
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral carbene–borane adducts: precursors for borenium catalysts for asymmetric FLP hydrogenations†
Jolie
Lam
a,
Benjamin A. R.
Günther
ab,
Jeffrey M.
Farrell
a,
Patrick
Eisenberger
*c,
Brian P.
Bestvater
c,
Paul D.
Newman
b,
Rebecca L.
Melen
*b,
Cathleen M.
Crudden
*cd and
Douglas W.
Stephan
*a
aDepartment of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario M5S 3H6, Canada. E-mail: dstephan@chem.utoronto.ca
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, Wales CF10 3AT, UK. E-mail: MelenR@cardiff.ac.uk
cDepartment of Chemistry, Queen's University, 90 Bader Lane, Kingston, Ontario, Canada K7L 3N6. E-mail: cruddenc@chem.queensu.ca
dInstitute of Transformative Bio-Molecules (ITbM–WPI), Nagoya University, Chikusa, Nagoya, Japan
First published on 23rd June 2016
Abstract
The carbene derived from (1R,3S)-camphoric acid was used to prepare the borane adduct with Piers’ borane 7. Subsequent hydride abstraction gave the borenium cation 8. Adducts with 9-BBN and the corresponding (1R,3S)-camphoric acid-derived carbene bearing increasingly sterically demanding N-substituents (R = Me 9, Et 10, i-Pr 11) and the corresponding borenium cations 12–14 were also prepared. These cations were not active as catalysts in hydrogenation, although 9–11 were shown to undergo carbene ring expansion reactions at 50 °C to give species 15–17. The IBOX-carbene precursors 18 and 19 derived from amino alcohols (S)-valinol and (S)-tert-leucinol (R = i-Pr, t-Bu) were used to prepare borane adducts 20–23. Reaction of the carbenes 1,3-dimethylimidazol-2-ylidene (IMe), 1,3-di-iso-propylimidazol-2-ylidene (IPr) 1-benzyl-3-methylimidazol-2-ylidene (IBnMe), 1-methyl-3-phenylimidazol-2-ylidene (IPhMe) and 1-tert-butyl-3-methylimidazol-2-ylidene (ItBuMe) with diisopinocampheylborane (Ipc2BH) gave chiral adducts: (IMe)(Ipc2BH) 24, (IPr)(Ipc2BH) 25, (IBnMe)(Ipc2BH) 26, (IPhMe)(Ipc2BH) 27, and (ItBuMe)(Ipc2BH) 28. Triazolylidene-type adducts including the (10)-phenyl-9-borabicyclo [3.3.2]decane adduct of 1,3,4-triphenyl-1H-1,2,3-triazolium, rac-29 and the 9-BBN derivative of (S)-2-amino-2′-methoxy-1,1′-binaphthalene-1,2,3-triazolium 34a/b were also prepared. In catalytic studies of these systems, while several species were competent catalysts for imine reduction, in general, low enantioselectivities, ranging from 1–20% ee, were obtained. The implications for chiral borenium cation catalyst design are considered.
Introduction
Following the advent of the concept of “frustrated Lewis pairs” (FLPs) a decade ago,1,2 the initial application that emerged from the metal-free activation of H2 was the use of these in hydrogenation catalysis,3–7 resulting in a burgeoning and exciting area of new developments.8–13 The scope of substrates that can be reduced by an FLP protocol has broadened dramatically from the original application to imines, protected nitriles and aziridines.7 Indeed protocols for the reduction of enamines,14 silylenol-ethers,15 alkenes,16–18 alkynes,19 ketones and aldehydes20–23 have emerged. These advances have been reviewed in several places.8–10,13,24–26Analogous to the development of homogeneous transition-metal based catalysts, following the discovery of FLP catalysts, attention has turned to the development of stereoselective systems. The first to report such an advance was Klankermayer, who described the use of a chiral electrophilic borane derived from (+)-α-pinene in ketimine reduction affording 13% ee in the reduced amine product.27 Since then, other chiral catalyst systems have emerged (Fig. 1). In some cases, stereoselectivity has climbed to give ee's as high as 99%.28–32 These systems (Fig. 1) all employ electron withdrawing pentafluorophenyl-groups to enhance the Lewis acidity at the boron centre that enables H2 activation and catalytic turnover in imine reduction. Recently however, the chemistry of borocations has gained significant momentum.33 In these systems the tri-coordinate boron possesses a formal positive charge, enhancing the Lewis acidity at boron without the need for electron withdrawing fluorinated groups. Such systems have been employed in 1,n-carboboration,34 hydroboration,35 haloboration36 and alkyne/arene borylation reactions.37 In addition, borenium cations have been found to act as the Lewis acid constituent of FLPs for catalytic hydrogenation.38–40 For example, borenium cations of the form [(L)BBN][B(C6F5)4] (L = N-heterocyclic carbene (NHC), BBN = 9-boreniumbicyclo[3.3.1]-nonane), were shown to be effective in the hydrogenation of imines and enamines.38,39 These catalysts offer the advantage of being readily derived from robust and easily prepared carbene adducts of 9-BBN. In a subsequent effort, we showed that these particular systems could be optimised for reactivity by modification of the carbene. Subsequently Crudden, Eisenberger et al. employed related triazolium–borenium cations in hydrogenation catalysis.40
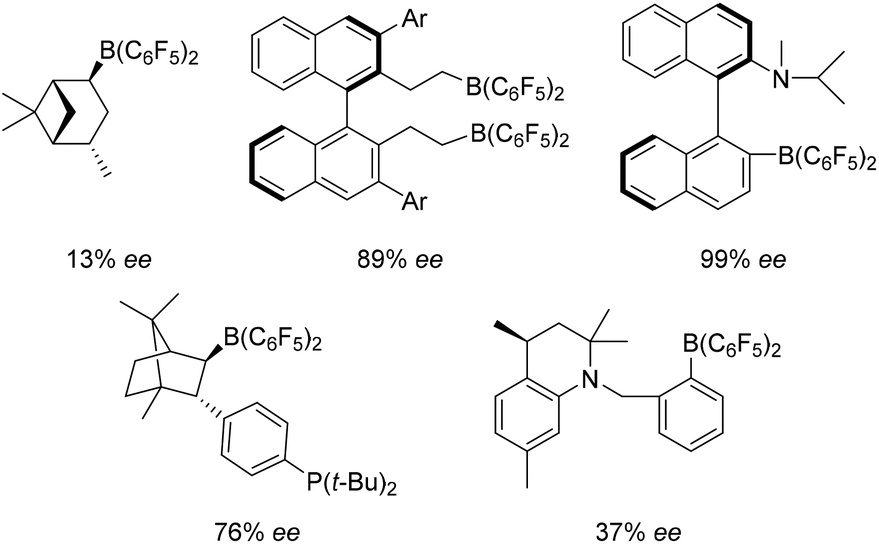 | ||
| Fig. 1 Selected chiral electrophilic boranes used in FLP hydrogenations. The highest enantioselectivities reported for each catalyst are shown. | ||
Imine reductions by borenium catalysts typically occur by delivery of hydride from the borane adduct to the iminium cation (Scheme 1).38,39 Thus, introducing chirality in the borane adduct provides potential for controlling stereochemistry in the products. The use of NHCs and triazolylidene derivatives as supporting ligands for borenium ions provides a straightforward approach to incorporate chirality through this ligand. In this manuscript, we describe the synthesis of chiral carbene adducts of borenium ions and preliminary evaluation of these systems in hydrogenation catalysis.
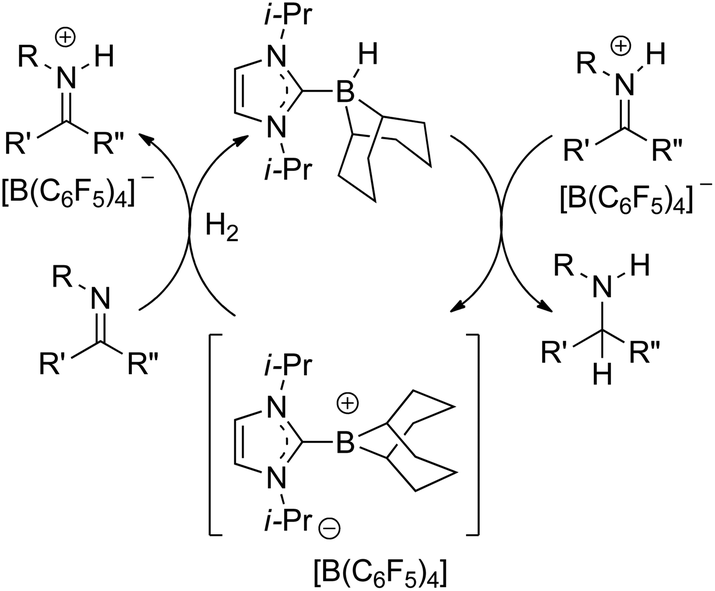 | ||
| Scheme 1 Mechanism of borenium-based hydrogenation of imines. | ||
Experimental section
General remarks
Unless otherwise stated, all reactions and manipulations were performed under an atmosphere of dry, oxygen-free, nitrogen in a glove box (Innovative Technology or MBraun). All solvents (including deuterated) were dried and stored over molecular sieves under a nitrogen atmosphere before use. 1H, 13C, 11B, 19F and 31P NMR data were recorded on a Bruker Avance III or Bruker 500 MHz spectrometer. All NMR spectra were recorded at 298 K unless otherwise stated and these data are reported in ppm. NMR assignments are further detailed in the ESI.† A Perkin-Elmer analyser was used for carbon, hydrogen, and nitrogen elemental analysis. (3S,7S)-3,7-Diisopropyl-2,3,7,8-tetrahydro-imidazo[4,3-b:5,1-b′]-bis (oxazole)-4-ium triflate (18), and (3S,7S)-3,7-di-tertbutyl-2,3,7,8-tetrahydroimidazo [4,3-b:5,1-b′]bis(oxazole)-4-ium triflate, [(3S,7S)-3,7-t-Bu2-2,3,7,8-C7H7O2N2][O3SCF3] (19),41,42N-(1-phenylethylidene) aniline,43 (+)-diisopinocampheylborane44 and 1,3-dimethylimidazolium iodide,45 1-methyl-3-phenylimidazolium iodide,46 1-benzyl-3-methylimidazolium iodide47 and (3S,7S)-3,7-di-isopropyl-2,3,7,8-tetrahydroimidazo[4,3-b:5,1-b′]yilidene-9-borabicyclo[3.3.1]nonane48 were prepared by literature procedures. In some cases repeated attempts to secure satisfactory elemental analyses lead to low C content, despite the use of additional oxidant, and is attributed to the formation of boron–carbide during combustion.5: Yield: 0.89 g (3.00 mmol, 96%). 1H NMR (CDCl3): 1.03 (s, 3H), 1.15 (s, 3H), 1.30 (t, 3JHH = 7 Hz, 3H), 1.34 (s, 3H), 1.38 (t, 3JHH = 7 Hz, 3H), 1.94–2.01 (m, 1H), 2.10–2.17 (m, 1H), 2.26–2.31 (m, 1H), 2.49–2.55 (m, 1H), 3.19 (d, 3JHH = 5 Hz, 1H), 3.52–3.60 (m, 1H), 3.62–3.69 (m, 1H), 3.70–3.77 (m, 1H), 3.79–3.86 (m, 1H), 9.05 (s, 1H). 13C{1H} NMR (CDCl3): 14.1, 14.3, 17.2, 17.3, 21.9, 32.1, 40.6, 40.8, 45.3, 48.7, 66.3, 69.9, 153.2. HRMS (ESI+): C13H25N2, [M+] m/z (calc.): 209.2012, m/z (obs.): 209.2018.
