DOI:
10.1039/C6DT02025A
(Paper)
Dalton Trans., 2016,
45, 13161-13168
A multi-target caffeine derived rhodium(I) N-heterocyclic carbene complex: evaluation of the mechanism of action†
Received
20th May 2016
, Accepted 7th June 2016
First published on 8th June 2016
Abstract
A rhodium(I) and a ruthenium(II) complex with a caffeine derived N-heterocyclic carbene (NHC) ligand were biologically investigated as organometallic conjugates consisting of a metal center and a naturally occurring moiety. While the ruthenium(II) complex was largely inactive, the rhodium(I) NHC complex displayed selective cytotoxicity and significant anti-metastatic and in vivo anti-vascular activities and acted as both a mammalian and an E. coli thioredoxin reductase inhibitor. In HCT-116 cells it increased the reactive oxygen species level, leading to DNA damage, and it induced cell cycle arrest, decreased the mitochondrial membrane potential, and triggered apoptosis. This rhodium(I) NHC derivative thus represents a multi-target compound with promising anti-cancer potential.
Introduction
Increasing evidence has proven N-heterocyclic carbene (NHC) metal complexes to be promising drug candidates for the treatment of cancer or infectious diseases.1 Silver, gold, and platinum are the most frequently used metals regarding bioactive metal NHC complexes. Surprisingly, the anti-cancer properties of ruthenium(II) or rhodium(I) NHC complexes have rarely been studied. Rh(I) is isoelectronic with Pt(II), and Rh(I) NHC complexes have a square-planar geometry like cisplatin, a clinically used anti-cancer drug. Ruthenium complexes are currently undergoing clinical trials as anticancer drugs and represent, in general, a strongly investigated group of metallodrugs.2 In recent reports we and others demonstrated that Rh(I) and Ru(II) NHC complexes show promising anti-cancer potential.3 However, their mechanism of anti-cancer action has not been fully resolved. Their influence on either cellular DNA or mitochondria has been shown, and some of the complexes also inhibited the enzyme thioredoxin reductase (TrxR), a common anti-cancer target for metallodrugs.
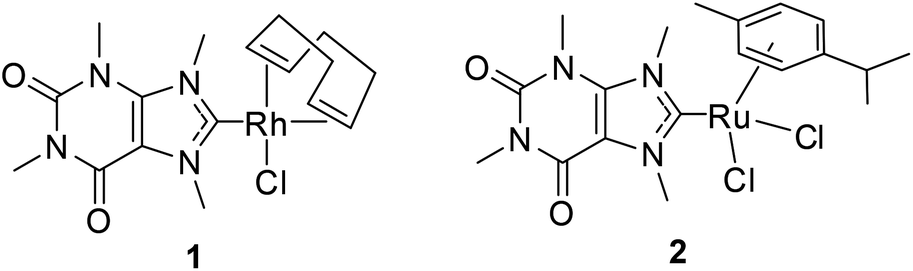
In this work, caffeine derived Rh(I) or Ru(II) complexes 1 and 2 have been prepared and investigated (Fig. 1). The xanthine derivative caffeine showed anti-angiogenic effects in the zebrafish model, and its analogue, pentoxifylline, displayed anti-metastatic activity.4 Moreover, a caffeine derived gold(I) NHC complex was found to stabilise DNA G-quadruplexes while silver(I) derivatives showed promising antibacterial activities.5a,b Thus, complexes 1 and 2 were expected to retain both the cytotoxicity of Rh(I)/Ru(II) NHC complexes and the anti-angiogenic and anti-metastatic properties of xanthine derivatives. This rationale was encouraged by our previous work on a Pt(II) complex containing the same NHC ligand, which displayed both cytotoxic and anti-angiogenic activities in vitro.5c The advantage of these multiple anti-cancer activities is that the risks of systemic toxicity caused by administration of more than one anti-cancer drug would be strongly decreased.6
 |
| | Fig. 1 Caffeine derived metal NHC complexes of this report. | |
Experimental
Materials and instrumentation
All chemicals and reagents were purchased from Sigma-Aldrich unless otherwise noted. 1H NMR spectra were recorded on a Bruker DRX-400 AS NMR spectrometer; positive-ion ESI (electrospray ionization) mass spectra were recorded on a LTQ-Orbitrap Velos linear ion-trap coupled with an Orbitrap mass analyser (Thermo Fisher Scientific); elemental analyses were conducted using Flash EA112.
Synthesis
Chlorido(η2,η2-cycloocta-1,5-diene)(1,3,7,9-tetramethylxanthine-8-ylidene)rhodium(I) (1).
