
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of Bi2Te3 and (BixSb1−x)2Te3 nanoparticles using the novel IL [C4mim]3[Bi3I12]†
M.
Loor
a,
G.
Bendt
a,
U.
Hagemann
b,
C.
Wölper
a,
W.
Assenmacher
c and
S.
Schulz
 *a
*a
aInstitute of Inorganic Chemistry and Center for Nanointegration Duisburg-Essen (CENIDE), University of Duisburg-Essen, Universitätsstr. 5-7, D-45117 Essen, Germany. E-mail: stephan.schulz@uni-due.de
bInterdisciplinary Center for Analytics on the Nanoscale (ICAN), NETZ, Carl-Benz-Str. 199, 47047 Duisburg, Germany
cInstitute of Inorganic Chemistry, University of Bonn, Römerstr. 164, D-53117 Bonn, Germany
First published on 1st September 2016
Abstract
The novel Bi-containing reactive ionic liquid [C4mim]3[Bi3I12], which was synthesized in quantitative yield by equimolar reaction of BiI3 and [C4mim]I, was used as a novel Bi-source for the ionothermal synthesis of Bi2Te3 nanoparticles by reaction with (Et3Si)2Te in the ionic liquid [C4mim]I. The solid state structure of [C4mim]3[Bi3I12] was determined by single crystal X-ray diffraction. In addition, the ionothermal synthesis of the single source precursor (Et2Sb)2Te and [C4mim]3[Bi3I12] yielded the ternary (BixSb1−x)2Te3 (x = 0.25, 0.5, 0.75) nanoparticles. The chemical composition and phase purity of the tetradymite-type materials were determined by EDX and XRD and the surface composition of the nanoparticles was further investigated by IR and XPS. In addition, the morphology of the nanoparticles was investigated by SEM and TEM.
Introduction
The synthesis of highly efficient thermoelectric materials for technical application as thermoelectric generators TEG, which allow the conversion of waste thermal energy into electric energy,1 has attracted considerable attention in recent years.2 Even though radioisotope thermoelectric generators (RTG), which were used in specific technical applications such as energy supply of satellites and space probes,3 have been known since the 1950's, their low thermoelectric efficiency, which is given by the dimensionless figure of merit ZT = (α2σ/λ)T (α = Seebeck coefficient, σ = specific electrical conductivity, λ = thermal conductivity = sum of electronic λel and lattice λla contribution, T = absolute temperature [K]), inhibited their broad technical application.Semiconducting materials with heavy elements are very promising materials due to their high effective masses. Among many different systems, which have been investigated in the last decades, Sb2Te3, Bi2Te3 and the solid ternary solutions (SbxBi1−x)2Te3 are still some of the most effective materials for technical applications operating near room temperature. These materials show high electrical conductivities and high Seebeck coefficients combined with glass-like low thermal conductivities.4 They are isostructural and crystallize in the tetradymite-type structure, with the Te atoms building a closed packing structure perpendicular to the c-axis in R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m:H and the stacking sequence chh in Jagodzinski-symbols. The stacking sequence (AγB□AγBαC□BαCβA□Cβ) results in the formation of double layers of edge-sharing Bi(Sb)Te6 octahedra consisting of 5 atom layers (quintuple layer) with the sequence Te1–Bi(Sb)–Te2–Bi(Sb)–Te1 and the composition (Bi(Sb))2Te3. The bonding inside the quintuple layer can be described as mixed covalent-ionic, while only weak van der Waals bonding is observed between the quintuple layers.4
m:H and the stacking sequence chh in Jagodzinski-symbols. The stacking sequence (AγB□AγBαC□BαCβA□Cβ) results in the formation of double layers of edge-sharing Bi(Sb)Te6 octahedra consisting of 5 atom layers (quintuple layer) with the sequence Te1–Bi(Sb)–Te2–Bi(Sb)–Te1 and the composition (Bi(Sb))2Te3. The bonding inside the quintuple layer can be described as mixed covalent-ionic, while only weak van der Waals bonding is observed between the quintuple layers.4
Nanostructuring has been identified in the late nineties of the last century as a promising method for increasing the thermoelectric efficiency (zT) of a given material,5 resulting from the decreased thermal conductivity of the material due to an efficient phonon scattering at boundaries and interfaces, and a simultaneously increased Seebeck coefficient due to both quantum confinement effects and the modification of the electronic band structure.6 In addition, ternary solid solutions (BixSb1−x)2Te3 showed enhanced zT values compared to the corresponding binary materials (Bi2Te3, Sb2Te3) as was demonstrated for (BixSb1−x)2Te3 single crystals (z = 3.2 × 10−3 K−1 at room temperature7) and p-type (Bi0.2Sb0.8)2Te3 nanocomposites (z of 3.52 × 10−3 K−1 at room temperature8). Poudel et al. reported record-high zT values of 1.4 at 373 K for bulk (BixSb1−x)2Te3 with embedded nanostructures2a and Xie et al. observed a maximum zT value of 1.56 at 300 K for the p-type (Bi0.26Sb0.74)2Te3 material, which is roughly a 50% improvement compared to commercial Bi2Te3.9
As a consequence, the synthesis of nanoparticles and thin films of tetradymite-type materials has received increasing interest. Unfortunately, their strong tendency to form antisite defects, which refers to the occupation of Te sites by Bi atoms or Bi sites by Te atoms and, as a consequence, the formation of either p- or n-type doped materials, often diminish their thermoelectric performance.10 In addition, the facile incorporation of excess bismuth into Bi2Te3 yield Bi-rich material phases, which typically adopt sandwich-like structures of the general form (Bi2)n(Bi2Te3)m, in which the quintuple layers are separated by Bi-bilayers11 as can be observed in tsumoite BiTe, pilsenite Bi4Te3 and hedleyite Bi7Te3, respectively.
The synthesis of bulk and nanostructured p-type (BixSb1−x)2Te3 has been widely investigated.2a,12 Bismuth-rich Bi2Te3 and (SbxBi1−x)2Te3 particles were obtained from the reduction of bismuth/antimony acetate with oleylamine (OA) in dodecanethiole and subsequent reaction with trioctyltellurophosphorane (TOPTe)13 as well as by reaction of TOPTe and bismuth oleate.14 In addition, hexagonal Bi2Te3 plates were obtained upon thermolysis of bismuth nitrate and TOPTe in octadecene and oleic acid15 and by reaction of BiCl3 and TOPTe in thioglycolic acid in a microwave assisted synthesis.16 The resulting material, which was sub-atomically doped with sulfur, showed a remarkably high zT value (1.1). In addition, Reid et al. recently demonstrated that [BiCl3(TenBu2)3] is a suitable single source precursor for the MOCVD (metal organic chemical vapor deposition) deposition of high-quality Bi2Te3 thin films.17 In addition to the widely applied Te source TOPTe, bis(triethylsilyl)tellurane (Et3Si)2Te has been demonstrated to be a promising low-temperature Te-precursor. Sb2Te3 and Bi2Te3 thin films were deposited by ALD process (atomic layer deposition)18 and by low-temperature MOCVD process.19 In addition, (Et3Si)2Te was successfully applied for the wet chemical synthesis of multiple Bi–Te phases including Bi2Te3.20
Due to our general interest in thermoelectric materials, we started only recently to investigate both gas phase deposition of thin films using ALD18b,c and MOCVD processes19c,d as well as wet chemical approaches for the synthesis of Sb2Te3 and Bi2Te3 nanoparticles in organic solvents20,21 and in ionic liquids (ILs). ILs were shown to be very promising solvents for the synthesis of Sb2Te3 nanoparticles with very high zT values of up to 1.5.22 However, the limited thermal stability of metal organic bismuth precursors such as bismuth amides or bismuth alkyl compounds often resulted in the formation of elemental bismuth or Bi-rich material phases.20 Therefore, we became interested in the development of alternate Bi precursors, which should be thermally stable to avoid simple thermal decomposition but whose reactivity should still be high enough to produce the desired materials at rather low temperatures.