6: Yield: 2.46 g (6.43 mmol, 96%). 1H NMR (CDCl3): 0.90 (s, 3H), 1.12 (s, 3H), 1.20 (d, 3JHH = 6.7 Hz, 3H), 1.27 (d, 3JHH = 7 Hz, 3H), 1.32 (s, 3H), 1.36 (d, 3JHH = 7 Hz, 3H), 1.47 (d, 3JHH = 7 Hz, 3H), 1.92–1.98 (m, 1H), 2.04–2.09 (m, 1H), 2.13–2.20 (m, 1H), 2.36–2.42 (m, 1H), 3.28 (d, 3JHH = 5 Hz, 1H), 3.85 (sept, 3JHH = 7 Hz, 1H), 4.31–4.39 (m, 1H), 8.13 (s, 1H). 13C{1H} NMR (CDCl3): 14.9, 17.7, 20.7, 21.7, 22.2, 23.6, 25.1, 33.4, 40.4, 41.1, 51.1, 55.9, 61.4, 71.5, 151.1. HRMS (ESI+): C15H29N2, [M+] m/z (calc.): 237.2325, m/z (obs.): 237.2330.
10: Yield: 220 mg (0.67 mmol, 67%), 1H NMR (benzene-d6): 0.43 (s, 3H), 0.69 (s, 3H), 0.74 (s, 3 H), 0.94 (t, 3JHH = 7 Hz, 3H), 1.05 (t, 3JHH = 7 Hz, 3H), 1.22 (m, 2H), 1.35 (m, 1H), 1.49 (m, 3H), 1.67 (m, 1H), 1.93 (m, 1H), 2.09 (m, 6H), 2.27 (m, 3H), 2.41 (m, 1H), 2.54 (m, 2H), 3.14 (m, 1H), 3.60 (sept, 3JHH = 7 Hz, 1H), 3.67 (sept, 3JHH = 7 Hz, 1H), 4.30–4.39 (m, 1H). 11B NMR (benzene-d6): −14.3 (d, 1JBH = 84 Hz). 13C{]H} NMR (benzene-d6): 18.1, 18.3, 20.3, 20.9, 21.7, 22.6, 24.6, 25.4, 25.8, 30.8, 32.1, 32.8, 37.7, 39.7, 40.5, 40.6, 49.9, 52.8, 59.8, 69.7 HRMS (ESI+): C21H40BN2, [M+] m/z (calc.): 330.3321, m/z (obs.): 330.3325. Anal. Calcd for C21H40BN2: C 76.35, H 11.40, N 8.48%; Obs. C 76.53, H 10.57, N 8.00%.
11: Yield: 227 mg (0.63 mmol, 63%). 1H NMR (benzene-d6): 0.45 (s, 3H), 0.66 (s, 3H), 0.93–0.97 (m, 9H), 1.15 (d, 3JHH = 7.2 Hz, 3H), 1.24 (d, 3JHH = 7.2 Hz, 3H), 1.27 (bs, 2H), 1.41 (bs, 1H), 1.45–1.52 (bm, 2H), 1.93–2.04 (bm, 3H), 2.09–2.71 (m, 12H), 5.47 (sept, 3JHH = 7 Hz, 1H), 5.56 (sept, 3JHH = 7 Hz, 1H). 11B NMR (benzene-d6): −12.9 (d, 1JBH = 85 Hz). 13C{1H} NMR (benzene-d6): 18.2, 18.5, 20.5, 21.0, 21.9, 22.7, 24.8, 25.0 (bs), 25.6, 26.0, 27.1 (bs), 30.9, 32.3, 33.0, 37.8, 39.9, 40.7, 40.8, 50.1, 52.9, 59.9, 69.8. HRMS(ESI+): C23H44BN2, [M+] m/z (calc.): 358.3634, m/z (obs.): 358.3624. Anal. Calcd for C23H44BN2: C 77.08, H 12.09, N 7.82%; Obs. C 76.76, H 12.91, N 7.76%.
13: Yield: 299 mg (0.29 mmol, 98%). 1H NMR (CD2Cl2): 1.05(s, 3H), 1.16 (s, 3H), 1.27 (t, 3J = 7.1 Hz, 3H), 1.37 (t, 3J = 7.1 Hz, 3H), 1.41 (s, 3H), 1.64–1.70 (m, 3H), 1.81 (s, br, 1H), 1.89–2.02 (m, 6H), 2.07–2.12 (m, 1H), 2.14–2.18 (m, 1H), 2.21–2.25 (m, 1H), 2.27–2.37 (m, 5H), 2.42–2.47 (m, 1H), 2.75 (dq, 3JHH = 7 Hz, 2JHH = 16 Hz, 1H), 3.09 (dq, 3JHH = 7 Hz, 2JHH = 14.5 Hz, 1H), 3.22–3.29 (m, 2H), 3.53 (dq, 3JHH = 7 Hz, 2JHH = 16 Hz, 1H). 11B NMR (CD2Cl2): −16.7 (s), 89.1 (bs). 13C{1H} NMR (CD2Cl2): 13.3, 13.3, 16.3, 16.7, 20.4, 21.8, 22.0, 30.9 (bs), 31.2 (bs), 31.5, 35.7, 36.0, 36.3, 36.4, 39.5, 40.0, 46.4, 48.6, 66.2, 69.4, 136.5 (d, 1JCF = 237 Hz), 138.2 (d, 1JFC = 240 Hz), 148.4 (d, 1JFC = 238 Hz) 171.3 19F NMR (CD2Cl2): −133.1 (bs), −163.7 (t, 3JFF = 20 Hz), −167.6 (t, 3JFF = 18 Hz). HRMS(ESI+): C21H38BN2, [M+] m/z (calc.): 329.3123, m/z (obs.): 329.2739. Anal. Calcd for C21H38BN2: C 53.60, H 3.80, N 2.78%; Obs. C 54.21, H 2.60, N 2.50%.
14: Yield: 276 mg (0.26 mmol, 93%). 1H NMR (CD2Cl2): 1.09 (s, 3H), 1.15 (s, 3H), 1.26 (d, 3JHH = 6.5 Hz, 3H), 1.34 (d, 3JHH = 6.5 Hz, 3H), 1.46 (d, 3JHH = 6.8 Hz, 3H), 1.53 (s, 3H), 1.61 (d, 3JHH = 6.8 Hz, 3H), 1.64–1.70 (m, 1H), 1.78–1.81 (bm, 1H), 1.81–1.85 (bm, 2H), 1.87–1.89 (bm, 1H), 1.90–1.94 (bm, 2H), 2.03–2.13 (m, 3H), 2.23–2.39 (bm, 7H), 2.55–2.61 (m, 1H), 2.68 (sept, 3JHH = 6.8 Hz, 1H), 3.10 (sept, 3JHH = 7 Hz, 1H), 3.32 (d, 3JHH = 5 Hz, 1H). 11B NMR (CD2Cl2): −16.7 (s), 85.8 (bs). 13C NMR (CD2Cl2): 13.9, 16.2, 16.8, 19.3, 19.7, 20.3, 21.3, 21.6, 21.8, 29.2, 29.4, 33.0, 35.5, 35.7, 36.4, 36.5, 39.7, 40.6, 55.9, 59.8, 61.0, 71.4, 136.1 (d, 1JCF = 242 Hz), 138.0 (d, 1JCF = 240 Hz), 148.2 (d, 1JCF = 241 Hz), 171.6 19F NMR (CD2Cl2): −133.1 (bs), −163.8 (t, 3JFF = 21 Hz), −167.6 (t, 3JFF = 18 Hz). HRMS (ESI+): C23H42BN2, [M+] m/z (calc.): 356.3472, m/z (obs.): 356.3477.
16: Yield: 20 mg (0.06 mmol, >99%). 1H NMR (benzene-d6): 0.81 (s, 3H), 0.85 (s, 3H), 0.92 (t, 3JHH = 7 Hz, 3H), 1.20 (t, 3JHH = 7 Hz, 3H), 1.22 (s, 3H), 1.36–1.43 (m, 1H), 1.53–2.01 (m, 15H), 2.12–2.24 (m, 2H), 2.43–2.58 (m, 3H), 2.64 (d, 3JHH = 5 Hz, 1H), 2.69 (s, 1H), 3.20–3.35 (m, 2H). 11B NMR (benzene-d6): 44.0 (bs). 13C{1H} NMR (benzene-d6): 14.3, 15.7, 17.1, 18.2, 21.7, 29.8, 30.6, 30.8, 31.0, 31.5, 37.6, 38.8, 39.0, 39.3, 41.8, 42.5, 46.9, 47.3, 50.4, 51.9, 66.3, 76.2. HRMS (ESI+): C21H38BN2, [M+] m/z (calc.): 328.3164, m/z (obs.): 328.3175. Anal. Calcd for C21H38BN2: C 76.35, H 11.90, N 8.48%; Obs. C 76.57, H 11.63, N 8.09%.
17: Yield: 45 mg (0.13 mmol, >99%). 1H NMR (benzene-d6): 0.74 (s, 3H), 0.85 (s, 3H), 0.86 (d, 3JHH = 7 Hz, 3H), 1.00 (d, 3JHH = 7 Hz, 3H), 1.22 (d, 3JHH = 7 Hz, 6H), 1.25 (s, 3H), 1.49–1.55 (m, 2H), 1.71–1.81 (m, 6H), 1.86–1.97 (m, 6H), 2.23–2.32 (m, 3H), 2.94 (d, 3JHH = 6 Hz, 1H), 3.31 (s, 1H), 3.49 (sept, 3JHH = 7 Hz, 1H), 3.93 (sept, 3JHH = 7 Hz, 1H). 11B NMR (benzene-d6): 44.0 (bs). 13C{1H} NMR (benzene-d6): 20.7, 21.3, 21.9, 22.1, 23.8, 25.0 (bs), 25.4, 27.1, 28.2, 29.0, 31.3, 32.2, 33.2, 33.4, 42.0, 47.2, 48.1, 48.6, 49.6, 54.6 (bs), 66.9, 70.0. HRMS (ESI+): C23H42BN2, [M+] m/z (calc.): 356.3477, (obs.): 356.3484. Anal. Calcd for C23H42BN2: C 77.08, H 12.04, N 7.82%; Obs. C 77.10, H 12.36, N 7.75%.
