
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscrimination of chiral copper(II) complexes upon binding of galactonoamidine ligands†
Susanne
Striegler
* and
Jessica B.
Pickens
University of Arkansas, Department of Chemistry and Biochemistry, 345 N Campus Drive, Fayetteville, 72704, USA. E-mail: susanne.striegler@uark.edu
First published on 26th August 2016
Abstract
The coordination between N-p-methylbenzyl-D-galactonoamidine, a putative transition state analogue of the hydrolyis of glycosidic bonds, and symmetric and chiral binuclear copper(II) complexes was characterized by spectroscopic titration, isothermal titration calorimetry, circular dichroism spectroscopy, and DFT calculations to elucidate the binding sites in the carbohydrate upon coordination to selected metal complexes. For the formation of metal complex-glyconoamidine assemblies, contributions of the amidine site and of the hydroxyl group at C-2 in the glycon of the amidine are noted. The chiral complexes S- and R-Cu2bpdbo are discriminated by a third binding site in the carbohydrate that leads to higher stability of complexes derived from S-Cu2bpdbo (4–5 kcal mol−1) compared to those formed from R-Cu2bpdbo.
Introduction
Glycosidases are carbohydrate-hydrolyzing enzymes that are widely spread in nature and found in all organisms.1 The majority of these catalysts cleave oligosaccharides or glycoconjugates in agreement with the mechanisms proposed by Koshland in 1953 for retention or inversion of the configuration at the anomeric center.2,3 It is now generally accepted that oxocarbenium ion-like transition states are formed,4,5 which are obtained after flattening of the substrate chair conformation into a half-chair and accompanied by a change of the hybridization at the anomeric C-atom from sp3 to sp2. Additionally, the formation of a partial positive charge of the endocyclic O-atom and lengthening of the glycosidic bond are noted.3,6 Aside from the amino acids acting as nucleophile and proton donor in the active site of glycosidases during the hydrolysis, further supporting interactions via H-bond donor and/or acceptor interactions, π–π-stacking, hydrophobic or electrostatic interactions, or coordinative bonds to metal ions,7 may further support the transition state of the glycoside hydrolysis and thus render glycosidases extremely efficient catalysts.8 It was recently recognized that enzymes categorized in glycoside hydrolase family 4 have an absolute requirement for a divalent metal ion for catalytic activity, although the hydrolysis of their substrates deviates from the classic Koshland mechanisms and is not of interest here.9,10While interactions between carbohydrates and metal complexes are typically very weak in aqueous solution near neutral pH value,11 carbohydrates with anchoring groups, such as amino, thio, carboxyl or amidino groups, can still interact with metal ions under these conditions rendering glyconoamidines ideal for an evaluation of their ability to form assemblies with metal complexes and identify their binding sites.12,13 Very recent reports toward the evaluation of binding sites of carbohydrate ligands upon metal chelation include evaluations of metal chelation to 2-amino-2-deoxy derivatives of aldohexoses, their N-acetyl derivatives, and of glycose phosphates via trivalent binding sites.12–15 Further studies reveal formation of carbohydrate platinum(IV) complexes with thioglycoside ligands and their use to switch the selectivity of carbohydrate building blocks.16,17 More details and additional reports including complex formation between carbohydrates and transition metal or lanthanide ions may be found elsewhere.11,18–30
In this context, we disclose here for the first time a study to evaluate the coordination between a putative transition state analog of the glycoside hydrolysis and metal complexes. The assemblies are characterized by spectrophotometric titrations, isothermal titration calorimetry, CD spectroscopy and DFT methods. The binding sites of the carbohydrate ligand upon interaction with binuclear copper(II) complexes were elaborated, a significant differentiation of chiral complexes upon coordination was noted, and a different number of binding sites upon complex formation was identified as a rationale for the observed metal complex discrimination.