We herein report on the synthesis and solid state structure of the novel reactive IL [C4mim]3[Bi3I12] and its promising potential to serve as alternate Bi source for the synthesis of phase-pure binary (Bi2Te3) and – together with the single source precursor (Et2Sb)2Te – ternary ([BixSb1−x]2Te3) bismuth telluride nanoparticles in an IL-based wet chemical approach. The composition, phase purity and morphology of the resulting tetradymite-type nanoparticles were investigated by IR, EDX, XPS, XRD, SEM and TEM.
Results and discussion
Our previous studies on the synthesis of Bi2Te3 nanoparticles in organic solvents showed a strong tendency for the formation of Bi-rich materials, most likely resulting from the thermal lability of the Bi precursor. Therefore, we became interested in a more stable Bi precursor and began to investigate the use of complex bismuth halide anions such as [Bi3I12]3+, which have been synthesized in the past.23–26 Hence, we synthesized the novel reactive IL [C4mim]3[Bi3I12] 1 in high yield by reaction of [C4mim]I and BiI3 (Scheme 1).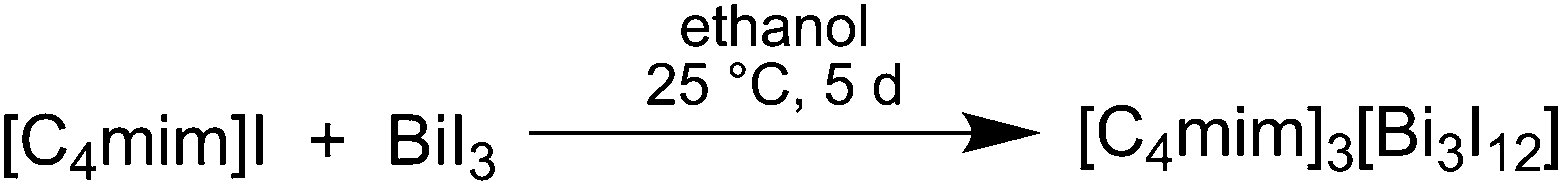 | ||
| Scheme 1 Synthesis of [C4mim]3[Bi3I12] 1. | ||
1, which shows a melting point of 98 °C, was obtained as a bright yellow powder at ambient temperature. The yellow color of [C4mim]3[Bi3I12] 1 was found to intensify upon cooling to −196 °C; whereas it turns red and finally becomes metallic-like, almost black-purple upon heating to 250 °C (Fig. S8†). Both processes are fully reversible. Even though the reason for this thermochromic behavior is not yet clear, we believe that the color change upon cooling and heating results from a phase transition of 1. Phase transition reactions are well known for bismuth(III)iodides, for which more than 60 compounds have been structurally characterized and which show a large structural diversity. To date, almost 20 different structural types have been reported.27 The structures were found to largely depend on the specific cation. A templating effect of the cations has been identified as the major structure determining factor that induces the formation of various metal–halide networks. In the case of (2-MIm)BiI4, a change of the electron lone pair activity of the bismuth atom was also found to play a major role in the phase transition mechanism.27b A DSC study revealed an exothermic process around −50 °C and the melting point was also observed (Fig. S9†).
1 is highly soluble in strong polar, aprotic solvents such as acetonitrile and DMSO and barely soluble in less polar solvents, e.g. ethanol and methanol, while it is insoluble in non-polar solvents such as pentane and hexane as well as in water. In comparison to the NMR spectra of [C4mim]I the aromatic signals of 1 are high-field shifted by 0.2 ppm in the 1H NMR and by 4 ppm in the 13C NMR spectra, while the resonances due to the aliphatic groups show a low-field shift by the same amounts (Fig. S1 and S2†), indicating stronger electronic interactions between the positively charged aromatic system with the [Bi3I12]−3 anion compared to the iodine anion, hence resulting in an increased electron density in the aromatic system but a decreased electron density in the aliphatic chain. The IR spectrum of 1 shows the typical absorption bands for the [C4mim]+ cation and additional sharp absorption bands between 1150 cm−1 and 930 cm−1, which can be ascribed to the Bi–I-stretching vibrations (Fig. S3†).‡
Single crystals of 1 were obtained by re-crystallization from ethanolic solution. [C4mim]3[Bi3I12] crystallises in the monoclinic space group P21/c. The asymmetric unit comprises three cations at general positions and two anions at special positions (centre of inversion) as was previously observed for the majority of the known [Bi3I12] polyanions. The two independent anions lead to an occupation of all vertices and face-centres of the unit cell similar to a cubic closest packing of spheres. However, their anisotropic shape – they are best described as a linear arrangement of three face-sharing BiI6 octahedra – prevents cubic symmetry. The cations fill the voids where the ones labelled X1x and X3x can be found in the pseudo-tetrahedral gap, while the X2x-labelled and its symmetry equivalent share the pseudo-octahedral gap.
