
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Adjustable coordination of a hybrid phosphine–phosphine oxide ligand in luminescent Cu, Ag and Au complexes†
Thuy Minh
Dau
a,
Benjamin Darko
Asamoah
a,
Andrey
Belyaev
a,
Gomathy
Chakkaradhari
a,
Pipsa
Hirva
*a,
Janne
Jänis
a,
Elena V.
Grachova
 b,
Sergey P.
Tunik
b and
Igor O.
Koshevoy
*a
b,
Sergey P.
Tunik
b and
Igor O.
Koshevoy
*a
aDepartment of Chemistry, University of Eastern Finland, Joensuu, 80101, Finland. E-mail: pipsa.hirva@uef.fi; igor.koshevoy@uef.fi
bSt. Petersburg State University, 7/9 Universitetskaya nab., 199034, St. Petersburg, Russia
First published on 17th August 2016
Abstract
A potentially tridentate hemilabile ligand, PPh2–C6H4–PPh(O)–C6H4–PPh2 (P3O), has been used for the construction of a family of bimetallic complexes [MM′(P3O)2]2+ (M = M′ = Cu (1), Ag (2), Au (3); M = Au, M′ = Cu (4)) and their mononuclear halide congeners M(P3O)Hal (M = Cu (5–7), Ag (8–10)). Compounds 1–10 have been characterized in the solid state by single-crystal X-ray diffraction analysis to reveal a variable coordination mode of the phosphine-oxide group of the P3O ligand depending on the preferable number of coordination vacancies on the metal center. According to the theoretical studies, the interaction of the hard donor P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety with d10 ions becomes less effective in the order Cu > Ag > Au. 1–10 exhibit room temperature luminescence in the solid state, and the intensity and energy of emission are mostly determined by the nature of metal atoms. The photophysical characteristics of the monometallic species were compared with those of the related compounds M(P3)Hal (11–16) with the non-oxidized ligand P3. It was found that in the case of the copper complexes 5–7 the P3O hybrid ligand introduces effective non-radiative pathways of the excited state relaxation leading to poor emission, while for the silver luminophores the PO group leads mainly to the modulation of luminescence wavelength.
O moiety with d10 ions becomes less effective in the order Cu > Ag > Au. 1–10 exhibit room temperature luminescence in the solid state, and the intensity and energy of emission are mostly determined by the nature of metal atoms. The photophysical characteristics of the monometallic species were compared with those of the related compounds M(P3)Hal (11–16) with the non-oxidized ligand P3. It was found that in the case of the copper complexes 5–7 the P3O hybrid ligand introduces effective non-radiative pathways of the excited state relaxation leading to poor emission, while for the silver luminophores the PO group leads mainly to the modulation of luminescence wavelength.
Introduction
Transition metal complexes, containing multidentate ligands with electronically different functionalities, have been very actively studied due to their versatile coordination chemistry and diverse reactivity. A major stimulus, which determines substantial progress observed in the area over the past few decades, is related to the notable success of such compounds in homogeneous catalysis1–3 and activation of small molecules,4 design of dynamic systems5,6 and sensing.7 The hybrid ligands, which comprise strongly and weakly bound moieties, were defined as hemilabile ones by Jeffrey and Rauchfuss in 1979.8 The labile functions of such ligands under favourable conditions are capable of easy and reversible coordination–dissociation to provide a vacancy on the metal center, accessible to accommodate a suitable substrate.9Among a variety of potentially bifunctional ligands, molecules with phosphorus donors as strongly bound anchors have been most widely used for the preparation of numerous late transition metal compounds. In particular, considerable interest has been focused on the ligands, combining soft (P atom) and hard donors (N or O atoms).2,3,10,11 The corresponding P–N (phosphine–amine/oxazoline/nitrile) and P–O (phosphine–ether/phosphine oxide) complexes of Ni, Pd, Co, Au, and Cu demonstrate impressive catalytic activity in many industrially important reactions, e.g. olefin oligo- and polymerization (including ring opening metathesis polymerization),12 amination,13,14 hydrogenation,15 annulation,16 hydroformylation,17 allylation,18 Suzuki3,19 and Heck20 coupling.
Alternatively, hemilabile phosphines have been extensively exploited in the construction of supramolecular systems, which exhibit geometry changes controlled by a weak-link approach (WLA).6,21 This general strategy developed by Mirkin resulted in a number of functional compounds, the behaviour of which can be effectively switched via binding of an incoming small guest molecule or ion.22
The mixed phosphine–phosphine oxide (P–PO) ligands have been less studied in comparison with other bifunctional phosphine congeners and are represented mainly by a family of bis(phosphine) monoxides (BPMO).11,23 The relatively limited development of this class of P–PO compounds could be attributed to a more challenging synthesis of unsymmetrical oxide derivatives in comparison with the preparation of the parent phosphines and the low selectivity of direct partial oxidation of the di- or oligophosphines. In this respect, a recently reported tridentate PP(O)P ligand, obtained via quite a facile route,24 opens a pathway to pincer POP complexes, which remain very poorly explored.
In our previous studies we have been extensively using the di-, tri- and tetraphosphines to support various coordination complexes and cluster compounds of coinage metals, which exhibit tunable photoluminescence characteristics and stimuli-responsive features.25–27 However, the employment of mixed P–PO type ligands for the design of luminescent transition metal compounds has been virtually neglected.28 Motivated by this lack of investigations, we attempted to study the coordination chemistry of a hybrid phosphine–phosphine oxide PPh2–C6H4–PPh(O)–C6H4–PPh2 (P3O) with respect to Cu subgroup metals. Utilizing potentially variable denticity of the ligand, which is able to adjust to a preferable coordination number of a given ion, herein we present a series of novel mono- and dinuclear d10 coinage complexes, their photoemissive properties and a computational analysis of the electronic structures to rationalize the photophysical behavior.
Results and discussion
Synthesis and structural characterization
In the course of our recent studies of d10 metal cyanide complexes supported by a triphosphine ligand, PPh2–C6H4–PPh–C6H4–PPh2 (P3),29 it was observed that heating a solution of a dinuclear complex [(P3)Ag–CN–Ag(P3)]+ on air resulted in some degradation and formation of a novel compound, crystallographically characterized as [(P3O)2Ag2]2+. The product contains the partially oxidized ligand PPh2–C6H4–PPh(O)–C6H4–PPh2 (P3O), which was apparently formed upon interaction with oxygen at elevated temperatures. Due to the low yield and slow rate of this transformation, we prepared independently the diphosphine–phosphine oxide P3O and investigated its coordination behavior with respect to coinage metal ions.The ligand, PPh2–C6H4–PPh(O)–C6H4–PPh2 (P3O), was reported earlier as a side product, obtained in 3% yield.30 Later a similar compound, P(iPr)2–C6H4–PPh(O)–C6H4–P(iPr)2, was obtained via a more practical route in 73% yield.24 Following the latter method, P3O was synthesized from (2-bromophenyl)diphenylphosphine31 in good yield (Scheme 1). The 31P NMR spectrum of P3O displays two resonances (31.9 and −13.6 ppm, 3JPP 12 Hz) with 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 integral relative intensities that are consistent with the proposed structure.
:2 integral relative intensities that are consistent with the proposed structure.
 | ||
| Scheme 1 Synthesis of the hybrid ligand P3O. | ||
 | ||
| Scheme 2 Synthesis of complexes 1–10. | ||
The crystal structures of 1–4 were determined by XRD analysis and are shown in Fig. 1 and 2, and selected structural parameters are given in Tables S4 and S5.† In the dication 1 the copper centers adopt a distorted tetrahedral coordination geometry. Both P3O ligands are bound to the metal ions in a tridentate mode via the terminal P atoms and the OPPh moiety to form a chair-like P3OCu2OP3 framework with the oxygen atoms in nearly symmetrical μ2-bridging positions between two Cu ions (Cu–O distances are 2.121 and 2.198 Å). In this structural pattern each of the oxygen centers donates both available electron pairs to complete the coordination vacancies at the metals. The copper–oxygen bond lengths are comparable to the values found for the previously reported Cu(I) complexes with coordinated phosphine-oxide function.13,32 The PO bond in 1 (1.520 Å) however is slightly longer than those in the abovementioned compounds (<1.503 Å)13,32 and in 2–4 (1.491, 1.494 and 1.507/1.506 Å, respectively; see Table 1) presumably due to the bridging coordination mode of oxygen and therefore more effective π-back donation from the metals to π*(PO) orbitals. The intramolecular Cu⋯Cu separation of 3.279 Å is noticeably longer than the sum of van der Waals radii (2.80 Å) and the range of common metallophilic Cu–Cu bonds.33,34 Together with the saturated coordination environment of Cu(I) ions these structural parameters point to the absence of appreciable cuprophilic interactions.
