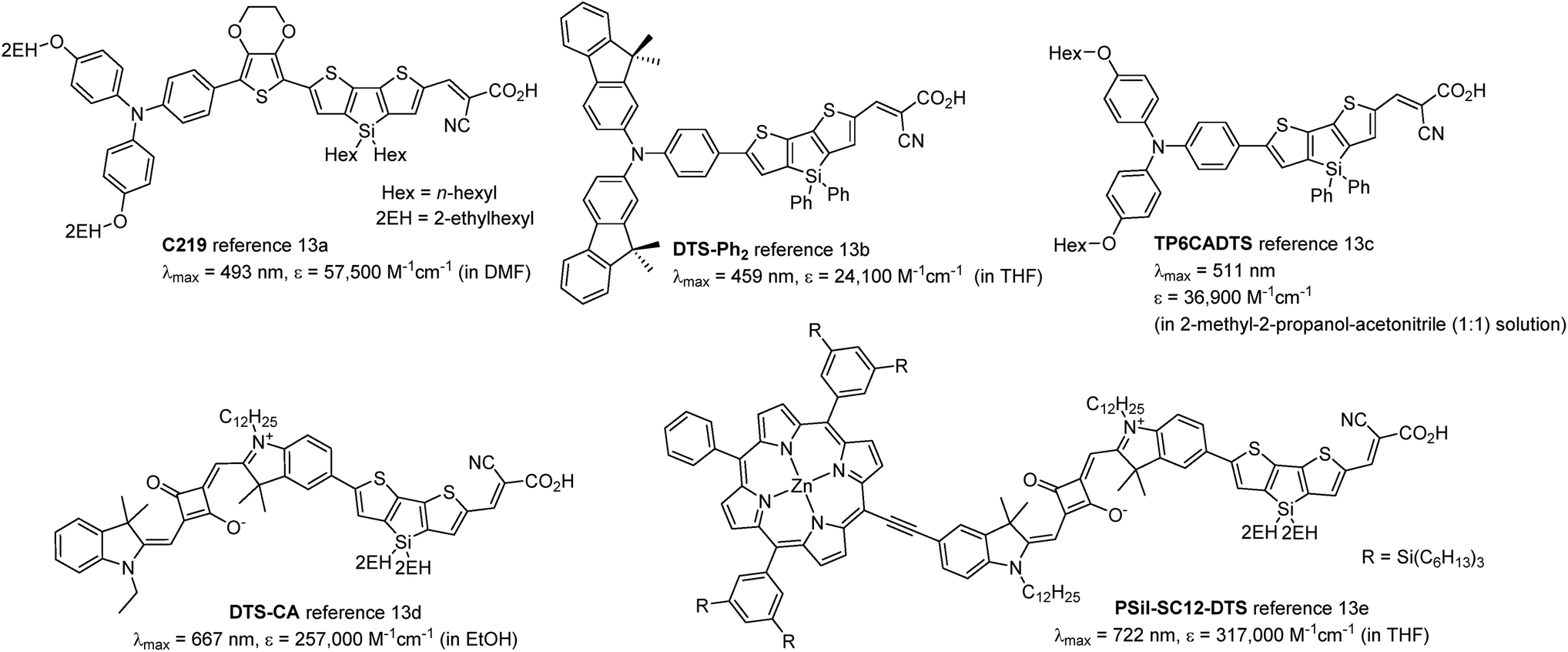
Dithienosilole (DTS) and dithienogermole (DTG) are silicon- and germanium-bridged bithiophenes (Chart 1) that exhibit efficient conjugation because of their high planarity, and are useful as building units of π-conjugated optoelectronic materials. DTS and DTG show interesting interactions between the Si–C or Ge–C σ*-orbitals and the π*-orbital of the bithiophene unit (σ*–π* interaction), which lowers LUMO energy levels and further reduces the HOMO–LUMO gaps of the compounds.9 DTS- and DTG-containing π-conjugated oligomers and polymers have been applied to organic photovoltaics10,11 and organic light emitting diodes.10,12 Ko et al. reported that photosensitizers with DTS as the π-conjugated linker exhibited electron injection readily from the photoexcited dyes to TiO2, due to the low-lying LUMO energy level (Chart 2).13b In addition, bulky substituents on the DTS silicon atom hindered not only contact between the electrolyte and the TiO2 surface, but also dye aggregation on TiO2. This furnished cells with high short-circuit current density (Jsc) and open circuit voltage (Voc). Jradi et al. reported that the introduction of 2-ethylhexyl-substituted DTS as the π-linker effectively suppressed dye aggregation on the TiO2 surface, which resulted in the high Voc and Jsc of the DSSCs.13d,e Zeng et al. also reported 7.6% PCE in a DSSC based on a DTS-containing dye with a simple D–π-A structure under standard global AM 1.5G solar irradiation of one sun (Chart 2).13a
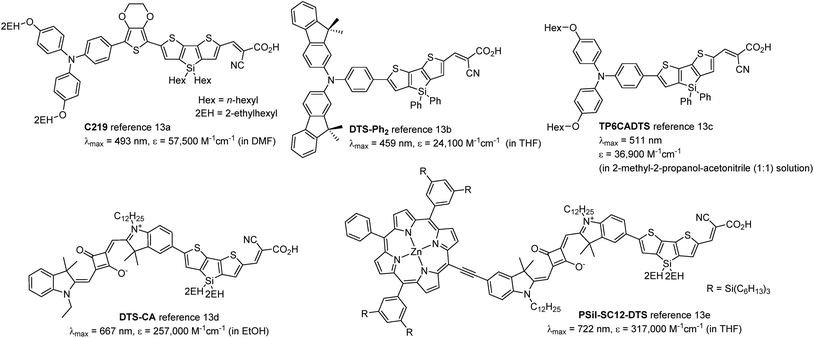 |
| | Chart 2 Structures of previously reported DTS-containing dyes. | |
In this report, we introduced PTz as the central acceptor unit of the D–π-A–π-A system to narrow the HOMO–LUMO gaps of the dyes. We also used DTG as the π-spacer unit for the first time. Previously, Welch, Bazan et al. synthesized molecular dyes with PTz as electron-acceptors and DTG or DTS as an electron-donor.14 These dyes had strong absorption in the visible region with the edges reaching 800 nm in solution, regardless of the bridging elements, i.e. Si or Ge. We therefore expected that the introduction of DTG in place of DTS may also yield an effective conjugation in the present dyes. As DTG has longer Ge–C (substituent) bonds than DTS, it is speculated that the longer bond would affect the dye aggregation on TiO2 by expanding inter-dye distances. It was also expected that the low polarization of Ge–C bonds compared with that of Si–C bonds would stabilize the π-systems towards hydrolysis, making hydrolytic synthetic transformation possible. This, for example, enabled the synthesis of a DTG-containing carboxylic acid derivative by alkali-catalyzed hydrolysis of the corresponding ester, in contrast to the analogous DTS compound whose silole ring underwent decomposition under the same conditions. Furthermore, DTG would be more stable than DTS towards oxidation, as Ge–O bonds (659.4 kJ mol−1) are usually weaker than Si–O bonds (799.6 kJ mol−1), while Ge–C and Si–C bonds have similar bond energies (460 kJ mol−1 and 451.5 kJ mol−1).15
Results and discussion