Bond lengths and angles of the cations show typical values. The conformation of the butyl group of the cations differs depending on the space available to fill (Cx5–Cx6–Cx7–Cx8: −170(2)° (x = 1), 66(2)° (x = 2), 175(2)° (x = 3)). The range of Bi–I bond lengths matches well with those previously reported by Carmalt et al. and others for [Bi3I12] and [Bi4I16] polyanions.23–25 These polyanions can be described as central BiI63− octahedral units with neutral BiI3 moieties capping two of its trans oriented faces. The independent anions are roughly perpendicular to one another (Bi11⋯Bi12/Bi21⋯Bi22 74.22(1)°) and parallel to the (100) plane. Inter-halide interactions lead to the formation of layers parallel to this plane (Table S1†). These layers are interconnected by non-classical hydrogen bonds between anion and cations (Table S2†).26
Synthesis of Bi2Te3 nanoparticles
Thermolysis of (Et3Si)2Te and [C4mim]3[Bi3I12] in a solution of 10 mL of [C4mim]I at 150 °C yielded a black precipitate, which was isolated by centrifugation and purified by repeatedly washing with acetonitrile and hexane (3x). The elemental composition of the material as determined by EDX proved the formation of slightly Te-rich Bi2Te3 materials (Table 1). NMR spectroscopic studies (1H, 13C) showed that the thermolysis not only occurred with the expected formation of Et3SiI but also with formation of hexaethyldisilane Si2Et6 (Fig. S4a–c†), whose formation points to a homolytic cleavage of the weak Te–Si bond. As formed Et3Si radicals then easily recombine to Si2Et6. These findings also explain the presence of small amounts of elemental Te in the resulting material, which can be removed by washing the material with TOP as was previously demonstrated.20 In contrast, thermolysis of (Et3Si)2Te and [C4mim]3[Bi3I12] in a solution of 10 mL [C4mim]I and 3 mL oleylamine (OA) at 150 °C yielded pure Bi2Te3 nanoparticles according to EDX studies (Scheme 2). We recently demonstrated that OA reacts with (Et3Si)2Te at ambient temperature with formation of silylamine (Et3SiOA) and tellurium polyanions,28 which then consequently react with [C4mim]3[Bi3I12] with the formation of Bi2Te3.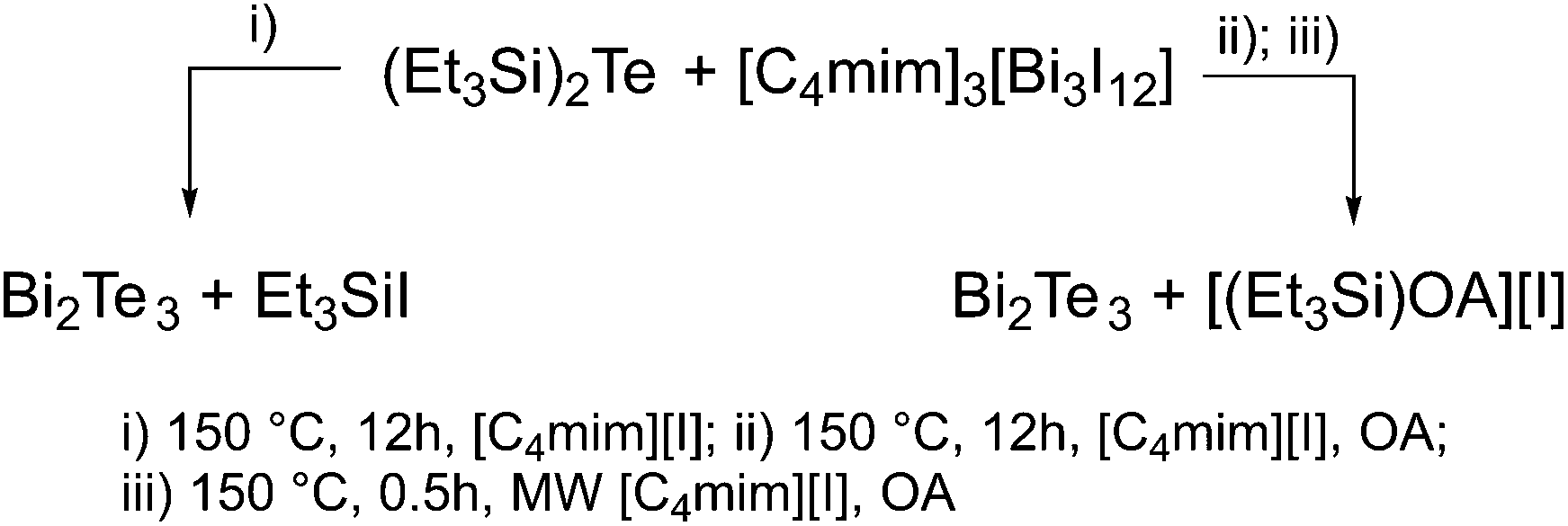 | ||
| Scheme 2 Synthesis of Bi2Te3 nanoparticles. | ||
We also investigated a microwave-assisted synthesis, since this technique was found to be very promising for the generation of pure Sb2Te3 nanoparticles.22 A solution of (Et3Si)2Te and [C4mim]3[Bi3I12] in 10 mL of [C4mim]I and 3 mL of OA was heated to 150 °C by microwave irradiation and the resulting particles were treated as mentioned before. However, slightly Te-rich materials were formed according to EDX studies (Table 1).
We further investigated the presence of any kind of organic molecules, i.e. solvent molecules or capping agents, on the surface of the materials as-obtained from thermolysis reactions of the precursors in [C4mim]I and OA. IR spectroscopy clearly demonstrated that the resulting Bi2Te3 nanoparticles do not contain OA (capping agent) on the surface as can clearly be seen by comparing the IR spectra of the nanoparticles and that of pure OA (Fig. 2). In addition, neither the IL nor acetonitrile (washing solvent) bind to the nanoparticles. According to these IR studies, the nanoparticles are considered to be almost capping-agent free.
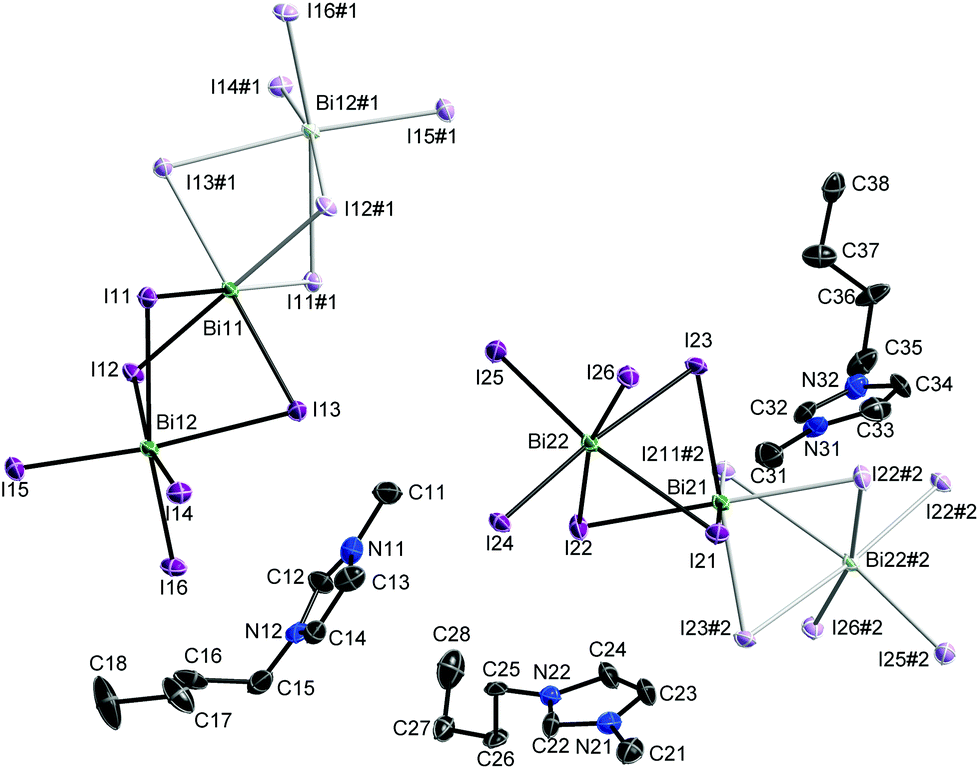 | ||
| Fig. 1 Solid state structure of [C4mim]3[Bi3I12] 1. Probability ellipsoids are displayed at 50% probability levels and hydrogen atoms are omitted for clarity. Symmetry generated parts are displayed in pale colours. #1 −x, 2 − y, −z; #2 1 − x, 1 − y, −z. | ||
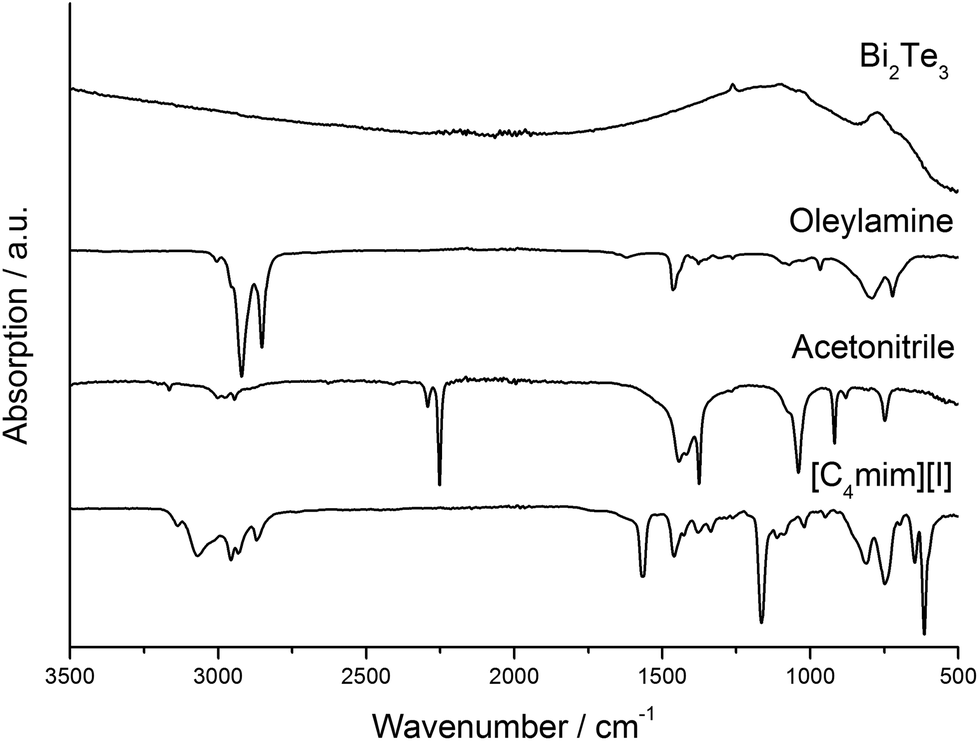 | ||
| Fig. 2 IR spectra of the Bi2Te3 nanoparticles synthesized with OA, pure OA, acetonitrile, and [C4mim]I. | ||
The resulting materials were investigated by powder X-ray diffraction (PXRD), which clearly proved the formation of Bi2Te3 in all experiments (Fig. 3). The peaks can be indexed on the basis of phase-pure Bi2Te3 (PDF 15-863), but the materials obtained in ionothermal syntheses in [C4mim]I in the absence of oleylamine as well as in microwave assisted reactions in [C4mim]I and OA showed additional reflexes at 23°, 38°, and 44°, respectively, which indicate the presence of small impurities of elemental tellurium. In contrast, the ionothermal synthesis in a mixture of [C4mim]I and oleylamine yielded phase pure Bi2Te3.