AgISO3CF3 (76.6 mg, 0.298 mmol) in CH3CN (8 mL) was added to a solution of 1,3,7,9-tetramethylxanthinium iodide (99.9 mg, 0.298 mmol) in CH3CN (20 mL). The mixture was stirred in the dark for 30 min and filtered. The residue resulting from the evaporation of the filtrate under vacuum, together with Ag2O (42.0 mg, 0.181 mmol), was suspended in CH2Cl2 (16 mL), and the mixture was stirred in the dark for 3.5 h. A solution of bis[chlorido(η2,η2-cycloocta-1,5-diene)rhodium(I)] (73.5 mg, 0.149 mmol) in CH2Cl2 (10 mL) was added, and the suspension was stirred in the dark for another 4 h. The final suspension was filtered over Celite, and the filtrate was washed twice with H2O (10 mL). The CH2Cl2 fraction was dried with Na2SO4, and evaporated under reduced pressure. The residue was dissolved in a small amount of CH2Cl2, and the precipitate was obtained by adding an excess of n-hexane. The crude product was isolated from the precipitate by column chromatography (eluent: ethyl acetate). The pure product was obtained by recrystallisation in CH2Cl2/n-hexane. Yield: 17.3 mg, 12.8%. 1H-NMR (400 MHz, CDCl3): δ (ppm) = 5.15 (m, 2H, CH COD), 4.59 (s, 3H, NCH3), 4.40 (s, 3H, NCH3), 3.77 (s, 3H, NCH3), 3.36 (s, 3H, NCH3), 3.32 (m, 2H, CH COD), 2.45 (m, 4 H, CH2 COD), 2.01 (m, 4 H, CH2 COD). ESI-MS (+ve, m/z): 419.1 [M − Cl−]+, 873.2 [2M − Cl−]+. Anal. Calcd for C17H24ClN4O2Rh: C 44.90, H 5.32, N 12.32. Found: C 44.48, H 5.26, N 11.89.
Dichlorido[1,3,7,9-tetramethylxanthine-8-ylidene](η6-p-cymene)ruthenium(II) (2).
A mixture of 1,3,7,9-tetramethylxanthinium iodide (200 mg, 0.597 mmol) and Ag2O (77.2 mg, 0.333 mmol) in CH2Cl2 (12 mL) was stirred in the dark for 3.5 h. A solution of bis[dichloride(η6-p-cymene)ruthenium(II)] (184.3 mg, 0.300 mmol) in CH2Cl2 (20 mL) was then added, and the resulting solution was stirred in the dark for 4 h, followed by filtration over Celite. The obtained filtrate was extracted twice with H2O (15 mL). After drying with Na2SO4, the CH2Cl2 solution was evaporated under reduced pressure. The residue was dissolved in a small amount of CH2Cl2, and an excess of n-hexane was added to form a precipitate. The pure product was obtained by recrystallisation of the precipitate from CH2Cl2/n-hexane. Yield: 70.8 mg, 23.1%. 1H-NMR (400 MHz, CDCl3): δ (ppm) = 5.49 (d, J = 6.1 Hz, 2H, CH3C6H4), 5.23 (d, J = 5.8 Hz, 2H, CH3C6H4), 4.34 (s, 3 H, NCH3), 4.29 (s, 3 H, NCH3), 3.79 (s, 3H, NCH3), 3.40 (s, 3H, NCH3), 2.98 (p, J = 7.0 Hz, 1H, (CH3)2CH), 2.15 (s, 3H, C6H4CH3), 1.29 (d, J = 7.0 Hz, 6H, CH(CH3)2). ESI-MS (+ve, m/z): 443.1 [M − 2Cl− − H+]+. Anal. Calcd for C19H26Cl2N4O2Ru: C 44.36, H 5.09, N 10.89. Found: C 44.15, H 4.89, N 10.72.
Cell culture
A human neuroblastoma cell line (IMR-32), a human hepatoma cell line (HepG2), human breast cancer cell lines (MCF-7 and MDA-MB-231), a human colon carcinoma cell line (HCT-116), a human pancreatic cell line (Panc-1), and a human prostate adenocarcinoma cell line (LNCaP) were maintained in Dulbecco's Modified Eagle's Medium (DMEM, Gibco) high glucose supplemented with 10% (v/v) Fetal Bovine Serum (FBS, Gibco) and 1% (v/v) penicillin–streptomycin (Gibco). The human pancreatic cell line JoPaca-1 was cultured in RPMI 1640 medium (Gibco) with the same supplements as those of DMEM. All cells were incubated under a humidified atmosphere at 37 °C under a 95% air and 5% CO2 atmosphere.
Cytotoxicity assay
The cytotoxicity of the test compounds towards the various cell lines was evaluated by the Sulforhodamine B (SRB) assay at different intervals according to an established procedure7 with some modifications. Cells were seeded in 100 μL aliquots into 96-well plates with a density of 5000 cells per well. After incubation overnight, different concentrations of compounds or respective amounts of DMF were added to the cells, which were further incubated for different time periods. When the treatment was completed, 50 μL of ice cold 10% trichloroacetic acid (TCA) in H2O were added to each well, and the plates were kept at 0 °C for 1 h. After that, the solution in the plates was discarded, and the plates were washed twice with H2O followed by drying. 100 μL of 1% acetic acid solution containing 0.054% of Sulforhodamine B Sodium Salt (SRB) were then added. The solution was discarded 30 min later, and the plates were washed twice by adding 200 μL of acetic acid (1%) into each well. The washing solution of acetic acid was discarded quickly every time after washing. The plates were further dried followed by addition of 200 μL of 10 mM Tris (pH 10.5) to each well to dissolve the SRB dye. After shaking the plates for 15 min, the dye was quantified with a plate reader (TECAN) by determining the absorption at 535 nm. The IC50 values were calculated as the concentrations of the compounds required to inhibit 50% of cell growth.