Synthesis
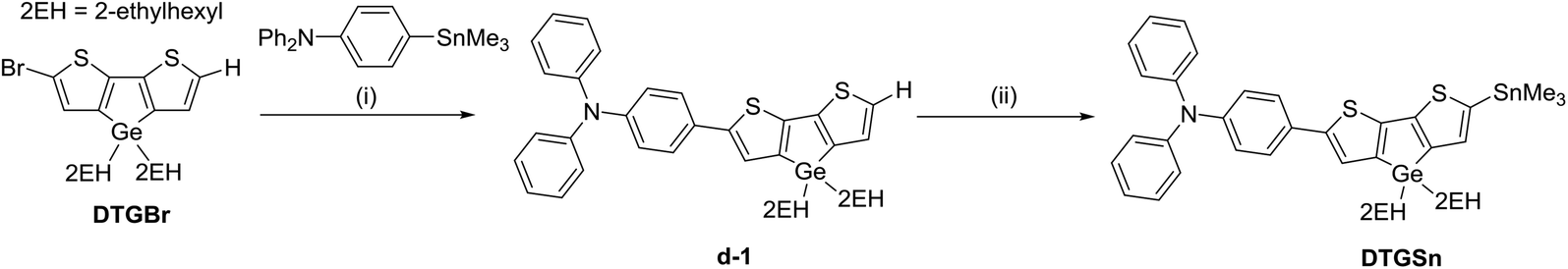
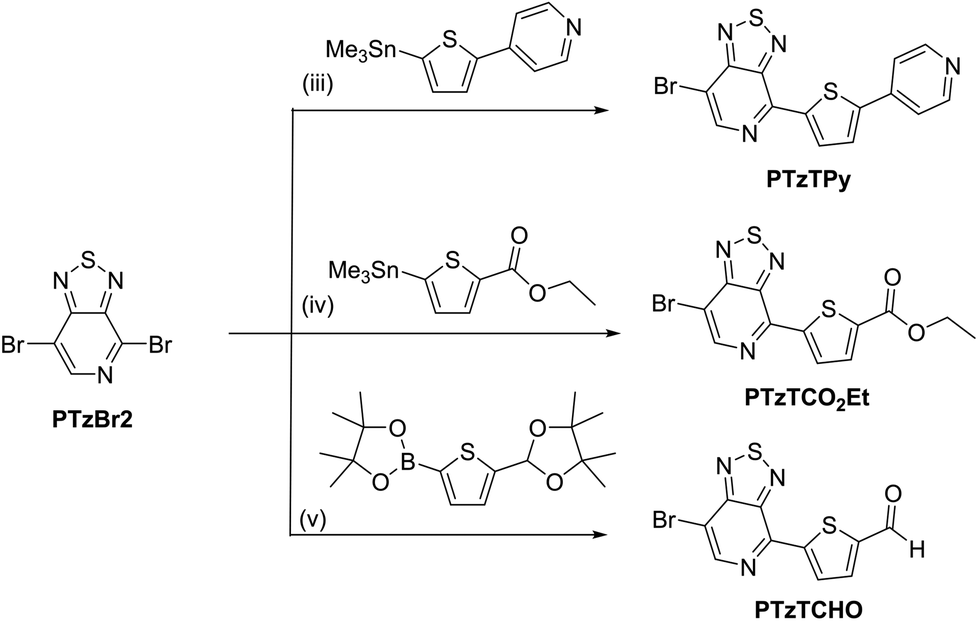
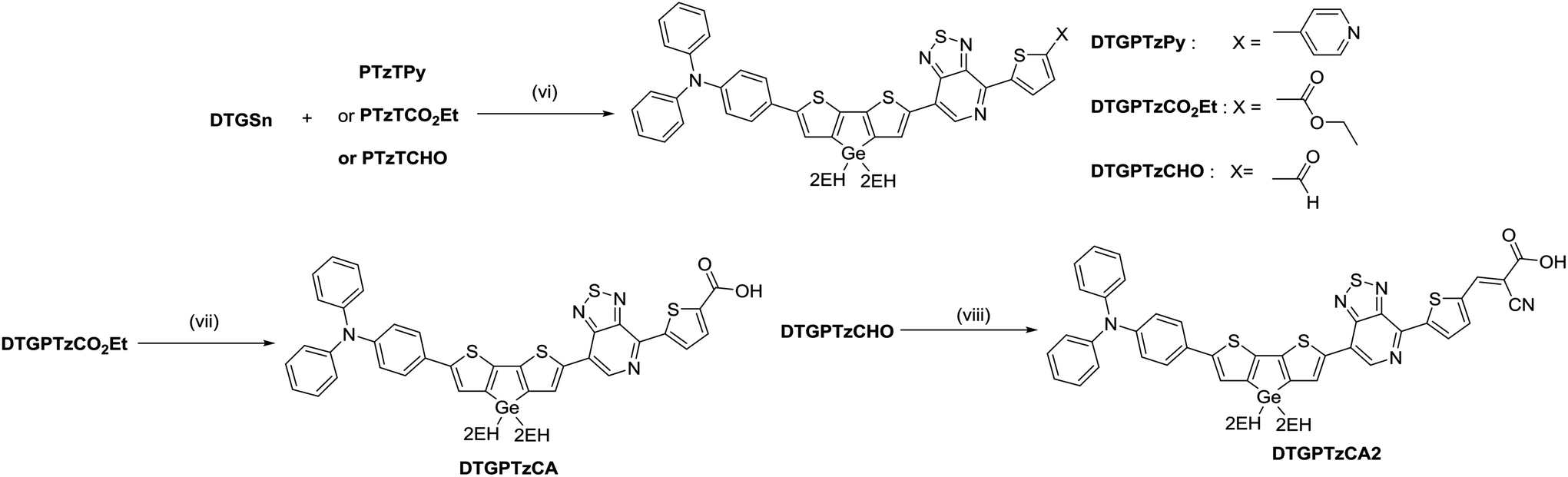
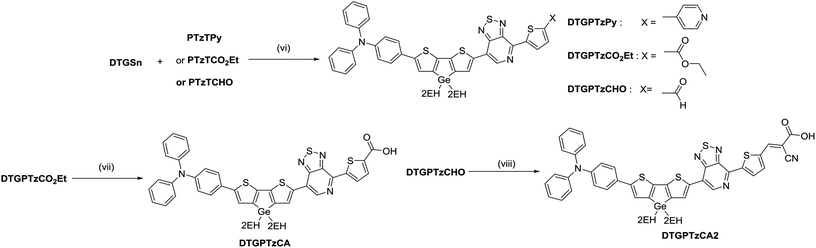
The procedures for the synthesis of the donor and acceptor units are shown in Schemes 1 and 2, respectively, using Stille and Suzuki cross coupling as the key reactions. DTGBr![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11f and PTzBr216 were synthesized according to the literature. Bulky 2-ethylhexyl groups were introduced on the Ge atom to enhance solubility and prevent aggregation upon adsorption to TiO2. As the Lewis basicity of PTz is too weak to allow coordination to the TiO2 surface, three anchor groups (pyridyl, carboxylic, and cyanoacrylic groups) were introduced. They were also expected to serve as additional acceptors. The D–π-A–π-A dye DTGPTzPy was synthesized by Stille cross coupling of DTGSn and PTzTPy (Scheme 3).
11f and PTzBr216 were synthesized according to the literature. Bulky 2-ethylhexyl groups were introduced on the Ge atom to enhance solubility and prevent aggregation upon adsorption to TiO2. As the Lewis basicity of PTz is too weak to allow coordination to the TiO2 surface, three anchor groups (pyridyl, carboxylic, and cyanoacrylic groups) were introduced. They were also expected to serve as additional acceptors. The D–π-A–π-A dye DTGPTzPy was synthesized by Stille cross coupling of DTGSn and PTzTPy (Scheme 3).
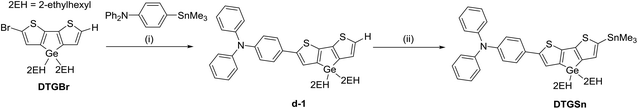 |
| | Scheme 1 Reagents and conditions: (i) toluene, Pd2(dba)3, P(o-tol)3, 100 °C, 1.5 d; (ii) (1) CH2Cl2, NBS, (2) THF, nBuLi, Me3SnCl, −78 °C, overnight. | |
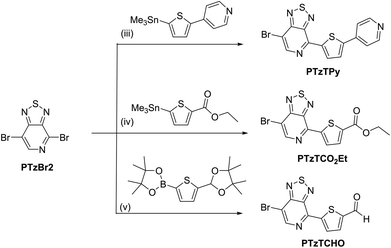 |
| | Scheme 2 Reagents and conditions: (iii) DMF, Pd(PPh3)4, 120 °C, 1 h; (iv) toluene, Pd2(dba)3, P(o-tol)3, 60 °C, 5 h; (v) (1) THF/H2O, Pd(PPh3)4, Na2CO3, 70 °C, overnight, (2) THF/H2O, HCl, reflux, overnight. | |
 |
| | Scheme 3 Reagents and conditions: (vi) toluene, Pd2(dba)3, P(o-tol)3, 60 °C, 4 h; (vii) THF/H2O, NaOH, reflux, 5 h; (viii) MeCN/CHCl3, cyanoacetic acid, piperidine, reflux, overnight. | |
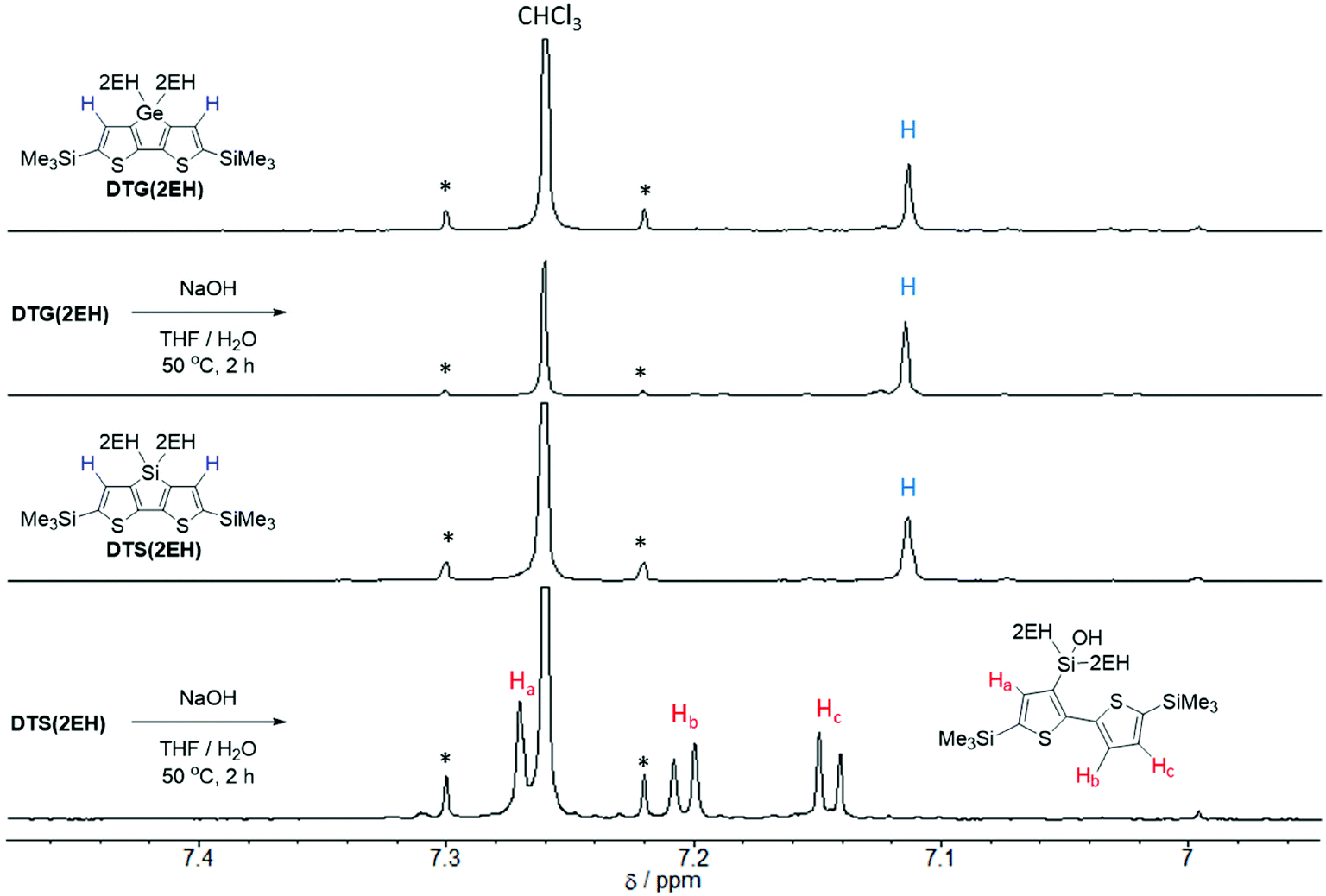
In general, to synthesize simple carboxylic acid derivatives, corresponding esters are hydrolyzed under strong alkali conditions. Thus, we carried out control experiments to evaluate the stability of DTG under alkali conditions. Treatment of DTG(2EH) with aq. NaOH caused no reaction, whereas treatment of DTS(2EH) provided a Si–C hydrolyzed product (Fig. 1 and S1†). This indicates the higher stability of DTG towards hydrolysis. As expected, the hydrolysis of DTGPTzCO2Et by aq. NaOH provided DTGPTzCA, although the yield was low (16%). This low yield may be attributed to the stronger reaction conditions than those of the control experiment. Under the same strong alkali conditions, the corresponding DTS derivative would not be prepared, as DTS is less stable than DTG. Cyanoacrylic acid derivative DTGPTzCA2 was synthesized by the Knoevenagel condensation in good yield (Scheme 3). All the new compounds were identified by 1H NMR, 13C NMR, and HR-MS analysis.