Results and discussion
Selection of the galactonoamidine ligand
In order to mimic the features of the transition state stabilization in glycoside hydrolyses and develop potent inhibitors of glycosidases, we and others evaluated galactonoamidines as inhibitors for the enzymatic hydrolysis of glycosides (Chart 1).31–34 Glyconoamidines resemble the above-discussed features associated with efficient transition state stabilization, and exist as tautomers with flattened half-chairs in their uncharged form. Glyconoamidines have a delocalized positive charge in aqueous solution at a pH value below their pKa (pH ∼ 8–10),33 and a delocalized negative charge in alkaline solution after deprotonation or metal complexation at pH 10.50 or above. The equilibrium of structures is dependent on solvent, pH and temperature and glycon configuration.35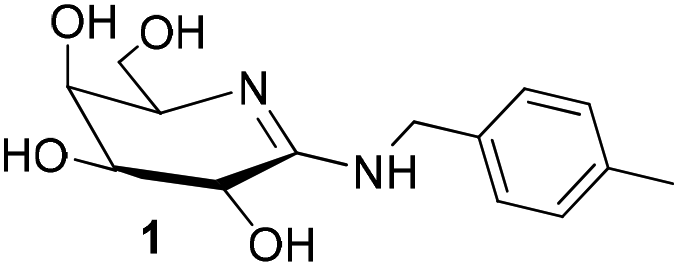 | ||
| Chart 1 Structure of N-p-methylbenzyl-D-galactonoamidine 1 in DMSO,35 putative TSA for the hydrolysis of glycosidic bonds. | ||
We initially developed a gram-scale synthesis of the precursor compound of 1,36 characterized all compounds as competitive inhibitors of β-galactosidase (A. oryzae) with inhibition constants in the low nanomolar concentration range (Ki = 8–63 nM),34,37 and prepared then a library of 22 galactonoamidines with alterations in their aglycon.38 Subsequently, N-p-methylbenzyl-D-galactonoamidine (1) was identified as a putative transition state analog (TSA) for the enzymatic hydrolysis of β-galactopyranosides by β-galactosidase (A. oryzae), while other members of the library were identified as fortuitous binders in the active site.37 Alteration in the glycon moiety revealed mandatory H-bond interactions of 1 over the hydroxyl groups at C2 and C4 with amino acids that act as proton donor and nucleophile in the active site of β-galactosidase (E. coli).39
Selection of binuclear copper(II) complexes
To evaluate galactonoamidine 1 upon coordination with binuclear complexes, Cu2bpdpo (2) was initially selected.40 The complex discriminates underivatized aldohexoses upon coordination in alkaline aqueous solution,41 and promotes glycoside hydrolysis in CAPS buffer at pH 10.50 about 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold over background.42
000-fold over background.42
Further investigations focused on the preparation of chiral analogs of 2, namely S-Cu2(bpdbo), S-(3), and R-Cu2(bpdbo), R-(3), that promoted remarkable discrimination of glycosidic bonds and epimeric substrates at near neutral conditions in aqueous solution (Chart 2).43
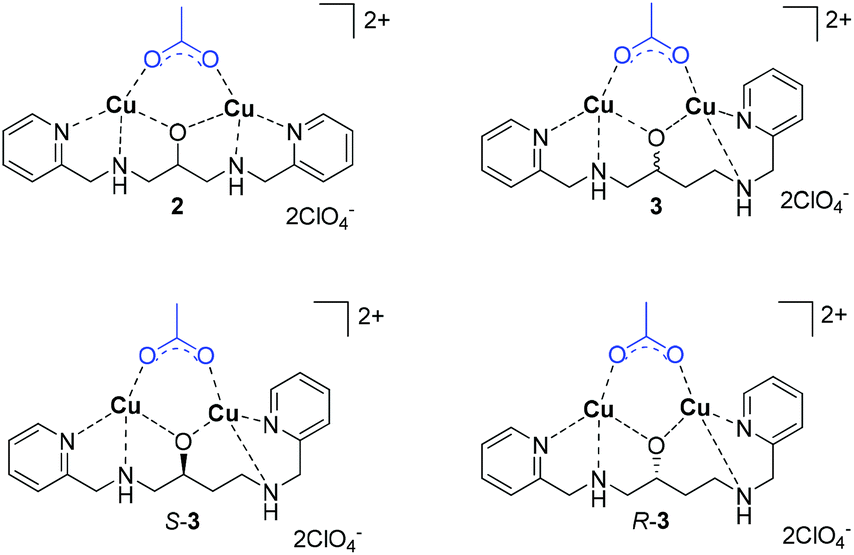 | ||
| Chart 2 Structures of selected binuclear copper(II) complexes. | ||
Structure and speciation of binuclear copper(II) complexes
All complexes were characterized in the solid state and in their composition in solution over an extended pH range.40,42–44 For the following discussion, only the speciation at pH 10.50 is relevant and thus briefly discussed.41,42,45 In short, spectrophometric titrations following the methods of Zuberbühler et al.,45 revealed that complex 2 exists primarily as [Cu2(L–H)(OH)]2+ species (88%) (Chart 3), whereas the remaining ligand is forming a Cu2[(L–H)(OH)2]+ species, with L (bpdpo) as backbone ligand of 2.36,37 By contrast, racemate 3 exists under the same conditions as equilibrium of a [Cu2(L–H)]3+ species (56%) and a [Cu2(L–H)(OH)]2+ species (44%), with L (bpdbo) as backbone ligand of 3. The speciation of chiral complexes S-3 and R-3 is identical to that of racemic 3 and thus derived from previous data.42 All species may interact with galactonoamidine 1.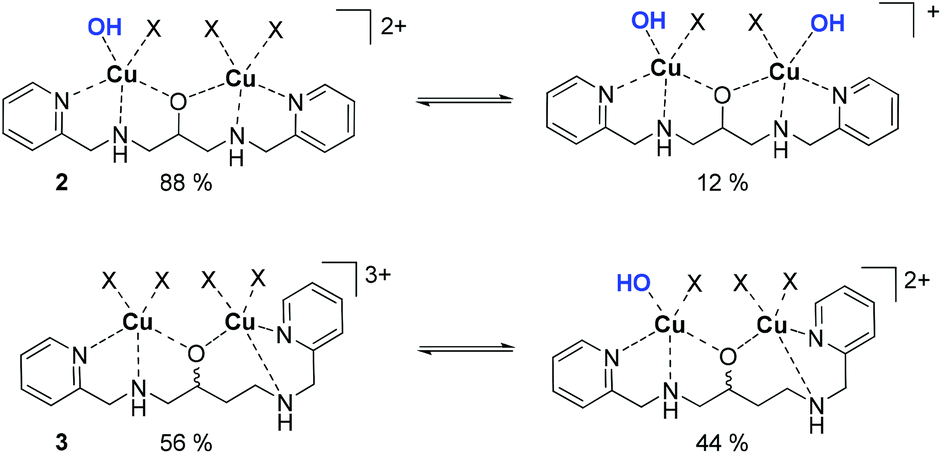 | ||
| Chart 3 Major species in aqueous solution at pH 10.50 derived from complexes 2 and 3; X = H2O. | ||
Speciation of assemblies derived from binuclear copper(II) complexes and the galactonoamidine ligand
To elucidate the interaction between 1 and the selected metal complexes between pH 6.0 and 11.5, their speciation was also determined using spectrophotometric titrations.45 Due to their basic character, amidines are predominately protonated in aqueous solution in the neutral pH range.46 While carbohydrate–metal complexes are typically very weak under these conditions,11 glyconoamidines can still coordinate to metal ions over the more basic and sterically less hindered imino N-atom in monodentate fashion in their neutral form or after further deprotonation above pH 10.0.47,48 Anchoring groups are known to act as the primary coordination sites that subsequently promote the coordination and deprotonation of the alcoholic hydroxyl groups of a carbohydrate.11 In their uncharged neutral form, amidines typically exist in equilibria of tautomers with endo- or exocyclic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bonds that may shift depending on the configuration of the glycon, the solvent composition, and pH value or temperature of the solution. As a representative example for all other associations, the formation of the metal complex–amidine assembly 2–1 is discussed in more detail.