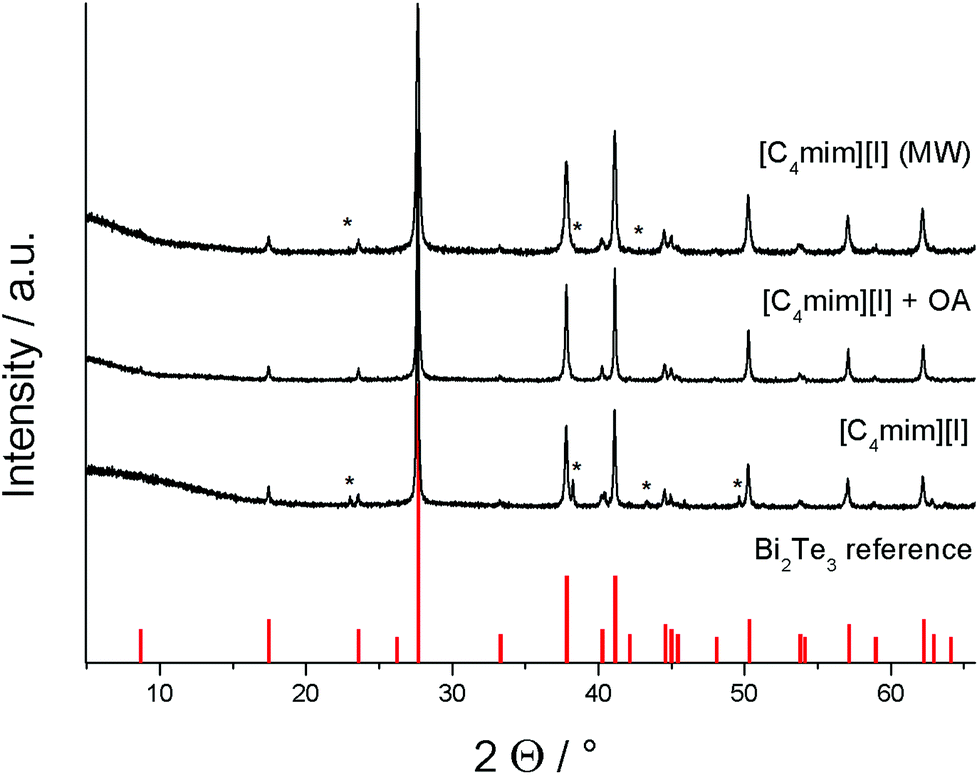 | ||
| Fig. 3 PXRD of the Bi2Te3 ionothermal and microwave (MW) approaches and reference for Bi2Te3 (PDF 15-863). Peaks marked with an * correspond to elemental Te.29 | ||
The lattice parameters for the nanosized Bi2Te3 particles, which were refined to a = 4.388(8) Å, c = 30.517(7) Å and V = 509.0(7) Å3, are in good agreement with the values reported for Bi2Te3 (PDF 15-863). The broadening of the full width half maximum (FWHM) of the peaks indicate an average crystal size of roughly 55 nm (Table 2).
We then investigated the stability of the Bi2Te3 nanoparticles toward oxidation reactions upon exposure to air by X-ray photoelectron spectroscopy (XPS).
A freshly prepared Bi2Te3 sample was shown to contain an oxygen-free surface. After expose to air for different period of times, the materials were again investigated by XPS. The changes of the Bi 4f and Te 3d signals are depicted in Fig. 4. It can clearly be seen, that the particles are fairly stable for an hour, showing only the metallic peaks at 157.2 eV for Bi 4f and 572 eV for Te 3d. These values agree with the literature values for Bi2Te3.30 After one day of exposure to air additional peaks due to the presence of both bismuth oxide at 159 eV and tellurium oxide at 576.6 eV binding energy can be observed. Oxide intensities increase dramatically after 1 month, so that more than 50% of the surface metal atoms (Bi 53%, Te 64%) are oxidised (Table 3). These findings agree with the results very recently observed for surface oxidation reactions of binary and ternary bismuth chalcogenides, in which Bi2Te3 and Bi2Te2Se were found to easily oxidise upon expose to air while Bi2Se3 was significantly more stable toward oxidation.31 As a consequence, the nanoparticles have to be stored and handled under inert gas conditions to avoid surface oxidation reactions.
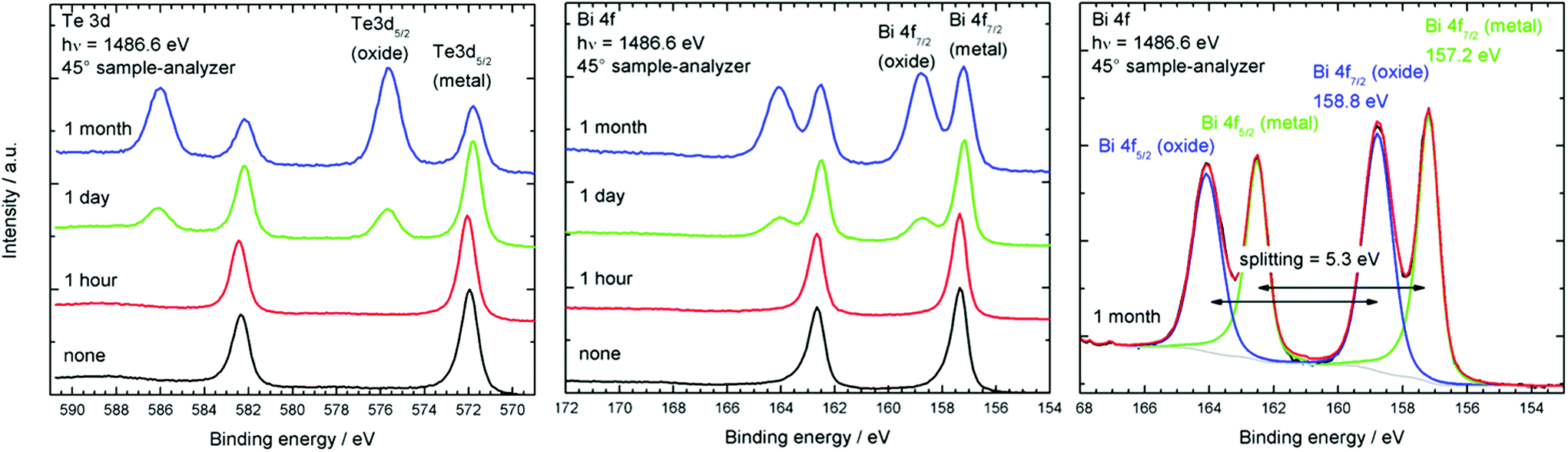 | ||
| Fig. 4 Change of the Bi 4f and Te 3d XPS signals of Bi2Te3 nanoparticles upon exposure to air. | ||
| Exposure to air | Bi-oxide/% | Te-oxide/% |
|---|---|---|
| None | 0 | 0 |
| 1 hour | 0 | 0 |
| 1 day | 22 | 27 |
| 1 month | 53 | 65 |
According to SEM studies, the Bi2Te3 materials as-obtained in a solution of [C4mim]I and oleylamine (OA) contain Bi2Te3 nanoplates with an average size of about 50 nm and a more or less hexagonal shape (Fig. 5c). These nanoplates are highly agglomerated as was expected due to the lack of any capping agents on the surface as was demonstrated by IR spectroscopy. Hence, TEM bright field images show plate-like particles with a size ranging from 20 to 100 nm (Fig. 5a and b), which are heavily inter-grown, reminiscent of sintered powder. The particles show diffraction contrast and are thus considered as crystalline. Selected area electron diffraction (SAED; Fig. 5d) taken from a crystal covered region gives a powder ring pattern with d-values as expected for Bi2Te3.