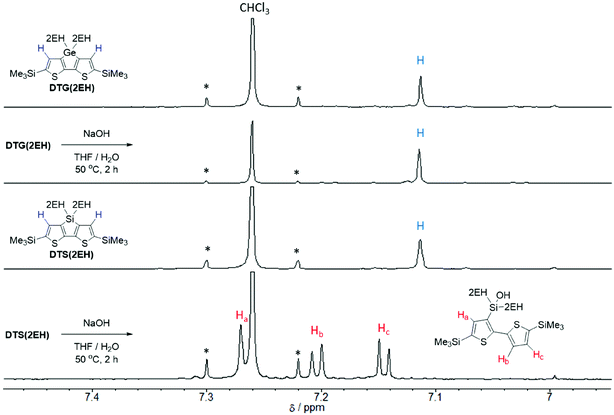 |
| | Fig. 1
1H NMR spectra of DTG(2EH) and DTS(2EH) before and after the control experiment (aromatic region). | |
Photophysical properties
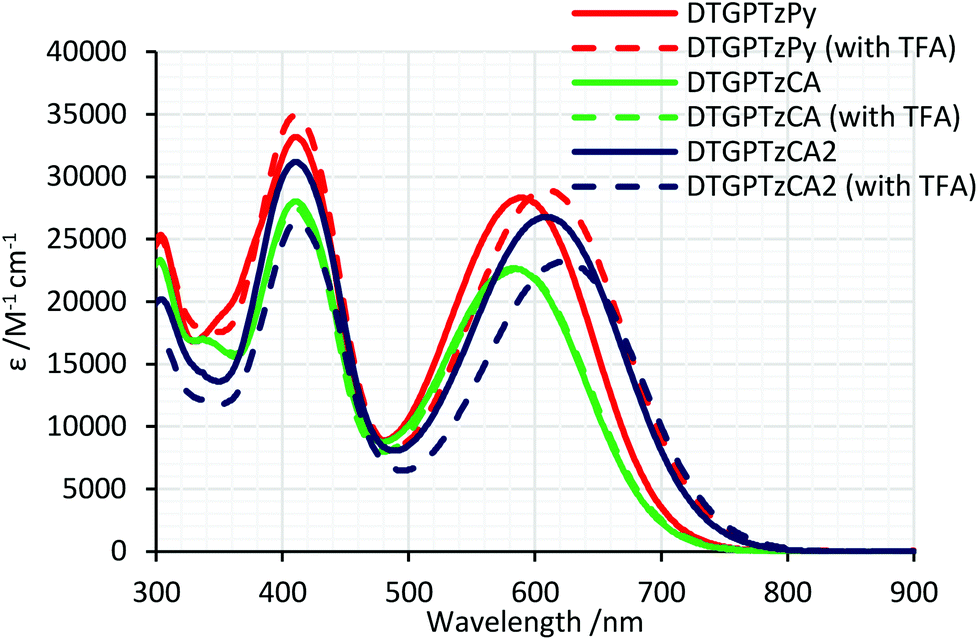
Fig. 2 shows the UV-vis absorption spectra of the dyes in THF. All the dyes exhibited two absorption bands around 410 nm and 600 nm. For the bands around 600 nm, that of DTGPTzCA2 appeared at the lowest energy of the three DTG-containing dyes, likely because of the strong electron-withdrawing properties of the cyanoacrylic group. The molar extinction coefficients were similar to those of the previously reported PTz-containing D–π-A–π-A dyes.7 When compared to previously reported DTS-based dyes, the present DTG-dyes had comparable13a–c or small13d,e molar extinction coefficients, presumably due to the difference in other π-systems in the molecules. Those bands are red-shifted by approximately 100 nm compared to the previously reported D–π-A type DTS analogues (Chart 2),13a–c which is obviously attributed to the strong electron-accepting properties of PTz. In THF containing 1 vol% trifluoroacetic acid (TFA), the absorption bands around 600 nm of DTGPTzPy and DTGPTzCA2 were red-shifted by 20 nm and 14 nm, respectively (Table 1). The shifts were likely due to the protonation of the anchor units. On the other hand, the spectrum of DTGPTzCA was unchanged by the TFA addition. These results indicated that the carboxylic group of DTGPTzCA was not deprotonated in neutral THF, whereas the cyanoacrylic group of DTGPTzCA2 formed a separated ion pair even in neutral THF, which was converted into neutral acid in TFA/THF. The absorption band shift of DTGPTzPy was ascribed to the formation of a pyridinium salt. As a consequence, it was found that the absorption bands of the neutral dyes shifted to the longer wavelength region in the following order: DTGPTzCA < DTGPTzPy < DTGPTzCA2.
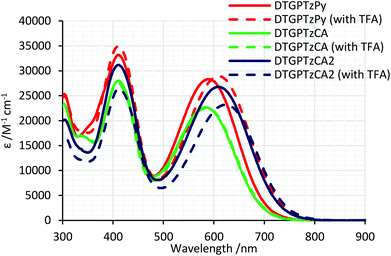 |
| | Fig. 2 UV-vis absorption spectra of DTG-containing dyes in THF with or without TFA. | |
Table 1 Photophysical and electrochemical properties of DTG-containing dyes
| Dye |
λ
max/nm |
ε
/M−1 cm−1 |
λ
max (on TiO2)d/nm |
HOMO/eV |
E
gg/eV |
LUMOh/eV |
|
Absorption maxima in neutral THF.
In THF containing 1 vol% TFA (0.0120 mM).
Molar extinction coefficient.
Absorption maxima on the TiO2 film without CDCA.
HOMO was determined from the oxidative potential in CV measurements in neutral DMF.
In DMF containing 1 vol% TFA (0.5 mM).
E
g (HOMO–LUMO gap) was determined from the onset of absorption spectra.