N bonds that may shift depending on the configuration of the glycon, the solvent composition, and pH value or temperature of the solution. As a representative example for all other associations, the formation of the metal complex–amidine assembly 2–1 is discussed in more detail.
Upon interaction of amidine 1 with 2, a neutralization reaction of the positively charged amidine is anticipated that results in its monodentate binding to the major species of 2, [Cu2(L–H)(OH)]2+, in the neutral pH range. The resulting 2–1 assembly is then strengthened by subsequent deprotonation of the amidine in increasingly alkaline solution leading to chelation and additional binding sites. The corresponding major species derived from 1 and 2 are [Cu2(L–H)1]3+ (82%) at pH 7.35, [Cu2(L–H)1–H]2+ (68%) at pH 9.13, and [Cu2(L–H)1–2H]+ (77%) at pH 10.50 (Fig. 1).
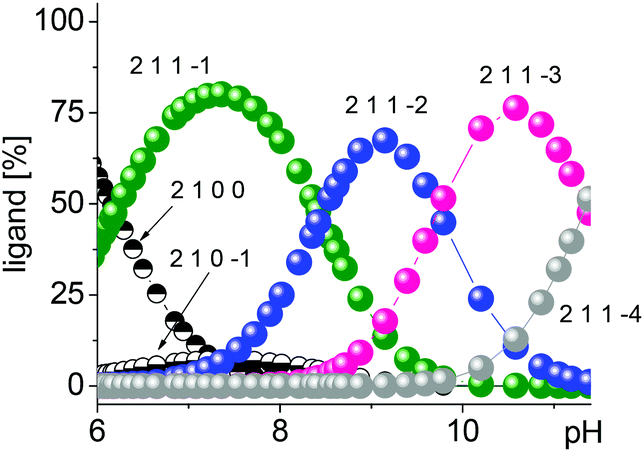 | ||
| Fig. 1 Speciation derived from complex 2 upon coordination to amidine 1. | ||
Identifying putative binding sites in galactonoamidine upon complex formation
The binding sites in galactonoamidine 1 during complex formation were subsequently elaborated indirectly by using galactonoamidine derivatives 1a–c (Chart 4) that will either show the same or diminished binding ability toward the metal complexes when compared to their parent 1.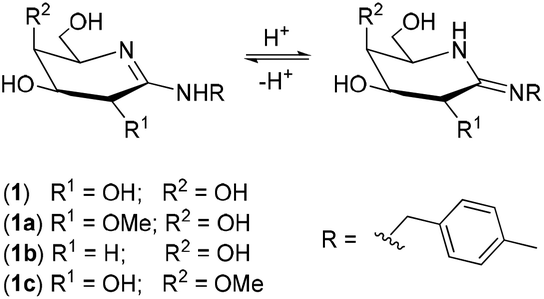 | ||
| Chart 4 Galactonoamidine ligand 1 and its derivatives 1a–c. | ||
All amidines have the same p-methylbenzyl aglycon eliminating any effect during coordination, but differ in the glycon at C-2 and C-4. The contributions of the hydroxyl groups at these positions in 1 toward complex formation will be apparent by the difference in metal complex binding ability, or the lack thereof, when using amidine derivatives 1a–c. The compounds were previously synthesized in this laboratory and characterized as potent competitive inhibitors of β-galactosidase (E. coli) in an unrelated study.39 For potent inhibition of the enzymatic hydrolysis, significant H-bond donor and acceptor interactions were identified at the hydroxyl groups in the glycon of 1 at C-2, and, to some lesser extent, at C-4 revealing the binding sites in the active site of the natural catalyst.39
Considering the reported acidity of hydroxyl groups in carbohydrates upon coordination to metal ions,47 deprotonation and binding sites at the hydroxyl groups at C-2, C-3, C-4 and/or C-6 of the glycon may be identified with decreasing binding strength in the given order. The amidino functional group will act as anchoring group, but may undergo further deprotonation upon coordination in alkaline solution above pH 10 as well.47 The stability of 1 against hydrolysis in alkaline condition was verified at pH 10.50 at 10 °C for at least 3 h; however, hydrolysis of 1 at higher temperatures was noted in the same time frame.49
With galactonoamidines 1 and 1a–c on hand, the binding sites of 1, and the strength of the resulting coordination compounds were determined using isothermal titration calorimetry (ITC) for the interaction of the amidines with complexes 2, S- and R-3, respectively. For this study, a pH of 10.50 was selected for three reasons: [Cu2(L–H)1–2H]+ was identified as a major species at this pH; 1 was shown to have sufficient stability against hydrolysis for at least 3 h, provided the temperature of the solution was kept at 10 °C or below;50 and a catalyst preparation envisaged in future efforts will benefit from strong chelating interactions between metal complexes and carbohydrates rather than monodentate binding.11,41,51
The isothermal titration of 1 and 1a–c into solutions of the metal complexes results in exothermic binding events (Fig. 2). As symmetric complex 2 was found to behave like S-3 throughout the study (Table 1), the following discussion focuses on the chiral complexes in order to streamline the discussion and highlight unusual differences in their coordination behaviour.