21: Crystals for a single crystal X-ray study were obtained by slow evaporation from toluene. Yield: (44 mg, 0.176 mmol, 62%). 1H NMR (benzene-d6): 4.06–3.91 (m, 4H), 3.86–3.74 (m, 2H), 2.89 (ddq, 3JHH = 10 Hz, 4JHH = 7 Hz, 3JHH = 4 Hz, 2H), 2.40–1.65 (m, 1H), 0.65 (d, 3JHH = 7 Hz, 6H), 0.44 (d, 3JHH = 7 Hz, 6H). 11B NMR (benzene-d6): −35.6 (q, 1JBH = 88 Hz). 13C NMR (benzene-d6): 76.1, 61.1, 29.0, 22.7, 18.3, 14.3. MS (DART) m/z: 249 [M − H]+. HRMS (ESI): (calc.) for C13H22BN2O2 249.1774, (obs): 249.1771. Anal. Calc. for C13H23BN2O2: C 62.42, H 9.27, N 11.20%. Found: C 62.02, H 9.27, N 11.10%.
22: Crystals for a single crystal X-ray study were obtained by slow evaporation of toluene. Yield: (50 mg, 0.111 mmol, 35%). 1H NMR (benzene-d6): 4.56 (dd, 3JHH = 7 Hz, 2JHH = 2 Hz, 2H), 3.91 (dd, 3JHH = 9 Hz, 3JHH = 1 Hz, 2H), 3.71 (ddd, 4JHH = 9 Hz, 3JHH = 7 Hz, 2JHH = 1 Hz, 2H), 3.02 (dtd, 3JHH = 14 Hz, 3JHH = 7 Hz, 4JHH = 3 Hz, 2H), 2.39 (bs, 1H), 2.36 (bs, 1H), 2.09–0.95 (m, 18H), 0.68 (d, 3JHH = 7 Hz, 6H), 0.55 (d, 3JHH = 7 Hz, 6H). 11B NMR (benzene-d6): 3.2 (bs). 13C NMR (benzene-d6): 75.9, 64.5, 32.8, 32.4, 31.4, 31.2, 31.1, 30.2, 29.7, 29.6, 29.5, 28.4, 28.3, 28.2, 28.0, 18.7, 14.4. MS (DART) m/z: 413 [M − Cl]+. HRMS (ESI): calc. for C25H42BN2O2 413.33393, found 413.33381.
23: Crystals for a single crystal X-ray study were obtained by slow evaporation of pentane. Yield: (88 mg, 0.228 mmol, 80%). 1H NMR (benzene-d6): 4.15 (d, 3JHH = 5 Hz, 2H), 4.04 (d, 3JHH = 9 Hz, 2H), 3.78 (dd, 3JHH = 9 Hz, 3JHH = 6 Hz, 2H), 2.82–1.74 (m, 14H), 1.55 (bs, 1H), 1.16 (bs, 1H), 0.88 (s, 18H). 11B NMR (benzene-d6): −16.4 (d, 1JBH = 86 Hz), 13C NMR (benzene-d6): 124.3, 78.6, 68.3, 39.4, 35.3, 33.1, 32.4, 27.5, 26.6, 25.9. MS (ESI+) m/z: 385 [M − H]+. HRMS (ESI): (calc.) for C23H38BN2O2+ 385.3026, (obs): 386.3032. Anal. Calc. for C23H39BN2O2: C 71.50, H 10.17, N 7.25%. Found: C 70.45, H 10.16, N 6.79%.
25: Yield: (198 mg, 34%). 1H NMR (CDCl3): 6.97 (d, 3JHH = 2 Hz, 1H), 6.92 (d, 3JHH = 2 Hz, 1H), 5.84 (septet, 3JHH = 7 Hz, 1H), 5.13 (septet, 3JHH = 7 Hz, 1H), 2.25 (m, 1H), 2.19–2.04 (m, 4H), 1.85, (m, 1H), 1.75–1.67 (m, 2H), 1.65–1.54 (m, 3H), 1.46 (d, 3JHH = 7 Hz, 3H), 1.459 (d, 3JHH = 7 Hz, 3H), 1.459 (d, 3JHH = 7 Hz, 3H), 1.37 (d, 3JHH = 7 Hz, 3H), 1.36 (d, 3JHH = 7 Hz, 3H), 1.12 (s, 3H), 1.11 (s, 3H), 1.10 (d, 3JHH = 7 Hz, 3H), 1.08 (s, 3H), 1.06 (s, 3H), 0.95 (d, 3JHH = 9 Hz, 1H), 0.84 (d, 3JHH = 9 Hz, 1H) 0.48 (d, 3JHH = 7 Hz, 3H). 11B NMR (toluene-d8): −8.2 (d, 1JBH = 86 Hz). 13C{1H} NMR (CDCl3, partial): 115.8, 115.4, 50.6, 49.9, 49.3, 48.9, 44.9, 43.3, 43.1, 42.1, 39.0, 38.9, 37.1, 35.4, 33.8, 32.7, 28.38, 28.36, 24.10, 24.08, 23.82, 23.70, 23.38, 23.33, 23.03, 22.9. Anal. Calc. for C29H51BN2: C 79.43, H 11.72, N 6.39%. Found: C 79.30, H 11.90, N 6.39%.
26: Yield: (667 mg, 83% yield). 1H NMR (CD2Cl2): 7.42–7.21 (bm, 5H), 6.88 (bs, 1H, minor), 6.80 (bs, 1H, major), 6.76 (bs, 1H, major), 6.66 (bs, 1H, minor), 5.74 (d, 2JHH = 15 Hz, 1H, minor), 5.66 (d, 2JHH = 15 Hz, 1H, major), 5.57 (d, 2JHH = 15 Hz, 1H, major), 5.17 (d, 2JHH = 15 Hz, 1H, minor), 3.94–3.88 (overlapping singlets, 3H, minor/major), 2.25–0.60 (br, 34H), (No B–H peak found). 11B NMR (CD2Cl2): −8.7 (obscured, minor), 9.11 (d, 1JBH = 85 Hz, major). 13C{1H} NMR (CDCl3, partial, major only): 136.6, 129.0, 128.5, 128.2, 120.6, 119.8, 52.8, 50.8, 49.94, 44.7, 43.4, 43.0, 42.7, 39.4, 39.1, 37.6, 36.3, 35.3, 33.5, 33.4, 28.6, 28.5, 23.9, 23.4, 23.4. Anal. Calc. for C31H47BN2: C 81.20, H 10.33, N 6.11%. Found: C 80.72, H 10.81, N 6.16%.
27: Yield: (407 mg, 56% yield). 1H NMR (CDCl3): 7.45–7.40 (bm, 5H), 6.95 (d, 3JHH = 2 Hz, 1H,), 6.90 (br, 1H), 4.01 (s, 3H) 2.15–1.55 (br, 10H), 1.15–0.60 (br, 15H), 1.11 (s, 3H), 1.08 (s, 3H), 0.72 (d, 3JHH = 8 Hz, 3H). 11B NMR (CD2Cl2): δ −9.74 (d, 1JBH = 75 Hz). 13C{1H} NMR (CD2Cl2, partial): 140.6, 129.0, 128.8, 128.4, 122.4, 121.2, 51.2, 50.3, 44.5, 43.7, 43.5, 42.8, 39.6, 39.4, 38.3, 36.4, 35.3, 33.6, 33.3, 28.7, 28.5, 23.9, 23.8, 23.2, 23.1. Anal. Calc. for C30H45BN2: C 81.06, H 10.20, N 6.30%. Found: C 80.63, H 10.66, N 6.22%.
28: (498 mg, 60% yield). 1H NMR (CD2Cl2): 7.09 (d, 3JHH = 2 Hz, 1H), 6.73 (d, 3JHH = 2 Hz, 1H), 3.87 (s, 6H), 2.41–2.32 (m, 1H), 2.27–2.15 (m, 1H), 2.11–1.95 (m, 3H), 1.82 (s, 9H), 1.77–1.60 (m, 3H), 1.58–1.48 (m, 2H), 1.22–1.00 (br, 20H), 0.63 (d, 3JHH = 7 Hz, 3H). 11B NMR (CD2Cl2): −6.85 (d, 1JBH = 87 Hz). 13C{1H} NMR (CDCl3, partial): 119.5, 118.1, 60.9, 52.0, 50.2, 43.5, 43.2, 43.0, 42.6, 39.7, 39.5, 38.6, 38.6, 34.1, 32.8, 32.2, 32.2, 28.5, 28.3, 24.7, 24.2, 23.5, 23.4. Anal. Calc. for C28H49BN2: C 79.22, H 11.63, N 6.20%. Found: C 78.64, H 11.51, N 6.65%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Combined yield: 4.44 g, (13.7 mmol, 82%). 1H NMR (CDCl3): 8.06 (d, 3JHH = 9 Hz, 1H), 8.03 (d, 3JHH = 9 Hz, 1H), 7.93 (d, 3JHH = 8 Hz, 2H), 7.53 (d, 3JHH = 9 Hz, 1H), 7.50 (d, 3JHH = 9 Hz, 1H), 7.45 (t, 3JHH = 7 Hz, 1H), 7.38 (t, 3JHH = 7 Hz, 1H), 7.34–7.25 (m, 2H), 7.34–7.25 (m, 2H), 7.09 (d, 3JHH = 8 Hz, 1H), 3.83 (s, 3H). 13C{1H} NMR (CDCl3): 155.0, 136.0, 134.0, 133.8, 131.2, 130.4, 129.8, 129.1, 128.3, 128.2, 127.1, 126.9, 126.0, 125.3, 125.0, 124.1, 123.9, 118.7, 117.8, 113.7, 56.8. HRMS(EI+): (calc.) for C21H15N3O 325.1215, (obs.): 325.1206.
:0 → 100:5). Yield: 589 mg (1.39 mmol, 90%). 1H NMR (CDCl3): 8.15 (d, 3JHH = 9 Hz, 1H), 8.05 (d, 3JHH = 8 Hz, 1H), 8.00 (d, 3JHH = 9 Hz, 1H), 7.99 (d, 3JHH = 9 Hz, 1H), 7.86 (d, 3JHH = 8 Hz, 1H), 7.58 (t, 3JHH = 7 Hz, 1H), 7.26–7.44 (m, 5H), 7.12 (d, 3JHH = 8 Hz, 1H), 7.05 (s, 1H), 3.67 (s, 3H), 0.10 (s, 9H). 13C{1H} NMR (CDCl3): 155.0, 145.0, 134.5, 133.8, 133.5, 133.1, 130.8, 130.2, 129.5, 128.8, 128.4, 128.3, 128.1, 127.4, 127.2, 127.1, 126.9, 124.5, 124.0, 123.4, 117.6, 113.0, 56.4, −1.4. HRMS(EI+): (calc.) for C26H25N3OSi 423.1767, (obs.): 423.1771.
:1 → 20:1) to give a slightly ochre solid. Yield: 1.121 g (2.17 mmol, 89%). 1H NMR (CDCl3): 8.87 (s, 1H), 8.51 (s, 1H), 8.23 (d, 3JHH = 9 Hz, 1H), 8.08 (d, 3JHH = 8 Hz, 1H), 8.00 (m, 2H), 7.85 (d, 3JHH = 8 Hz, 1H), 7.68 (t, 3JHH = 8 Hz, 1H), 7.54–7.24 (m, 10H), 7.02 (d, 3JHH = 8.4 Hz, 1H), 3.75 (s, 3H). 13C{1H} NMR (CDCl3): 154.7, 134.8, 134.4, 133.5, 132.7, 132.6, 132.1, 132.0, 131.9, 131.8, 130.6, 130.3, 128.7, 128.6, 128.6, 128.4, 128.3, 127.7, 127.3, 124.1, 124.1, 121.8, 121.3, 114.7, 112.9, 56.3. HRMS(EI+): (calc.) for C29H22N3O 428.1757, (obs.) 428.1739.