 | ||
| Fig. 5 TEM bright field images (a, b); SEM image (c) and contrast inverted selected area electron diffraction pattern of Bi2Te3 (Philips CM30 T @300 keV) and simulated ring pattern as overlay SAED (d) of Bi2Te3 nanoparticles.32 | ||
The synthesis of phase-pure Bi2Te3 nanoparticles proves the promising potential of the novel reactive IL [C4mim]3[Bi3I12] 1 to serve as a Bi-source in materials synthesis. The formation of Bi-rich materials such as BiTe or Bi4Te3, which was often observed in reactions of metal organic precursors as a result of the often thermally rather unstable Bi precursors, is completely avoided in the reaction with the reactive Te-source (Et3Si)2Te, indicating a constant release of Bi under the specific reaction conditions.
Synthesis of ternary (BixSb1−x)2Te3 nanoparticles
After the successful synthesis of phase-pure Bi2Te3 nanoparticles, we became interested in the synthesis of ternary solid solutions of (BixSb1−x)2Te3. Since the thermolysis of the single source precursor (Et2Sb)2Te was previously established for the synthesis of highly stoichiometric Sb2Te3 nanoplates,21,22 we investigated its thermolysis in the presence of different amounts of the reactive IL [C4mim]3[Bi3I12] 1 in 10 mL of [C4mim]I. After thermolysis at 150 °C, the resulting black solid was isolated by centrifugation and purified by repeatedly washing with acetonitrile (Scheme 3). | ||
| Scheme 3 Thermolysis of the single source precursor (Et2Sb)2Te; reaction of (Et2Sb)2Te and [C4mim]3[Bi3I12] 1 for the synthesis of (BixSb1−x)2Te3 nanoparticles. | ||
Ternary materials of the general type (BixSb1−x)2Te3 were obtained in all cases, even though they showed a slight tellurium deficit. In addition, a general trend for bismuth-rich particles was observed (Table 4). The reactions probably proceed with the initial formation of small Sb2Te3 nanoparticles, in which the antimony content is consequently substituted by bismuth successively provided from the reactive IL 1.
| x | Bi (%) | Sb (%) | Te (%) |
|---|---|---|---|
| 0.25 | 12.4 ± 0.6 | 28.3 ± 0.9 | 59.3 ± 1.8 |
| 0.5 | 22.5 ± 0.9 | 19.3 ± 0.5 | 58.2 ± 1.5 |
| 0.75 | 30.1 ± 1.2 | 11.3 ± 0.5 | 58.7 ± 1.6 |
PXRD studies proved the formation of phase pure ternary materials (BixSb1−x)2Te3 (Fig. 6). The successful substitution of bismuth by antimony in the tetradymite-type material can be monitored by a slight shift of the reflections toward higher angles with increasing antimony concentration, resulting in decreasing lattice parameters as was expected due to the replacement of the large bismuth atoms by smaller antimony atoms. This is confirmed by the refinement of the lattice parameters, which show a monotonically decreasing cell volume (Table 5).
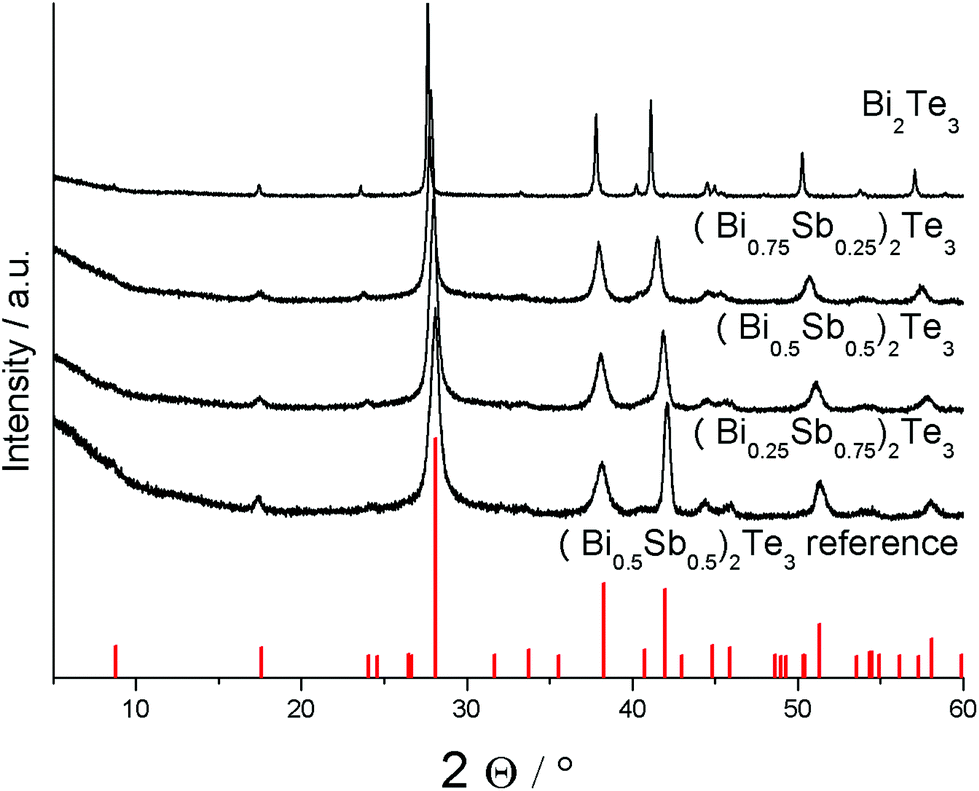 | ||
| Fig. 6 Powder X-ray diffractogram of (BixSb1−x)2Te3 (x = 0.25, 0.5, 0.75, 1) nanoparticles and reference for (Bi0.5Sb0.5)2Te3 (PDF 072-1853).33 | ||
| x | a [Å] | c [Å] | V [Å3] | Sizea [nm] |
|---|---|---|---|---|
| a The determination of the crystal size of anisotropic particles such as thin plates by using the Scherrer equation is always problematic. Values should therefore be taken with care. | ||||
| 1 | 4.388(8) | 30.517(7) | 509.0(7) | 38.3(9) |
| 0.75 | 4.360(6) | 30.449(2) | 501.4(1) | 33.6(6) |
| 0.5 | 4.319(9) | 30.496(2) | 492.8(6) | 25.7(2) |
| 0.25 | 4.291(9) | 30.580(4) | 487.8(3) | 22.9(3) |
Compared to the phase-pure Bi2Te3 nanoplates, the XRDs of the as-prepared ternary solid solutions (BixSb1−x)2Te3 showed increasing peak broadening, clearly indicating a steadily decreasing size of the crystalline domains with increasing bismuth concentration (Fig. 6). In addition, anisotropic peak broadening becomes more dominant, underlining the preferential growth along the ab-plane perpendicular to the c-axis, which manifests itself in a relatively sharp (110) reflection at 41.12° (2θ) compared to the other reflections, i.e. the neighboring (1010) reflex at 37.8°.