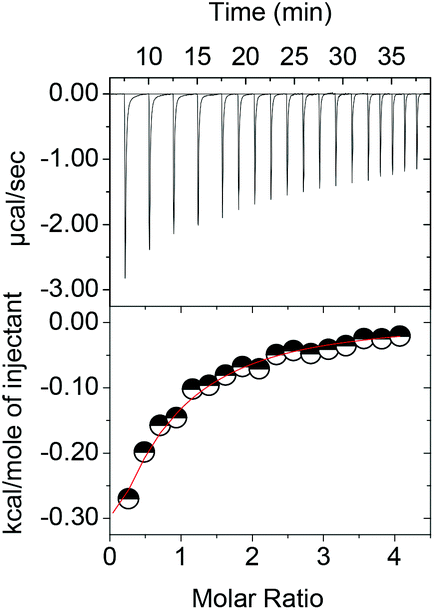 | ||
| Fig. 2 Isothermal titration of 1 into R-3 in 5 mM CAPS buffer at pH 10.50 and 10 °C as representative example. | ||
| Complex | 1 | 1a | 1b | 1c |
|---|---|---|---|---|
| a — No binding. The Gibb's free energy ΔG was calculated from the experimental data as ΔG = ΔH − TΔS, T = 283.15 K) | ||||
| Cu2bpdpo, 2 | ||||
| K 1 | 47 ± 21 | —a | 484 ± 39 | 46.3 ± 13 |
| ΔH1 | 493 ± 387 | —a | −1645 ± 55.5 | 1829 ± 541 |
| ΔS1 | 9.4 | —a | 10.8 | 14.1 |
| K 2 | 1130 ± 390 | —a | —a | 469 ± 140 |
| ΔH2 | −1529 ± 817 | —a | —a | −6920 ± 2.25 × 103 |
| S 2 | 8.6 | —a | —a | −12.2 |
| K 3 | 1720 ± 520 | —a | —a | 3.49 × 103 ± 8.3 × 102 |
| ΔH3 | 313 ± 71.2 | —a | —a | 4247 ± 2.24 × 103 |
| ΔS3 | 15.9 | —a | —a | 31.2 |
| ΔG total | −10.3 | — | −4.7 | −10.2 |
| S-Cu2bpdbo, S-3 | ||||
| K 1 | 559 ± 92 | 243 ± 195 | 792 ± 60 | 394 ± 47 |
| ΔH1 | −576.9 ± 22.0 | −535.8 ± 70.4 | −1185 ± 30.9 | −973.1 ± 45.3 |
| ΔS1 | 10.5 | 9.0 | 11.7 | 8.4 |
| K 2 | 152 ± 51 | —a | —a | 366 ± 49 |
| ΔH2 | 1628 ± 483 | —a | —a | 1527 ± 89.5 |
| ΔS2 | 15.7 | —a | —a | 17.1 |
| K 3 | 2900 ± 730 | —a | —a | 809 ± 150 |
| ΔH3 | −1888 ± 578 | —a | —a | −1564 ± 190 |
| ΔS3 | 9.2 | —a | —a | 7.8 |
| ΔG total | −10.8 | −3.1 | −4.5 | −10.4 |
| R-Cu2bpdbo, R-3 | ||||
| K 1 | 209 ± 94 | 377 ± 61 | 1610 ± 120 | 686 ± 240 |
| ΔH1 | −505 ± 48 | −423.0 ± 31.5 | −1402 ± 27 | −400.3 ± 36.6 |
| ΔS1 | 8.8 | 15.0 | 10.6 | 11.6 |
| K 2 | 230 ± 120 | —a | —a | 536 ± 230 |
| ΔH2 | −923 ± 679 | —a | —a | 111.3 ± 98.0 |
| ΔS2 | 7.6 | —a | —a | 12.9 |
| ΔG total | −6.1 | −4.7 | −4.4 | −6.5 |
Most remarkably, the coordination of amidine 1 to complex S-3 occurs over three binding sites, while only two different sites are identified for its coordination to R-3. The corresponding Gibb's free energies of the amidine–metal complex assemblies reflect this observation and differ by about 4–5 kcal mol−1 revealing a significant differentiation of the chiral complexes by amidine 1 upon coordination.