:1). Combined yield: 4.44 g, (13.7 mmol, 82%). 1H NMR (CDCl3): 8.06 (d, 3JHH = 9 Hz, 1H), 8.03 (d, 3JHH = 9 Hz, 1H), 7.93 (d, 3JHH = 8 Hz, 2H), 7.53 (d, 3JHH = 9 Hz, 1H), 7.50 (d, 3JHH = 9 Hz, 1H), 7.45 (t, 3JHH = 7 Hz, 1H), 7.38 (t, 3JHH = 7 Hz, 1H), 7.34–7.25 (m, 2H), 7.34–7.25 (m, 2H), 7.09 (d, 3JHH = 8 Hz, 1H), 3.83 (s, 3H). 13C{1H} NMR (CDCl3): 155.0, 136.0, 134.0, 133.8, 131.2, 130.4, 129.8, 129.1, 128.3, 128.2, 127.1, 126.9, 126.0, 125.3, 125.0, 124.1, 123.9, 118.7, 117.8, 113.7, 56.8. HRMS(EI+): (calc.) for C21H15N3O 325.1215, (obs.): 325.1206.
:0 → 100:5). Yield: 589 mg (1.39 mmol, 90%). 1H NMR (CDCl3): 8.15 (d, 3JHH = 9 Hz, 1H), 8.05 (d, 3JHH = 8 Hz, 1H), 8.00 (d, 3JHH = 9 Hz, 1H), 7.99 (d, 3JHH = 9 Hz, 1H), 7.86 (d, 3JHH = 8 Hz, 1H), 7.58 (t, 3JHH = 7 Hz, 1H), 7.26–7.44 (m, 5H), 7.12 (d, 3JHH = 8 Hz, 1H), 7.05 (s, 1H), 3.67 (s, 3H), 0.10 (s, 9H). 13C{1H} NMR (CDCl3): 155.0, 145.0, 134.5, 133.8, 133.5, 133.1, 130.8, 130.2, 129.5, 128.8, 128.4, 128.3, 128.1, 127.4, 127.2, 127.1, 126.9, 124.5, 124.0, 123.4, 117.6, 113.0, 56.4, −1.4. HRMS(EI+): (calc.) for C26H25N3OSi 423.1767, (obs.): 423.1771.
:1 → 20:1) to give a slightly ochre solid. Yield: 1.121 g (2.17 mmol, 89%). 1H NMR (CDCl3): 8.87 (s, 1H), 8.51 (s, 1H), 8.23 (d, 3JHH = 9 Hz, 1H), 8.08 (d, 3JHH = 8 Hz, 1H), 8.00 (m, 2H), 7.85 (d, 3JHH = 8 Hz, 1H), 7.68 (t, 3JHH = 8 Hz, 1H), 7.54–7.24 (m, 10H), 7.02 (d, 3JHH = 8.4 Hz, 1H), 3.75 (s, 3H). 13C{1H} NMR (CDCl3): 154.7, 134.8, 134.4, 133.5, 132.7, 132.6, 132.1, 132.0, 131.9, 131.8, 130.6, 130.3, 128.7, 128.6, 128.6, 128.4, 128.3, 127.7, 127.3, 124.1, 124.1, 121.8, 121.3, 114.7, 112.9, 56.3. HRMS(EI+): (calc.) for C29H22N3O 428.1757, (obs.) 428.1739.
General procedure for high pressure hydrogenation
In an inert atmosphere glovebox, NHC-borane (0.0261 mmol, 1 equiv.), trityl tetrakis(pentafluorophenyl)borate (24 mg, 0.0261 mmol, 1 equiv.) and substrate (0.522 mmol, 20 equiv.) were weighed into vials. 0.4 mL of solvent was used to dissolve the trityl tetrakis(pentafluorophenyl)borate and the orange solution was transferred to the vial with the NHC-borane, which turns colourless upon mixing. The colourless solution was transferred to the vial with the substrate with an additional 0.2 mL solvent and equipped with a stir bar and placed in a Parr pressure reactor. The reactor was sealed, removed from the glovebox, and purged 10 times with hydrogen and ultimate pressurised to 102 atm. The reactor was stirred magnetically at the specified temperature for the specified time and then vented slowly. NMR samples were taken in CDCl3. Conversions were determined by 1H NMR spectroscopy from an aliquot. The sample was then concentrated in vacuo, dissolved in 9:1 hexanes:ethyl acetate and passed through a short silica plug. The sample was concentrated in vacuo and enantiomeric excess was determined by chiral HPLC (Chiralcel OD-H, 98.0 hexanes, 1.0 isopropanol) with comparison to a racemic sample prepared by 1,3-dimethylimidazol-2-ylidene-9-borabicyclo[3.3.1]nonane in the same hydrogenation process.
Racemisation experiment
In a glove box 9-(1,3-diphenyl-1,2,3-triazol-5-ylidene)-9-borabicyclo[3.3.1]nonane (21 mg, 0.060 mmol) and trityl tetrakis(pentafluorophenyl)borate (55 mg, 0.060 mmol) were placed in a vial and dissolved in CD2Cl2 (0.5 mL) generating borenium salt 36.40 The resulting solution was transferred to a J. Young NMR tube equipped with a Teflon stopper. To this solution (+)-bis[(R)-1-phenylethyl]amine (35, 140 μL, 0.612 mmol) was added by syringe and the NMR tube was capped and shaken. A separate NMR tube was charged with a mixture of B(C6F5)3 (31 mg, 0.060 mmol) and (+)-bis[(R)-1-phenylethyl]amine (35, 140 μL, 0.612 mmol) in CD2Cl2 (0.5 mL). The reaction progress was monitored by 1H NMR spectroscopy.X-ray diffraction studies
Crystals were coated in paratone oil and mounted in a cryo-loop. Data were collected on a Bruker APEX2 X-ray diffractometer using graphite monochromated Mo-Kα radiation (0.71073 Å). The temperature was maintained at 150(2) K using an Oxford cryo-stream cooler for both, initial indexing and full data collection. Data were collected using Bruker APEX-2 software and processed using SHELX and Olex2 an absorption correction applied using multi-scan within the APEX-2 program. All structures were solved by direct methods within the SHELXTL package54,55 and refined with Olex2.56,57Results and discussion
For the synthesis of our first, camphor-derived borenium ion, the commercially available, enantiomerically pure (1R,3S)-camphoric acid was converted to the corresponding diamine 2 in an acid-mediated reaction with sodium azide to give the product in a yield of 58% (Scheme 2). Diamine 2 was then transformed into carbene precursor 3 in a ring-closing reaction with triethyl orthoformate (HC(OEt)3) and ammonium tetrafluoroborate (NH4BF4) in a yield of 79%. Finally, the introduction of R-groups by nucleophilic substitution reactions with halogenated alkanes affords carbene precursors salts 4–6 in yields ranging from 60 to 95% (Scheme 2).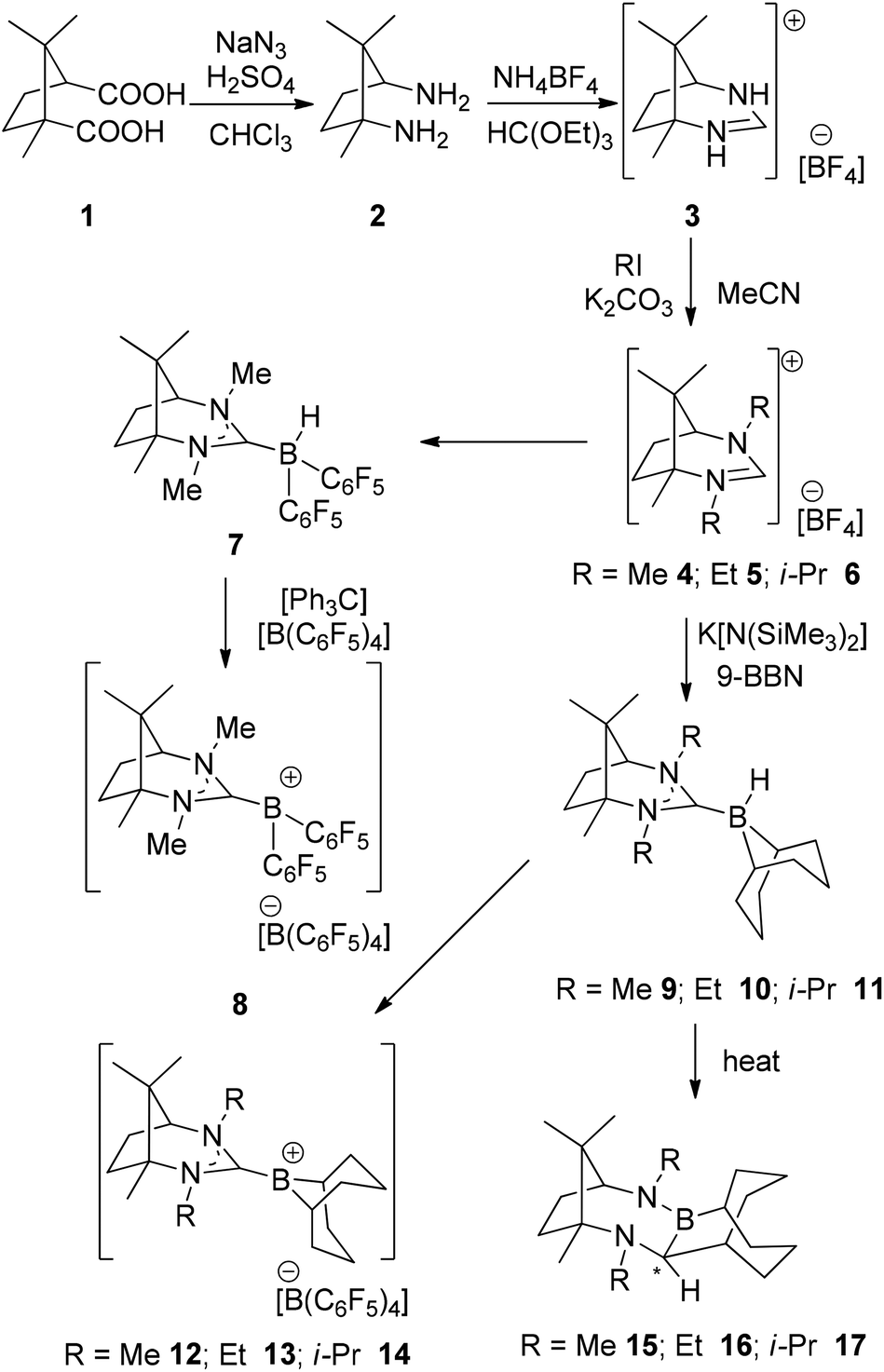 | ||
| Scheme 2 Synthesis of 2–17. | ||
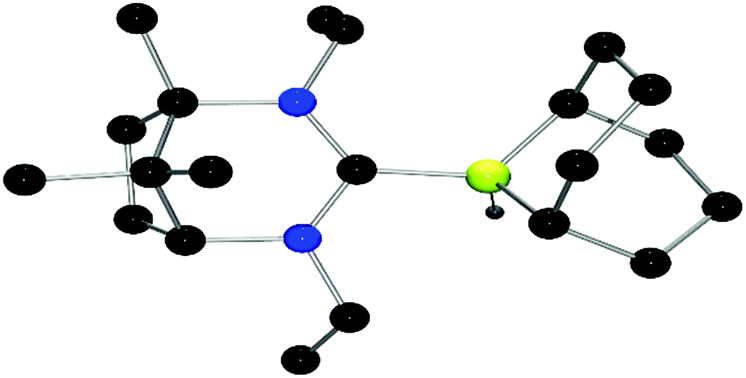
Reaction of the salt 4 with K[N(SiMe3)2] and subsequent addition of Piers’ borane (HB(C6F5)2) permitted the formation and isolation of adduct 7 in 60% yield. Recrystallisation by slow evaporation of the solvent afforded crystals suitable for an X-ray diffraction study. These data confirmed the structure of 7 (Fig. 2). As expected, the structure shows a pseudo tetrahedral geometry at the boron centre with a B–CNHC bond distance of 1.654(2) Å. Subsequent treatment of 7 with trityl tetrakis(pentafluorophenyl)borate resulted in hydride abstraction from boron and the isolation of the corresponding borenium cation 8 in 72% yield. 19F NMR resonances are consistent with diastereotopic C6F5 rings, and the appearance of 11B NMR signals at −16.7 and 56.0 ppm is consistent with the presence of the anionic and cationic boron centres, respectively. Unfortunately, attempts to employ 8 in the catalytic hydrogenation of N-benzylidene-tert-butylamine using a 5 mol% catalyst loading at 25 °C and 4 atm H2 were unsuccessful. Indeed, analysis of the reaction by NMR spectroscopy was consistent with the stoichiometric formation of 7 and the iminium cation. These data demonstrate that while the FLP derived from the borenium cation of 8 and imine effectively split H2, the carbene adduct 7 is insufficiently hydridic to deliver hydride to the iminium cation, thus inhibiting catalysis.