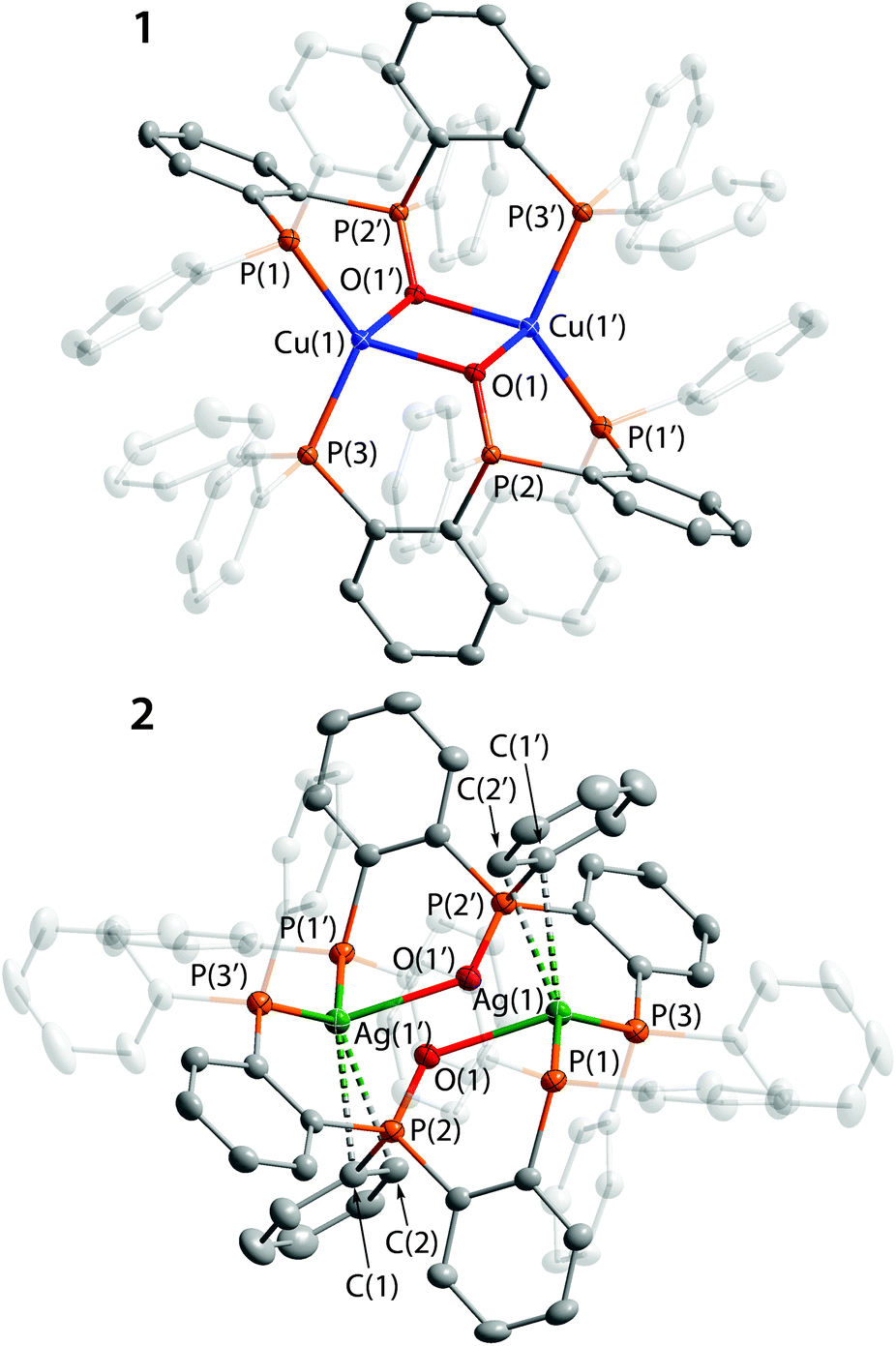 | ||
| Fig. 1 Molecular views of complexes 1 and 2. Thermal ellipsoids are shown at the 50% probability level. Counterions and H atoms are omitted for clarity. Symmetry transformations used to generate equivalent atoms (′) in 1: 1 − x, −y, 1 − z; in 2: 2 − x, 2 − y, 2 − z. | ||
 | ||
| Fig. 2 Molecular views of complexes 3 and 4. Thermal ellipsoids are shown at the 50% probability level. Counterions and H atoms are omitted for clarity. Symmetry transformations used to generate equivalent atoms (′) in 3: 1 − x, y, 0.5 − z. | ||
| BCP | ρ (e Å−3) | |V|/Gb |
E
INTc (kJ mol−1) |
δ(A, B)d | q (M) | |
|---|---|---|---|---|---|---|
| a Local electron density at the BCP. b Ratio of potential energy density and kinetic energy density. c Interaction energy between two interacting atoms. d Delocalization index between A and B (bonding) atoms. e Total atomic charge of metal atoms. | ||||||
| 1 | Cu(1)⋯O(1) | 0.325 | 1.04 | −87.8 | 0.30 | 0.501 |
| Cu(1)⋯O(2) | 0.310 | 1.02 | −82.2 | 0.28 | ||
| Cu(2)⋯O(1) | 0.310 | 1.02 | −82.2 | 0.28 | ||
| Cu(2)⋯O(1) | 0.325 | 1.04 | −87.8 | 0.30 | ||
| P(2)–O(1) | 1.374 | 1.35 | −741.1 | 0.77 | ||
| Cu–P | 0.532 | 1.41 | −120.1 | 0.67 | ||
| 2 | Ag(1)⋯O(1) | 0.210 | 1.03 | −42.9 | 0.20 | 0.400 |
| Ag(1)⋯O(2) | 0.219 | 1.01 | −46.3 | 0.21 | ||
| Ag(2)⋯O(1) | 0.219 | 1.01 | −46.3 | 0.21 | ||
| Ag(2)⋯O(1) | 0.210 | 1.03 | −42.8 | 0.20 | ||
| P(2)–O(1) | 1.441 | 1.33 | −808.2 | 0.82 | ||
| Ag–P | 0.481 | 1.36 | −94.8 | 0.63 | ||
| 3 | Au(1)⋯O(1) | 0.174 | 0.99 | −29.9 | 0.21 | 0.062(Au1) |
| Au(1)⋯O(2) | 0.174 | 0.99 | −29.9 | 0.21 | 0.024(Au2) | |
| Au(2)⋯O(1) | 0.111 | 0.97 | −15.8 | 0.11 | ||
| Au(2)⋯O(1) | 0.111 | 0.97 | −15.8 | 0.11 | ||
| P(2)–O(1) | 1.466 | 1.33 | −831.9 | 0.85 | ||
| Au–P | 0.716 | 1.69 | −148.3 | 0.88 | ||
| 4 | Cu(1)⋯O(1) | 0.336 | 1.04 | −92.1 | 0.32 | 0.514(Cu) |
| Cu(1)⋯O(2) | 0.336 | 1.04 | −92.1 | 0.32 | 0.014(Au) | |
| Au(2)⋯O(1) | 0.116 | 0.99 | −17.2 | 0.12 | ||
| Au(2)⋯O(1) | 0.116 | 0.99 | −17.2 | 0.12 | ||
| P(2)–O(1) | 1.427 | 1.34 | −792.8 | 0.82 | ||
| Cu–P | 0.524 | 1.39 | −118.8 | 0.65 | ||
| Au–P | 0.730 | 1.73 | −149.6 | 0.87 | ||
In 2, the equivalent silver ions adopt a highly distorted tetrahedral arrangement of the ligand sphere with Ag⋯Ag distance (4.475 Å) significantly exceeding the values typical for argentophilic bonding.35 Each metal center is connected to two terminal P-donors and to the O atom of the phosphine oxide fragment. In contrast to 1, oxygen occupies a visibly unsymmetrical position between the Ag ions, being predominantly coordinated to only one metal (Ag–O distances are 2.457 and 2.885 Å) that is indicative of terminal oxygen coordination in 2 in contrast to the bridging one in 1. The Ag–O bond length of 2.457 Å fits in the range found for other structurally characterized phosphine-oxide silver complexes,36 which are quite rare. However, the tendency of silver(I) to have a tetracoordinate geometry results in a visible π-interaction of the metal ions with adjacent phenyl rings of the OPPh group (see Fig. 1). The distances Ag(1)–C(1) and Ag(1)–C(2) are 2.954 and 2.822 Å, respectively, that point to a moderately weak Ag–η2(CC) bonding37 as the effective interaction of this type requires a characteristic bond length less than 2.9 Å.38
The dimeric complex 3 contains two gold(I) ions, which are held together by the P3O ligands. The phosphorus atoms of the PPh2 functions form a two-coordinate environment of the metal centers, typical of Au(I) compounds.39 The deviation of the P–Au–P angles (164.3 and 159.8°) from the ideal value of 180° could be attributed to some weak Au–O interactions in the solid state (Au–O distances are 2.986 and 2.745 Å), which are much less efficient than M–O bonds in 1 and 2, reflecting generally a higher coordination number of copper(I) and silver(I) than that of gold(I)40 and a lower affinity of Au(I) centers to hard electron donors. Due to the stereochemical arrangement of 3 the large Au⋯Au separation of 4.477 Å suggests no intramolecular metal–metal bonding.41 Interestingly, a mononuclear arrangement was suggested for the gold(I) complex [Au(i-Pr-P3O)]+ with a closely related ligand (i-Pr-P3O = (o-iPr2P–(C6H4))2P(O)Ph) on the basis of the computationally optimized structure and NMR spectroscopic data.42
In the heterobimetallic complex 4 a tridentate mode of P3O provides two types of ligand spheres (“P2O2” and “P2”), which successfully saturate four and two coordination vacancies of Cu(I) and Au(I) ions, respectively. The structural parameters of 4 (Table S5†) are somewhat comparable with the related values determined for homometallic congeners 1 and 3.