Derivatizations of the binding sites at C-2 of the glycon in 1 by methylation of the hydroxyl group eliminate the coordination ability of the resulting amidine 1a. The methylation is likely to cause a combination of steric hindrance around the amidine anchoring group and removal of the alcoholate formation that indicates strong participation of a deprotonated hydroxyl group at C-2 in the parent amidine 1 during coordination to metal complexes. Likewise, decreased binding ability is noted for deoxyamidine 1b in comparison to 1 that is ascribed to monodentate binding over the amidine anchor group and a lack of support for chelation over a binding site at C-2. By contrast, the complex formation between metal complexes and amidine 1c does not differ significantly from the association of 1 indicating that the hydroxyl group at C-4 in the parent is not participating in the binding events.
To further elucidate the discrimination of S- or R-3 upon binding to 1, the molar ellipticities of the resulting coordination compounds were determined by circular dichroism (CD) spectroscopy, and correlated to those of the original complexes (Fig. 3). The buffer, amidine 1 and its derivatives do not show contributions to the circular dichroism under the selected conditions.
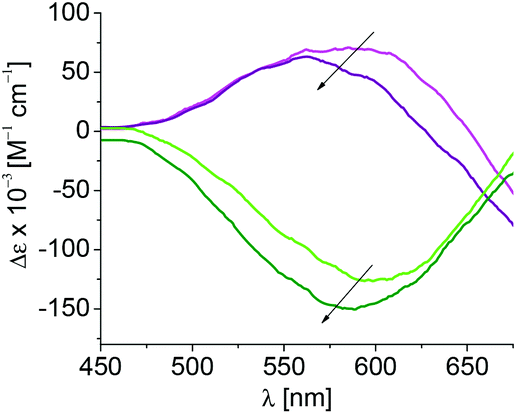 | ||
| Fig. 3 Circular dichroism of S-3 (magenta) and R-3 (green) upon coordination of 1 resulting in formation of complexes S-3–1 (purple) and R-3–1 (olive) in 5 mM CAPS buffer at pH 10.50 and 10 °C; λmax = 603 nm. | ||
Complex S-3 shows positive and R-3 negative molar ellipticity with an absorption maximum at 603 nm. A blue-shift of 40 nm is observed upon coordination of 1 to S-3 and of 15 nm for coordination of 1 to R-3, reflecting stronger binding of 1 to S-3 than toward R-3 (Table 2). Smaller shifts of the maxima are observed during coordination of 1a indicating steric hindrance during coordination imposed by the methoxy group at C-2 in 1a, and in turn participation of the free hydroxyl group at C-2 in parent 1 during association with S-3 and R-3. The complex formation of 1c with both chiral complexes results in shifts comparable to those observed during the coordination of the parent 1 indicating a lack of contribution of the hydroxyl group at C-4 to complex formation.
| Amidine | λ max(S-3) [nm] | λ max (R-3) [nm] |
|---|---|---|
| — | 603 | 603 |
| 1 | 563 | 588 |
| 1a | 572 | 590 |
| 1c | 563 | 584 |
The combined experimental results emphasize coordination of 1 over the amidino anchoring group, a deprotonated hydroxyl group at C-2 of the glycon, and a lack of contributions of the hydroxyl group at C-4 for association toward all complexes. Additionally, the experimental data give evidence for binding of 1 over three binding sites and chelation toward S-3 that is by 4–5 kcal mol−1 stronger than the complex formation from R-3 over two binding sites. The experimental results thus suggest coordination of 1 to S-3via a deprotonated amidine binding site, a deprotonated hydroxyl group at C-2, and a third binding sites via the hydroxyl group at C-3 or C-6, which is absent during the association of 1 with R-3. A putative coordination mode of the chiral complexes to amidine 1 is deduced from these observations (Chart 5). Coordination of metal ions or metal complexes to carbohydrates and their derivatives in tridentate fashion has been described.11,12,14,18,22,41,52,53
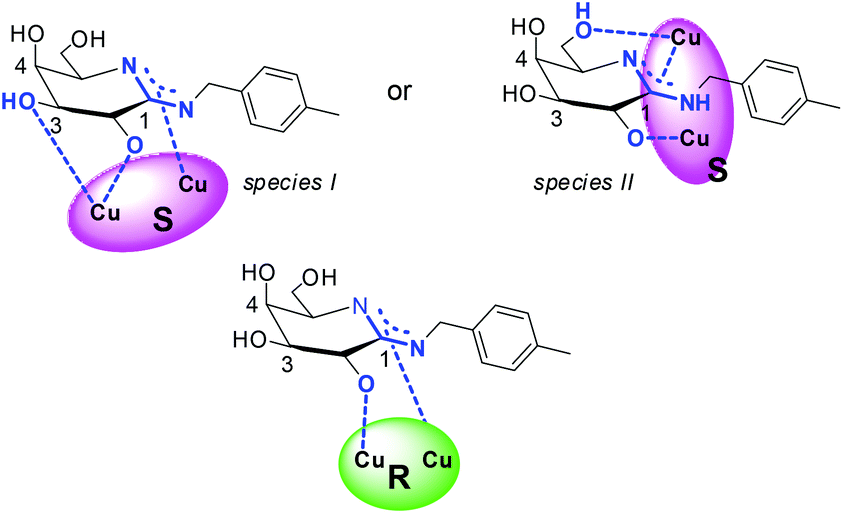 | ||
| Chart 5 Putative coordination of S-3 and R-3 to amidine 1. | ||
To obtain further evidence of the coordination mode of amidine 1 to S-3via the hydroxyl group at C-3 (species I) or at C-6 (species II), the difference in the Gibbs's free energy of the both assemblies was computed in the gas phase and in aqueous solution using DFT methods and the COSMO model.54,55
The resulting energies of the two putative assemblies derived from S-3 and 1 are by 25.2 kcal mol−1 (gas phase) and 28.3 kcal mol−1 (water) lower, when 1 coordinates over the hydroxyl group at C-3 (Chart 5, species I) instead of at C-6 (Chart 5, species II). The computed values agree well with the general preference for metal ion coordination to carbohydrates and are also in line with the tridentate metal-binding site model proposed by Klüfers et al.11,52
Conclusions