 | ||
| Fig. 2 POV-ray depictions of X-ray structure of 7. H-atoms are omitted for clarity. C: black; N: blue; B: yellow-green, F: pink. | ||
To circumvent this issue, carbene adducts of the weaker Lewis acid 9-BBN were targeted. The reaction of the salts 4–6 with K[N(SiMe3)2] and 9-BBN affords the corresponding adducts 9–11 which give rise to 11B NMR signals that are sharp doublets at 14.2 ppm (1JBH = 83 Hz), 14.3 ppm (1JBH = 84 Hz) and 12.9 ppm (1JBH = 84 Hz), respectively. These compounds were isolated in yields of 81% (9), 67% (10) and 63% (11) and gave the expected 1H NMR spectra. The structure of 10 was also confirmed by X-ray diffraction (Fig. 3). The B–CNHC distance is 1.693(5) Å which is significantly larger than those seen in NHC–borane adducts (ca. 1.63–1.65 Å).38,39
 | ||
| Fig. 3 POV-ray depiction of 10 (one of the two molecules in the asymmetric unit is shown). H-atoms have been omitted for clarity. C: black; N: blue; B: yellow-green, H: gray. | ||
By analogy to previous reports,39 reaction of adducts 9–11 with trityl tetrakis(pentafluorophenyl)borate gave the corresponding borenium cations 12–14, which were isolated in 83–93% yields. 1H NMR data show the concurrent formation of Ph3CH while the 11B NMR spectra show signals attributable to the cation as broad peaks at 89.1 ppm (12, 13) and 85.8 ppm (14). Crystals of 13 suitable for X-ray diffraction (Fig. 4) revealed an average B–CNHC bond length of 1.609(4) Å. This is slightly longer than the analogous B–C distances previously described in previously characterised borenium cations (1.5768(3) Å,38 1.580(3) Å (ref. 39)). This may be caused by the increase in steric demand of the present carbene relative to less demanding NHC ligands. Although the borenium centres in compounds 12–14 were expected to be more active than that in 8, exposure of a range of imines to 100 bar of H2 at 25 and 110 °C gave no reduction. Equimolar reactions of 12–14 with t-Bu3P and H2 also showed no reactivity at 25 °C, although upon heating to 110 °C for 24 h a slow formation of [HPt-Bu3][B(C6F5)4] in 26% yield was observed as evidenced by 31P NMR spectroscopy. Interestingly, the corresponding 11B NMR spectrum did not show the expected formation of the borane adduct but rather a broad signal at 44.0 ppm. These observations suggest the possibility of a thermal rearrangement of the borane adducts 9–11 (Scheme 2).
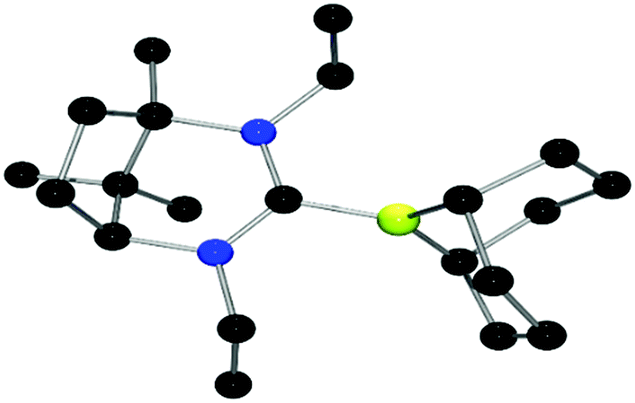 | ||
| Fig. 4 POV-ray depictions of the cation of 13 (one of the two molecules in the asymmetric unit is shown). H-atoms and the anion are omitted for clarity. C: black; N: blue; B: yellow-green. | ||
To examine this question, compounds 9–11 were allowed to stand in solution for 24 h. Each species was observed to react further, affording a new signal in the 11B NMR spectra at ca. 44 ppm. In the case of 9, mild heating at 50 °C for a few hours led to complete conversion to compound 15. Interestingly, the analogous diethyl-substituted derivative gave 16, only after overnight heating whereas the bulkiest adduct 11 was fully converted to 17 upon heating to 110 °C overnight. NMR data were consistent with three-coordinated boron centres in 15–17. An X-ray structure of 17 (Fig. 5) confirmed that this compound results from cleavage of a C–N bond in the carbene and formation of a new B–N-bond with a hydride shift from boron to the carbene carbon atom. This affects the net ring expansion of the carbene by the B–C link originating from the 9-BBN framework. The C–B bonds were found to average 1.581(3) Å, which is significantly shorter than the carbene C–B-bond in the precursor adduct. The newly formed B–N-bonds average 1.401(3) Å in the two molecules in the asymmetric unit. These are considerably shorter than typical B–N bonds.58 It is interesting to note that a single diastereomer of 17 was obtained demonstrating that the ring exapansion is stereoselective, presumably a result of the existing chiral centres in the carbene in 11en route to 17.
 | ||
| Fig. 5 POV-ray depiction of 17, one of the molecules in the asymmetric unit is shown. C: black; N: blue; B: yellow-green, H: gray. H-atoms, except those on chiral carbons are omitted for clarity. | ||
The formation of 15–17 suggests that thermal degradation is a significant factor in explaining the inability of 9–11 to act as the Lewis acid component of an FLP in the activation of dihydrogen. While activation of hydrogen may proceed to some extent, ring-expansion may out-compete hydride delivery and thus inhibit catalytic activity. Alternatively, the substituents on the carbene may act to sterically inhibit access to the vacant p-orbital on boron. This latter view is also consistent with the well-known increase in steric demands resulting from the expanded NCN-angle in carbene ligands in 9–11 compared to those seen in the imidazole-based NHC analogues.
In an effort to circumvent the steric demands in the above borenium cations, a second group of chiral borane adducts were selected. In this case, fused oxazole rings were employed to deter ring expansion and hopefully provide viable borenium catalysts for FLP hydrogenations. The triflate salts 18 and 19 were prepared by literature methods.41,42 Reaction of the former species with 9-BBN, BH3 and Cy2BCl afforded the anticipated carbene adducts 20–22 (Scheme 3). In a similar fashion, reaction of the tert-butyl-substituted ligand 19 and 9-BBN afforded adduct 23. The spectroscopic data were as expected. The 11B NMR signals of 20 and 23 were found to be similar, appearing at −16.9 and −16.4 ppm as doublets with 1JBH of 83 and 86 Hz, respectively. These resonances are consistent with four coordinate boron centres and thus are consistent with the formation of 20–23 as chiral carbene adducts. It is noteworthy that compounds 20, 21 and 23 have been exploited by Lindsay and McArthur as reagents for the stoichiometric, enantioselective reduction of ketones.48
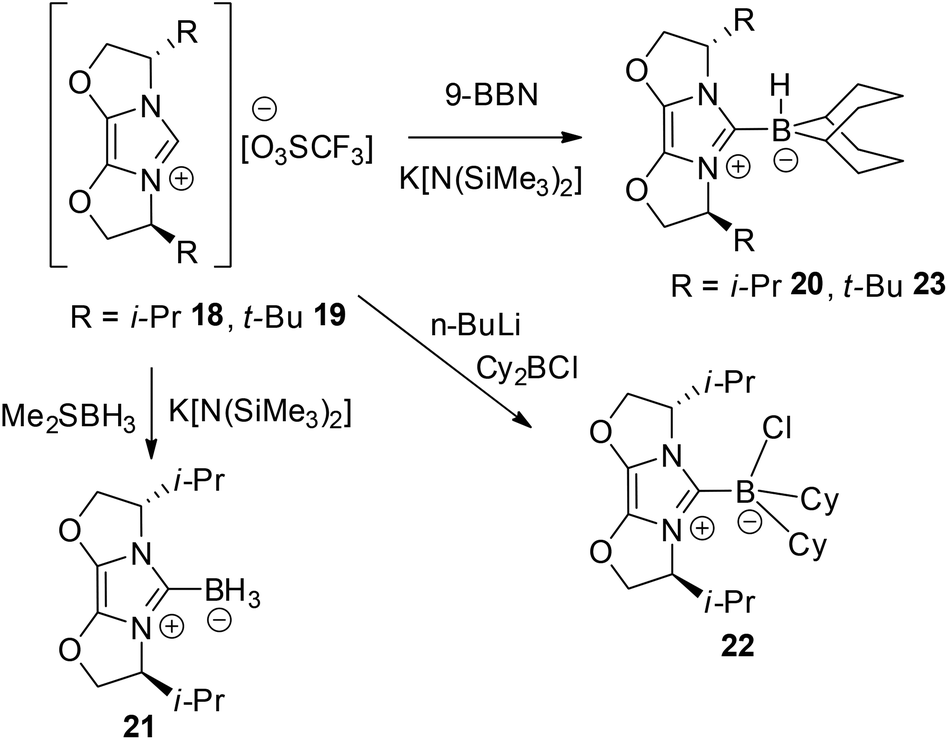 | ||
| Scheme 3 Synthesis of 20–23. | ||
The structures of these compounds were subsequently confirmed with crystallographic studies (Fig. 6–9). The corresponding B–Ccarbene bond distances in 20–23 were found to be 1.610(3) Å, 1.593(2) Å, 1.647(2) Å and 1.641(2) Å, respectively. These data reflect the steric impact of the substituents at the boron centre. The shortest B–C bond distance is seen for the borane adduct 21, while the longest of these four derivatives occurs for the Cy2BCl derivative 22. Comparing the difference between 20 and 23 indicates that steric conflict between the t-butyl substituents and the 9-BBN fragment results in a slight lengthening of the B–C bond.