Mammalian thioredoxin reductase (TrxR) inhibition assay
The mammalian TrxR inhibition assay was performed according to an established microplate reader based assay with minor modifications.8 TrxR from rat liver (Sigma) was diluted with distilled water to achieve a concentration of 3.5 U mL−1. DMF was used to prepare stock solutions of the compounds. To each well of a 96-well plate 25 μL of the enzyme solution and 25 μL of phosphate-buffered saline (PBS, pH 7.0) solution, which contained stock solutions of the complexes in different concentrations, were added. After incubation at 37 °C with moderate shaking for 75 min, 225 μL of the reaction mixture (1.0 mL consisted of 500 μL of PBS pH 7.0, 80 μL of 100 mM EDTA solution pH 7.5, 20 μL of BSA solution 0.2%, 100 μL of 20 mM β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (NADPH) solution and 300 μL of distilled water) were added to each well. The enzyme catalysed reaction was initiated by adding 25 μL of 20 mM dithio-bis-2-nitrobenzoic acid (DTNB) solution in ethanol. The formation of 5-thio-2-nitrobenzoic acid (5-TNB) was monitored by using a microplate reader (Perkin Elmer Victor X4) at 405 nm at 35 s intervals for 350 s. A negative control experiment using enzyme free solution was performed to confirm that the tested complexes do not interfere with the assay components. The residual enzyme activity was determined as the percentage of vehicle control (100 × k2/k1, where k1 and k2 are the initial rates of enzyme catalysed reaction in the absence and presence of the tested complexes, respectively).
Escherichia coli (E. coli) TrxR inhibition assay
The E. coli TrxR inhibition assay was also performed based on a reported method with minor modifications.9 To each well of a 96-well plate, 100 μL of a 200 μM NADPH solution in TE buffer (50 mM Tris-HCl, 1 mM EDTA, pH 7.5), 10 μL of TrxR from E. coli (35.4 U mL−1, Sigma) in distilled water, 20 μL of PBS (pH 7.0) solution containing the complexes in different concentrations or DMF as a vehicle control, and 10 μL of Trx from E. coli (156 μg mL−1, Sigma) in water as the substrate solution were added. The final mixture was incubated at 25 °C with moderate shaking for 75 min, and 100 μL of the reaction mixture (200 μM NADPH in TE buffer, 5 mM DTNB in TE buffer) were added. The absorption of the formed TNB was then monitored by using a microplate reader at 412 nm. A negative control experiment using enzyme free solution was conducted to confirm that the tested complexes had no interference with the assay components. The residual enzyme activity was determined in the same way as described in the mammalian TrxR inhibition assay.
Antibacterial activity
The following strains were used and maintained at 37 °C in MH (21 g L−1 Müller–Hinton, 1% glucose, pH 7.4) or TSY (30 g l−1 trypticase soy broth, 3 g l−1 yeast extract, pH 7.0–7.2) medium: Acinetobacter baumannii (DSM 30007, ATCC 19606) in MH, Enterobacter cloacae (DSM46481, ATCC 23355) in MH, Escherichia coli (DSM 1116, ATCC 9637) in TSY, Klebsiella pneumoniae (DSM 11678, ATCC 33495) in MH, Pseudomonas aeruginosa PA7 (DSM24068) in MH, Enterococcus faecium (DSM 20477, ATCC 19434) in TSY, Staphylococcus aureus MRSA (DSM 11822, ICB 25701) in TSY, and Staphylococcus aureus MRSA (clinical isolate, RKI 11-02670) in TSY. The determination of minimum inhibitory concentration (MIC) values was carried out following a standardized protocol in broth dilution assays. Compounds were serially diluted starting from 100 μM. The starting inocula of 2–8 × 105 colony forming units per ml (CFU per ml) in MH or TSY medium at 37 °C were used and serial dilutions were carried out in 96-well microtiter plates in duplicate. After incubation of the plates for 20 h at 37 °C, the absorbance at 600 nm was measured to determine the MIC value.
Intracellular ROS formation assay
HCT-116 cells were seeded into 12-well plates with a density of 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well in 1.0 mL DMEM. After incubation overnight, the cells were treated with complex 1 or DMF in different concentrations for 1 h, 3 h, 6 h and 24 h, respectively. At the end of treatment, the cells were washed with 1.0 mL of PBS, trypsinized, and collected by centrifugation (1600 rcf, 3 min). The cell pellets were resuspended and incubated in 500 μL of DMEM without phenol red containing 30 μM of dihydroethidium (DHE) in the dark for 15 min at room temperature. Cell pellets were obtained by centrifugation at 1600 rcf for 3 min, and resuspended in 500 μL of DMEM without phenol red. The single cell suspensions were measured with a FACS Calibur flow-cytometer (Becton Dickinson) at 488 nm excitation and 564–606 nm emission wavelengths, and analysed with CellQuest Pro (Becton Dickinson) analysis software.