Galactonoamidines were previously identified as potent inhibitors of glycosidases by us and others, and are studied here as ligands of binuclear metal complexes. The coordination between N-p-methylbenzyl-D-galactonoamidine, a putative transition state analogue of the hydrolysis of glycosidic bonds, and symmetric and chiral binuclear copper(II) complexes was characterized by experimental and theoretical methods. In more detail, the interactions between galactonoamidine and selected metal complexes were characterized by spectroscopic titration, isothermal titration calorimetry, circular dichroism spectroscopy, and DFT calculations, and elucidated the binding sites in the carbohydrate ligand upon coordination. Different contributions of the hydroxyl groups in the glycon were also noted that lead to significantly different binding strengths of the resulting galactonoamidine- metal complex assemblies. For the formation of these assemblies, contributions of the amidine site and of the hydroxyl group at C-2 in the glycon of the amidine were experimentally identified. Further evidence of the experimental data suggested that the chiral complexes S- and R-Cu2bpdbo are discriminated by a third binding site that leads to S-Cu2bpdbo-galactonoamidine assemblies that are 4–5 kcal mol−1 more stable than those formed from R-Cu2bpdbo. Computational studies at DFT level then revealed interactions of the hydroxyl group at C-3, but not at C-6, in the galactonoamidine glycon as cause for the noted higher stability upon coordination of the carbohydrate ligand to S-Cu2bpdbo.Aside from the immediate coordination results and characterization of resulting complexes, our results provide insights for the interaction of mechanism-based inhibitors with metal ions. These results are likely to impact further studies toward the optimization of structure–activity relationships when targeting inhibition of metalloenzymes, such as β-galactosidase (E. coli), that are not accessible in solution otherwise.
Experimental
Instrumentation
All ITC studies were performed on a Microcal ITC200 from GE Healthcare; the ITC200 v1.26.1 software supplied by the manufacturer was used to record the data and Origin 7 v7.0552 was used for data analysis. CD spectra were recorded on a Jasco J-1500 CD Spectrometer Model 150 with EXOS liquid cooling system from Koolance Inc. with the supplied Spectra Manager software Version 2.12.00 using a Fisher Scientific Suprasil quartz microcell with 500 μL capacity, 45 mm height and an optical path length l of 10 mm at 10.00 ± 0.01 °C. UV-vis spectra were recorded at 30.0 ± 0.1 °C over a range of 200–800 nm on a Varian Cary 50 with WinUV Analysis Suite software, version 3.0, using disposable Brandtech macro cells (220–900 nm) of 1 cm thickness and 4.5 mL volume. The pH values were measured using a Beckman Φ 250 pH meter equipped with a refillable ROSS Orion combination pH electrode with a 165 mm long epoxy body, a 95 mm long semi-micro tip and an 8 mm diameter. The pH meter was calibrated before each set of readings. Nanopure water at a resistance of 18.2 mΩ was obtained from a ThermoScientific Barnstead E-pure™ water purification system.Methods and materials
Spectrophotometric titrations
In a typical experiment, a 2 mL aqueous solution that is 1 mM in Cu2bpdpo 2, 5 mM in amidine 1, and 50 mM in NaClO4 was titrated with 10 μL aliquots of freshly prepared aqueous sodium hydroxide solution after equilibration at 30.0 ± 0.1 °C. After each aliquot addition, UV/Vis spectra were recorded as a function of the pH value. The resulting data were computed by the global fitting model implemented in the analysis program Specfit.45 The concentration of the sodium hydroxide solution was increased gradually over the course of the titration from 0.005 M to 5 M.Isothermal titration calorimetry
All experiments were conducted at a nominal temperature of 10 °C (10.08033 ± 0.00005 °C) in a 5 mM CAPS buffer solution. The buffer solution was prepared at ambient temperature with a pH of 10.25 resulting in a pH of 10.50 at 10 °C.Circular dichroism
All experiments were conducted at 10 °C in a 5 mM CAPS buffer solution. The buffer solution was prepared at ambient temperature with a pH of 10.25 resulting in a pH of 10.50 at 10 °C. The total volume of all experiments was kept constant at 400 μL.| Δε = θ × 100/(c × l × 3298.2) | (1) |
The resulting data were plotted against the wavelength λ (nm) after application of a smoothing 25-point Savitzky–Golay filter.
Computational analysis
All electronic structure calculations in gas phase and solution were performed with PQSmol.54 Low energy conformers of the complexes derived from amidine 1 and S-3 were calculated with density functional theory at the B3LYP/m6-31G(d) theory level.57,58 All stationary points were examined with vibrational analyses and confirmed as minima with zero imaginary frequency. The solvation free energies are computed at the COSMO/B3LYP/m6-31+G(d) level of theory in water at 298.15 K and 1 atm.55,59Acknowledgements
Support of this research to S. S. by the National Science Foundation (CHE-1305543), the Arkansas Biosciences Institute, access to PQSmol,54 and invaluable discussions with Dr Pulay are gratefully acknowledged. The authors thank Natasha Zimmerman and James D. Barnett for conducting the spectrophotometric titrations. The facilities used in this study were supported by Grant Number P30 GM103450 from the National Institute of General Medical Sciences (NIGMS) of the National Institutes of Health (NIH).Notes and references
- V. Kren and J. Thiem, Chem. Soc. Rev., 1997, 26, 463–473 RSC.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS.
- M. L. Sinnott, Chem. Rev., 1990, 90, 1171–1202 CrossRef CAS.
- L. Bohe and D. Crich, Nat. Chem., 2016, 8, 99–100 CrossRef CAS PubMed.
- A. Martin, A. Arda, J. Désiré, M. A. Mingot, N. Probst, P. Sinaÿ, J. Jiménez Barbero, S. Thibaudeau and Y. Blériot, Nat. Chem., 2016, 8, 186–191 CAS.
- D. L. Zechel and S. G. Withers, Acc. Chem. Res., 2000, 33, 11–18 CrossRef CAS PubMed.
- D. H. Juers, R. H. Jacobson, D. Wigley, X. J. Zhang, R. E. Huber, D. E. Tronrud and B. W. Matthews, Protein Sci., 2000, 9, 1685–1699 CrossRef CAS PubMed.
- T. M. Gloster and G. J. Davies, Org. Biomol. Chem., 2010, 8, 305–320 CAS.
- V. L. Y. Yip and S. G. Withers, Biochemistry, 2006, 45, 571–580 CrossRef CAS PubMed.
- A. C. Elliott, B. F. Li, C. A. J. Morton, J. D. Pownall, T. Selwood, M. L. Sinnott, I. J. L. Souchard and A. K. Stuarttilley, J. Mol. Catal., 1988, 47, 255–263 CrossRef CAS.
- B. Gyurcsik and L. Nagy, Coord. Chem. Rev., 2000, 203, 81–149 CrossRef CAS.
- T. Schwarz, D. Heß and P. Klüfers, Dalton Trans., 2010, 39, 5544–5555 RSC.
- M. Steinborn, M. Suhanji and P. Klüfers, Dalton Trans., 2013, 42, 5749–5754 RSC.
- K. Gilg, T. Mayer, N. Ghaschghaie and P. Klüfers, Dalton Trans., 2009, 7934–7945 RSC.
- P. Grimminger and P. Klüfers, Dalton Trans., 2010, 39, 715–719 RSC.
- C. Vetter, P. Pornsuriyasak, J. Schmidt, N. P. Rath, T. Ruffer, A. V. Demchenko and D. Steinborn, Dalton Trans., 2010, 39, 6327–6338 RSC.
- A. K. Kayastha, X. G. Jia, J. P. Yasomanee and A. V. Demchenko, Org. Lett., 2015, 17, 4448–4451 CrossRef CAS PubMed.
- S. Yano, Coord. Chem. Rev., 1988, 92, 113–156 CrossRef CAS.
- S. J. Angyal, Adv. Carbohydr. Chem. Biochem., 1989, 47, 1–43 CrossRef CAS.
- D. M. Whitfield, S. Stojkovski and B. Sarkar, Coord. Chem. Rev., 1993, 122, 171–225 CrossRef CAS.
- R. P. Bandwar and C. P. Rao, Curr. Sci., 1997, 72, 788–796 CAS.
- D. Steinborn and H. Junicke, Chem. Rev., 2000, 100, 4283–4318 CrossRef CAS PubMed.
- V. Benessere, R. Del Litto, A. De Roma and F. Ruffo, Coord. Chem. Rev., 2010, 254, 390–401 CrossRef CAS.
- L. Yang, X. Hua, J. Xue, Q. Pan, L. Yu, W. Li, Y. Xu, G. Zhao, L. Liu, K. Liu, J. E. Chen and J. Wu, Inorg. Chem., 2012, 51, 499–510 CrossRef CAS PubMed.
- H. Hu, J. Xue, X. Wen, W. Li, C. Zhang, L. Yang, Y. Xu, G. Zhao, X. Bu, K. Liu, J. E. Chen and J. Wu, Inorg. Chem., 2013, 52, 13132–13145 CrossRef CAS PubMed.
- Y. Jiang, J. Xue, X. Wen, Y. Zhai, L. Yang, Y. Xu, G. Zhao, K. Kou, K. Liu, J. E. Chen and J. Wu, J. Mol. Struct., 2016, 1109, 179–191 CrossRef CAS.
- T. Bauer and M. Majdecki, Curr. Org. Chem., 2014, 18, 1749–1767 CrossRef CAS.
- M. Boege, C. Fowelin, P. Bednarski and J. Heck, Organometallics, 2015, 34, 1507–1521 CrossRef CAS.
- A. S. Henderson, J. F. Bower and M. C. Galan, Org. Biomol. Chem., 2016, 14, 4008–4017 CAS.
- C. Shen and P.-F. Zhang, Curr. Org. Chem., 2013, 17, 1507–1524 CrossRef CAS.
- Y. Bleriot, A. Genre-Grandpierre and C. Tellier, Tetrahedron Lett., 1994, 35, 1867–1870 CrossRef CAS.
- Y. Bleriot, T. Dintinger, A. Genre-Grandpierre, M. Padrines and C. Tellier, Bioorg. Med. Chem. Lett., 1995, 5, 2655–2660 CrossRef CAS.
- B. Ganem, Acc. Chem. Res., 1996, 29, 340–347 CrossRef CAS.
- R. Kanso, E. A. Yancey and S. Striegler, Tetrahedron, 2012, 68, 47–52 CrossRef CAS.
- J. B. Pickens, S. Striegler and Q.-H. Fan, Bioorg. Med. Chem., 2016, 24, 3371–3377 CrossRef CAS PubMed.
- R. Kanso and S. Striegler, Carbohydr. Res., 2011, 346, 897–904 CrossRef CAS PubMed.
- Q.-H. Fan, S. Striegler, R. G. Langston and J. D. Barnett, Org. Biomol. Chem., 2014, 12, 2792–2800 CAS.
- Q.-H. Fan, K. A. Claunch and S. Striegler, J. Med. Chem., 2014, 57, 8999–9009 CrossRef CAS PubMed.
- Q.-H. Fan, J. B. Pickens, S. Striegler and C. D. Gervaise, Bioorg. Med. Chem. Lett., 2016, 24, 661–671 CrossRef CAS PubMed.
- T. Gajda, M. S. Jancsó, H. Lönnberg and H. Sirges, J. Chem. Soc., Dalton Trans., 2002, 1757–1763 RSC.
- S. Striegler and M. Dittel, J. Am. Chem. Soc., 2003, 125, 11518–11524 CrossRef CAS PubMed.
- S. Striegler, N. A. Dunaway, M. G. Gichinga, J. D. Barnett and A.-G. D. Nelson, Inorg. Chem., 2010, 49, 2639–2648 CrossRef CAS PubMed.
- S. Striegler, Q.-H. Fan and N. P. Rath, J. Catal., 2016, 338, 349–364 CrossRef CAS.
- S. Striegler, Curr. Org. Chem., 2003, 7, 81–102 CrossRef CAS.
- R. A. Binstead, B. Jung and A. D. Zuberbühler, SPECFIT/32™ Global Analysis System, Vers. 3.0, Spectrum Software Associates, Marlborough, MA, USA, 2000 Search PubMed.
- R. C. Neuman and G. S. Hammond, J. Phys. Chem., 1963, 67, 1659–1665 CrossRef CAS.
- D. P. Fairlie and W. G. Jackson, Inorg. Chem., 1990, 29, 140–143 CrossRef CAS.
- J. Barker, Coord. Chem. Rev., 1994, 133, 219–300 CrossRef CAS.
- Unpublished results.
- The stability of galactonoamidines has been determined in decomposition studies and will be disclosed in the context of a related study soon. At pH 10.5, the compounds are stable at 0 or 10 °C for more than 3 h, while at ambient temperature or 30 °C immediate hydrolysis is noted.
- S. Striegler and E. Tewes, Eur. J. Inorg. Chem., 2002, 487–495 CrossRef CAS.
- N. Ghaschghaie, T. Hoffmann, M. Steinborn and P. Klüfers, Dalton Trans., 2010, 39, 5535–5543 RSC.
- Y. Mikata, K. Takahashi, Y. Noguchi, M. Naemura, A. Ugai, S. Itami, K. Yasuda, S. Tamotsu, T. Matsuo and T. Storr, Eur. J. Inorg. Chem., 2012, 2012, 217–225 CrossRef CAS.
- Parallel Quantum Solutions, Vers. 4.1, Fayetteville, AR 72702-0293, 2013 Search PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- S. Striegler and M. G. Gichinga, Chem. Commun., 2008, 5930–5932 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev., 1988, 37, 785–789 CAS.
- Y. Takano and K. N. Houk, J. Chem. Theory Comput., 2005, 1, 70–77 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopic titrations between 1 and 2, coordinates and energies for the complex formation between 1 and S-3, and 1 and R-3 obtained by DFT methods. See DOI: 10.1039/c6dt02153k |
| This journal is © The Royal Society of Chemistry 2016 |