As was observed for the Bi2Te3 nanoplates, the as-prepared (BixSb1−x)2Te3 materials also consist of hexagonal nanoplates, which again are highly agglomerated. The average size of the nanoparticles according to SEM studies ranges from roughly 50 nm to 100 nm (Fig. 7).
 | ||
| Fig. 7 SEM image of (Bi0,5Sb0,5)2Te3. | ||
TEM studies confirm the hexagonal plate-like shape of the (BixSb1−x)2Te3 materials with the growth direction perpendicular to the c-axis and a broad size distribution. Although some of the larger hexagonal plates consist of an intergrowth of several crystalline domains, crystals from 10 to 200 nm in size are present in each of the (BixSb1−x)2Te3 materials (Fig. 8a–c). EDX spot analyses in the STEM mode on single crystals (Table S3†) confirm the composition corresponding to the sum formula (BixSb1−x)2Te3 with x = 0.25, 0.5 and 0.75 and agree with those summarized in Table 4, which were obtained from big agglomerates.
 | ||
| Fig. 8 TEM bright field images of (BixSb1−x)2Te3 with (a) x = 0.25; (b) x = 0.5; (c) x = 0.75. (d) Electron diffraction pattern of a (Bi0.25Sb0.75)2Te3 crystal along c*. | ||
Fig. 8d shows an electron diffraction (ED) pattern of (Bi0.25Sb0.75)2Te3 in [001] orientation. The d-values of the {100} and {030} reflections are 2.19 Å and 1.22 Å, respectively, and agree with the lattice parameters from PXRD and with the reflection conditions for space group Rm:H. The presence of Bi and Sb in small volumes proven by spot EDX and the absence of any super-structure reflections in the ED-patterns reveal the random distribution of antimony and bismuth on the cation positions of the tetradymite-type structure.
Unfortunately, HRTEM studies of (BixSb1−x)2Te3 perpendicular to the c-axis, which enables the depiction of the stacking of the quintuple Bi2Te3 layers, was difficult to perform since the plate-like crystals were predominantly found lying in [001] orientation or they were most often too thick for HRTEM, when the orientation was perpendicular to the c-axis. Fig. 9 shows a HRTEM image of (Bi0.75Sb0.25)2Te3 in [100] orientation. Although the atom columns are not well resolved, the stacking of the quintuple layers of 10.2 Å is found in all areas. This indicates that no additional Bi-bilayers are present and confirms the Bi2Te3-type of structure.
 | ||
| Fig. 9 (a) HRTEM bright field image of (Bi0.75Sb0.25)2Te3 in [100] orientation; (b): Fourier filtered cutout with ball and stick model of (BiSb)2Te3 as overlay (green: Te, orange: Bi, Sb). | ||
Experimental
All synthetic procedures including the synthesis of the IL as well as all thermolysis experiments were performed under inert gas conditions (Ar atmosphere) in a glovebox or using standard Schlenk techniques. Acetonitrile (99.9+%, Extra Dry, Acros organics), ethanol (J. T. Baker 98%) and 1-iodobutane (99%, Acros organics) were commercially available and used as received, while ethyl acetate (J. T. Baker) was redistilled prior to use. Solvents were carefully dried according to standard procedures and degassed prior to use. (Et2Sb)2Te was prepared according to the literature method.34 Melting points were determined with an Electrothermal IA9300 digital melting point instrument from ThermoFisher Scientific.Synthesis of 1-butyl-3-methyl-imidazolium iodide [C4mim]I
22 mL (0.276 mol) 1-methylimidazole (99%, Sigma Aldrich) was dissolved in 100 mL of acetonitrile and 1-iodobutane (0.308 mol, 35 mL) was added dropwise in the dark at 0 °C. The solution was stirred at ambient temperature for 12 h and then all volatiles were removed under dynamic vacuum. The resulting residue was washed with 150 mL of ethyl acetate. After removal of the solvent, the remaining yellowish oil was dried for 72 h under dynamic vacuum at 50 °C.Yield: 59.47 g (81%). 1H NMR (300 MHz, 25 °C, DMSO-d6): δ (ppm) 9.14 (s, 1H), 7.76 (dt, 1JH–H = 13.5 Hz, 2JH–H = 1.7 Hz, 2H), 4.19 (t, 3JH–H = 7.2 Hz, 2H), 3.88 (s, 3H), 1.79 (dt, 1JH–H = 14.8 Hz, 2JH–H = 7.5 Hz, 2H), 1.29 (m, 2H), 0.93 (t, 3JH–H = 7.3 Hz, 3H).
Synthesis of [C4mim]3[Bi3I12] 1
14.91 g (0.561 mol) [C4mim]I and 27.57 g (0.468 mol) BiI3 were added to 500 mL ethanol and the resulting suspension was stirred at ambient temperature for 5 d. The resulting bright yellow solid was separated via filtration, washed with 100 mL of ethanol and carefully dried for 72 h under dynamic vacuum at ambient temperature.Yield: 30.87 g (77.16%). Melting point: 98 °C. Elemental analysis (EDX): Bi: 19.79 ± 0.95 at%, I: 80.21 ± 1.74 at%. 1H NMR (300 MHz, 25 °C, DMSO-d6): δ (ppm) 9.11 (s, 1H), 7.73 (dt, 1JH–H = 20.2 Hz, 2JH–H = 1.8 Hz, 2H), 4.16 (t, 3JH–H = 7.2 Hz, 2H), 3.85 (s, 3H), 1.76 (m, 2H), 1.29 (m, 2H), 0.93 (t, 3JH–H = 7.3 Hz, 3H).
Synthesis of Bi2Te3 nanoparticles
3.4 g (3.9 mmol) of 1 was dissolved in 10 mL of [C4mim]I or in a mixture of 10 mL of [C4mim]I and 3 mL of oleylamine in a centrifuge tube and the mixture was stirred for 1 h at 100 °C. 1.24 g (3.47 mmol) (Et3Si)2Te were then added and the resulting black suspension was stirred for 12 h at 150 °C. The resulting colloidal solutions were centrifuged (2500 rpm), washed with 6 × 15 mL acetonitrile and 2 × 15 mL hexane, and dried at room temperature under vacuum.Synthesis of (BixSbi1−x)2Te3 nanoparticles
In a centrifuge tube, an appropriate amount (see Table 6) of [C4mim][Bi3I12] was dissolved in 2 mL [C4mim]I and stirred for 1 h at 100 °C. 137 μl (1.44 mmol) (Et2Sb)2Te were then added and the solution was stirred for 12 hours at 150 °C. The resulting black particles were washed with 6 × 15 mL acetonitrile.| x | m 1 [mg] | n 1 [mmol ] |
|---|---|---|
| 0.25 | 103 | 0.04 |
| 0.5 | 205 | 0.08 |
| 0.75 | 308 | 0.12 |
Single crystal X-ray diffraction
Crystallographic data of 1 were collected on a Bruker D8 Kappa APEX2 diffractometer (MoKα radiation, λ = 0.71073 Å) at 100(1) K: [C24H45Bi3I12N6], M = 2567.40, red crystal, (0.145 × 0.094 × 0.047 mm); monoclinic, space group P21/c; a = 17.5900(9) Å, b = 15.9186(7) Å, c = 19.3836(9) Å; α = 90°, β = 96.503(3)°, γ = 90°, V = 5392.6(4) Å3; Z = 4; μ = 16.658 mm−1; ρcalc = 3.162 g cm−3; 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 677 reflexes (θmax = 28.408°), 13458 unique (Rint = 0.1193); 415 parameters; largest max./min. in the final difference Fourier synthesis 2.683 e Å−3/−2.694 e Å−3; max./min. transmission 0.41/0.75; R1 = 0.0423 (I > 2σ(I)), wR2 = 0.0864 (all data). The solid-state structure of 1 is shown in Fig. 1. The structure was solved by direct methods (SHELXS-97)35 and refined anisotropically by full-matrix least-squares on F2 (SHELXL-2014).36 Absorption corrections were performed numerical based on indexed faces (Bruker AXS APEX2). Hydrogen atoms were refined using a riding model or rigid methyl groups.