000 cells per well in 1.0 mL DMEM. After incubation overnight, the cells were treated with complex 1 or DMF in different concentrations for 1 h, 3 h, 6 h and 24 h, respectively. At the end of treatment, the cells were washed with 1.0 mL of PBS, trypsinized, and collected by centrifugation (1600 rcf, 3 min). The cell pellets were resuspended and incubated in 500 μL of DMEM without phenol red containing 30 μM of dihydroethidium (DHE) in the dark for 15 min at room temperature. Cell pellets were obtained by centrifugation at 1600 rcf for 3 min, and resuspended in 500 μL of DMEM without phenol red. The single cell suspensions were measured with a FACS Calibur flow-cytometer (Becton Dickinson) at 488 nm excitation and 564–606 nm emission wavelengths, and analysed with CellQuest Pro (Becton Dickinson) analysis software.
Measurement of mitochondrial membrane potential
HCT-116 cells were seeded into 12-well plates with a density of 200000 cells per well in 1.0 mL of DMEM, and incubated overnight. Different concentrations of complex 1 or DMF were then added, and the plates were further incubated for 1 h, 3 h, 6 h and 24 h. Afterwards, cells were washed with PBS, trypsinized, and collected by centrifugation at 1600 rcf for 3 min. The cell pellets were washed with 1 mL of PBS, and the cell suspensions were spun down at 1600 rcf for 3 min. The cell pellets were then resuspended in 500 μL of DMEM without phenol red containing 2 μM of 5,5,6,6-tetrachloro-1,1,3,3-tetraethylbenzimidazolylcarbocyanineiodide (JC-1) and incubated for 30 min at room temperature in the dark. Afterwards, the cells were collected by centrifugation at 1600 rcf for another 3 min, and suspended in 500 μL of DMEM without phenol red for FACS analysis at 488 nm excitation and 515–545 nm (FL1 channel) and 564–606 nm (FL2 channel) emission wavelengths for JC-1 monomers (green) and JC-1 aggregates (red), respectively.
Western blot analysis
HCT-116 cells (400000 cells per well in 2.0 mL DMEM) were seeded into 6-well plates and incubated overnight. Afterwards, the medium was replaced with a fresh medium containing DMF or different concentrations of the complexes. After incubation for a further 24 h, the floating cells were collected and washed with PBS, while the attached cells were also washed with PBS. Then, both fractions of the cells were combined and lysed in 80 μL of lysis buffer (6 M urea, 1 mM EDTA, 5 mM NaF, 0.5% Triton X-100, 10 μg mL−1 pepstatin, 0.1 mM PMSF, 10 μg mL−1 aprotinin, 2.5 mM Na4P2O7, and 1 mM Na3VO4 in PBS) on ice for 5 min. The supernatant was collected after centrifugation at 13200 rpm for 30 min. The protein content in the supernatant was measured by the standard Bradford protein assay. After denaturation at 95 °C for 10 min, 40 μg of total protein were separated by Tris-glycine SDS-PAGE in a 10% gel, and transferred onto a polyvinylidene fluoride (PVDF) membrane (GE Healthcare). The membrane was then washed with TBST buffer (24.8 mM Tris, 137 mM NaCl, 2.68 mM KCl, 0.1% Tween-20 (ROTH) (v/v), pH 7.4), and blocked in TBST containing 5% nonfat milk (ROTH) (m/v) at room temperature for 1–2 h. Afterwards, the membrane was incubated with primary antibodies at a 1:1000 dilution in TBST containing 5% nonfat milk (w/v) at 4 °C overnight. After washing 3 times with TBST, the membrane was further incubated with the corresponding secondary antibodies at a 1:5000 dilution in TBST containing 5% nonfat milk (w/v) at room temperature for 1–2 h. The protein bands were visualized with Western Lightning™ Plus-ECL Oxidizing Reagent Plus and Enhanced Luminol Reagent Plus and detected using a LAS 3000 Imaging System (Fuji). The phospho-p53 (Ser15) antibody (#9284), caspase-9 antibody (Human Specific) (#9502), caspase-3 antibody (#9662) and PARP antibody (#9542) were purchased from Cell Signaling. The vinculin (7F9) antibody (sc-73614) was purchased from Santa Cruz Biotechnology, and goat anti-rabbit [IgG (H + L)] antibody (#111-035-003) and goat anti-mouse IgG (H + L)] antibody (#115-035-003) were purchased from Dianova GmbH.
Cell cycle analysis
HCT-116 cells were seeded into 6-well plates with a density of 400000 cells per well in 2.0 mL DMEM. After incubation overnight, the cells were treated with complex 1 in different concentrations for 24 h. At the end of treatment, the floating cells were collected by centrifugation at 200g for 5 min. The attached cells were washed with PBS, trypsinized, and collected by centrifugation at 200g for 5 min at 4 °C. The total amount of cells was washed with PBS. Cell pellets were obtained after centrifugation at 200g for 5 min at 4 °C, resuspended in ice-cold 70% ethanol (300 μL) for fixation and incubated at −20 °C for at least 24 h. Afterwards, the suspension was centrifuged at 500g for 5 min at 4 °C. The pellets were washed with ice cold PBS containing 1% BSA (m/v), and the suspension was centrifuged at 500g for 5 min at 4 °C. The collected cell pellets were then resuspended in 100 μL of PBS containing 1% BSA and 50 μg mL−1 of RNase at 37 °C for 30 min, after which 100 μL of PBS containing 1% BSA and 0.1 mg mL−1 propidium iodide (PI) was added. The final suspension was mixed well and incubated in the dark for 30 min at room temperature, followed by centrifugation at 500g for 5 min at 4 °C. The cell pellets were washed with PBS containing 1% BSA, and suspended in 500 μL of PBS. The single cell suspension was analyzed by FACS at 488 nm excitation and 564–606 nm (FL2 channel) emission.