LUMO was calculated from HOMO + Eg.
|
|
DTGPTzPy
|
589, 411a |
28300, 33200 |
601, 426 |
−5.03e |
1.77 |
−3.26 |
|
DTGPTzPy (TFA added) |
609, 410b |
28900, 34900 |
— |
−5.05f |
1.69 |
−3.36 |
|
DTGPTzCA
|
583, 410a |
22800, 28100 |
577, 419 |
−5.04e |
1.78 |
−3.26 |
|
DTGPTzCA2
|
609, 411a |
26800, 31300 |
649, 453 |
−5.00e |
1.71 |
−3.29 |
|
DTGPTzCA2 (TFA added) |
623, 412b |
23300, 26400 |
— |
−5.05f |
1.67 |
−3.38 |
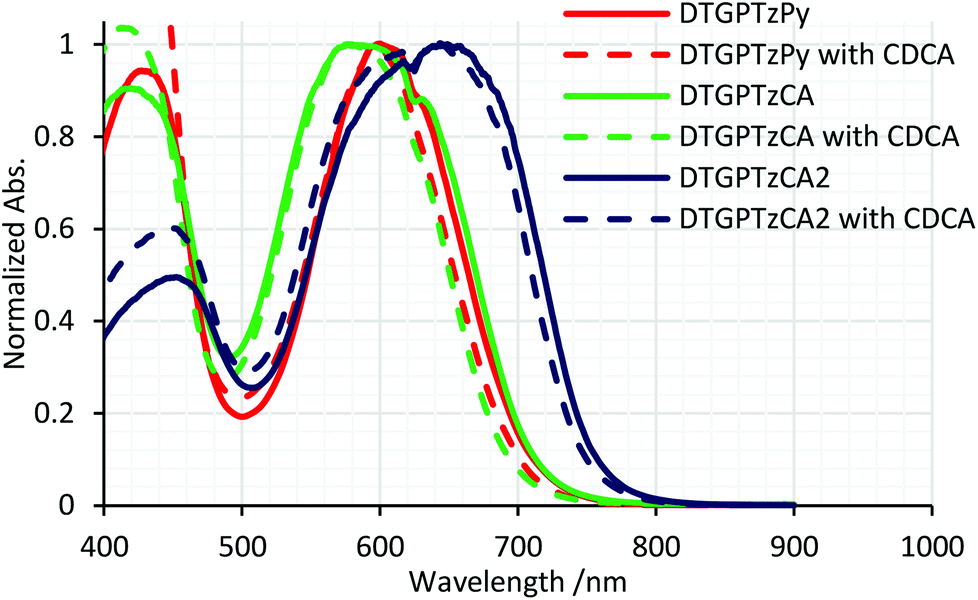
The normalized and non-normalized absorption spectra of DTG-containing dyes attached to the TiO2 thin film are shown in Fig. 3 and S2,† respectively. The spectrum of DTGPTzPy showed a slight red shift of the absorption band around 600 nm relative to that in solution. In contrast, the spectrum of DTGPTzCA exhibited a slight blue shift. For DTGPTzPy, the pyridyl moiety would interact with the Lewis acidic site on the TiO2 surface to become more cationic than that in solution, which would enhance the electron-withdrawing properties of the pyridyl moiety and narrow the HOMO–LUMO gap. On the other hand, the carboxylic group would become more anionic by forming CO2–Ti bonds on the TiO2 surface, which would lead to the blue-shifted spectrum of DTGPTzCA. We expected a similar blue shift in the case of DTGPTzCA2. However, we obtained a broadened and red-shifted absorption compared with that in solution. This tendency may be attributed to dye aggregation. We speculated that the dipole moment of DTGPTzCA2 should be larger than that of DTGPTzCA, because of the electron-withdrawing properties of the cyano group. In general, a large dipole moment enhances intermolecular interaction. In addition, the amount of dye loading of DTGPTzCA2 was larger than that of DTGPTzCA (Table 2, vide infra). We also speculated that the large amount of dye-loading induced the dye aggregation. However, we could not find solid evidence of the dye aggregation. When co-adsorbed with chenodeoxycholic acid (CDCA), the absorbance was decreased and dye loading was minimized (Fig. S2† and Table 2). However, the shape of the spectra was unchanged (Fig. 3). Hence, we speculated that inter-dye interactions on the TiO2 surface were not strong. This seemed to be caused by the long Ge–C (substituent) bonds, which sterically separated the dye molecules.
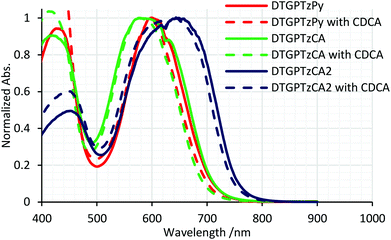 |
| | Fig. 3 Normalized UV-vis absorption spectra of DTG-containing dyes on TiO2 films; DTGPTzPy, DTGPTzCA, and DTGPTzCA2 were also co-adsorbed with 0.5 mM, 1.0 mM, and 2.0 mM CDCA, respectively. | |
Table 2 Photovoltaic properties of DSSCs based on the three dyes in full sunlight (AM 1.5G, 100 mW cm−2)
| Dye |
|
J
sc/mA cm−2 |
V
oc/V |
FF |
PCE/% |
Dye loading/1016 mol cm−2 |
|
DTGPTzPy
|
Without CDCA |
5.16 |
0.41 |
0.62 |
1.31 |
3.39 |
| With 0.5 mM CDCA |
9.52 |
0.43 |
0.67 |
2.76 |
1.65 |
|
DTGPTzCA
|
Without CDCA |
4.93 |
0.46 |
0.64 |
1.45 |
7.65 |
| With 1.0 mM CDCA |
7.12 |
0.48 |
0.66 |
2.29 |
4.48 |
|
DTGPTzCA2
|
Without CDCA |
4.23 |
0.35 |
0.59 |
0.88 |
8.12 |
| With 2.0 mM CDCA |
6.11 |
0.37 |
0.60 |
1.36 |
6.41 |
Electrochemical properties
To evaluate electron injection ability and regeneration of the present dyes, cyclic voltammogram (CV) measurements were performed in DMF containing tetrabutylammonium perchlorate (0.1 M) under nitrogen (Fig. S3†). For DTGPTzPy and DTGPTzCA2, measurements under acidic conditions with 1 vol% TFA were also carried out. The CVs have pseudo-reversible properties. It may be considered that the DTG-based dyes are not stable in the DSSC system. The irreversible properties, however, were likely due to the addition of nucleophiles, such as a perchlorate anion, to oxidized molecules. Thereafter, in the absence of nucleophiles, the DTG units would be stable, and in fact, heating DTG(2EH) in a DSSC redox solution, i.e. in CD3CN containing 0.05 M I2/0.1 M LiI, at 75 °C, for 2 days caused no decomposition of the compound, indicating the stability of DTG units under these conditions. The HOMO energy levels of the dyes were estimated from the anodic onsets (Eox, HOMO = −4.8 − Eox) (Table 1). They were more negative than the iodide/triiodide redox potential (−4.8 eV), indicating that the dye regeneration in DSSC may readily proceed by electron transfer from the redox solution. The HOMO–LUMO energy gaps (Eg) were obtained from the onset of absorption spectra in solution. The LUMO energy levels were estimated by HOMO + Eg. The small Eg of the dyes in the range of 1.60–1.80 eV seemed to reflect the effective conjugation in the D–π-A–π-A system. The LUMO energy levels were more positive than the conduction band edge of TiO2 (−3.9 eV), which was desirable for photoelectron injection. The HOMO–LUMO gaps of DTGPTzPy and DTGPTzCA were nearly the same, whereas that of DTGPTzCA2 was slightly smaller. This difference may be due to the stronger electron-withdrawing properties of the cyanoacrylic group than those of the pyridyl group and the simple carboxylic group.
Under acidic conditions, Eox of DTGPTzPy and DTGPTzCA2 was slightly shifted to the positive potentials. This meant that the HOMO energy levels were lowered by the protonation of the anchors.
Theoretical calculations
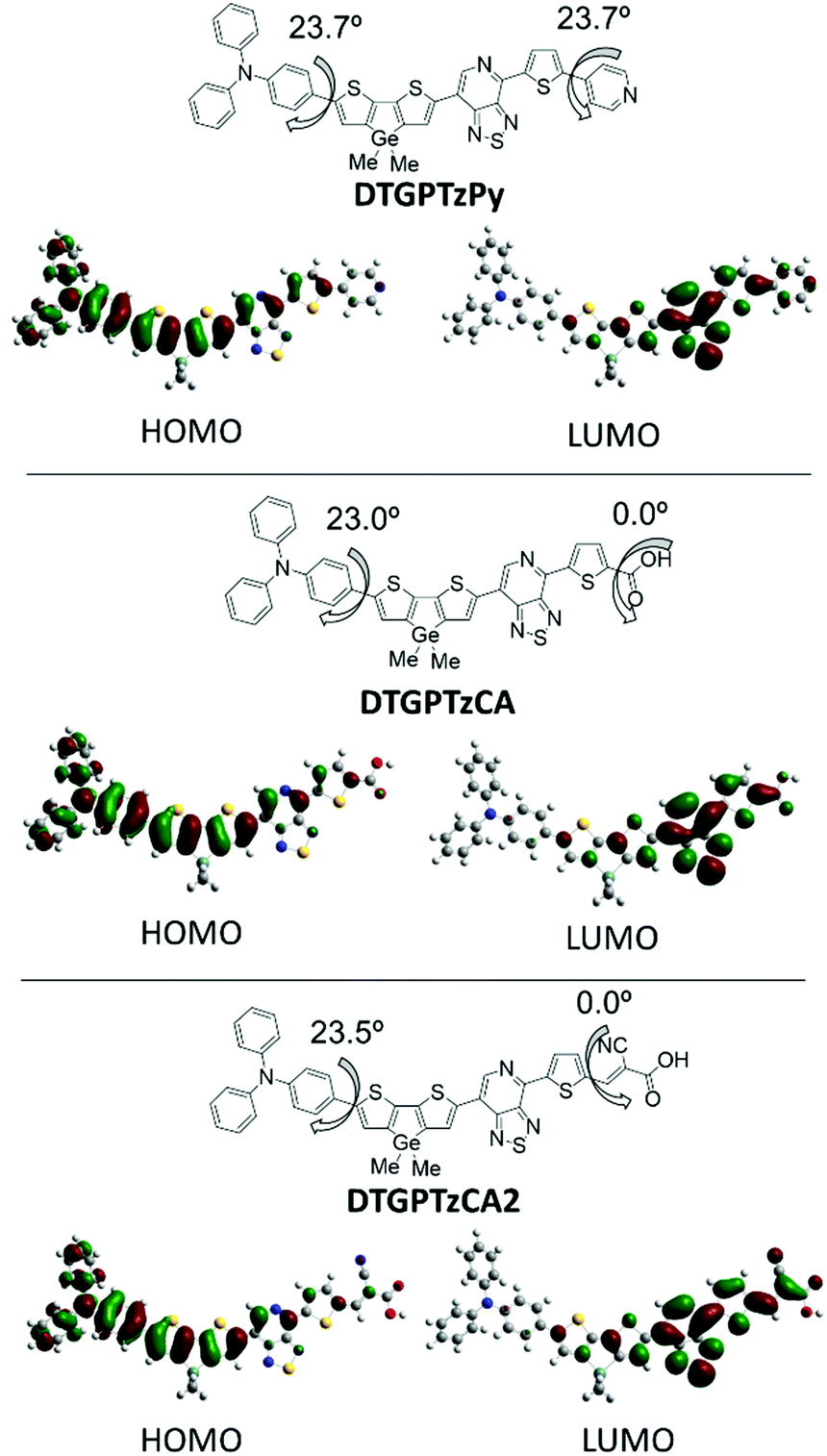
To elucidate the geometrical and electronic properties, density functional theory (DFT) calculations of the three dye models that bear methyl groups in place of 2-ethylhexyls on the Ge atom were carried out by using the Gaussian 09 program at the B3LYP/6-31G(d) level of theory. The optimized geometries in the gas phase and the frontier orbital profiles are shown in Fig. 4. The HOMOs were delocalized mainly on the donor units of triphenylamine and the π-spacer of DTG, and the LUMOs were distributed over the PTz, thiophene π-spacer, and anchor acceptor units. These separated HOMO and LUMO distributions would contribute to effective intramolecular charge-transfer (ICT), consistent with the strong absorption bands around 600 nm of the real compounds. Despite that, the phenylene and DTG moieties were twisted by approximately 23° with respect to the interplane angles, and the delocalized HOMOs indicated efficient conjugation between the triphenylamine and DTG units. For DTGPTzCA and DTGPTzCA2, the high planarity of the A–π-A fragment, including the PTz ring and adjacent π-conjugated units, also led to enhanced conjugation. In the case of DTGPTzPy, the torsional angle between the thiophene π-spacer and the pyridyl moiety was computed to be 23.7°. This twisting may bring about a decline of the photoelectron injection efficiency, because the LUMO orbital is not sufficiently spread to the pyridyl anchor moiety.
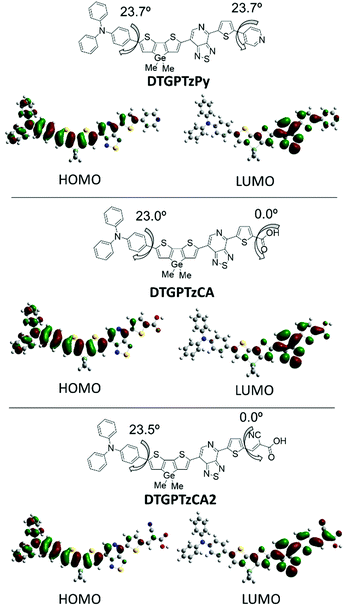 |
| | Fig. 4 Frontier orbitals of DTG-containing dyes calculated by the DFT at B3LYP/6-31G(d) level. | |
Photovoltaic properties
We fabricated DSSCs based on the present three DTG-containing dyes as sensitizers. The electrolyte was composed of 0.6 M 1,2-dimethyl-3-propylimidazolium/0.05 M I2/0.1 M LiI and the cell effective area was 0.25 cm2. TiO2 electrodes were modified with 0.1 mM dye solution for 15 hours. Co-adsorption with CDCA was also examined to prevent aggregation of the dyes on TiO2.
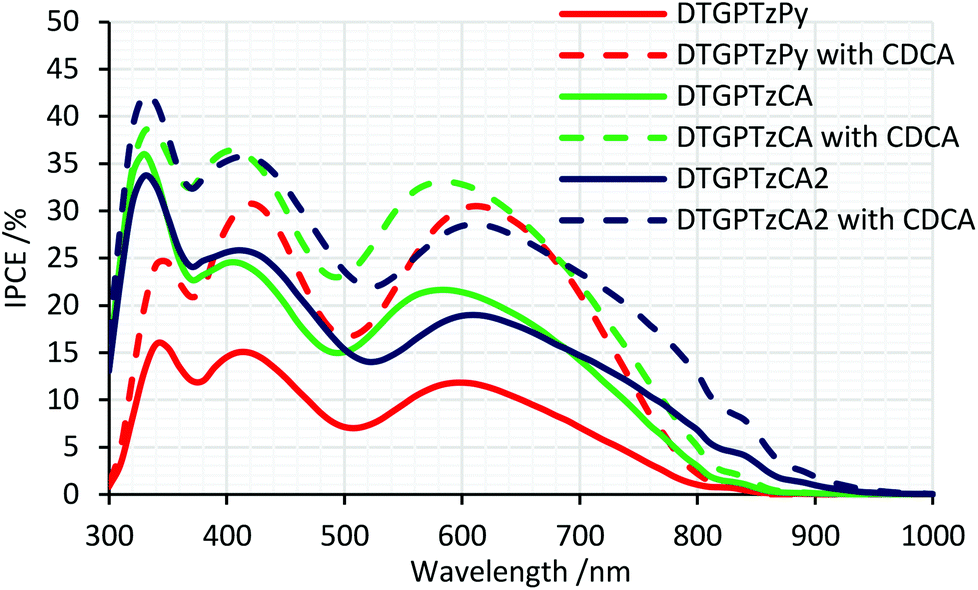
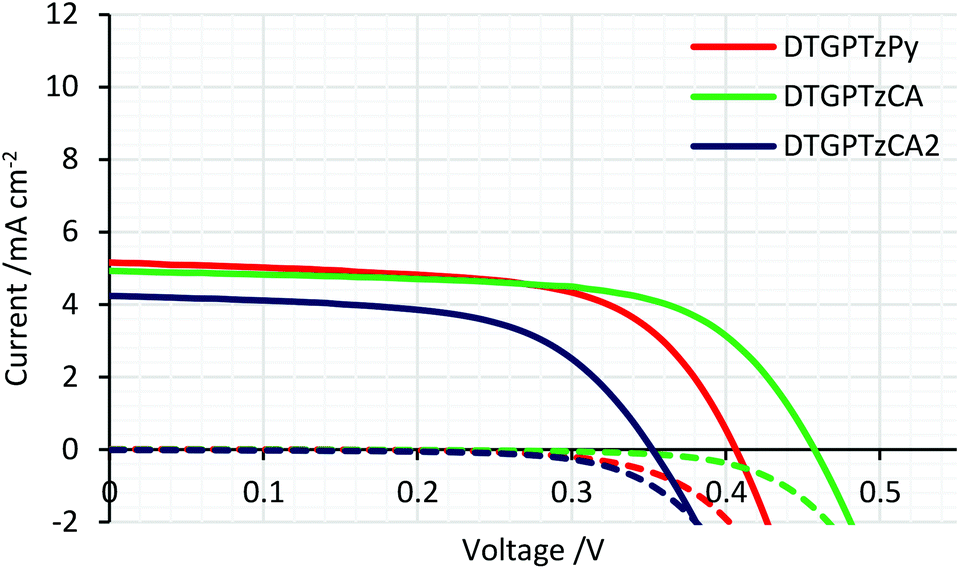
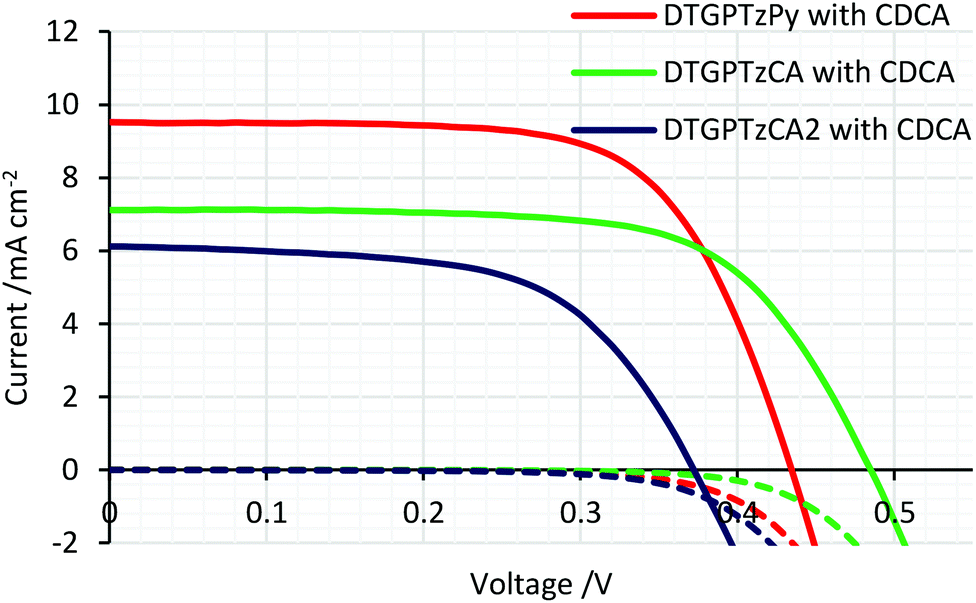
The IPCE spectra of the fabricated cells are shown in Fig. 5. (IPCE = LHE × φinj × φreg × ηcc, where LHE is light-harvesting efficiency, LHE = 1–10−A, A is the absorbance of the film, φinj and φreg are electron injection and dye regeneration efficiencies, respectively, and ηcc is the charge collection efficiency.17) As shown in Fig. 5, the IPCE values were not very high. LHEs were estimated to be higher than 90% in the range of 400–700 nm from the absorbance of the dye-adsorbed TiO2 film (Fig. S2†). Therefore, the relatively low IPCE would be attributed to some other factors. For example, the evaluated HOMO and LUMO energy levels of the dyes in solution would not be always consistent with those on the TiO2 surface because the dyes interact with TiO2 strongly; thus, the HOMO energy levels of the dyes might be too close to the potential of the iodide/triiodide redox couples on TiO2, which would reduce dye regeneration efficiency. The onsets of the IPCE spectra reached NIR wavelengths. Particularly, DSSC based on DTGPTzCA2 showed photoelectronic conversion up to 900 nm. The J–V curves of the fabricated DSSCs without CDCA are shown in Fig. 6. Although the amount of dye loading of DTGPTzPy was less than half of that of DTGPTzCA, the Jsc values were nearly the same (Table 2). This result is consistent with the previous report that DSSCs with dyes containing a pyridyl anchor showed superior performance to analogous dyes with a carboxylic anchor, in terms of electron injection efficiency.18 Next, we optimized the cells by co-adsorption with CDCA (Fig. 7). Cell performance was improved and maximized with 0.5 mM CDCA for DTGPTzPy, 1.0 mM CDCA for DTGPTzCA, and 2.0 mM CDCA for DTGPTzCA2. In particular, the performance of the device with DTGPTzPy was dramatically improved with high Jsc of 9.52 mA cm−2 to reach a PCE of 2.76%.
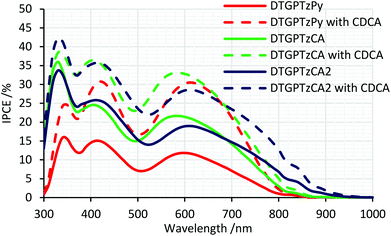 |
| | Fig. 5 IPCE spectra of DSSCs based on DTG-containing dyes. | |
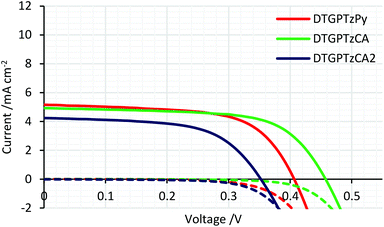 |
| | Fig. 6
J–V curves of DSSCs based on DTG-containing dyes without CDCA. Dark current/potential relationships are shown by dashed lines. | |
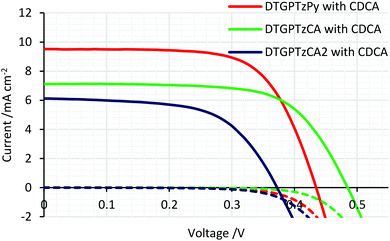 |
| | Fig. 7
J–V curves of DSSCs based on DTG-containing dyes with CDCA. Dark current/potential relationships are shown by dashed lines. | |
Electrochemical impedance spectroscopy
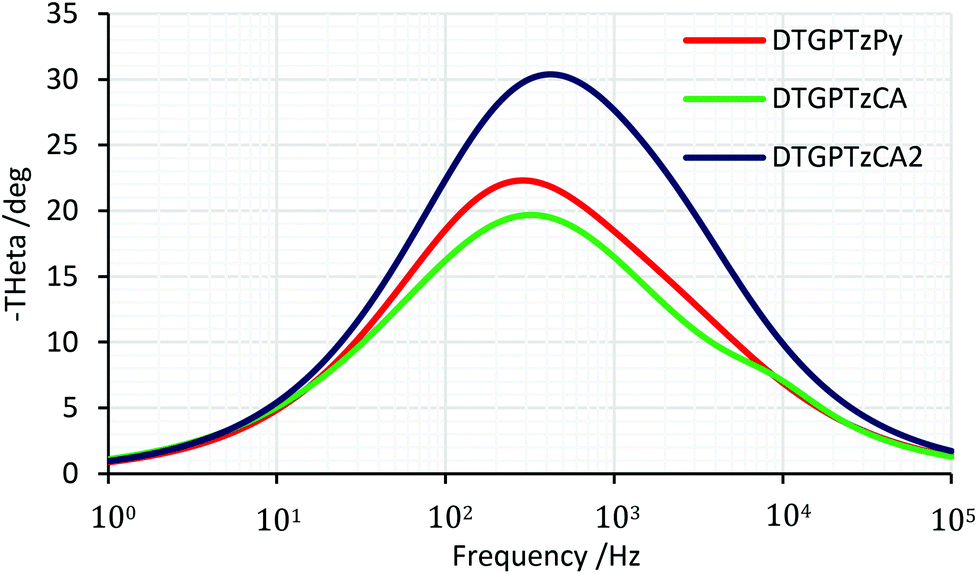
We carried out electrochemical impedance spectroscopy (EIS) measurements to evaluate the interfacial charge recombination process in the DSSCs. The measurements were performed under a forward bias of Voc in the dark. The Bode plots and the Nyquist plots are shown in Fig. 8 and S4,† respectively, and the electron lifetimes (τe) and the electron transport resistances (Rrec) are summarized in Table 3. A long electron lifetime leads to a high charge collection efficiency and the larger semicircle in the Nyquist plots indicates higher charge transport resistance and thus improved Voc. The electron lifetimes of the DSSCs based on the three dyes were estimated to be 0.4–0.5 ms, which were much shorter than that of the previously reported DTS-containing dye.13c This short lifetime indicates a fast back electron transfer from TiO2 to the dye, lowering Voc.
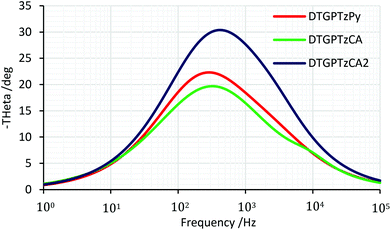 |
| | Fig. 8 EIS Bode plots of DSSCs based on DTG-containing dyes. | |
Table 3 Electron lifetimes and electron transport resistances of DSSCs based on DTG-containing dyes
| Dye |
τ
ea/ms |
R
recb/ohm cm−1 |
|
Electron lifetime in the TiO2 film.
Electron transport resistance among the TiO2/dye/electrolyte.
|
|
DTGPTzPy
|
0.50 |
63.8 |
|
DTGPTzTCA
|
0.50 |
59.2 |
|
DTGPTzTCA2
|
0.40 |
102.5 |
Conclusions
In summary, we synthesized new D–π-A–π-A type organic sensitizers with DTG and PTz as a π-conjugated spacer and a strong acceptor, respectively. DTGPTzCA was synthesized via the hydrolysis of the ester group, which proved that the DTG unit was stable to reactions under strong basic conditions. The synthesized DTG-containing dyes showed strong absorption in the visible and NIR regions, of which the absorption onset of DTGPTzCA2 on TiO2 reached 900 nm. The effective D–A interaction was supported by DFT calculations. The synthesized dyes were applicable to DSSCs and the best PCE of 2.76% was obtained for the DSSC based on DTGPTzPy co-adsorbed with CDCA. Although the PCEs of DSSCs based on DTG-containing dyes were not very high, the photoelectric conversion ranges were excellent as compared to previously reported DSSCs based on PTz-containing D–π-A–π-A type dyes7 and DTS-based dyes,13 which have the IPCE onsets from 700 to 850 nm.
Experimental
General
NMR spectra were recorded on Varian System 500 and Varian 400MR spectrometers. HR-MS spectral data were acquired on a Thermo Fisher Scientific LTQ Orbitrap XL spectrometer. Melting points were measured with a Yanaco MP-500P system in air. UV-Vis absorption spectra in solution were obtained with a Hitachi U-2910 spectrometer and those on TiO2 films were recorded with a Shimadzu UV-3150 with an ISR-3100 integrating sphere by the reflection method and converted into absorption using the Kubelka–Munk formula. Cyclic voltammograms were measured with an AMETEK VersaSTAT 4 potentiostat/galvanostat in a solution of 0.1 M tetrabutylammonium perchlorate (TBAP) in DMF using a three-electrode system with a Pt plate counter electrode, a Pt wire working electrode, and an Ag/Ag+ reference electrode.
All reactions were carried out under dry argon. As the reaction solvents, THF, toluene, and MeCN were purchased from Kanto Chemical Co., Ltd. They were distilled from calcium hydride and stored over activated molecular sieves until use. Chloroform was purchased from Kanto Chemical Co., Ltd and distilled from calcium hydride immediately before use. Usual workup mentioned below included hydrolysis of the reaction mixture with water and separation of the organic layer. The aqueous layer was extracted with toluene, chloroform, and ethyl acetate. The organic layer and the extract were combined and washed with water. After drying over anhydrous magnesium sulfate, the solvent was evaporated.
Synthesis of DTGPTzPy.
A mixture of 118 mg (0.136 mmol) of DTGSn, 50.2 mg (0.134 mmol) of PTzTPy, 7.0 mg (5 mol%) of Pd2(dba)3, 8.7 mg (20 mol%) of P(o-tol)3, and 5 mL of toluene was heated at 60 °C for 4 h with stirring. After usual workup using chloroform for extraction, the residue was purified by preparative GPC with toluene as an eluent to give a diastereomeric mixture of 60.8 mg (60.7 μmol, 45% yield) of DTGPTzPy: dark greenish powder, m.p.: 73–76 °C. 1H NMR (in CD2Cl2): δ = 0.80–0.92 (12H, m, CH3), 1.18–1.44 (20H, m, CH2), 1.57–1.67 (2H, m, CH), 7.01–7.09 (4H, m, p-Ph and phenylene), 7.12 (4H, dd, J = 8.8, 1.2 Hz, o-Ph), 7.26 (1H, s, DTG), 7.27–7.32 (4H, m, m-Ph), 7.45 (2H, d, J = 8.4 Hz, phenylene), 7.51–7.56 (2H, m, pyridyl), 7.54 (1H, d, J = 4.0 Hz, thiophene), 8.16 (1H, m, DTG), 8.51 (1H, d, J = 4.0 Hz, thiophene), 8.55–8.59 (2H, m, pyridyl), 8.70 (1H, m, PTz). 13C NMR (in CD2Cl2): δ = 11.1, 11.2, 14.4, 21.3, 23.50, 23.52, 29.25, 29.28, 29.5, 36.0, 37.55, 37.57, 120.1, 122.0, 123.6, 124.0, 125.0, 125.7, 126.8, 127.3, 128.9, 129.8, 131.9, 132.5, 137.3, 139.9, 141.1, 143.9, 144.5, 144.88, 144.90, 145.0, 145.66, 145.68, 145.72, 146.58, 146.60, 146.62, 147.63, 147.67, 147.70, 147.9, 148.4, 149.7, 150.9, 154.8. HR-MS (APCI) Calcd for C56H57GeN5S4: M+: 1001.27031, Found: 1001.27148.
Synthesis of DTGPTzCO2Et.
Ester DTGPTzCO2Et was obtained in a fashion similar to that of DTGPTzPy from DTGSn and PTzTCO2Et in 77% yield as a diastereomeric mixture: dark greenish powder, m.p.: 39–40 °C. 1H NMR (in CD2Cl2): δ = 0.78–0.85 (12H, m, CH3 in 2EH), 1.16–1.43 (23H, m, CH2 in 2EH and CH3 in EtO), 1.50–1.60 (2H, m, CH in 2EH), 4.38 (2H, q, J = 6.8 Hz, CH2 in EtO), 7.04–7.09 (4H, m, p-Ph and phenylene), 7.12 (4H, dd, o-Ph, J = 8.8, 1.2 Hz), 7.26–7.33 (5H, m, DTG and m-Ph), 7.51 (2H, d, J = 8.8 Hz, phenylene), 7.88 (1H, d, J = 4.0 Hz, thiophene), 8.25 (1H, m, DTG), 8.61 (1H, d, J = 4.0 Hz, thiophene), 8.87 (1H, m, PTz). 13C NMR (in CD2Cl2): δ = 11.17, 11.22, 14.42, 14.43, 14.6, 21.22, 21.25, 21.29, 23.51, 23.53, 29.25, 29.29, 29.5, 36.0, 37.55, 37.57, 61.8, 122.6, 123.6, 123.9, 125.0, 125.71, 126.74, 128.9, 129.8, 131.0, 132.3, 134.4, 136.9, 137.2, 139.7, 144.0, 144.8, 145.70, 145.72, 145.8, 146.71, 146.73, 146.8, 147.6, 147.78, 147.85, 147.89, 147.93, 148.0, 148.4, 150.0, 154.7, 162.5. HR-MS (APCI) Calcd for C54H58GeN4O2S4: [M + H]+: 997.27271, Found: 997.27105.
Synthesis of DTGPTzCHO.
Aldehyde DTGPTzCHO was obtained in a fashion similar to that of DTGPTzPy from DTGSn and PTzTCHO in 91% yield as a diastereomeric mixture: dark greenish powder, m.p.: 67–69 °C. 1H NMR (in CD2Cl2): δ = 0.80–0.91 (12H, m, CH3), 1.19–1.48 (20H, m, CH2), 1.59–1.69 (2H, m, CH), 7.02 (2H, d, J = 8.8 Hz, phenylene), 7.04–7.09 (2H, m, p-Ph), 7.12 (4H, dd, J = 8.8, 1.2 Hz, o-Ph), 7.25 (1H, s, DTG), 7.26–7.32 (4H, m, m-Ph), 7.42 (2H, d, J = 8.8 Hz, phenylene), 7.75 (1H, d, J = 4.0 Hz, thiophene), 8.21 (1H, m, DTG), 8.53 (1H, d, J = 4.0 Hz, thiophene), 8.70 (1H, m, PTz), 9.90 (1H, s, CHO). 13C NMR (in CD2Cl2): δ = 11.1, 11.2, 14.41, 14.42, 21.10, 21.13, 21.2, 23.48, 23.50, 29.18, 29.22, 29.4, 36.0, 37.47, 37.49, 123.0, 123.6, 123.8, 125.0, 125.6, 126.7, 128.7, 129.7, 131.2, 132.43, 132.45, 132.48, 137.00, 137.01, 137.4, 139.5, 143.3, 144.65, 144.67, 144.69, 145.5, 145.72, 145.74, 145.8, 146.85, 146.88, 146.90, 147.6, 147.8, 148.0, 148.0, 148.1, 148.4, 150.46, 150.49, 154.4, 183.6. HR-MS (APCI) Calcd for C52H54GeN4OS4: [M + H]+: 953.24650, Found: 953.24707.
Synthesis of DTGPTzCA.
In a mixture of 20 mL of THF, 3 mL of 1 M aq. NaOH, and 12 mL of water was dissolved 95.2 mg (95.6 μmol) of DTGPTzCO2Et. After the solution was heated to reflux for 5 h with stirring, the reaction mixture was neutralized by adding 10% aq. HCl. After usual workup using ethyl acetate for extraction, the residue was subjected to silica gel column chromatography using a mixture of hexane/acetic acid = 99/1 as an eluent to give a diastereomeric mixture of 16.0 mg (16.5 μmol, 17% yield) of DTGPTzCA: dark greenish solid, m.p.: 81–84 °C. 1H NMR (in CD2Cl2): δ = 0.75–1.66 (34H, m), 6.99 (2H, br d, J = 8.4 Hz), 7.02–7.13 (6H, m, o-Ph and p-Ph), 7.22 (1H, s, DTG), 7.23–7.30 (4H, m, m-Ph), 7.40 (2H, br d, J = 8.4 Hz, phenylene), 7.85 (1H, br s, thiophene), 8.13 (1H, br s, DTG), 8.42 (1H, br s, thiophene), 8.72 (1H, br s, PTz). 13C NMR (in CD2Cl2): δ = 11.15, 11.20, 14.41, 14.42, 21.2, 21.3, 23.51, 23.53, 29.25, 29.29, 29.5, 36.1, 37.5, 37.6, 122.8, 123.6, 123.9, 125.0, 125.7, 126.8, 129.0, 129.8, 131.1, 132.1, 136.0, 137.1, 139.6, 143.8, 144.9, 145.7, 146.8, 147.6, 147.9, 148.4, 149.3, 150.4, 154.6. HR-MS (APCI) Calcd for C52H54GeN4O2S4: [M + H]+: 969.24141, Found: 969.24146.
Synthesis of DTGPTzCA2.
A mixture of 112 mg (0.118 mmol) of DTGPTzCHO, 22.8 mg (0.268 mmol) of cyanoacetic acid, 6 μL of piperidine, and 20 mL of a mixed solvent of acetonitrile/chloroform = 1/1 was heated to reflux overnight. The solvent was evaporated and the residue was subjected to silica gel chromatography with ethyl acetate to elute unreacted ester and byproducts. Then, DTGPTzCA2 was eluted with methanol to give a diastereomeric mixture of 104 mg (0.102 mmol, 86%) of DTGPTzCA2: dark greenish powder, m.p.: 145–146 °C. 1H NMR (in DMSO-d6): δ = 0.70–0.82 (12H, m, CH3), 1.00–1.44 (20H, m, CH2), 1.44–1.57 (2H, m, CH), 6.96 (2H, d, J = 8.4 Hz, phenylene), 7.03–7.12 (6H, m, Ph), 7.33 (4H, t, J = 8.0 Hz, m-Ph), 7.48 (1H, br s, DTG), 7.54 (2H, d, J = 8.4 Hz, phenylene), 7.81 (1H, d, J = 4.0 Hz, thiophene), 8.14 (1H, s, olefin), 8.31 (1H, br s, DTG), 8.55 (1H, d, J = 4.0 Hz, thiophene), 8.94–8.99 (1H, m, PTz). 13C NMR spectrum was not obtained because of the low solubility of the product. HR-MS (ESI) Calcd for C55H55GeN5O2S4: M+: 1019.24449, Found: 1019.24554.
Cell fabrication and characterization
A layer of TiO2 paste (PST-18NR, JGC Catalysts and Chemicals Ltd, Japan) was formed on an FTO substrate by the doctor blade technique. The electrodes were sintered for 45 min at 450 °C to coat the surface with an approximately 9 μm thick TiO2 film, which was shaved to 0.5 × 0.5 cm2 as the photoactive area. The sintered electrodes were immersed in 0.1 mM dye solution in THF for 15 h. Dye loading was estimated by subtracting the concentration of the dye solution after immersion from that before immersion, on the basis of UV-vis absorbance measurements. The cells were fabricated with the modified TiO2 electrode with Pt-vapored glass as the counter electrode, a spacer, and an electrolyte composed of 0.05 M iodine, 0.1 M lithium iodide, and 0.6 M 1,2-dimethyl-3-propylimidazolium iodide in acetonitrile.
The photocurrent–voltage (I–V) characteristics of the DSSCs were measured on a Keithley 2611A source meter under AM 1.5, 100 mW cm−2 (1 sun) irradiation by a solar simulator (Bunkoukeiki Co., Ltd, OTENTO-SUN III). The incident light intensity was calibrated with a reference Si solar cell equipped with a heat absorbing filter (Bunkoukeiki Co., Ltd, BS520BK). The IPCE spectra were recorded with an SM-250 (Bunkoukeiki Co., Ltd). The active areas of the TiO2 films were determined with a KEYENCE VHX-1000 digital microscope.
EIS measurements were performed on the same DSSCs in the dark under a forward bias of Voc using an AMETEK VersaSTAT 3 potentiostat/galvanostat.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research (B) (JSPS KAKENHI Grant No. 26288094).
Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
-
(a) M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed;
(b) M. Grätzel, J. Photochem. Photobiol., A, 2004, 164, 3–14 CrossRef.
-
(a) N. Koumura, Z.-S. Wang, S. Mori, M. Miyashita, E. Suzuki and K. Hara, J. Am. Chem. Soc., 2006, 128, 14256–14257 CrossRef CAS PubMed;
(b) S. Kim, J. Kwan Lee, S. O. Kang, J. Ko, J.-H. Yum, S. Fantacci, F. D. Angelis, D. D. Censo, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS PubMed;
(c) H. Choi, C. Baik, S. O. Kang, J. Ko, M.-S. Kang, M. K. Nazeeruddin and M. Grätzel, Angew. Chem., Int. Ed., 2008, 47, 327–330 CrossRef CAS PubMed.
- Y. Wu and W. Zhu, Chem. Soc. Rev., 2013, 42, 2039 RSC.
-
(a) Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261–8272 RSC;
(b) K. Pei, Y. Wu, W. Wu, Q. Zhang, B. Chen, H. Tian and W. Zhu, Chem. – Eur. J., 2012, 18, 8190–8200 CrossRef CAS PubMed.
-
(a) S. Haid, M. Marszalek, A. Mishra, M. Wielopolski, J. Teuscher, J.-E. Moser, R. Humphry-Baker, S. M. Zakeeruddin, M. Grätzel and P. Bäuerle, Adv. Funct. Mater., 2012, 22, 1291–1302 CrossRef CAS;
(b) S. Qu, C. Qin, A. Islam, Y. Wu, W. Zhu, J. Hua, H. Tian and L. Han, Chem. Commun., 2012, 48, 6972–6974 RSC.
-
(a) S. Chaurasia, C.-Y. Hsu, H.-H. Chou and J. T. Lin, Org. Electron., 2014, 15, 378–390 CrossRef CAS;
(b) W. Ying, X. Zhang, X. Li, W. Wu, F. Guo, J. Li, H. Ågren and J. Hua, Tetrahedron, 2014, 70, 3901–3908 CrossRef CAS;
(c) Y. Hua, J. He, C. Zhang, C. Qin, L. Han, J. Zhao, T. Chen, W.-Y. Wong, W.-K. Wong and X. Zhu, J. Mater. Chem. A, 2015, 3, 3103–3112 RSC.
- N. Blouin, A. Michaud, D. Gendron, S. Wakim, E. Blair, R. Neagu-Plesu, M. Belletête, G. Durocher, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2008, 130, 732–742 CrossRef CAS PubMed.
- J. Ohshita, M. Nodono, T. Watanabe, Y. Ueno, A. Kunai, Y. Harima, K. Yamashita and M. Ishikawa, J. Organomet. Chem., 1998, 553, 487–491 CrossRef CAS.
- J. Ohshita, Macromol. Chem. Phys., 2009, 210, 1360–1370 CrossRef CAS.
-
(a) T.-Y. Chu, J. Lu, S. Beaupré, Y. Zhang, J.-R. Poulio, S. Wakim, J. Zhou, M. Leclerc, Z. Li, J. Ding and Y. Tao, J. Am. Chem. Soc., 2011, 133, 4250–4253 CrossRef CAS PubMed;
(b) J. Ohshita, Y.-M. Hwang, T. Mizumo, H. Yoshida, Y. Ooyama, Y. Harima and Y. Kunugi, Organometallics, 2011, 30, 3233–3236 CrossRef CAS;
(c) C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS PubMed;
(d) Y.-M. Hwang, J. Ohshita, Y. Harima, T. Mizumo, Y. Ooyama, Y. Morihara, T. Izawa, T. Sugioka and A. Fujita, Polymer, 2011, 52, 3912–3916 CrossRef CAS;
(e) J. Ohshita, M. Miyazaki, D. Tanaka, Y. Morihara, Y. Fujita and Y. Kunugi, Polym. Chem., 2013, 4, 3116–3122 RSC;
(f) J. Ohshita, M. Miyazaki, F.-B. Zhang, D. Tanaka and Y. Morihara, Polym. J., 2013, 45, 979–984 CrossRef CAS;
(g) D. Tanaka, J. Ohshita, Y. Ooyama and Y. Morihara, Polym. J., 2013, 45, 1153–1158 CrossRef CAS;
(h) F.-B. Zhang, J. Ohshita, M. Miyazaki, D. Tanaka and Y. Morihara, Polym. J., 2014, 46, 628–631 CrossRef CAS;
(i) J. Ohshita, M. Miyazaki, M. Nakashima, D. Tanaka, Y. Ooyama, T. Sasaki, Y. Kunugi and Y. Morihara, RSC Adv., 2015, 5, 12686–12691 RSC.
-
(a) A. Kunai, J. Ohshita, T. Iida, K. Kanehara, A. Adachi and K. Okita, Synth. Met., 2003, 137, 1007–1008 CrossRef CAS;
(b) T. Lee, I. Jung, K. H. Song, H. Lee, J. Choi, K. Lee, B. J. Lee, J. Pak, C. Lee, S. O. Kang and J. Ko, Organometallics, 2004, 23, 5280–5285 CrossRef CAS;
(c) K.-H. Lee, J. Ohshita, K. Kimura, Y. Kunugi and A. Kunai, J. Organomet. Chem., 2005, 690, 333–337 CrossRef CAS;
(d) J. Ohshita, D. Hamamoto, K. Kimura and A. Kunai, J. Organomet. Chem., 2005, 690, 3027–3032 CrossRef CAS;
(e) J. Ohshita, K. Kimura, K.-H. Lee, A. Kunai, Y.-W. Kwak, E.-C. Son and Y. Kunugi, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 4588–4596 CrossRef CAS;
(f) J. Ohshita, Y. Kurushima, K.-H. Lee, A. Kunai, Y. Ooyama and Y. Harima, Organometallics, 2007, 26, 6591–6595 CrossRef CAS;
(g) J. Ohshita, M. Nakamura, K. Yamamoto, S. Watase and K. Matsukawa, Dalton Trans., 2015, 44, 8214–8220 RSC.
-
(a) W. Zeng, Y. Cao, Y. Bai, Y. Wang, Y. Shi, M. Zhang, F. Wang, C. Pan and P. Wang, Chem. Mater., 2010, 22, 1915–1925 CrossRef CAS;
(b) S. Ko, H. Choi, M.-S. Kang, H. Hwang, H. Ji, J. Kim, J. Ko and Y. Kang, J. Mater. Chem., 2010, 20, 2391–2399 RSC;
(c) L.-Y. Lin, C.-H. Tsai, K.-T. Wong, T.-W. Huang, L. Hsieh, S.-H. Liu, H.-W. Lin, C.-C. Wu, S.-H. Chou, S.-H. Chen and A.-I. Tsai, J. Org. Chem., 2010, 75, 4778–4785 CrossRef CAS PubMed;
(d) F. M. Jradi, X. Kang, D. O'Neil, G. Pajares, Y. A. Getmanenko, P. Szymanski, T. C. Parker, M. A. El-Sayed and S. R. Marder, Chem. Mater., 2015, 27, 2480–2487 CrossRef CAS;
(e) F. M. Jradi, D. O'Neil, X. Kang, J. Wong, P. Szymanski, T. C. Parker, H. L. Anderson, M. A. El-Sayed and S. R. Marder, Chem. Mater., 2015, 27, 6305–6313 CrossRef CAS.
- G. C. Welch, R. C. Bakus II, S. J. Teat and G. C. Bazan, J. Am. Chem. Soc., 2013, 135, 2298–2305 CrossRef CAS PubMed.
-
Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, 76th edn, 1995–1996 Search PubMed.
- Y. Sun, S.-C. Chien, H.-L. Yip, Y. Zhang, K.-S. Chen, D. F. Zeigler, F.-C. Chen, B. Lin and A. K.-Y. Jen, J. Mater. Chem., 2011, 21, 13247–13255 RSC.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429–7433 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, complemental electrochemical/optical spectra. See DOI: 10.1039/c6dt02469f |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b and
Joji
Ohshita
*a
b and
Joji
Ohshita
*a