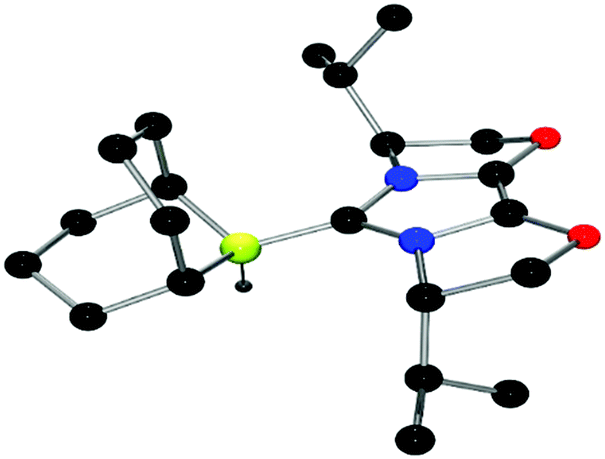 | ||
| Fig. 6 POV-ray depiction of 20, C: black; N: blue; B: yellow-green; O: red, H: gray. H-atoms except the BH are omitted for clarity. | ||
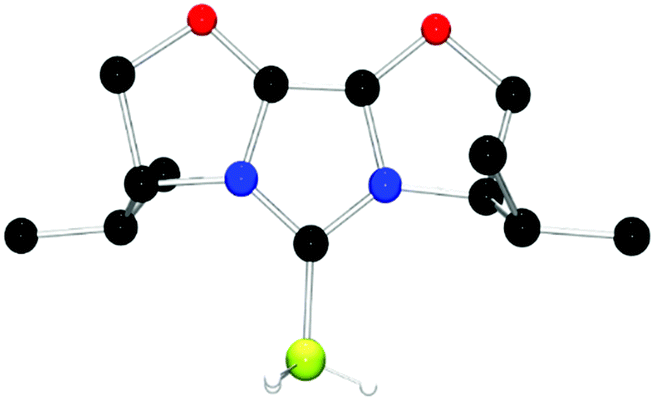 | ||
| Fig. 7 POV-ray depiction of 21, C: black; N: blue; B: yellow-green; O: red., H: gray. H-atoms except the BH are omitted for clarity. | ||
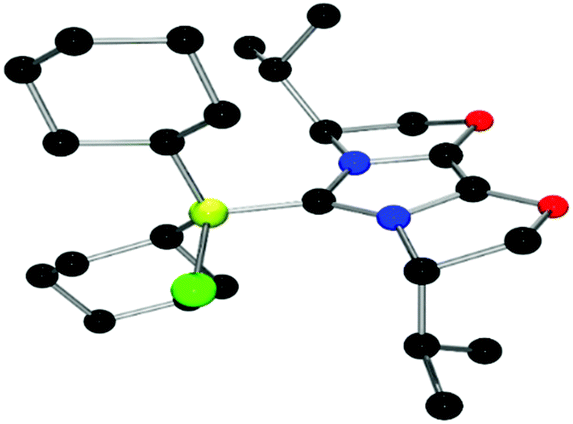 | ||
| Fig. 8 POV-ray depiction of 22, C: black; N: blue; B: yellow-green; O: red, Cl: green, H: gray. H-atoms are omitted for clarity. | ||
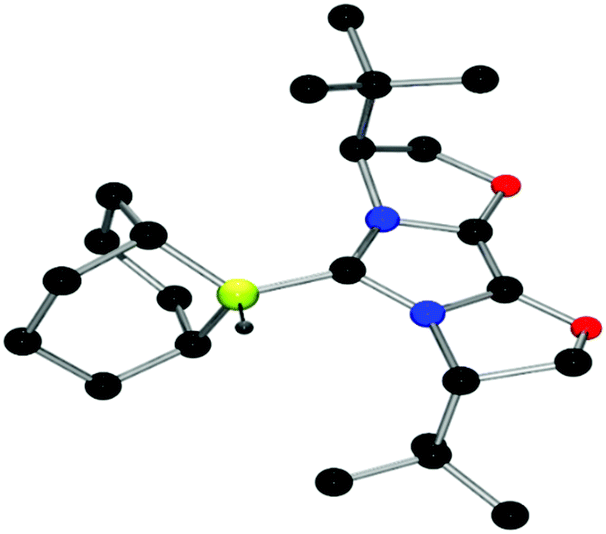 | ||
| Fig. 9 POV-ray depiction of 23, C: black; N: blue; B: yellow-green; O: red, H: gray. H-atoms except the BH are omitted for clarity. | ||
Chiral borenium cations derived from chiral boranes were also prepared. To this end, (+)-diisopinocampheylborane (Ipc2BH) which is accessible from the hydroboration of α-pinene, was combined with the carbenes 1,3-dimethylimidazol-2-ylidene and 1,3-di-iso-propylimidazol-2-ylidene to give 24 and 25 which could be isolated in 34% and 37% yield, respectively (Scheme 3). Similarly, reactions of unsymmetrically substituted NHCs 1-benzyl-3-methylimidazol-2-ylidene, 1-methyl-3-phenylimidazol-2-ylidene and 1-tert-butyl-3-methylimidazol-2-ylidene with Ipc2BH gave the corresponding adducts 26–28 in 83%, 56% and 60% yields, respectively (Scheme 4). 11B NMR spectra of each of these products show doublet resonances ranging from −6.8 to 9.7 ppm with B–H coupling constants of 77–86 Hz. Interestingly, 26 degrades in chloroform, presumably a result of the steric conflict between the NHC and isopinocampheyl groups. Crystals suitable for X-ray diffraction crystallography could be obtained for 24, 26 and 27 (Fig. 10–12). These data confirmed the formulations and the expected pseudotetrahedral geometry about boron. In the solid-state, B–CNHC bond lengths are 1.638(2) Å, 1.648(4) Å and 1.636(2) Å for 24, 26 and 27, respectively.
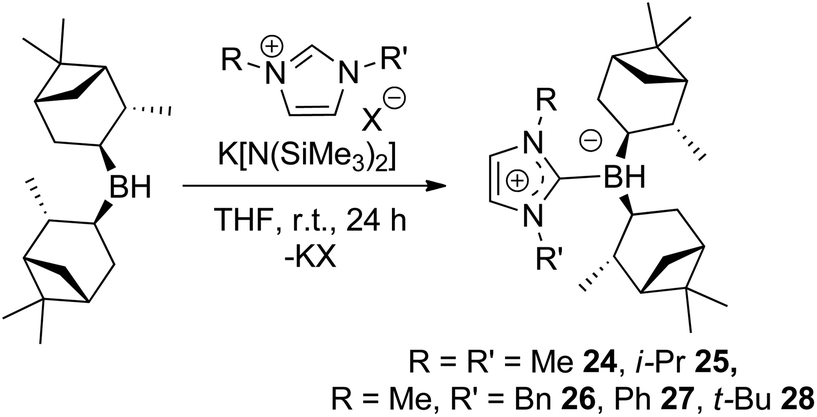 | ||
| Scheme 4 Synthesis of 24–28. | ||
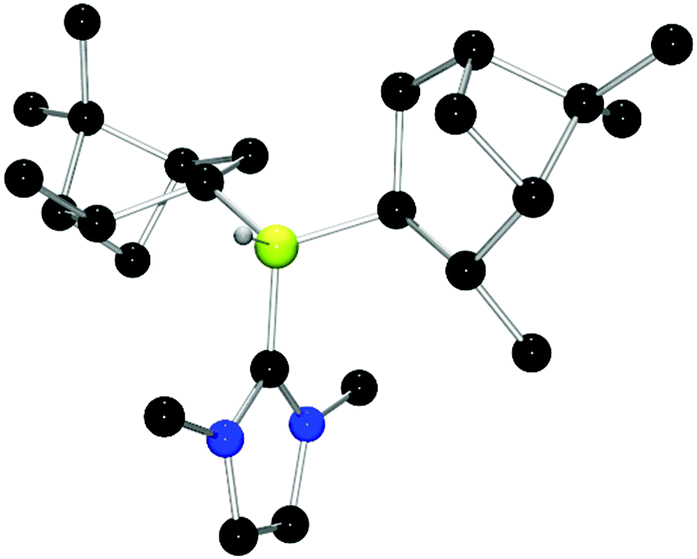 | ||
| Fig. 10 POV-ray depiction of 24. C: black; N: blue; B: yellow-green; O: red, H: gray. H-atoms except the BH are omitted for clarity. | ||
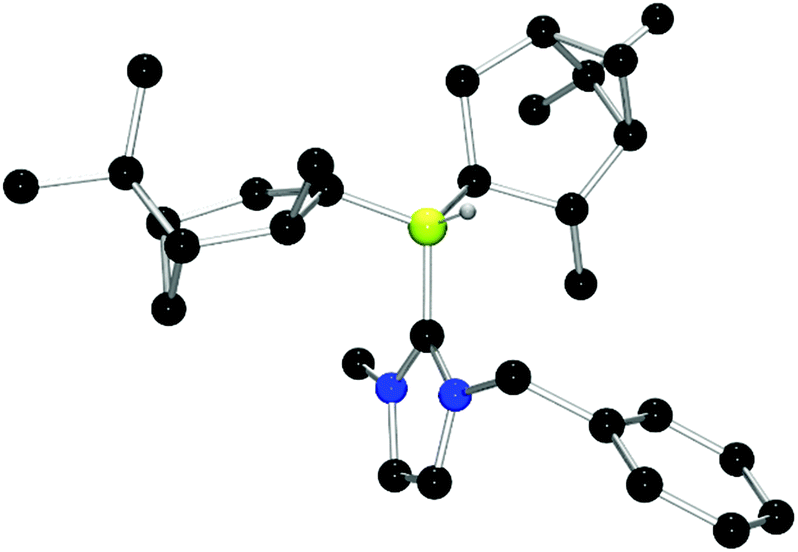 | ||
| Fig. 11 POV-ray depiction of 26. C: black; N: blue; B: yellow-green; O: red, H: gray. H-atoms except the BH are omitted for clarity. | ||
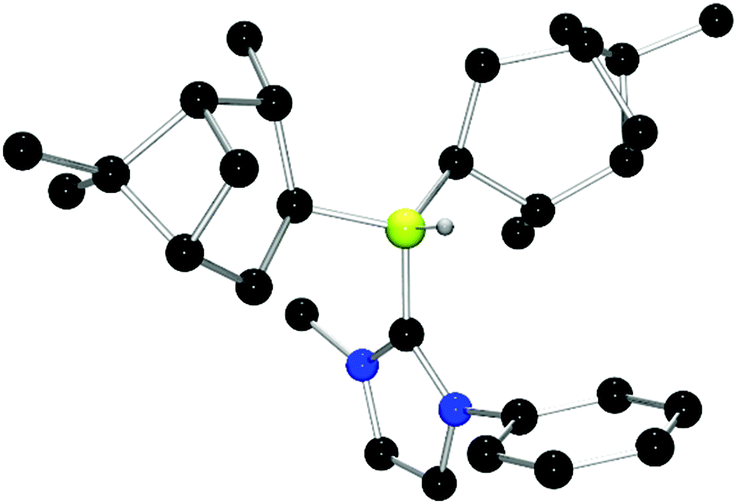 | ||
| Fig. 12 POV-ray depiction of 27. C: black; N: blue; B: yellow-green; O: red, H: gray. H-atoms except the BH are omitted for clarity. | ||
Triazolium-based carbene adducts of chiral boranes developed by Soderquist were also investigated49 as we speculated that the C1-symmetric environment about boron might improve selectivities in catalytic hydrogenations. To this end, lithium-B–H2-(10)-phenyl-9-borabicyclo[3.3.2]decane was prepared, however formation of the carbene adduct of the corresponding neutral borane (29) was isolated in low yield (Scheme 5). Presumably these difficulties are caused by the steric demands of both components. This view was consistent with the observation that use of the more hindered 1,3,4-triphenyl-1H-1,2,3-triazolium tetrafluoroborate failed to give an adduct. Furthermore, when rac-29 precatalyst was activated by [Ph3C][B(C6F5)4] in the hydrogenation of N-benzylidene tert-butylamine at elevated H2 pressure, no hydrogenation was seen, further indicating that excessive steric hindrance impedes effective hydride delivery even to otherwise reactive substrates.