ELISA microarray analysis
HCT-116 cells were treated and lysed in the same way as described in the western blot analysis section. The proteins were diluted 1:6 in dilution buffer (1 mM EDTA, 0.5% (v/v) Triton X-100, 5 mM NaF in PBS, pH 7.2), and the corresponding concentrations were determined by BCA assay using the PierceTM BCA Protein Assay kit (Thermo Scientific). The levels of phosphorylated proteins (ERK1/2, Hsp27) associated with the cellular stress response were quantified with an ELISA microarray based on the ArrayStrip™ platform (Alere Technologies GmbH) according to a published protocol.10
Chorioallantoic membrane (CAM) assay
Fertilised, specific pathogen-free (SPF) chicken eggs (VALO BioMedia) were bred in an incubator at 37 °C and 60% relative humidity. On day 5, windows (Ø 1.5–2 cm) were cut into the shell at the more rounded pole of the egg. The holes were sealed with tape (Leukosilk®, BSN medical), and incubation was continued overnight. Then rings of thin silicon foil (Ø 5 mm) were placed on the CAM with its developing blood vessels, and complex 1 (10 nmol) and a respective amount of DMF as a solvent control (all in a volume of 10 μL H2O) were added. The effects on the developing vasculature were documented after further incubation for 6 and 24 h using a light microscope (60× magnification, Traveler).11
Wound healing assay
MDA-MB-231 cells were seeded into 24-well plates. When the cells were almost confluent, a narrow artificial wound was created in the cell monolayer by using a 20–200 μL pipette tip. The cell monolayer was washed extensively with DMEM followed by treatment with complex 1 or DMF in different concentrations. The scratch areas were monitored by microscopy (EVOSxl digital microscope) after 24 h and 45 h of incubation.12
Results and discussion
Synthesis and characterization
Complexes 1 and 2 were prepared based on reported methods (Scheme 1).3b,d,13 1,3,7,9-Tetramethylxanthinium triflate or 1,3,7,9-tetramethylxanthinium iodide was first activated by Ag2O, followed by a transmetalation reaction with bis[chlorido(η2,η2-cycloocta-1,5-diene)rhodium(I)] or bis[dichloride(η6-p-cymene)ruthenium(II)] to form 1 and 2, respectively. Of note, an iodido analogue of 1 has been described by Herrmann et al. recently.13 Both complexes were characterized using 1H NMR and mass spectrometry (Fig. S1–S4, ESI†), and their purity was confirmed by elemental analysis. Complex 1 showed comparable patterns regarding the COD ligand in the 1H NMR spectrum as reported for [Rh(I)Cl(COD)(NHC)] complexes (COD = 1,5-cyclooctadiene) (Fig. S1, ESI†).3b Two signals ascribed to the cis- and trans-olefin protons of CHCH groups in COD were observed. Moreover, the two characteristic signals of the equatorial and axial CH2 protons in COD were monitored.
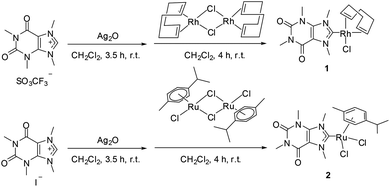 |
| | Scheme 1 Synthesis of complexes 1 and 2. | |
Cytotoxicity
The in vitro cytotoxicity of 1, 2 and the 1,3,7,9-tetramethylxanthinium iodide ligand against various cancer cell lines, including a human hepatoma cell line (HepG2), human breast cancer cell lines (MCF-7 and MDA-MB-231), a human colon carcinoma cell line (HCT-116), human pancreatic cell lines (Panc-1 and JoPaca-1), and a human prostate adenocarcinoma cell line (LNCaP), has been investigated by the Sulforhodamine B (SRB) assay.7 As depicted in Fig. 2, complex 1 showed a broad spectrum of cytotoxicity with IC50 values ranging from 8.8 to 76.7 μM after 96 h of treatment. It was particularly active against HCT-116 cells with an IC50 value 2–8 times lower than that in the other cancer cell lines. However, complex 1 did not reach submicromolar IC50 values as reported for some examples of highly cytotoxic gold(I) NHC complexes.8 As a further comparison, the cytotoxicity of cisplatin was investigated. Cisplatin was used as a reference and did not show specificity towards any cancer cell line, with narrower IC50 values ranging from 5.5 to 21.0 μM after 96 h (Fig. S5, ESI†). A time-dependent SRB assay was also performed with 24 h, 48 h and 96 h incubation periods for complexes 1 and 2. An increased cytotoxicity over time was only observed in the case of Panc-1 cells, while in the other cell lines, the cytotoxicity either hardly changed at all or slightly decreased after longer incubation periods. Complex 2 and the 1,3,7,9-tetra-methylxanthinium iodide ligand did not show any cytotoxicity up to 100 μM under the same conditions, which indicates that the presence of Rh(I) is crucial for a high cytotoxicity.
 |
| | Fig. 2 IC50 values (μM) of 1 towards various cell lines as determined by SRB assays (n = 3). | |
Inhibition of mammalian TrxR
To study the potential mechanism of the cytotoxic action, the inhibitory activity against purified mammalian thioredoxin reductase (TrxR) was investigated, since a different Rh(I) NHC complex inhibited TrxR in our previous report.3b Rh(I) complex 1 inhibited half of the TrxR activity at 3.64 μM, while neither 2 nor the 1,3,7,9-tetramethylxanthinium iodide ligand showed any inhibitory effect at 10 μM. This result suggests that inhibition of TrxR might contribute to the biological effects triggered by 1.