677 reflexes (θmax = 28.408°), 13458 unique (Rint = 0.1193); 415 parameters; largest max./min. in the final difference Fourier synthesis 2.683 e Å−3/−2.694 e Å−3; max./min. transmission 0.41/0.75; R1 = 0.0423 (I > 2σ(I)), wR2 = 0.0864 (all data). The solid-state structure of 1 is shown in Fig. 1. The structure was solved by direct methods (SHELXS-97)35 and refined anisotropically by full-matrix least-squares on F2 (SHELXL-2014).36 Absorption corrections were performed numerical based on indexed faces (Bruker AXS APEX2). Hydrogen atoms were refined using a riding model or rigid methyl groups.
The crystallographic data of 1 (excluding structural factors) have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1479636.
Instrumentation
Conclusions
The novel Bi source [C4mim]3[Bi3I12] 1 containing the complex [Bi3I12] trianion was synthesized and structurally characterized. 1 is a promising Bi source for the preparation of phase pure Bi2Te3 material in ILs using ionothermal synthesis. The formation of Bi-rich material phases, as has been often described for other synthetic routes, is completely avoided. The Bi2Te3 nanoparticles are surfactant free according to IR spectroscopic studies and are easily oxidised upon exposure to air.In addition, co-thermolysis of 1 with the single source precursor (Et2Sb)2Te in different molar ratios in [C4mim]3I allowed for the synthesis of a whole range of ternary solid solutions of the general formula (BixSb1−x)2Te3, which were characterized by XRD, SEM, EDX and TEM.
Acknowledgements
S. Schulz gratefully acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG) within the Priority Program SPP 1708 “Material Synthesis near Room Temperature” and the University of Duisburg-Essen.Notes and references
- M. H. Elsheikh, D. A. Shnawah, M. F. M. Sabri, S. B. M. Said, M. H. Hassan, M. B. A. Bashir and M. Mohamad, Renewable Sustainable Energy Rev., 2014, 30, 337 CrossRef.
- (a) B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634 CrossRef CAS PubMed; (b) F. Xiao, C. Hangarter, B. Yoo, Y. Rheem, K. H. Lee and N. V. Myungj, Electrochim. Acta B, 2008, 53, 8103 CrossRef CAS; (c) I. S. Beloborodov, A. V. Lopatin and V. M. Vinokur, Rev. Mod. Phys., 2007, 79, 469 CrossRef CAS; (d) G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105 CrossRef CAS PubMed; (e) E. L. Bell, Science, 2008, 321, 1457 CrossRef PubMed; (f) G. Chen, M. S. Dresselhaus, G. Dresselhaus, J. P. Fleurial and T. Caillat, Int. Mater. Rev., 2003, 48, 45 CrossRef CAS.
- J. Sommerlatte, K. Nielsch and H. Böttner, Phys. J., 2007, 6, 35 CAS.
- (a) D. M. Rowe, in CRC Handbook of Thermoelectrics, CRC Press, Boca Raton, FL, 1995 Search PubMed; (b) J. R. Drabble and C. H. L. Goodman, J. Phys. Chem. Solids, 1958, 5, 142 CrossRef CAS; (c) W. J. Xie, J. He, H. Jung Kang, X. F. Tang, S. Zhu, M. Laver, S. Wang, J. R. D. Copley, C. M. Brown, Q. Zhang and T. M. Tritt, Nano Lett., 2010, 10, 3283 CrossRef CAS PubMed; (d) L. D. Zhao, B. P. Zhang, J. F. Li, H. L. Zhang and W. S. Liu, Solid State Sci., 2008, 10, 651 CrossRef CAS.
- (a) Z. Wang, J. E. Alaniz, W. J. Jang, E. Garay and C. Dames, Nano Lett., 2011, 11, 2206 CrossRef CAS PubMed; (b) G. Q. Zhang, Q. X. Yu, W. Wang and X. G. Li, Adv. Mater., 2010, 22, 1959 CrossRef CAS PubMed; (c) J. P. Heremans, V. Jovovic, E. S. Toberer, A. Saramat, K. Kurosaki, A. Charoenphakdee, S. Yamanaka and G. J. Snyder, Science, 2008, 321, 554 CrossRef CAS PubMed; (d) J. P. Heremans, C. M. Thrush and D. T. Morelli, Phys. Rev. B: Condens. Matter, 2004, 70, 115334 CrossRef; (e) A. J. Minnich, M. S. Dresselhaus, Z. Ren and G. Chen, Energy Environ. Sci., 2009, 466 RSC.
- (a) C. J. Vineis, A. Shakouri, A. Majumdar and M. G. Kanatzidis, Adv. Mater., 2010, 22, 3970 CrossRef CAS PubMed; (b) Y. C. Lan, A. J. Minnich, G. Chen and Z. F. Ren, Adv. Funct. Mater., 2010, 20, 357 CrossRef CAS.
- (a) T. Caillat, M. Carle, P. Pierrat, H. Scherrer and S. J. Scherrer, Phys. Chem. Solids, 1992, 53, 1121 CrossRef CAS; (b) T. Caillat, L. Gailliard, H. Scherrer and S. J. Scherrer, Phys. Chem. Solids, 1993, 54, 575 CrossRef CAS.
- M. Y. Kim, Y. H. Yeo, D. H. Park and T. S. Oh, Ceram. Int., 2012, 38, 529 CrossRef.
- W. Xie, X. Tang, Y. Yan, Q. Zhang and T. M. Tritt, J. Appl. Phys., 2009, 105, 113713 CrossRef.
- Z. Stary, J. Horak, M. Stordeur and M. Stölzer, J. Phys. Chem. Solids, 1988, 49, 29 CrossRef CAS.
- J. W. G. Bos, H. W. Zandbergen, M.-H. Lee, N. P. Ong and R. J. Cava, Phys. Rev. B: Condens. Matter, 2007, 75, 195203 CrossRef.
- (a) X. Fan, X. Cai, Z. Rong, F. Yang, G. Li and Z. Gan, J. Alloys Compd., 2014, 607, 91 CrossRef CAS; (b) Y. Ma, Q. Hao, B. Poudel, Y. Lan, B. Yu, D. Wang, G. Chen and Z. Ren, Nano Lett., 2008, 8, 2580 CrossRef CAS PubMed; (c) W. Xie, X. Tang, Y. Yan, Q. Zhang and T. M. Tritt, Appl. Phys. Lett., 2009, 94, 102111 CrossRef; (d) W. Xie, S. Wang, S. Zhu, J. He, X. Tang, Q. Zhang and T. M. Tritt, J. Mater. Sci., 2013, 48, 2745 CrossRef CAS; (e) Y. Xiao, J. Yang, G. Li, M. Liu, L. Fu, Y. Luo, W. Li and J. Peng, Intermetallics, 2014, 50, 20 CrossRef CAS; (f) Y. Zhang and G. D. Stucky, Chem. Mater., 2014, 26, 837 CrossRef CAS.
- M. Scheele, N. Oeschler, I. Veremchuk, K.-G. Reinsberg, A.-M. Kreuziger, A. Kornowski, J. Broekaert, C. Klinke and H. Weller, ACS Nano, 2010, 4, 4283 CrossRef CAS PubMed.
- V. Stavila, D. B. Robinson, M. A. Hekmaty, R. Nishimoto, D. L. Medlin, S. Zhu, T. M. Tritt and P. A. Sharma, ACS Appl. Mater. Interfaces, 2013, 5, 6678 CAS.