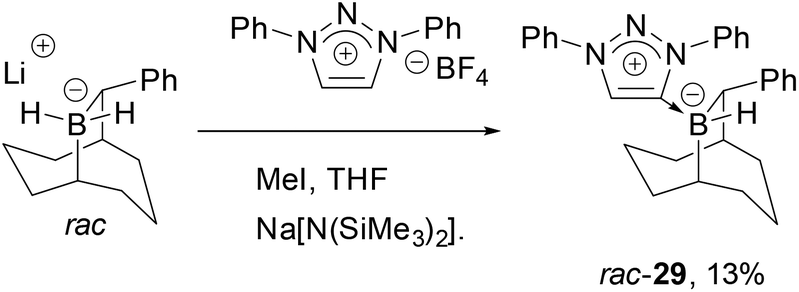 | ||
| Scheme 5 Synthesis of meso-ionic carbene 9-BBD adduct rac-29. | ||
An alternative strategy based on triazolium derivatives was based on use of a chiral triazolium derivatives. To this end, the known (S)-2-amino-2′-methoxy-1,1′-binaphthalene50 was reacted with tert-butylnitrite and trimetylsilyl azide to give the corresponding binaphthyl azide 30 in 82% yield. While sluggish, the Cu-catalysed Huisgen cyclo-addition of 31 with ethynyltrimethylsilane was achieved in 90% yield of 32 using [Cu(PPh3)Br] as the catalyst. Following deprotection, 32 was arylated to give the 1,2,3-triazolium tetrafluoroborate 33 in 89% yield. Subsequent reaction with Na[N(SiMe3)2] and 0.5 equivalents of (9-BBN)2 gave a 10:1 mixture of the two regio-isomeric carbene boranes 34a/b which was isolated in 82% combined yield (Scheme 6).
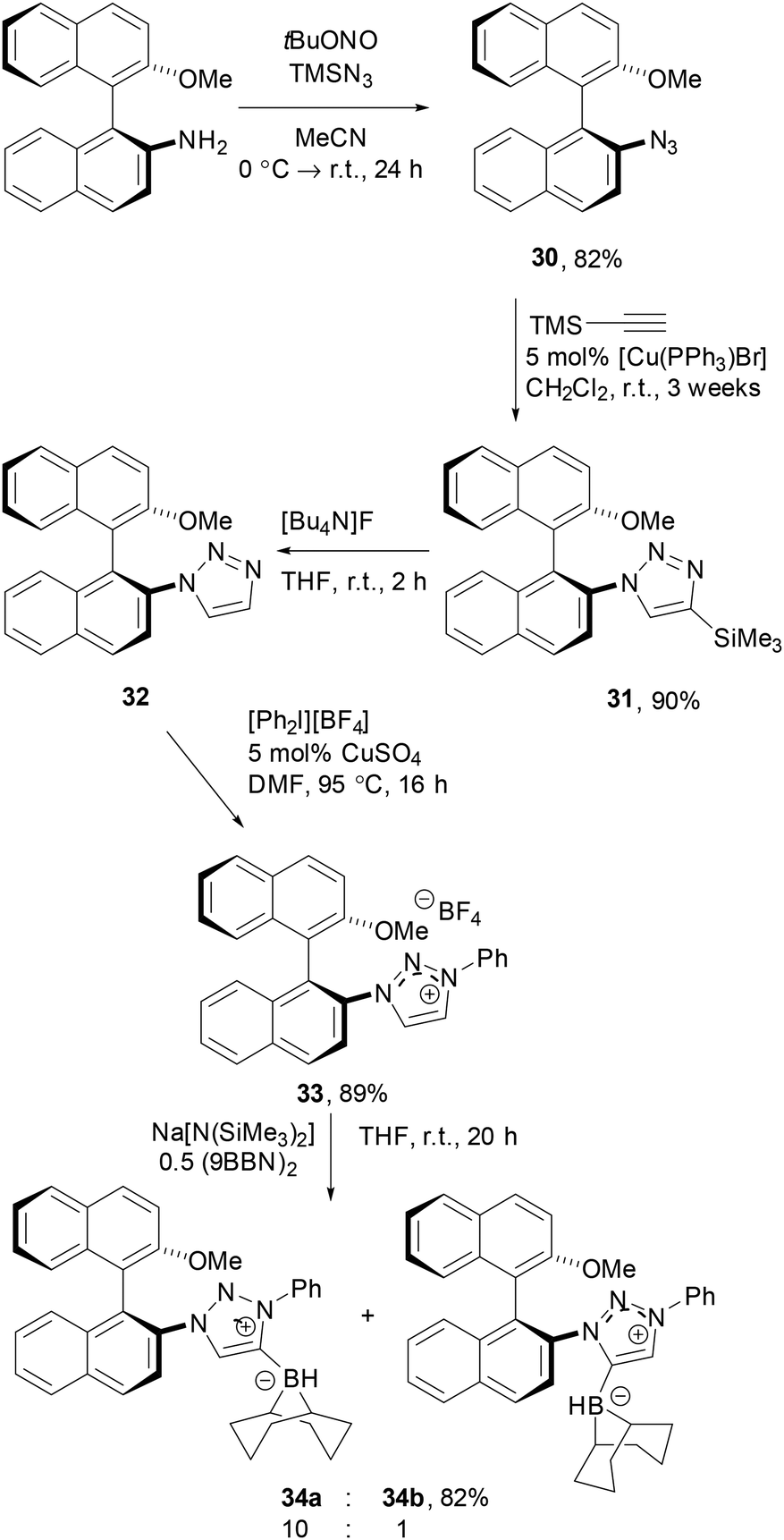 | ||
| Scheme 6 Synthesis of axial chiral carbene borane 34. | ||
The potential of 20, 21, and 23 to act as precursors for borenium cation-based catalysts was probed via a protocol in which the borane precursors were treated with trityl tetrakis(pentafluorophenyl)borate in CH2Cl2 and the in situ catalyst (5 mol%) was exposed to a solution of the prochiral imine substrate Ph(Me)C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh and pressurised under 102 atm of H2. Following the reaction, the conversion and products were characterised by NMR spectroscopy and by chiral HPLC. Interestingly, 21 and 23 are inactive giving 6% reduction at best even upon heating to 50 °C. In contrast, compound 20 serves as a competent catalyst precursor, affording effective imine reduction after 24 h at temperatures ranging from 0–50 °C. However, in all cases, the enantioselectivity for the ketamine reduction is low, ranging from 1–12% ee (Table 1). In a similar trial, reduction of MeOC6H4(Me)CNPh using the catalyst precursor 20 occurred after 18 h giving the product in 91% yield with 11% ee. In contrast, Ph(Me)CNCH2Ph as well as the ketones Et2CO and PhMeCO resulted in no reaction at all (see ESI†).
NPh and pressurised under 102 atm of H2. Following the reaction, the conversion and products were characterised by NMR spectroscopy and by chiral HPLC. Interestingly, 21 and 23 are inactive giving 6% reduction at best even upon heating to 50 °C. In contrast, compound 20 serves as a competent catalyst precursor, affording effective imine reduction after 24 h at temperatures ranging from 0–50 °C. However, in all cases, the enantioselectivity for the ketamine reduction is low, ranging from 1–12% ee (Table 1). In a similar trial, reduction of MeOC6H4(Me)CNPh using the catalyst precursor 20 occurred after 18 h giving the product in 91% yield with 11% ee. In contrast, Ph(Me)CNCH2Ph as well as the ketones Et2CO and PhMeCO resulted in no reaction at all (see ESI†).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Pre-catalyst | t (h) | T (°C) | Yield (%) | eec |
| a Toluene. b Chlorobenzene, all other catalysis done in CH2Cl2. Yield determined by 1H NMR spectroscopy. c Enantiomeric excess determined by chiral HPLC. | |||||
| 1 | 20 | 24 | 25 | 100 | 7 |
| 2 | 20 | 6 | 25 | 88 | 9 |
| 3 | 20 | 3 | 25 | 50 | 12 |
| 4 | 20 | 12 | 0 | 94 | 1 |
| 5 | 20 | 3 | 50 | 71 | 8 |
| 6 | 21 | 24 | 25 | 3 | 5 |
| 7 | 21 | 3 | 50 | 6 | 7 |
| 8 | 23 | 24 | 25 | 0 | — |
| 9 | 23 | 48 | 25 | 0 | — |
| 10 | 24 | 4 | 25 | 55 | 12 |
| 11 | 24 | 24 | −30 | 5 | 20 |
| 12 | 24 | 24 | 25 | 0 | — |
| 13 | 24 | 24 | 25 | 12 | 15 |
| 14 | 25 | 24 | −30 | <5 | 8 |
| 15 | 26 | 4 | 25 | 47 | 13 |
| 16 | 26 | 24 | −30 | 5 | 13 |
| 17 | 27 | 20 | −30 | <5 | — |
| 18 | 28 | 4 | 25 | 0 | — |
| 19 | 28 | 20 | −30 | 0 | — |
| 20 | 34 | 18 | 25 | 100 | 6 |
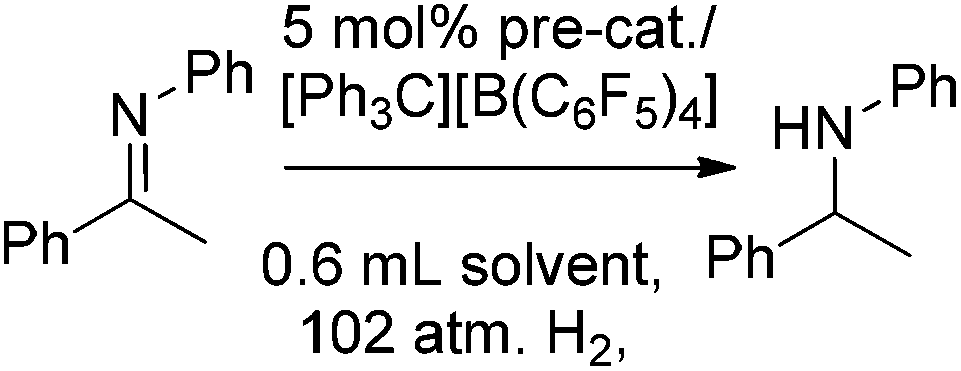
Similarly catalytic studies of 24–28 and 34 at the 5 mol% catalyst loading under 102 atm of hydrogen were studied in the reduction of N-(1-phenylethylidene)aniline. For 27, no reaction was observed after 4 h and for 28, only traces of hydrogenation to N-phenyl-1-phenylethylamine were observed. Compounds 24 and 26 gave conversions of 55% and 47%, respectively, reflecting the observation that high steric demands inhibit hydrogenation catalysis for NHC-stabilised borenium catalysts. Hydrogenation products of 24 and 26 showed enantiomeric excesses of 12% and 13%, respectively. Reduced temperature (−30 °C) hydrogenation under the similar conditions for 20 hours severely diminished conversion and offered only a modest improvement in stereoselectivity (22% ee). Under these latter conditions, 25 showed limited conversion and an enantioselectivity of only 8% (Table 1). The two isomers of 34 could not be separated by crystallisation and were therefore subsequently tested in hydrogenations as mixtures. At 10 mol% catalyst loading and 90 atm H2 pressure in CH2Cl2, full conversion to the corresponding amine was observed after less than 18 h reaction time. However, the product was obtained in only 6% ee.