Inhibition of E. coli TrxR and anti-bacterial activity
Besides the anti-cancer properties, the growth inhibitory effect towards E. coli TrxR, which represents an anti-bacterial target, was investigated. It should be mentioned that Rh(I) NHC complexes were the first metal NHC complexes to be reported with antibacterial activity.14 Complex 1 displayed 15 times higher inhibitory activity against E. coli TrxR (IC50 value: 0.23 μM) when compared to mammalian TrxR, which suggests a strong potential as an antibacterial agent. In contrast, complex 2 was not active as an inhibitor of E. coli TrxR (IC50 value > 10 μM).
Both complexes were screened for their antibacterial activities against a panel of Gram-positive and Gram-negative bacterial strains (E. coli, P. aeruginosa, E. faecium, E. cloacae, E. faecium, K. pneumoniae, A. baumannii, methicillin-resistant S. aureus). However, both 1 and 2 failed to trigger antibacterial effects up to a concentration of 100 μM.
Increased ROS formation
Based on the results described above, our study was further focused on clarifying the anti-cancer mechanisms triggered by Rh(I) complex 1. Inhibition of TrxR can result in the accumulation of reactive oxygen species (ROS) by decreasing the reduced form of Trx that acts as a ROS scavenger.15 Thus, the influence of 1 (12 μM and 15 μM) on ROS formation was investigated in HCT-116 cells after 1 h, 3 h, 6 h and 24 h of treatment. A significant increase of ROS levels (2.0–2.4 fold of the control level) was observed after 1 h. After 3 h and 6 h of incubation the ROS levels were further elevated up to 3.6–4.6 fold, followed by a decrease to 1.6–1.8 fold at 24 h (Fig. 3). The decrease might result from the antioxidant defense response of cancer cells as observed recently also for gold NHC complexes, which activated ROS formation at lower tested exposure levels (2–5 μM).16 To confirm the accumulation of ROS, the expression of several cellular stress related proteins including phosphorylated ERK1/2 and HSP72 was analysed by an ELISA microarray,17 as increased ROS levels always result in oxidative stress in cells. As expected, the expression of all these proteins was concentration-dependently increased in HCT-116 cells after 3 h of treatment with 1 and decreased afterwards, which is in agreement with the changes of ROS levels (Fig. S6, ESI†). Similar results have been observed for other Rh(I)NHC complexes.3a,b All these results reveal that 1 can induce a significant accumulation of ROS in a time- and concentration-dependent manner, which is most likely mediated by the inhibition of TrxR by 1.
 |
| | Fig. 3 Complex 1 induces an increase of ROS in HCT-116 cells (n = 3). | |
DNA damage
ROS have been reported to cause DNA damage by initiating oxidative DNA lesions.18 In agreement with this, 1 induced DNA damage in HCT-116 cells, which was confirmed by a concentration-dependent increase in the expression of the DNA damage marker phospho-p53 (Ser15) after 24 h of treatment, as ascertained by western blot analysis (Fig. S6, ESI†). Additionally, a lower cytotoxicity against HCT-116 Chk2 knockout cells was observed. Chk2 is a DNA damage signal transducer.19 In HCT-116 Chk2 knockout cells an IC50 value of 17.7 μM was observed, while in wild type HCT-116 cells an IC50 value of 8.76 μM after 96 h of treatment had been determined (Fig. S8, ESI†).
Cell cycle arrest
For the repair of damaged DNA, cell cycle checkpoints are usually activated. Thus, extensive DNA damage will result in cell cycle arrest followed by apoptosis.20 A concentration-dependent cell cycle arrest was also observed in HCT-116 cells treated with 1 for 24 h (Fig. 4). Cell cycle arrest in the G2-phase was observed at 12 μM as cells at the G2-phase increased by 10%, while G0/G1-phase arrest happened at 15 and 18 μM with 22% and 12% increase, respectively. At 18 μM, 1 also induced an S-phase arrest as is apparent from the 6% increase of cells in this phase. The population at the sub-G1 fraction, which indicates apoptosis, increased by around 30% following treatment with 15 and 18 μM of compound 1 (Fig. S9, ESI†). Taken together, these findings indicate that a high accumulation of ROS caused by 1 might in turn lead to DNA damage followed by cell cycle arrest and eventually apoptosis.