- L. Chen, Q. Zhao and X. Ruan, Mater. Lett., 2012, 82, 112 CrossRef CAS.
- R. J. Mehta, Y. Zhang, C. Karthik, B. Singh, R. W. Siegel, T. Borca-Tasciuc and G. Ramanath, Nat. Mater., 2012, 11, 233 CrossRef CAS PubMed.
- S. L. Benjamin, C. H. de Groot, C. Gurnani, A. L. Hector, R. Huang, E. Koukharenko, W. Levason and G. Reid, J. Mater. Chem. A, 2014, 2, 4865 CAS.
- (a) K. Knapas, T. Hatanpää, M. Ritala and M. Leskelä, Chem. Mater., 2010, 22, 1386 CrossRef CAS; (b) S. Zastrow, J. Gooth, T. Boehnert, S. Heiderich, W. Toellner, S. Heimann, S. Schulz and K. Nielsch, Semicond. Sci. Technol., 2013, 28, 035010 CrossRef; (c) C. Bae, T. Bohnert, J. Gooth, S. Lim, S. Lee, H. Kim, S. Heimann, S. Schulz, H. Shin and K. Nielsch, Semicond. Sci. Technol., 2014, 29, 064003 CrossRef.
- (a) T. J. Groshens, R. W. J. Gedridge and C. K. Lowe-Ma, Chem. Mater., 1994, 6, 727 CrossRef CAS; (b) T. Groshens, R. Gedridge, R. Scheri and T. Cole, Fifteenth International Conference on Thermoelectrics Proceedings ICT ‘96, IEEE, 1996, p. 430 Search PubMed; (c) G. Bendt, S. Zastrow, K. Nielsch, P. S. Mandal, J. Sánchez-Barriga, O. Rader and S. Schulz, J. Mater. Chem. A, 2014, 2, 8215 RSC; (d) S. Schulz, G. Bendt, J. Sonntag, A. Lorke, U. Hagemann and W. Assenmacher, Semicond. Sci. Technol., 2015, 30, 085021 CrossRef.
- G. Bendt, A. Weber, S. Heimann, W. Assenmacher, O. Prymak and S. Schulz, Dalton Trans., 2015, 44, 14272 RSC.
- S. Schulz, S. Heimann, J. Friedrich, M. Engenhorst, G. Schierning and W. Assenmacher, Chem. Mater., 2012, 24, 2228 CrossRef CAS.
- S. Heimann, S. Schulz, J. Schaumann, A. Mudring, J. Stötzel and G. Schierning, J. Mater. Chem. C, 2015, 3, 10375 RSC.
- (a) S. A. Adonin, E. V. Peresypkina, M. N. Sokolov and V. P. Fedin, J. Struct. Chem., 2015, 56, 795 CrossRef CAS; (b) S. A. Adonin, E. V. Peresypkina, M. N. Sokolov and V. P. Fedin, Russ. J. Coord. Chem., 2014, 40, 867 CrossRef CAS.
- (a) G. A. Fisher and N. C. Norman, Adv. Inorg. Chem., 1994, 41, 233 CrossRef CAS; (b) A. Okrut and C. Feldmann, Z. Anorg. Allg. Chem., 2006, 632, 409 CrossRef CAS.
- (a) C. J. Carmalt, L. J. Farrugia and N. C. Norman, Z. Naturforsch., 1995, 50b, 1591 Search PubMed; (b) C. J. Carmalt, L. J. Farrugia and N. C. Norman, Z. Anorg. Allg. Chem., 1995, 621, 47 CrossRef CAS.
- (a) G. R. Desiraju and T. Steiner, in The Weak Hydrogen Bond In Structural Chemistry And Biology, Oxford University Press, 1999 Search PubMed; (b) B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS PubMed; (c) A. D. Buckingham, J. E. Del Bene and S. A. C. McDowell, Chem. Phys. Lett., 2008, 463, 1 CrossRef CAS; (d) G. Gilli and P. Gilli, in The Nature of the Hydrogen Bond, Oxford University Press, Oxford, UK, 2009 Search PubMed; (e) E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg and P. Hobza, Pure Appl. Chem., 2011, 83, 1637 CAS; (f) U. Adhikari and S. Scheiner, J. Phys. Chem. A, 2013, 117, 10551 CrossRef CAS PubMed; (g) X. Zhang, H. Dai, H. Yan, W. Zou and D. Cremer, J. Am. Chem. Soc., 2016, 138, 4334 CrossRef CAS PubMed.
- (a) W.-X. Chai, L.-M. Wu, J.-Q. Li and L. Chen, Inorg. Chem., 2007, 46, 8698 CrossRef CAS PubMed; (b) A. Gągor, M. Węcławik, B. Bondzior and R. Jakubas, CrystEngComm, 2015, 17, 3286 RSC.
- S. Schulz, S. Heimann, K. Kaiser, O. Prymak, W. Assenmacher, J. T. Brüggemann, B. Mallik and A.-V. Mudring, Inorg. Chem., 2013, 52, 14326 CrossRef CAS PubMed.
- Y. Feutelais, B. Legendre, N. Rodier and V. Agafonov, Mater. Res. Bull., 1993, 28, 591 CrossRef CAS.
- (a) C. D. Wagner, A. V. Naumkin, A. Kraut-Vass, J. W. Allison, C. J. Powell and J. R. Rumble Jr., NIST Standard Reference Database 20, Version 3.4. (web version) (http:/srdata.nist.gov/xps/), 2003 Search PubMed; (b) H. Bando, K. Koizumi, Y. Oikawa, K. Daikohara, V. A. Kulbachinskii and H. Ozaki, J. Phys.: Condens. Matter, 2000, 12, 5607 CrossRef CAS; (c) C. R. Thomas, M. K. Vallon, M. G. Frith, H. Sezen, S. K. Kushwaha, R. J. Cava, J. Schwartz and S. L. Bernasek, Chem. Mater., 2016, 28, 35 CrossRef CAS; (d) E. J. Menke, M. A. Brown, Q. Li, J. C. Hemminger and R. M. Penner, Langmuir, 2006, 22, 10564 CrossRef CAS PubMed.
- C. R. Thomas, M. K. Vallon, M. G. Frith, H. Sezen, S. K. Kushwaha, R. J. Cava, J. Schwartz and S. L. Bernasek, Chem. Mater., 2016, 28, 35 CrossRef CAS.
- Simulation: Jems software java version V3, JEMS-SAAS, P. Stadelmann, Saas-Fee, Switzerland, Input: Bi2Te3 ICSD 42546, Only the strongest reflections are shown as rings in the overlay.
- M. M. Stasova and N. Abrikosov, Izv. Akad. Nauk SSSR, Neorg. Mater., 1970, 6, 1090 CAS.
- (a) H. J. Breunig and H. Jawad, Z. Naturforsch., 1982, 37b, 1104 CAS; (b) S. Heimann, S. Schulz, D. Bläser and C. Wölper, Eur. J. Inorg. Chem., 2013, 28, 4909 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, 467 CrossRef.
- (a) G. M. Sheldrick, SHELXL-2014, Program for the Refinement of Crystal Structures, University of Göttingen, Göttingen, Germany, 2014 Search PubMed; (b) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed; (c) C. B. Hübschle, G. M. Sheldrick and B. Dittrich, J. Appl. Crystallogr., 2011, 44, 1281 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1479636. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02361d |
| ‡ 1H, 13C NMR and IR spectra of [C4mim]I and [C4mim]3[Bi3I12] 1; in situ NMR spectroscopic studies of the reaction of (Et3Si)2Te and 1; thermal behavior of 1 upon heating and cooling; central structural parameters of 1 and EDX results from STEM spot analyses on single crystals. |
| This journal is © The Royal Society of Chemistry 2016 |