While in some cases, the above borenium-based hydrogenation catalysts were effective for reduction of ketamines, in general, poor enantioelectivities were seen. In an effort to understand this, we speculated that the delivery of the hydride to the prochiral carbon, may be reversible leading to epimerisation of the resulting chiral amine. To probe this question, the chiral, enantiopure secondary amine (+)-bis[(R)-1-phenylethyl]amine (35) was exposed to a catalytic amount of the previously reported achiral MIC-borenium ion 36.40 While at ambient temperature, only trace amounts of meso-35 were detected by 1H NMR spectroscopy after 16 h, a significant degree of epimerisation was seen after 42 h at 65 °C. This stands in contrast, to the more rapid epimerisation of 35 with B(C6F5)359 and supports the view that it should be possible to exploit that borenium cations for asymmetric reductions.
Conclusions
In conclusion, exploiting both NHC and triazolium-based carbenes, we have reported the synthesis of a number of novel chiral carbene–borane adducts in which chirality is located either on the carbene or alternatively on the borane. While some of these species are effective precursors for imine reduction in hydrogenation catalysis, in general these systems exhibit poor enantioselectivity. Species substituted with C6F5 groups on boron preclude effective hydride delivery, while for 9-BBN and 9-BBD derivatives steric demands appear to be problematic under mild conditions. In some cases, elevated temperatures were shown to afford a thermally induced ring-expansion resulting from C–N bond cleavage in the carbene. Bisoxazoline-derived NHC borenium cations, the analogous axially chiral triazolium–borenium derivatives as well as borenium cations derived from the chiral borane, Ipc2BH, provide active hydrogenation catalysts however these systems provided limited stereoselectivity. Nonetheless, the low selectivities observed for these chiral borenium cation catalysts are not caused by selectivity erosion by epimerisation of the chiral product. This supports the notion that further catalyst design considerations may be fruitful. Our efforts to this end will be reported in due course.Acknowledgements
BARG is grateful to ERASMUS+ for funding. RLM would like to acknowledge the EPSRC (grant number EP/N02320X/1) and Leverhulme Trust for funding. DWS is grateful for the support from the National Sciences and Engineering Research Council of Canada (NSERC, Canada). DWS is grateful for the award of a Canada Research Chair. CMC acknowledges NSERC for Discovery and Accelerator Awards and the Canada Foundation for Innovation for instrumental support. The authors wish to acknowledge the Canadian Foundation for Innovation, project number 19119, and the Ontario Research Fund for support of the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers.Notes and references
- J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 4968–4971 CrossRef CAS PubMed.
- G. C. Welch, R. R. S. Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- L. J. Hounjet and D. W. Stephan, Org. Process Res. Dev., 2014, 18, 385–391 CrossRef CAS.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 85–110 CrossRef CAS PubMed.
- D. W. Stephan, Org. Biomol. Chem., 2012, 10, 9747–9747 Search PubMed.
- P. A. Chase, T. Jurca and D. W. Stephan, Chem. Commun., 2008, 1701–1703 RSC.
- P. A. Chase, G. C. Welch, T. Jurca and D. W. Stephan, Angew. Chem., Int. Ed., 2007, 46, 8050–8053 CrossRef CAS PubMed.
- D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS PubMed.
- D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Frustrated Lewis Pairs I: Uncovering and Understanding, 2013, vol. 332, pp. V-V Search PubMed.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 334 Search PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2010, 49, 46–76 CrossRef CAS PubMed.
- P. Spies, S. Schwendemann, S. Lange, G. Kehr, R. Fröhlich and G. Erker, Angew. Chem., Int. Ed., 2008, 47, 7543–7546 CrossRef CAS PubMed.
- H. D. Wang, R. Fröhlich, G. Kehr and G. Erker, Chem. Commun., 2008, 5966–5968 RSC.
- J. Paradies, Synlett, 2013, 777–780 CrossRef CAS.
- L. Greb, C. G. Daniliuc, K. Bergander and J. Paradies, Angew. Chem., Int. Ed., 2013, 52, 5876–5879 CrossRef CAS PubMed.
- L. Greb, P. Oña-Burgos, B. Schirmer, S. Grimme, D. W. Stephan and J. Paradies, Angew. Chem., Int. Ed., 2012, 51, 10164–10168 CrossRef CAS PubMed.
- K. Chernichenko, Á. Madarász, I. Pápai, M. Nieger, M. Leskelä and T. Repo, Nat. Chem., 2013, 5, 718–723 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, Angew. Chem., Int. Ed., 2015, 54, 8511–8514 CrossRef CAS PubMed.
- T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809–15812 CrossRef CAS PubMed.
- D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813–15816 CrossRef CAS PubMed.
- Frustrated Lewis Pairs II, Expanding the Scope, ed. D. W. Stephan and G. Erker, Springer Press, 2013, p. 334, Top. Curr. Chem Search PubMed.
- Frustrated Lewis Pairs I, ed. D. W. Stephan and G. Erker, Springer Press, 2013, p. 332, Top. Curr. Chem Search PubMed.
- D. W. Stephan, Top. Curr. Chem., 2013, 332, 1–44 CrossRef CAS PubMed.
- D. J. Chen and J. Klankermayer, Chem. Commun., 2008, 2130–2131 RSC.
- D. J. Chen and J. Klankermayer, Top. Curr. Chem., 2013, 334, 1–26 CrossRef PubMed.
- Y. B. Liu and H. F. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- M. Lindqvist, K. Borre, K. Axenov, B. Kotai, M. Nieger, M. Leskela, I. Papai and T. Repo, J. Am. Chem. Soc., 2015, 137, 4038–4041 CrossRef CAS PubMed.
- G. Ghattas, D. Chen, F. Pan and J. Klankermayer, Dalton Trans., 2012, 41, 9026–9028 RSC.
- V. Sumerin, K. Chernichenko, M. Nieger, M. Leskelä, B. Rieger and T. Repo, Adv. Synth. Catal., 2011, 353, 2093–2110 CrossRef CAS.
- M. J. Ingleson, Top. Organomet. Chem., 2015, 49, 39–71 CrossRef CAS.
- I. A. Cade and M. J. Ingleson, Chem. – Eur. J., 2014, 20, 12874–12880 CrossRef CAS PubMed.
- J. S. McGough, S. M. Butler, I. A. Cade and M. J. Ingleson, Chem. Sci., 2016, 7, 3384–3389 RSC.
- J. R. Lawson, E. R. Clark, I. A. Cade, S. A. Solomon and M. J. Ingleson, Angew. Chem., Int. Ed., 2013, 52, 7518–7522 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. A. Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474–487 CrossRef CAS PubMed.
- J. M. Farrell, R. T. Posaratnanathan and D. W. Stephan, Chem. Sci., 2015, 6, 2010–2015 RSC.
- J. M. Farrell, J. A. Hatnean and D. W. Stephan, J. Am. Chem. Soc., 2012, 134, 15728–15731 CrossRef CAS PubMed.
- P. Eisenberger, B. P. Bestvater, E. C. Keske and C. M. Crudden, Angew. Chem., Int. Ed., 2015, 54, 2467–2471 CrossRef CAS PubMed.
- S. E. Denmark, R. A. Stavenger, A. M. Faucher and J. P. Edwards, J. Org. Chem., 1997, 62, 3375–3389 CrossRef CAS PubMed.
- F. Glorius, G. Altenhoff, R. Goddard and C. Lehmann, Chem. Commun., 2002, 22, 2704–2705 RSC.
- M. C. Hansen and S. L. Buchwald, Org. Lett., 2000, 2, 713–715 CrossRef CAS PubMed.
- S. N. Greszler, J. T. Malinowski and J. S. Johnson, Org. Lett., 2011, 13, 3206–3209 CrossRef CAS PubMed.
- A. M. Oertel, V. Ritleng, L. Burr and M. J. Chetcuti, Organometallics, 2011, 30, 6685–6691 CrossRef CAS.
- C.-H. Hsieh, F.-I. Wu, C.-H. Fan, M.-J. Huang, K.-Y. Lu, P.-Y. Chou, Y.-H. O. Yang, S.-H. Wu, I. C. Chen, S.-H. Chou, K.-T. Wong and C.-H. Cheng, Chem. – Eur. J., 2011, 17, 9180–9187 CrossRef CAS PubMed.
- A. Rojas, L. Gomez-Hortiguela and M. A. Camblor, Dalton Trans., 2013, 42, 2562–2571 RSC.
- D. M. Lindsay and D. McArthur, Chem. Commun., 2010, 46, 2474–2476 RSC.
- A. Z. González, J. G. Román, E. Alicea, E. Canales and J. A. Soderquist, J. Am. Chem. Soc., 2009, 131, 1269 CrossRef PubMed.
- J. J. V. Veldhuizen, S. B. Garber, J. S. Kingsbury and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 4954–4955 CrossRef PubMed.
- K. Barral, A. D. Moorhouse and J. E. Moses, Org. Lett., 2007, 9, 1809–1811 CrossRef CAS PubMed.
- T. Lv, Z. Wang, J. You, J. Lan and G. Gao, J. Org. Chem., 2013, 78, 5723–5730 CrossRef CAS PubMed.
- M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 73, 4602–4607 CrossRef CAS PubMed.
- SHELX-TL, Bruker AXS Inc., Madison, WI, 2000 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2015, 71, 59–75 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, 1–19 RSC.
- J. M. Farrell, Z. M. Heiden and D. W. Stephan, Organometallics, 2011, 30, 4497–4500 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopy details. CCDC 1482172–1482179. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt02202b |
| This journal is © The Royal Society of Chemistry 2016 |