 |
| | Fig. 4 Complex 1 induces a concentration-dependent cell cycle arrest in HCT-116 cells (n = 3). | |
Decreased mitochondrial membrane potential
Besides leading to DNA damage, high ROS levels can also trigger mitochondrial membrane permeabilization, which can be evaluated by measuring the mitochondrial membrane potential (ΔΨm).18 There was a strong decrease of more than 85% to 95% of the ΔΨm in HCT-116 cells treated with 1 for up to 6 h as indicated by a decrease in the ratio of red (J-aggregates) to green (monomers) JC-1 fluorescence observed at 12 μM and 15 μM, respectively (Fig. 5). The subsequent increase of the red-to-green ratio after 24 h at both concentrations is in line with the decrease of ROS levels under the same conditions. This result indicates that an increased accumulation of ROS induced by 1 might result in a decrease of the mitochondrial membrane potential.
 |
| | Fig. 5 Complex 1 interferes with the mitochondrial membrane potential in HCT-116 cells (n = 3). | |
Apoptotic cell death
Loss of the mitochondrial membrane potential has been reported to trigger the intrinsic apoptotic pathway including activation of caspases and cleavage of poly-ADP-ribose polymerase (PARP).15 The results of western blot analyses revealed that 1 decreased the protein levels of pro-caspase 9 and pro-caspase 3, and induced cleavage of PARP, which is in good agreement with previous reports15 (Fig. 6).
 |
| | Fig. 6 Decreases in the protein levels of pro-caspase 9 and pro-caspase 3 and cleavage of PARP were observed in HCT-116 cells treated with 1 for 24 h. | |
The anti-metastatic and anti-vascular effects
The anti-metastatic and anti-vascular activities of 1 were further investigated to see whether these xanthine-related properties were maintained upon complexation of the caffeine derived NHC ligand to Rh(I). A wound healing assay was performed to identify any anti-metastatic effect by using MDA-MB-231 cells as an example of highly invasive cells. In comparison to the control cells, the wound closed much more slowly after 45 h of treatment with 1 at a sub-toxic concentration (10 μM) (Fig. S10, ESI†). This result corroborates a previous report on a Rh(I) NHC complex inhibiting the migration.3c The effect of 1 on the developing vasculature was investigated using the chorioallantoic membrane (CAM) assay as an in vivo model.11 Strong vascular-disruptive effects of 1 (10 nmol in 10 μL H2O) were observed after 6 h of treatment (Fig. 7) as small blood vessels in the CAM were completely destroyed and a distinct blood leakage from bigger vessels occurred. Comparable antivascular/vascular-disruptive effects have already been observed for other NHC metal complexes with a gold(I) or platinum(II) centre.11c,d The observation of different anti-cancer effects together with an anti-metastatic and an anti-vascular activity of 1 underlines the high potential for creating complexes with multiple biological activities by combining bioactive ligands with metal centres.
 |
| | Fig. 7 Effects of 1 (each 10 nmol in 10 μL H2O) on the vasculature in the chorioallantoic membrane of fertilised chicken eggs after 6 h. DMF was used as the control; images are representative of at least three separate experiments. | |
Conclusions
In conclusion, a Rh(I) complex (1) with promising anti-cancer effects has been designed and prepared. Multiple anti-cancer properties including cytotoxicity against various tumour cell lines, anti-metastatic and anti-angiogenic effects have been combined in this complex. The proposed mechanism of action for complex 1 is as follows (Fig. 8): complex 1 inhibits TrxR, which in turn leads to an accumulation of ROS that most likely damage DNA and thereby initiate cell cycle arrest and apoptosis. A decrease of the mitochondrial membrane potential was also induced as a consequence of the increased ROS level. This led to intrinsic apoptotic processes manifesting in a decrease of the levels of pro-caspase 9 and pro-caspase 3 and in the cleavage of PARP. Future studies will be required to identify possible additional mechanisms of drug action of 1 and other rhodium(I) NHC complexes.
 |
| | Fig. 8 Proposed mechanism of the cytotoxic action of 1. | |
Acknowledgements
The financial support by the Humboldt Foundation (postdoctoral fellowship to J. J. Z) is strongly appreciated. We would like to acknowledge C. Kraemer, Dr H. Alborzinia and A. C. Heit for their assistance in some of the biological assays and thank Dr X. Cheng for his advice.
Notes and references
-
(a) L. Oehninger, R. Rubbiani and I. Ott, Dalton Trans., 2013, 42, 3269 RSC;
(b) S. A. Patil, S. A. Patil, R. Patil, R. S. Keri, A. Budagumpi, G. R. Balakrishna and M. Tacke, Future Med. Chem., 2015, 7, 1305 CrossRef CAS PubMed;
(c) F. Cisnetti and A. Gautier, Angew. Chem., Int. Ed., 2013, 52, 11976 CrossRef CAS PubMed.
-
(a) C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201 RSC;
(b) M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183 RSC;
(c) Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Chem. Commun., 2005, 4764 RSC.