The ESI-MS of the dimeric 1 and 2 display dominating signals at m/z 709.1 and 753.1, respectively, which correspond to the monocations as a result of fragmentation under the conditions of the ESI experiment. The spectra of complexes 3 and 4, however, show the signals of the doubly charged dinuclear cations at m/z of 843.1 and 776.1. The observed isotopic patterns completely match the calculated ones for the proposed [Cu(P3O)]+, [Ag(P3O)]+, [Au2(P3O)2]2+ and [AuCu(P3O)2]2+ stoichiometry (Fig. S1, ESI†). It has to be noted that in the case of 4 additional strong signals assigned to the presence of 1 and 3 were detected. This denotes the formation of a mixture of 1, 3 and 4 upon dissolving 4, which was also confirmed by NMR spectroscopic studies (vide infra).
The 31P{1H} NMR spectra of 1 and 2 display two signals each and agree with idealized symmetrical structures of these complexes. The low field resonances at 42.0 ppm (1) and 36.1 ppm (2) can be assigned to the OPPh moieties. The high field signals (−11.3 ppm and −1.3 for 1 and 2, respectively) of double integral intensities correspond to the metal-coordinated PPh2 groups, which become essentially equivalent in solution. The presence of well resolved signals of the two isotopomers with P–107, 109Ag couplings (J(P–109Ag) 473 Hz and J(P–107Ag) 412 Hz) in the high field resonance of 2 additionally confirms coordination of both diphenylphosphine functions to silver(I) ions. The 1H NMR data of 1 and 2 are also in accordance with the symmetrical arrangement of the complexes in solution. The corresponding spectra (Fig. S2 and S3†) are very much alike, showing poorly resolved resonances of the aromatic protons that prevent their complete assignment, but the number of the signals and their relative intensities indicate the equivalence of the P3O ligands in fluid medium or possible dissociation of the dimers into the monomeric species, as suggested by the ESI-MS measurements.
Complex 3 demonstrates higher rigidity in solution compared to the Cu and Ag relatives. Its phosphorus NMR spectrum shows 3 resonances with 1:1:1 relative intensities (Fig. 3). The low field multiplets (37.5 and 32.5 ppm, dd JPPca. 6 Hz) from the nonequivalent PPh2 moieties testify to the retaining of the structure found in the crystal state, in which two types of terminal phosphorus atoms are identified. The P centres apparently become different due to the chiral twisting of the 16-membered metallocycle, induced by the arrangement of the phenyl rings to minimise intramolecular repulsion. The proton NMR of 3 (Fig. S4†) is in line with this structural arrangement and supports the unsymmetrical coordination of P3O to the gold centers.
 | ||
| Fig. 3 162 MHz 31P{1H} NMR spectrum of the equilibrated mixture of complexes formed upon dissolution of 4 (dmso-d6, 298 K). The relative molar ratio of 1:3:4 is ca. 1:1:1. | ||
NMR investigation of 4 revealed that in solution it produces a mixture of three compounds existing in dynamic equilibrium. The 31P{1H} NMR spectrum of 4 (Fig. 3) exhibits two sets of signals, which can be easily assigned to compounds 1 and 3 by comparing them with spectroscopic patterns obtained for the individual homometallic species. The third suite of resonances of equal intensities is completely compatible with the structure of heterometallic cation 4, and contains three signals, corresponding to phosphine oxide (45.3 dd, JPP 11.2, 10.7 Hz) and diphenylphosphine groups coordinated to gold (42.8 s) and copper (−12.3 br) ions.
The freshly dissolved recrystallized complex 4 shows the presence of a dominant heterometallic form found in the crystal state, and gradually growing amounts of the homometallic complexes 1 and 3, until the ratio of the species becomes roughly equimolar in ca. 2 h (room temperature). Recrystallization of the mixture results in recovery of a uniform crystalline material of 4, as confirmed by the XRD analysis of several crystals. Additionally, the crystalline form of 4 demonstrates unique photophysical characteristics different from those of 1 and 3 (see below) that clearly indicate the difference in the nature of these solid materials and supports phase purity of 4. A similar behavior of heterometallic phosphine coinage metal complexes, which involves migration of the metal ions and appearance of multicomponent mixtures in solution, was described earlier by us for some other multidentate ligand complexes.26,29,43
The nature of the intramolecular interactions was investigated via topological charge density analysis utilizing the Quantum Theory of Atoms in Molecules (QTAIM). Table 1 presents selected properties of the electron density at the corresponding bond critical points for the bimetallic complexes 1–4. The interaction energies, which were calculated to be half of the potential energy density according to the method of Espinosa et al.,44 show a clear trend in the strength of the M⋯O interactions, Cu > Ag > Au. The difference in the bond strength is most pronounced in the heterobimetallic complex 4, where the interaction energies are −92 kJ mol−1 and −17 kJ mol−1 at the Cu⋯O and Au⋯O bond critical points, respectively. The strength of the interaction is directly proportional to the amount of local electron density at the bond critical point.
However, even though the interaction energies are rather notable, at least in the case of copper complex 1, the M⋯O interactions are mostly non-covalent in nature, which can be seen in the ratio of the potential energy density and kinetic energy density, |V|/G ∼ 1, and in the small amount of shared electrons given by the delocalization index, δ(A, B). Because of the non-covalent nature of the M⋯O interactions, the original properties of the P(2)–O(1) interaction are only slightly changed, when compared to the values in the free ligand. For example, the ρ, |V|/G, EINT and δ(A, B) values are 1.500 e Å−3, 1.32, −868.2 kJ mol−1, and 0.88, respectively, for the freely optimized P3O. On the other hand, the weaker contacts between metal and oxygen were found to increase the phosphorus–metal interaction, thus leading to an opposite trend in the M–P BCPs, Au > Ag > Cu, which can be seen in Table 1, where average values at the M–P interactions are presented for complexes 1–4. As can be seen in Fig. S5,† no metal–metal interactions were found in the bimetallic compounds. It should be noted that although the optimized geometry of 2 was found to be more symmetrical than the experimental one, for which the charge density analysis led to two stronger Ag⋯O interactions and two weaker ones, this did not change either the nature of the Ag⋯O contacts or the trend in the strength of bonding in the bimetallic species.
Complexes 5–10 were isolated by crystallization as colourless to pale yellow materials in good yields (Scheme 2). The XRD analysis revealed an essentially similar geometry for these mononuclear compounds (Fig. 4 and S6†). The structural motif implies a quasi-tetracoordinate environment of the metal ions, which involves the regular metal–phosphine/halide bonding45 along with some metal–OPPh interactions (Table S6†).
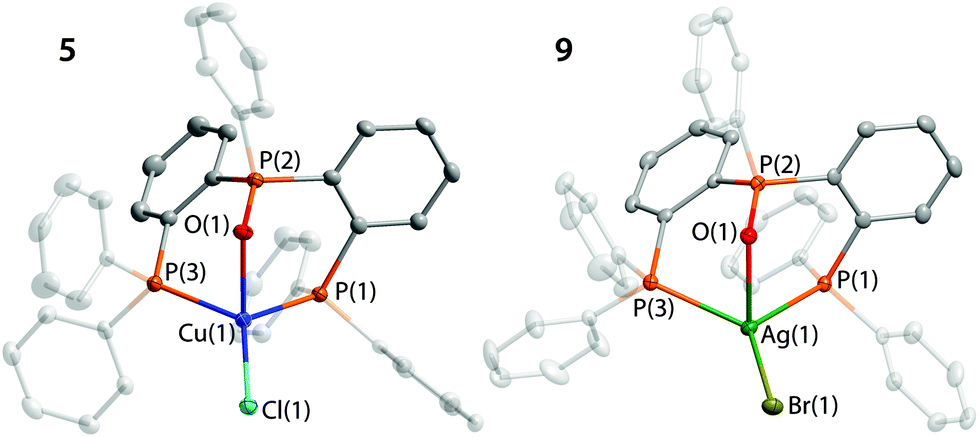 | ||
| Fig. 4 Molecular views of complexes 5 and 9. Thermal ellipsoids are shown at the 50% probability level. H atoms are omitted for clarity. | ||
The weakness of phosphine oxide coordination to Cu and Ag ions evidently determines trigonal planar geometry of the P2X ligand at the metal centers that is similar to that found for the congener Ag(i-Pr-P3O)X.42 The Cu–O distances particularly in 5 and 6 are visibly longer than those in 1, 4 and other related copper species,13,32 but are comparable to the value reported for the [Cu(κ2-DPEphos)(κ2-DPEphosO)]+ complex.46 In the silver species 9 and 10 the Ag–O separations are similarly longer than those in 2 and congener compounds,36 which points to a rather insignificant interaction of the OPPh moiety with silver ions.
The solution NMR spectroscopic data of the halide complexes 5–10 are in complete agreement with the molecular arrangement found in the crystal. Their 31P{1H} spectra display two signals with a 1:2 intensity ratio, which correspond to the P–oxide fragment and the metal-coordinated equivalent PPh2 groups, respectively (see the Experimental section).
We have also synthesized the non-oxidized triphosphine halide compounds M(P3)X (M = Cu, X = Cl (11), Br (12), I (13); M = Ag, X = Cl (14), Br (15), I (16), P3 = bis(2-diphenylphosphinophenyl)phenyl phosphine) to compare their photophysical characteristics with those of M(P3O)X complexes 5–10. The species 11 and 14–16 were employed earlier in the fabrication of luminescent membranes and electroluminescent devices,47 and 14 was characterized spectroscopically and crystallographically.48 Complexes 11, 12, 15 and 16 were studied by means of XRD analysis (Fig. S7†), which revealed that 11, 12, and 15 are isomorphous to 14 (space group P21/n) with small alterations of the unit cell parameters. Additionally, M(P3)X compounds closely resemble the related cyanide complexes M(P3)CN we described recently.29 Complex 16 crystallizes in the P21/c type space group with somewhat different cell dimensions, but its molecular geometry is virtually identical to that of 11, 12, and 15 (Fig. S6†). The tetracoordinate ligand sphere of the metal ions in M(P3)X compounds is completed by the tridentate phosphine P3 and the halide X that is in line with previous reports.29,48 The selected structural parameters are listed in Table S7.† The NMR spectra are consistent with the structural data and indicate that all the P donors are bound to the corresponding metal centers (see the Experimental section).
Selected mononuclear halides were also optimized and the intramolecular interactions were investigated. The compounds studied were copper complexes 7 and 13, and silver complexes 10 and 16. The results of the QTAIM analysis are given in Table S8 of the ESI.† The relative trends in the properties of the electron density in the monomeric compounds were essentially the same as those observed in the bimetallic species, the Cu⋯O interaction with the P3O ligand being stronger than the corresponding Ag⋯O contact. The same trend was also found for M⋯P(2) interactions with the P3 ligand. Interestingly, in the complexes with P3 ligands, the Cu⋯P(2) is similar to the other Cu⋯P interactions, but the Ag⋯P in 16 is notably weaker, which becomes apparent from the larger optimized distance.
Solid state photophysical properties
The title compounds do not demonstrate appreciable photoluminescence in solution, and their photophysical behavior was investigated in the solid state only. The relevant data are listed in Table 2 and the spectroscopic patterns are shown in Fig. 5–7 and S8–S10.† | ||
| Fig. 5 Normalized solid state excitation (dotted lines) and emission (solid lines) spectra of 1–4 at 298 K. | ||
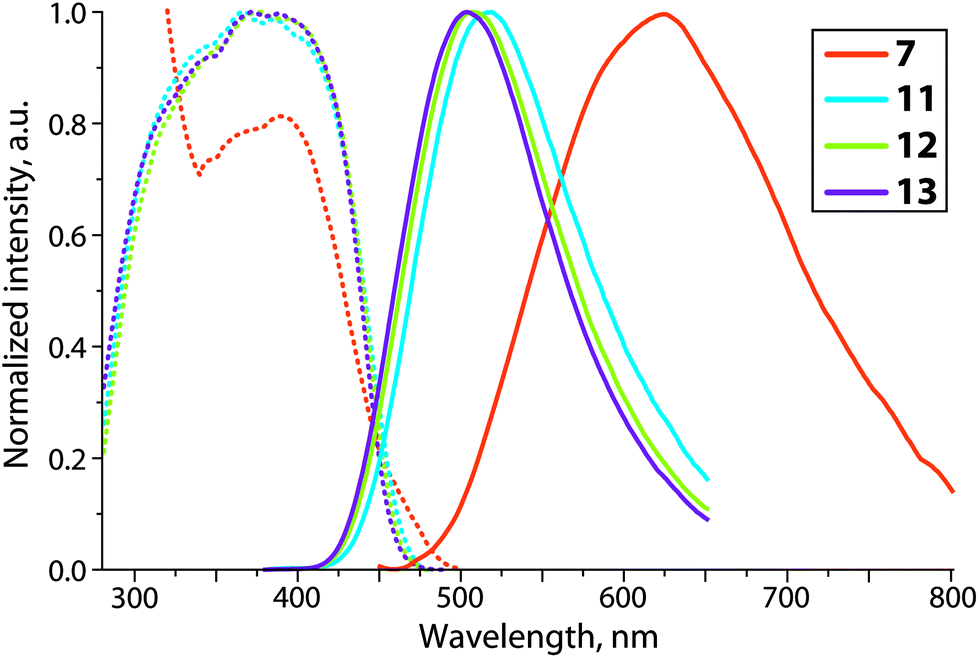 | ||
| Fig. 6 Normalized solid state excitation (dotted lines) and emission (solid lines) spectra of copper complexes 7 and 11–13 at 298 K. | ||
 | ||
| Fig. 7 Normalized solid state excitation (dotted lines) and emission (solid lines) spectra of copper complexes 8–10 and 14–16 at 298 K. | ||
| λ ex, nm | λ em, nm |
τ
obsa, μs |
Φ em, % |
k
rb, s−1 |
k
nrc, s−1 |
||||
|---|---|---|---|---|---|---|---|---|---|
| 298 K | 77 K | 298 K | 77 K | 298 K | 77 K | 298 K | |||
| a Average emission lifetimes for 1, 4 and 7 for the two-exponential decay determined using the equation τav = (A1τ12 + A2τ22)/(A1τ1 + A2τ2), where Ai is the weight of the i-exponent. b k r were estimated using Φ/τobs. c k nr were estimated using kr(1 − Φ)/τobs. | |||||||||
| 1 | 364 | 365 | 604 | 604 | 0.35 | 174.1 | 0.5 | 1.4 × 104 | 2.0 × 106 |
| 2 | 332 | 324 | 500 | 485 | 10.9 | 2354.3 | 5.9 | 5.0 × 103 | 8.5 × 104 |
| 3 | 330 | 325 | 548 | 564 | 6.7 | 82.3 | 28.9 | 4.3 × 104 | 1.0 × 105 |
| 4 | 360 | 363 | 538 | 546 | 5.5 | 777.5 | 25.8 | 4.7 × 104 | 1.3 × 105 |
| 7 | 394 | 362, 380 | 621 | 624 | 0.5 | 30.7 | 0.8 | 1.7 × 104 | 2.1 × 106 |
| 8 | 310, 372 | 340 | 480 | 495 | 64.8 | 482.0 | 17.7 | 2.7 × 103 | 1.3 × 104 |
| 9 | 362 | 340 | 467 | 464 | 19.0 | 529.9 | 7.5 | 3.9 × 103 | 4.7 × 104 |
| 10 | 362 | 353 | 488 | 486 | 38.1 | 95.7 | 20.4 | 5.4 × 103 | 2.1 × 104 |
| 11 | 370 | 363, 397 | 517 | 520 | 9.0 | 565.8 | 21.0 | 2.3 × 104 | 8.7 × 104 |
| 12 | 370 | 363, 397 | 507 | 523 | 11.7 | 794.4 | 36.3 | 3.1 × 104 | 5.4 × 104 |
| 13 | 370 | 311 | 504 | 515 | 10.5 | 136.9 | 37.2 | 3.5 × 104 | 6.0 × 104 |
| 14 | 335, 367 | 329, 375 | 521 | 535 | 27.0 | 595.7 | 39.5 | 1.5 × 104 | 2.2 × 104 |
| 15 | 335, 395 | 329, 375 | 521 | 535 | 25.6 | 537.7 | 40.5 | 1.6 × 104 | 2.3 × 104 |
| 16 | 335, 396 | 329, 375 | 513 | 525 | 13.5 | 179.9 | 38.9 | 2.8 × 104 | 4.5 × 104 |
The dinuclear copper (1) and silver (2) complexes are weakly luminescent at room temperature and exhibit broad emission peaks centered at 604 (Φem 0.5%) and 500 nm (Φem 5.9%), respectively. Cooling the samples to 77 K doesn't affect the emission maximum of 1 and causes some blue shift for 2 (Table 2, Fig. S7†) that allows excluding for these species a phenomenon of thermally activated delayed fluorescence, for which a small red shift of the emission band is typically observed upon lowering the temperature.34,49 The gold-containing compounds show the emission bands at 548 (3) and 538 (4) nm with considerably larger quantum efficiencies (Φem 28.9 and 25.8%), which is reflected by increased radiative rate constants (kr) with respect to the rates of radiationless deactivation (knr, Table 2). The effect evidently results from larger spin orbit coupling in gold complexes and a higher ISC rate compared to analogous compounds of the first and second transition row metals. The lifetimes for 1–4 are found to be in the microsecond domain at 298 K and show a dramatic growth upon lowering the temperature, reaching the value of 2354.3 μs (2, 216-fold increase) that is comparable to the demeanor of some [Ag(diphosphine)2]+ compounds.50 The emission profile of 4 does not reveal any detectable shoulders to evidence the presence of homometallic complexes 1 and 3 (vide supra), which were identified in the solution of 4 by ESI-MS and NMR spectroscopy. This observation additionally supports the very selective crystallization of heterobimetallic complex 4.
The mononuclear copper chloride and bromide complexes 5 and 6 are not luminescent either at 298 K or at the temperature of liquid nitrogen. Their iodide congener 7 however exhibits weak orange emission (621 nm, Φem 0.8%, Fig. 6). A comparison of 7 with non-oxidized complexes Cu(P3)X (11–13) reveals the dramatic effect of changing the phosphine PPh group to phosphine oxide OPPh on the photophysical properties of copper halides. Unlike 7, the Cu(P3)X species demonstrate a moderately strong blue-green luminescence (504–517 nm, Φem 21–37%), which is only slightly sensitive to the nature of the X ligand (Fig. 6 and S9†). The significant decrease of emission energy for 7 with respect to 11–13 can be tentatively assigned to a possibly larger flexibility of the Cu(P3O)X framework, which could facilitate the formation of a planar metal geometry in the MLCT excited state. The latter, as generally accepted, is able to provide nonradiative ways of relaxation.51
The two families of silver complexes Ag(P3O)X (8–10) and Ag(P3)X (14–16) display a very different phosphine ligand effect on the photophysics of the solid materials compared to the copper relatives. The triphosphine compounds 14–16 show rather intense room temperature emission with quantum yields of around 40% (Table 2). The broad bands with maxima in the range 513–521 nm are slightly red shifted with respect to the corresponding Cu species 11–13 at 298 K (Fig. 7), and in a similar way experience a small decrease of luminescence energies at 77 K (Fig. S10†). The emission bands for oxygen-modified complexes 8–10 show a visible hypsochromic shift of 25–54 nm, accompanied by at least 2-fold drop of intensities, which, however, is not as drastic as in the case of copper congeners. The observed difference of the photophysical characteristics between the series 8–10 and 14–16 could be attributed to apparently weak interaction of Ag–OPPh2 and thus formally a lower coordination number of silver ions in Ag(P3O)X complexes that evidently affects the frontier orbitals and the energies of electronic transitions.
In order to rationalize the difference in photophysical behavior between the complexes with P3O ligands and P3 ligands, we studied the emission properties by optimizing the first excited triplet state for copper compounds 7 and 13 as well as for silver complexes 10 and 16. The calculated emission wavelengths are well consistent with the experimental spectra for the monometallic compounds. However, the emission energies were considerably overestimated in the case of the bimetallic complexes, even though the general trend could be correctly represented. Obviously, the DFT method was not able to describe the electron distribution in the triplet state for the dimeric compounds. This can be seen as an example of complex 1 in Fig. S11,† which shows the HOMO and LUMO orbitals of the singlet state, where the MOs are evenly distributed in the vicinity of both copper atoms, leading to correct absorption energies. On the contrary, in the triplet state the singly occupied MOs are very unsymmetrically expanded around different metals, leading to a less stable triplet state, and hence overestimated emission energies. Because of this inconsistency we focused on the electronic features of the monometallic complexes. The appearance of the singly occupied molecular orbitals (SOMOs, HSOMOs) is compared along with the HOMOs of the S0 state in Fig. 8 and 9. In the singlet ground state, the HOMO orbitals mainly consist of a combination of metal d-orbitals with the halogen p-orbital. There is a notable difference between the complexes with different ligands, since in the ground state, the oxygen and P(2) orbitals of the P3O ligand do not participate in HOMO (complexes 7 and 10), which leads to its higher energy compared to the complexes 13 and 16, where p-orbitals of all phosphorus atoms of P3 interact with the metal center and contribute to HOMO.
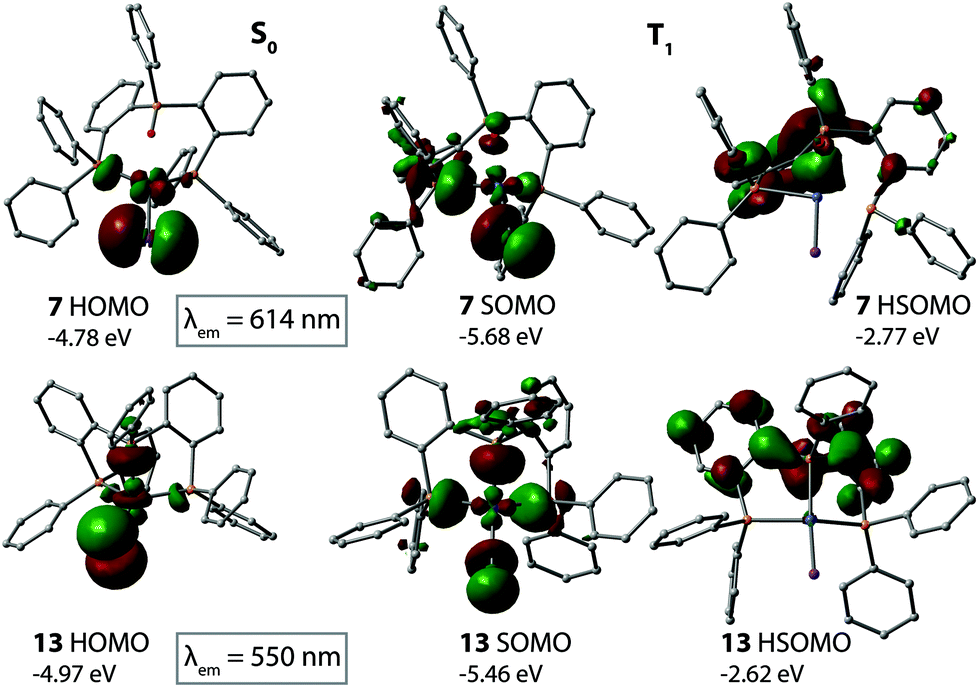 | ||
| Fig. 8 The appearance of the highest occupied orbitals in the singlet ground state and the first excited triplet state of copper complexes 7 and 13. The emission wavelength was estimated from the total energy difference of the states. | ||
 | ||
| Fig. 9 The appearance of the highest occupied orbitals in the singlet ground state and the first excited triplet state of silver complexes 10 and 16. The emission wavelength was estimated from the total energy difference of the states. | ||
When one electron is excited from HOMO, the remaining electron forms a SOMO orbital (singly occupied molecular orbital), the energy of which is considerably lowered in the case of the oxygen containing ligand P3O, as much as 0.90 eV in 7 and 0.55 eV in 10. In both cases, the stabilization results from the substantial increase of the contribution of metal d-orbitals in SOMO, which was not observed in the ground state (18% → 31% in 7 and 11% → 21% in 10). On the contrary, 16 showed slight destabilization of the energy of SOMO compared to the singlet state HOMO, probably due to decreasing contribution from the iodide ligand, which is also observed, though not so extensively, for other compounds upon excitation. The full fragment analysis of the frontier MOs is presented in Table S9.† This discrepancy can also account for the different trends in the emission energies between the series of copper and silver compounds.
The highest singly occupied MO (HSOMO) is rather similar in all cases; it is formed as a combination of the phenylene ring orbitals. Because of the larger flexibility of the framework with the oxygen containing ligand, the optimized geometry in the triplet state was less symmetrical for P3O-containing complexes than that found for their P3-based relatives. Therefore, the HSOMO orbital is less evenly distributed along the ligand. The rather notable contribution of oxygen p-orbitals leads to the localization of HSOMO more in the central part of the ligand, which has a small stabilizing effect. However, this has a minimal effect on the energetics of the HSOMOs and, consequently, on the photophysical characteristics. Basically, the emission can be assigned to the MXLCT type for all mononuclear compounds. Nevertheless, the variations in the metal–ligand interactions in the series with P3O and P3 ligands lead to the observed differences in luminescence behavior.
Conclusions
The phosphine–phosphine oxide hybrid ligand P3O, which shows variable binding ability with respect to d10 coinage metal ions, was used for the preparation of a series of novel mono- and dinuclear complexes. The combination of soft (P) and hard (O) donor functions allows for an efficient adaptation of the ligand environment to the preferable coordination number of the CuI, AgI and AuI centers that preserves the nuclearity of the title dicationic compounds [MM′(P3O)2]2+ (M = M′ = Cu (1), Ag (2), Au (3); M = Au, M′ = Cu (4)). The nature of metal–ligand interactions was elucidated using the QTAIM computational approach, which revealed a clear trend in the strength of the M⋯O bonds, Cu > Ag > Au. A similar tendency was estimated for the neutral monometallic halide complexes M(P3O)Hal (M = Cu (5–7), Ag (8–10)). The studied complexes exhibit weak to moderate room temperature photoluminescence in the solid state except 5 and 6, which are virtually not emissive even at 77 K. The bimetallic compounds demonstrate quantum efficiency up to 28.9% (3, λem = 548 nm), while the mononuclear ones reach a Φem value of 20.4% (10, λem = 488 nm). The effect of partial phosphine oxidation on the photophysical properties was analyzed comparing the M(P3O)Hal species with their M(P3)Hal congeners. For the copper complexes the presence of the phosphine-oxide group is apparently detrimental, leading to a dramatic decrease of quantum yields that is accompanied by a 117 nm bathochromic shift (7vs.13). On the contrary, for silver complexes the emission intensity is much less influenced by the introduction of oxygen, but the wavelength shows a substantial blue shift (up to 54 nm for 9vs.15) that illustrates a convenient possibility of fine tuning the luminescence characteristics of this class of inorganic material.Experimental
General comments
(2-Bromophenyl)diphenylphosphine,31 bis(2-diphenylphosphinophenyl)phenyl phosphine (P3)27 and the complex AuCl(tht) (tht = tetrahydrothiophene)52 were synthesized according to published procedures. Tetrahydrofuran (THF) was distilled over Na-benzophenone ketyl under a nitrogen atmosphere prior to use. Other reagents and solvents were used as received. The solution 1H, 31P{1H} and 1H–1H COSY NMR spectra were recorded on a Bruker Avance 400 spectrometer. Mass spectra were recorded on a Bruker micrOTOF 10223 instrument in the ESI+ mode. Microanalyses were carried out in the analytical laboratory of the University of Eastern Finland.:1 v/v mixture) solution of 4 at 278 K to give a yellow crystalline material (72 mg, 84%). 31P{1H} NMR (dmso-d6, 298 K, δ): the spectrum corresponds to the mixture of the 3 complexes, see discussion; 45.3 (dd, JPP 11.2, 10.7 Hz, 1P, P(O)Ph), 44.4 (3), 42.8 (s, 1P, Au–PPh2), 42.0 (1), 37.5 (3), 32.5 (3), −11.3 (1), −12.3 (br s, 1P, PPh2), −144.6 (sept, 1P, PF6). ES MS (m/z): [AuCu(P3O)2]2+: 776.12 (calc. 776.13). Anal. calc. for AuCuC42H33OP4F6 (%): C 54.72; H 3.61. Found: C 54.56; H 3.65.
General procedure for the synthesis of CuX(P3O) (X = Cl, Br, I) complexes (5–7)
CuX (0.123 mmol) and P3O (80 mg; 0.123 mmol) were suspended in dichloromethane (10 ml). The reaction mixture was stirred for 5 hours to give a transparent light yellow solution. The solvent was evaporated and the solid residue was recrystallized.:1 v/v mixture) at 278 K to give a yellow crystalline material (86 mg, 89%). 31P{1H} NMR (dmso-d6, 298 K, δ): 39.6 (br s, 1P, P(O)Ph), −14.5 (br s, 2P, PPh2). 1H NMR (dmso-d6, 298 K, δ): 7.64 (m, 1H), 7.44–7.59 (m, 12H), 7.31–7.44 (m, 10H), 7.25 (dd, JHHca. 7.6 Hz, 4H), 6.97 (m, 4H), 6.89 (m, 2H). Anal. calc. for CuC42H33OP3Br (%): C 63.85; H 4.21. Found: C 63.82; H 4.18.
:2 v/v mixture) at 278 K to give a yellow crystalline material (93 mg, 90%). 31P{1H} NMR (dmso-d6, 298 K, δ): 37.63 (br s, 1P, P(O)Ph), −14.27 (br s, 2P, PPh2). 1H NMR (dmso-d6, 298 K, δ): 7.65 (m, 1H), 7.44–7.55 (m, 12H), 7.37–7.43 (m, 4H), 7.35 (dd, JHHca. 7.2 Hz, 6H), 7.25 (dd, JHHca. 7.5 Hz, 4H), 7.00 (m, 4H), 6.86 (m, 2H). Anal. calc. for CuC42H33OP3I (%): C 60.26; H 3.97. Found: C 59.98; H 4.00.
General procedure for the synthesis of AgX(P3O) (X = Cl, Br, I) complexes (8–10)
AgPF6 (25.3 mg, 0.1 mmol) was dissolved in acetone (5 ml) and a solution of the corresponding tetrabutyl ammonium halide NBu4X (0.1 mmol) in dichloromethane (10 ml) was added. The suspension was stirred for 10 min in the absence of light. Then P3O (64.6 mg, 0.1 mmol) was added to the reaction mixture, which was stirred for 1 hour to give a colorless transparent solution. The solvents were evaporated and the solid residue was recrystallized by a gas-phase diffusion of diethyl ether into a dichloromethane solution of a crude complex at 278 K to afford a colorless crystalline material.General procedure of CuXP3 (X = Cl, Br, I) complexes (11–13)
CuX (0.123 mmol) and P3 (77.5 mg, 0.123 mmol) were suspended in dichloromethane (10 ml). The reaction mixture was stirred overnight to give a transparent light yellow solution. The solvent was evaporated and the solid residue was recrystallized by a gas-phase diffusion of diethyl ether into a dichloromethane solution of a crude complex at 278 K to give a yellow crystalline material.General procedure of AgXP3 (X = Cl, Br, I) complexes (14–16)
AgPF6 (25.3 mg, 0.1 mmol) was dissolved in acetone (5 ml) and a solution of the corresponding tetrabutyl ammonium halide NBu4X (0.1 mmol) in dichloromethane (10 ml) was added. The suspension was stirred for 10 min in the absence of light. Then P3 (63.0 mg, 0.1 mmol) was added to the reaction mixture, which was stirred for 1 hour to give a colorless transparent solution. The solvents were evaporated and the solid residue was recrystallized by a gas-phase diffusion of diethyl ether into a dichloromethane solution of a crude complex at 278 K to afford a colorless crystalline material.X-ray structure determination
The crystals of 1–12, 15, and 16 were immersed in cryo-oil, mounted in a Nylon loop, and measured at a temperature of 120 K. The X-ray diffraction data were collected with Bruker SMART APEX II or Bruker Kappa Apex II Duo diffractometers using MoKα radiation (λ = 0.71073 Å). The APEX253 program package was used for cell refinements and data reductions. The structures were solved by direct methods using the SHELXS-2013/201454 programs with the WinGX55 graphical user interface. A semiempirical absorption correction (SADABS)56 was applied to all data. Structural refinements were carried out using SHELXL-2013/2014.54The PF6− counterion in 1 was disordered over two positions and was refined with occupancies 0.78/0.22. The displacement parameters of the fluorine atoms in both components were constrained to be equal and were restrained, so that their Uij components approximate isotropic behaviour.
The contribution of the missing solvent to the calculated structure factors in complexes 1, 3, 4, and 8 was taken into account by using a SQUEEZE routine of PLATON.57 The missing solvent was not included in the cell content.
The crystal of 8 was of low quality due to the partial loss and disorder of crystallization solvent molecules. Therefore, high-quality refinement could not be achieved and only the structural data of 8 are presented in the ESI†.
All H atoms in 1–12, 15, and 16 were positioned geometrically and constrained to ride on their parent atoms, with C–H = 0.95–0.99 Å and Uiso = 1.2–1.5Ueq. (parent atom). The crystallographic details are summarized in Tables S1–S3.† CCDC 1484959–1484962, 1484966–1484970, 1484973, 1484974, 1484978 (1–12), 1484979 (15), and 1484980 (16) contain the supplementary crystallographic data for this paper.
Photophysical measurements
The steady-state emission and excitation spectra of complexes 1–16 in the solid state at room temperature and at 77 K were recorded on a FluoroMax 4 and a FluoroLog 3 Horiba spectrofluorimeters. The xenon lamps (300 W and 450 W) were applied as light sources to excite luminescence. Two LEDs (maxima of emission at 340 and 390 nm) were used in pulse mode to pump luminescence in lifetime measurements at room temperature (pulse width 1.2 nm, repetition rate 100 Hz to 10 kHz). A pulse laser, DTL-399QT “Laser-Export Co. Ltd” (maximum of emission at 351 nm, 50 mW, pulse width 6 ns, repetition rate 1 kHz); a digital oscilloscope, Tektronix DPO3034 (bandwidth 300 MHz); a MUM monochromator (LOMO, interval of wavelengths 10 nm); and a photomultiplier tube from Hamamatsu were used for lifetime measurements at 77 K. The absolute emission quantum yield of the powders was determined using a FluoroLog 3 Horiba spectrofluorimeter and a Quanta-phi integration sphere.Computational details
All models were fully optimized with the Gaussian 09 program package58 at the DFT level of theory. The hybrid density functional PBE059 was utilized together with the basis set consisting of the def2-TZVPPD60 effective core potential basis set with the triple-zeta-valence basis set with two sets of polarization and diffuse basis functions for Cu, Ag, and Au atoms, the standard all-electron basis set 6-31G(d,p) for C and H atoms, and a slightly more flexible basis set 6-311+G(d) for the interacting P and O atoms. Halogens were described with the def2-TZVPPD basis, which for iodine includes also ECP for the core electrons. To study the intramolecular interactions of the complexes in detail, we performed topological charge density analysis with the QTAIM (Quantum Theory of Atoms in Molecules)61 method, which allowed us to access the nature of the bonding via calculating different properties of the electron density at the bond critical points. The analysis was performed with the AIMAll program62 using the wave functions obtained from the DFT calculations.Emission properties were studied by optimizing the corresponding models in the triplet state, and studying the changes in the appearance of the frontier molecular orbitals. Emission wavelengths were estimated by the total energy difference of the molecules in T1 and S0 electronic states, which seriously underestimates the emission wavelength in the bimetallic complexes 1–4, but is able to reproduce reasonable values for monomer complexes.
Single molecules were used as models for structures 1–4, 7, 10, 13 and 16. The geometries of all models were fully optimized in the singlet and triplet electronic states. The counteranions were not included for the cationic bimetallic models 1–4. The optimized coordinates of all calculated compounds are included in the ESI.† Generally, the optimized geometries were very well in line with the experimental crystal structures, except for the silver dimer 2, which was somewhat symmetrized in the computational optimization, and therefore, all computational properties were also calculated for the non-optimized experimental structure.
Acknowledgements
The financial support from the Academy of Finland (grant 268993, I. O. K.) and the Russian Science Foundation (grant 16-13-10064, E. V. G.) is gratefully acknowledged. The computational work has been facilitated and made available by the Finnish Grid Infrastructure (FGI) resources. The photophysical measurements were performed using core facilities of St. Petersburg State University Research Park: Center for Optical and Laser Materials Research.Notes and references
- C. S. Slone, D. A. Weinberger and C. A. Mirkin, in Progress in Inorganic Chemistry, ed. K. D. Karlin, John Wiley & Sons, New York, 1999, vol. 48, pp. 233–250 CrossRef CAS; M. V. Jiménez, J. J. Pérez-Torrente, M. I. Bartolomé, V. Gierz, F. J. Lahoz and L. A. Oro, Organometallics, 2008, 27, 224–234 CrossRef CAS; M. Bierenstiel and E. D. Cross, Coord. Chem. Rev., 2011, 255, 574–590 CrossRef; L. V. A. Hale, K. A. McGarry, M. A. Ringgold and T. B. Clark, Organometallics, 2015, 34, 51–55 CrossRef; G. S. Nyamato, S. O. Ojwach and M. P. Akerman, Organometallics, 2015, 34, 5647–5657 CrossRef; A. M. Lifschitz, R. M. Young, J. Mendez-Arroyo, C. L. Stern, C. M. McGuirk, M. R. Wasielewski and C. A. Mirkin, Nat. Commun., 2015, 6, 6541 CrossRef PubMed.
- P. Braunstein and F. Naud, Angew. Chem., Int. Ed., 2001, 40, 680–699 CrossRef CAS.
- Z. Weng, S. Teo and T. S. A. Hor, Acc. Chem. Res., 2007, 40, 676–684 CrossRef CAS PubMed.
- X.-L. Pei, Y. Yang, Z. Lei, S.-S. Chang, Z.-J. Guan, X.-K. Wan, T.-B. Wen and Q.-M. Wang, J. Am. Chem. Soc., 2014, 137, 5520–5525 CrossRef CAS PubMed; N. Frank, K. Hanau and R. Langer, Inorg. Chem., 2014, 53, 11335–11343 CrossRef PubMed; B. N. Sanchez-Eguia, M. Flores-Alamo, M. Orio and I. Castillo, Chem. Commun., 2015, 51, 11134–11137 RSC.
- J. S. Park, A. M. Lifschitz, R. M. Young, J. Mendez-Arroyo, M. R. Wasielewski, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2013, 135, 16988–16996 CrossRef CAS PubMed; M. H. Al-Afyouni, E. Suturina, S. Pathak, M. Atanasov, E. Bill, D. E. DeRosha, W. W. Brennessel, F. Neese and P. L. Holland, J. Am. Chem. Soc., 2015, 137, 10689–10699 Search PubMed.
- A. M. Lifschitz, M. S. Rosen, C. M. McGuirk and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 7252–7261 CrossRef CAS PubMed.
- S. E. Angell, C. W. Rogers, Y. Zhang, M. O. Wolf and W. E. J. Jones, Coord. Chem. Rev., 2006, 250, 1829–1841 CrossRef CAS; C. W. Tate, A. deMello, A. D. Gee, S. Kealey, R. Vilar, A. J. P. White and N. J. Long, Dalton Trans., 2012, 41, 83–89 RSC; A. M. Lifschitz, C. M. Shade, A. M. Spokoyny, J. Mendez-Arroyo, C. L. Stern, A. A. Sarjeant and C. A. Mirkin, Inorg. Chem., 2013, 52, 5484–5492 CrossRef PubMed.
- J. C. Jeffrey and T. B. Rauchfuss, Inorg. Chem., 1979, 18, 2658–2666 CrossRef CAS.
- M. Bassetti, Eur. J. Inorg. Chem., 2006, 4473–4482 CrossRef CAS.
- A. Bader and E. Lindner, Coord. Chem. Rev., 1991, 108, 27–110 CrossRef CAS; R. Lindner, B. van den Bosch, M. Lutz, J. N. H. Reek and J. I. van der Vlugt, Organometallics, 2011, 30, 499–510 CrossRef; D. C. Babbini and V. M. Iluc, Organometallics, 2015, 34, 3141–3151 CrossRef.
- V. V. Grushin, Chem. Rev., 2006, 104, 1629–1662 CrossRef PubMed.
- I. Brassat, W. Keim, S. K. Illat, M. Mothrath, P. Mastrorilli, C. F. Nobile and G. P. Suranna, J. Mol. Catal. A: Chem., 2000, 157, 41–58 CrossRef CAS; A. Hamada and P. Braunstein, Inorg. Chem., 2009, 48, 1624–1637 CrossRef PubMed; B. P. Carrow and K. Nozaki, J. Am. Chem. Soc., 2012, 134, 8802–8805 CrossRef PubMed; N. D. Contrella, J. R. Sampson and R. F. Jordan, Organometallics, 2014, 33, 3546–3555 CrossRef; N. D. Contrella and R. F. Jordan, Organometallics, 2014, 33, 7199–7208 CrossRef; Y. Mitsushige, B. P. Carrow, S. Ito and K. Nozaki, Chem. Sci., 2016, 737–744 RSC.
- A. Cote and A. B. Charette, J. Org. Chem., 2005, 70, 10864–10867 CrossRef CAS PubMed.
- N. Nakagawa, E. J. Derrah, M. Schelwies, F. Rominger, O. Trapp and T. Schaub, Dalton Trans., 2016, 45, 6856–6865 RSC.
- A. A. Boezio, J. Pytkowicz, A. Cote and A. B. Charette, J. Am. Chem. Soc., 2003, 125, 14260–14261 CrossRef CAS PubMed.
- K. Skoch, I. Cisarova and P. Stepnicka, Chem. – Eur. J., 2015, 21, 15998–16004 CrossRef CAS PubMed.
- R. Weber, W. Keim, M. Mothrath, U. Englert and B. Ganter, Chem. Commun., 2000, 1419–1420 RSC.
- J. Bayardon, M. Maronnat, A. Langlois, Y. Rousselin, P. D. Harvey and S. Jugé, Organometallics, 2015, 34, 4340–4358 CrossRef CAS.
- K. H. Chung, C. M. So, S. M. Wong, C. H. Luk, Z. Zhou, C. P. Lau and F. Y. Kwong, Chem. Commun., 2012, 48, 1967–1969 RSC.
- M. Oestreich, Angew. Chem., Int. Ed., 2014, 53, 2282–2285 CrossRef CAS PubMed; A. Luquin, N. Castillo, E. Cerrada, F. L. Merchan, J. Garrido and M. Laguna, Eur. J. Org. Chem., 2016, 789–798 CrossRef.
- C. G. Oliveri, P. A. Ulmann, M. J. Wiester and C. A. Mirkin, Acc. Chem. Res., 2008, 41, 1618–1629 CrossRef CAS PubMed.
- C. W. Machan, A. M. Spokoyny, M. R. Jones, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2011, 133, 3023–3033 CrossRef CAS PubMed; J. Mendez-Arroyo, J. Barroso-Flores, A. M. Lifschitz, A. A. Sarjeant, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 10340–10348 CrossRef PubMed.
- Y. Ji, R. E. Plata, C. S. Regens, M. Hay, M. Schmidt, T. Razler, Y. Qiu, P. Geng, Y. Hsiao, T. Rosner, M. D. Eastgate and D. G. Blackmond, J. Am. Chem. Soc., 2015, 137, 13272–13281 CrossRef CAS PubMed.
- E. J. Derrah, S. Ladeira, G. Bouhadir, K. Miqueu and D. Bourissou, Chem. Commun., 2011, 47, 8611–8613 RSC.
- J. R. Shakirova, E. V. Grachova, A. A. Melekhova, D. V. Krupenya, V. V. Gurzhiy, A. J. Karttunen, I. O. Koshevoy, A. S. Melnikov and S. P. Tunik, Eur. J. Inorg. Chem., 2012, 4048–4056 CrossRef CAS; J. R. Shakirova, E. V. Grachova, A. S. Melnikov, V. Gurzhiy, S. P. Tunik, M. Haukka, T. Pakkanen and I. O. Koshevoy, Organometallics, 2013, 32, 4061–4069 CrossRef.
- T. M. Dau, J. R. Shakirova, A. J. Karttunen, E. V. Grachova, S. P. Tunik, A. S. Melnikov, T. A. Pakkanen and I. O. Koshevoy, Inorg. Chem., 2014, 53, 4705–4715 CrossRef PubMed.
- G. Chakkaradhari, A. A. Belyaev, A. J. Karttunen, V. Sivchik, S. P. Tunik and I. O. Koshevoy, Dalton Trans., 2015, 44, 13294–11330 RSC.
- S. Eller, B. Trettenbrein, D. Oberhuber, C. Strabler, R. Gutmann, W. E. van der Veer, M. Ruetz, H. Kopacka, D. Obendorf and P. Brüggeller, Inorg. Chem. Commun., 2012, 23, 41–45 CrossRef CAS PubMed; Z.-C. Fu, Q. Yin, Z.-F. Yao, C. Li and W.-F. Fu, J. Coord. Chem., 2015, 67, 3282–3294 CrossRef.
- G. Chakkaradhari, Y.-T. Chen, A. J. Karttunen, M. T. Dau, J. Jänis, S. P. Tunik, P.-T. Chou, M.-L. Ho and I. O. Koshevoy, Inorg. Chem., 2016, 55, 2174–2184 CrossRef CAS PubMed.
- N. J. Farrer, R. McDonald, T. Piga and J. S. McIndoe, Polyhedron, 2010, 29, 254–261 CrossRef CAS.
- X. Luo, H. Zhang, H. Duan, Q. Liu, L. Zhu, T. Zhang and A. Lei, Org. Lett., 2007, 9, 4571–4574 CrossRef CAS PubMed.
- G. Pilloni, B. Corain, M. Degano, B. Longato and G. Zanotti, J. Chem. Soc., Dalton Trans., 1993, 1777–1778 RSC; D. Saravanabharathi, M. Nethaji and A. G. Samuelson, Polyhedron, 2002, 21, 2793–2800 CrossRef CAS; Q.-Y. Cao, X. Gan and W.-F. Fu, Z. Anorg. Allg. Chem., 2007, 633, 176–179 CrossRef; W. Sun, Q. Zhang, L. Qin, Y. Cheng, Z. Xie, C. Lu and L. Wang, Eur. J. Inorg. Chem., 2010, 4009–4017 CrossRef.
- A. Sundararaman, L. N. Zakharov, A. L. Rheingold and F. Jäkle, Chem. Commun., 2005, 1708–1710 RSC; L. Maini, D. Braga, P. P. Mazzeo and B. Ventura, Dalton Trans., 2012, 41, 531–539 RSC; D. Yadav, R. K. Siwatch, S. Sinhababu, S. Karwasara, D. Singh, G. Rajaraman and S. Nagendran, Inorg. Chem., 2015, 54, 11067–11076 CrossRef CAS PubMed; E. Cariati, E. Lucenti, C. Botta, U. Giovanella, D. Marinotto and S. Righetto, Coord. Chem. Rev., 2016, 306, 566–614 CrossRef.
- J. Nitsch, F. Lacemon, A. Lorbach, A. Eichhorn, F. Cisnetti and A. Steffen, Chem. Commun., 2016, 52, 2932–2935 RSC.
- H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- P. Perez-Lourido, J. A. Garcia-Vazquez, J. Romero, A. Sousa, E. Block, K. P. Maresca and J. Zubieta, Inorg. Chem., 1999, 38, 538–544 CrossRef CAS PubMed; M. C. Gimeno, P. G. Jones, A. Laguna, C. Sarroca and M. D. Villacampa, Inorg. Chim. Acta, 2001, 316, 89–93 CrossRef; A. M. Kirillov, S. W. Wieczorek, A. Lis, M. F. C. Guedes da Silva, M. Florek, J. Król, Z. Staroniewicz, P. Smoleński and A. J. L. Pombeiro, Cryst. Growth Des., 2011, 11, 2711–2716 Search PubMed; D. A. Padron and K. K. Klausmeyer, Eur. J. Inorg. Chem., 2013, 299–308 CrossRef.
- A. N. Khlobystov, A. J. Blake, N. R. Champness, D. A. Lemenovskii, A. G. Majouga, N. V. Zyk and M. Schröder, Coord. Chem. Rev., 2001, 222, 155–192 CrossRef CAS.
- M. Munakata, L. P. Wu, G. L. Ning, T. Kuroda-Sowa, M. Maekawa, Y. Suenaga and N. Maeno, J. Am. Chem. Soc., 1999, 121, 4968–4976 CrossRef CAS.
- M. C. Gimeno, in Modern Supramolecular Gold Chemistry, ed. A. Laguna, Wiley-VCH, Weinhem, 2008, pp. 1–64 Search PubMed.
- M. A. Carvajal, J. J. Novoa and S. Alvarez, J. Am. Chem. Soc., 2004, 126, 1465–1477 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Chem. Soc. Rev., 2012, 41, 370–412 RSC.
- E. J. Derrah, C. Martin, S. Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Dalton Trans., 2012, 41, 14274–14280 RSC.
- I. O. Koshevoy, J. R. Shakirova, A. S. Melnikov, M. Haukka, S. P. Tunik and T. A. Pakkanen, Dalton Trans., 2011, 40, 7927–7933 RSC.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molins, J. Chem. Phys., 2002, 117, 5529–5542 CrossRef CAS.
- A. Tsuboyama, K. Kuge, M. Furugori, S. Okada, M. Hoshino and K. Ueno, Inorg. Chem., 2007, 46, 1992–2001 CrossRef CAS PubMed; M. Osawa, M. Hoshino, M. Hashimoto, I. Kawata, S. Igawa and M. Yashima, Dalton Trans., 2015, 44, 8369–8378 RSC.
- R. Venkateswaran, M. S. Balakrishna, S. M. Mobin and H. M. Tuononen, Inorg. Chem., 2007, 46, 6535–6541 CrossRef CAS PubMed.
- N. Kobayashi, H. Higashimura and T. Kaikoh, Japan Pat., WO2011152358A1, 2011 Search PubMed; N. Kobayashi, H. Higashimura and T. Kaikoh, Japan Pat., WO2012144530A1, 2012 Search PubMed.
- J. Zank, A. Schier and H. Schmidbaur, J. Chem. Soc., Dalton Trans., 1999, 415–420 RSC.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622–2652 CrossRef CAS; M. Osawa, I. Kawata, R. Ishii, S. Igawa, M. Hashimoto and M. Hoshino, J. Mater. Chem. C, 2013, 1, 4375–4383 RSC; C. L. Linfoot, M. J. Leitl, P. Richardson, A. F. Rausch, O. Chepelin, F. J. White, H. Yersin and N. Robertson, Inorg. Chem., 2014, 53, 10854–10861 CrossRef PubMed; T. Hofbeck, U. Monkowius and H. Yersin, J. Am. Chem. Soc., 2015, 137, 399–404 CrossRef PubMed; A. Kobayashi, T. Hasegawa, M. Yoshida and M. Kato, Inorg. Chem., 2016, 55, 1978–1985 CrossRef PubMed.
- K. Matsumoto, T. Shindo, N. Mukasa, T. Tsukuda and T. Tsubomura, Inorg. Chem., 2010, 49, 805–814 CrossRef CAS PubMed.
- C. T. Cunningham, J. J. Moore, K. L. H. Cunningham, P. E. Fanwick and D. R. McMillin, Inorg. Chem., 2000, 39, 3638–3644 CrossRef CAS PubMed; A. Lavie-Cambot, M. Cantuel, Y. Leydet, G. Jonusauskas, D. M. Bassania and N. D. McClenaghan, Coord. Chem. Rev., 2008, 252, 2572–2584 CrossRef.
- R. Uson, A. Laguna and M. Laguna, Inorg. Synth., 1989, 26, 85–91 CrossRef CAS.
- APEX2 - Software Suite for Crystallographic Programs, Bruker AXS, Inc., Madison, WI, USA, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- G. M. Sheldrick, SADABS-2008/1 - Bruker AXS Area Detector Scaling and Absorption Correction, Bruker AXS, Madison, Wisconsin, USA, 2008 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1.17 edn, 2013 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396–1396 CrossRef CAS.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, Oxford, UK, 1990 Search PubMed.
- T. A. Keith, AIMAll, TK Gristmill Software, Overland Park KS, USA, 12.06.03 edn, 2003 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of the optimized geometries, and additional NMR, ESI-MS, computational and crystallographic data for compounds 1–12, 15, 16. CCDC 1484959–1484962, 1484966–1484970, 1484973, 1484974 and 1484978–1484980. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02435a |
| This journal is © The Royal Society of Chemistry 2016 |