-
(a) L. Oehninger, S. Spreckelmeyer, P. Holenya, S. M. Meier, S. Can, H. Alborzinia, J. Schur, B. K. Keppler, S. Wölfl and I. Ott, J. Med. Chem., 2015, 58, 9591 CrossRef CAS PubMed;
(b) L. Oehninger, L. N. Küster, C. Schmidt, A. Munoz-Castro, A. Prokop and I. Ott, Chem. – Eur. J., 2013, 19, 17871 CrossRef CAS PubMed;
(c) J. R. McConnell, D. P. Rananaware, D. M. Ramsey, K. N. Buys, M. L. Cole and S. R. McAlpine, Bioorg. Med. Chem. Lett., 2013, 23, 2527 CrossRef CAS PubMed;
(d) L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Munoz-Castro, R. W. Köster, K. Baumann, S. Wölfl, W. S. Sheldrick and I. Ott, Dalton Trans., 2013, 42, 1657 RSC.
-
(a) C.-H. Yeh, Y.-F. Liao, C.-Y. Chang, J.-N. Tsai, Y.-H. Wang, C.-C. Cheng, C.-C. Wen and Y.-H. Chen, Drug Chem. Toxicol., 2012, 35, 361 CrossRef CAS PubMed;
(b) A. Ratheesh, M. Jain and R. P. Gude, World J. Oncol., 2010, 1, 194 CAS.
-
(a) B. Bertrand, L. Stefan, M. Pirrotta, D. Monchaud, E. Bodio, P. Richard, P. L. Gendre, E. Warmerdam, M. H. de Jager, G. M. M. Groothuis, M. Picquet and A. Casini, Inorg. Chem., 2014, 53, 2296 CrossRef CAS PubMed;
(b) A. Kascatan-Nebioglu, A. Melaiye, K. Hindi, S. Durmus, M. J. Panzner, L. A. Hogue, R. J. Mallett, C. E. Hovis, M. Coughenour, S. D. Crosby, A. Milsted, D. L. Ely, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2006, 49, 6811 CrossRef CAS PubMed;
(c) J.-J. Zhang, C.-M. Che and I. Ott, J. Organomet. Chem., 2015, 782, 37 CrossRef CAS.
- J.-J. Zhang, R. W.-Y. Sun and C.-M. Che, Chem. Commun., 2012, 48, 3388 RSC.
- X. Cheng, P. Rasque, S. Vatter, K. H. Merz and G. Eisenbrand, Bioorg. Med. Chem., 2010, 18, 4509 CrossRef CAS PubMed.
- R. Rubbiani, S. Can, I. Kitanovic, H. Alborzinia, M. Stefanopoulou, M. Kokoschka, S. Mönchgesang, W. S. Sheldrick, S. Wölfl and I. Ott, J. Med. Chem., 2011, 54, 8646 CrossRef CAS PubMed.
- R. Zhao, H. Masayasu and A. Holmgren, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 8579 CrossRef CAS PubMed.
- P. Holenya, I. Kitanovic, F. Heigwer and S. Wölfl, Proteomics, 2011, 11, 2129 CrossRef CAS PubMed.
-
(a) K. Norrby, J. Cell. Mol. Med., 2006, 10, 588 CrossRef CAS PubMed;
(b) B. Nitzsche, C. Gloesenkamp, M. Schrader, M. Ocker, R. Preissner, M. Lein, A. Zakrzewicz, B. Hoffmann and M. Hçpfner, Br. J. Cancer, 2010, 103, 18 CrossRef CAS PubMed;
(c) J. K. Muenzner, B. Biersack, H. Kalie, I. C. Andronache, L. Kaps, D. Schuppan, F. Sasse and R. Schobert, ChemMedChem, 2014, 9, 1195 CrossRef CAS PubMed;
(d) J. K. Muenzner, T. Rehm, B. Biersack, A. Casini, I. A. de Graaf, P. Worawutputtapong, A. Noor, R. Kempe, V. Brabec, J. Kasparkova and R. Schobert, J. Med. Chem., 2015, 58, 6283 CrossRef CAS PubMed.
- C.-C. Liang, A. Y. Park and J.-L. Guan, Nat. Protoc., 2007, 2, 329 CrossRef CAS PubMed.
- J. Schütz and W. A. Herrmann, J. Organomet. Chem., 2004, 689, 2995 CrossRef.
- B. Cetinkaya, E. Cetinkaya, H. Kücükbay and R. Durmaz, Arzneim. Forsch./Drug Res., 1996, 46, 821 CAS.
- K. F. Tonissen and G. D. Trapani, Mol. Nutr. Food Res., 2009, 53, 87 CAS.
-
(a) X. Cheng, P. Holenya, S. Can, H. Alborzinia, R. Rubbiani, I. Ott and S. Wölfl, Mol. Cancer, 2014, 13, 221 CrossRef PubMed;
(b) R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608 CrossRef CAS PubMed.
- P. Holenya, F. Heigwer and S. Wölfl, J. Immunol. Methods, 2012, 380, 10 CrossRef CAS PubMed.
- O. A. Sedelnikova, C. E. Redon, J. S. Dickey, A. J. Nakamura, A. G. Georgakilas and W. M. Bonner, Mutat. Res., 2010, 704, 152 CAS.
- P. Holenya, S. Can, R. Rubbiani, H. Alborzinia, A. Jünger, X. Cheng, I. Ott and S. Wölfl, Metallomics, 2014, 6, 1591 RSC.
- N. S. Pellegate, R. J. Antoniono, J. L. Redpath and E. J. Stanbridge, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15209 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Examples of spectroscopic data and more details of biological results. See DOI: 10.1039/c6dt02025a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence