
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structural studies of Schiff-base [2 + 2] macrocycles derived from 2,2′-oxydianiline and the ROP capability of their organoaluminium complexes†
Wenxue
Yang
a,
Ke-Qing
Zhao
a,
Timothy J.
Prior
b,
David L.
Hughes
c,
Abdessamad
Arbaoui
c,
Mark R. J.
Elsegood
d and
Carl
Redshaw
*ab
aCollege of Chemistry and Materials Science, Sichuan Normal University, Chengdu, 610066, China. E-mail: C.Redshaw@hull.ac.uk
bDepartment of Chemistry, University of Hull, Hull, HU6 7RX, UK
cSchool of Chemistry, University of East Anglia, Norwich, NR4 7TJ, UK
dChemistry Department, Loughborough University, Loughborough, Leicestershire, LE11 3TU, UK
First published on 20th June 2016
Abstract
The molecular structures of a number of solvates of the [2 + 2] Schiff-base macrocycles {[2-(OH)-5-(R)-C6H2-1,3-(CH)2][O(2-C6H4N)2]}2 (R = Me L1H2, tBu L2H2, Cl L3H2), formed by reacting 2,6-dicarboxy-4-R-phenol with 2,2′-oxydianiline (2-aminophenylether), (2-NH2C6H4)2O, have been determined. Reaction of LnH2 with two equivalents of AlR′3 (R′ = Me, Et) afforded dinuclear alkylaluminium complexes [(AlR′2)2L1–3] (R = R′ = Me (1), R = tBu, R′ = Me (2), R = Cl, R′ = Me (3), R = Me, R′ = Et (4), R = tBu, R′ = Et (5), R = Cl, R′ = Et (6)). For comparative studies, reactions of two equivalents of AlR′3 (R′ = Me, Et) with the macrocycle derived from 2,2′-ethylenedianiline and 2,6-dicarboxy-R-phenols (R = Me L4H2, tBu L5H2) were conducted; the complexes [(AlMe)(AlMe2)L5]·2¼MeCN (7·2¼MeCN) and [(AlEt2)2L4] (8) were isolated. Use of limited AlEt3 with L3H2 or L5H2 afforded mononuclear bis(macrocyclic) complexes [Al(L3)(L3H)]·4toluene (9·4toluene) and [Al(L5)(L5H)]·5MeCN (10·5MeCN), respectively. Use of four equivalents of AlR′3 led to transfer of alkyl groups and isolation of the complexes [(AlR′2)4L1′–3′] (R = L2′, R′ = Me (11); L3′, R′ = Me (12); L1′, R′ = Et (13); L2′, R′ = Et (14); L3′, R′ = Et (15)), where L1′–3′ is the macrocycle resulting from double alkyl transfer to imine, namely {[2-(O)-5-(R)C6H2-1-(CH)-3-C(R′)H][(O)(2-(N)-2′-C6H4N)2]}2. Molecular structures of complexes 7·2¼MeCN, 8, 9·4toluene, 10·5MeCN and 11·1¾toluene·1¼hexane are reported. These complexes act as catalysts for the ring opening polymerisation (ROP) of ε-caprolactone and rac-lactide; high conversions were achieved over 30 min at 80 °C for ε-caprolactone, and 110 °C over 12 h for rac-lactide.
Introduction
Schiff-base compounds have attracted attention over the years primarily for their biological activity,1 whilst macrocyclic Schiff bases are of potential interest given their multiple binding sites.2 We have been investigating the simplest members of this Schiff-base macrocyclic family, so-called Robson type macrocycles, derived from the [2 + 2] condensation of a diamine with a dialdehyde, specifically herein 1,3-diformylphenol in combination with the diamine 2,2′-oxydianiline, 2-(2-aminophenoxy)aniline, (2-NH2C6H4)2O. The structural chemistry of this particular macrocycle is unexplored, indeed a search of the CSD revealed no hits,3a other than our recently reported manganese chemistry.3b Our interest stems primarily from their coordination chemistry and the potential to bind multiple metal centres in close proximity,3,4 particularly those which could be of use for ring opening polymerisation (ROP) of cyclic esters to produce biodegradable polymers.5 Poly(ε-caprolactone), PCL, and poly(lactide), PLA, are favoured polymers given both their biodegradability, and that their co-polymers are considered as potential environmentally friendly commodity plastic.6 Given the central role played by metal-complex induced coordination/insertion type ROP processes, investigations into new combinations of metals and ancillary ligands are pivotal when trying to identify structure–activity relationships. Indeed, in previous work,4a we communicated how remote alkylaluminium centres bound to a Schiff-base macrocycle derived from the dianiline [(CH2CH2)(2-C6H4NH2)2] exhibited beneficial cooperative effects in the ROP of ε-caprolactone, whereas the presence of aluminoxane type (Al–O–Al) bonding proved detrimental. Given this, we have re-focused our efforts on such Schiff-base systems and have extended our studies to [2 + 2] macrocycles derived from the dianiline (2-NH2C6H4)2O (see Chart 1). Herein, we report the molecular structures of a number of these [2 + 2] macrocycles, and find that they tend to adopt a taco-like, folded conformation. Interestingly, a series of zinc complexes bearing phenol compartmental type ligation were recently found to exhibit controllable photophysical properties by manipulation of the substituent (Me, tBu, Cl) positioned para to the phenolic group.7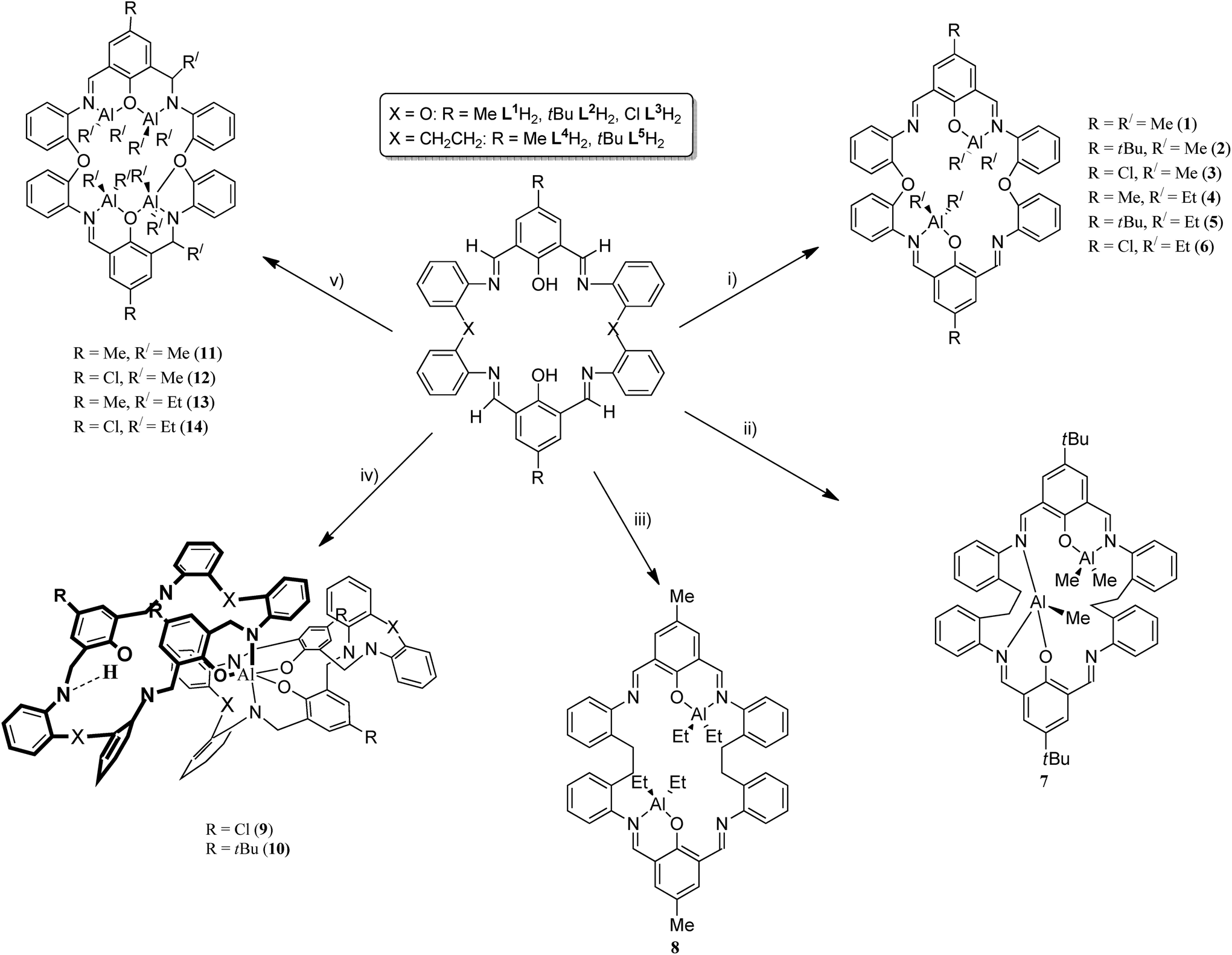 | ||
| Chart 1 Synthesis of aluminium complexes 1–14 prepared herein. Reagents and conditions: (i) 2R′3Al, hexane, Δ, 12 h; (ii) 2Me3Al, toluene, Δ, 12 h; (iii) 2Et3Al, toluene, Δ, 12 h; (iv) ½Et3Al, hexane, Δ, 12 h; (v) 4R′3Al, hexane, Δ, 12 h. | ||
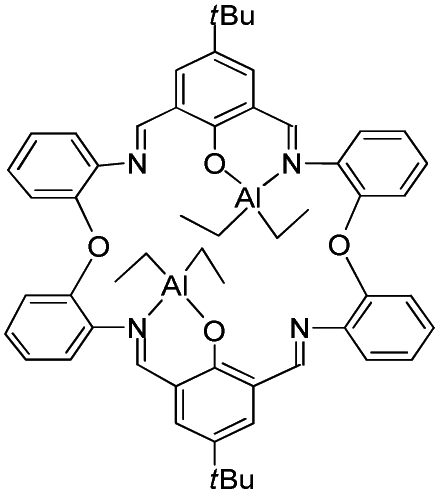
Furthermore, we have investigated the reaction chemistry of L1–3H2 towards the alkylaluminium reagents R3Al (R = Me, Et) and have isolated some unexpected products (Chart 1). Given this, related studies on macrocycles derived from the ethylene-bridged dianiline [(CH2CH2)(2-C6H4NH2)2] were conducted, and the ability of these complexes to act as catalysts for the ring opening polymerisation (ROP) of ε-caprolactone and rac-lactide has been investigated. The use of alkylaluminium complexes for the ROP of cyclic esters has recently been reviewed.8
Results and discussion
Preparation, structure and emission studies on LnH2
The [2 + 2] Schiff base macrocycles of type LnH2 are readily available in high yield via the reaction of 2,6-dicarboxy-4-R-phenol, where R = Me (n = 1), tBu (n = 2) or Cl (n = 3), with 2,2′-oxydianiline, (2-NH2C6H4)2O. In the IR spectra, v(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) for L1H2 (1626 cm−1), L2H2 (1630 cm−1) and L3H2 (1627 cm−1) bands are strong and are very similar to those reported for related ethylene (–CH2CH2–) bridged bis(imino)phenoxide macrocycles (1627–1629 cm−1),3b,4 and also lie within the range reported for other Schiff-base macrocycles.9 In the 1H NMR spectra, the imino hydrogen chemical shifts for L2H2 (8.40 ppm) and L3H2 (8.43 ppm) are comparable with those reported previously for bis(imino)phenol-based macrocycles [8.12 to 8.66 ppm],10 whilst that for L1H2 (8.87 ppm) is shifted slightly downfield.
N) for L1H2 (1626 cm−1), L2H2 (1630 cm−1) and L3H2 (1627 cm−1) bands are strong and are very similar to those reported for related ethylene (–CH2CH2–) bridged bis(imino)phenoxide macrocycles (1627–1629 cm−1),3b,4 and also lie within the range reported for other Schiff-base macrocycles.9 In the 1H NMR spectra, the imino hydrogen chemical shifts for L2H2 (8.40 ppm) and L3H2 (8.43 ppm) are comparable with those reported previously for bis(imino)phenol-based macrocycles [8.12 to 8.66 ppm],10 whilst that for L1H2 (8.87 ppm) is shifted slightly downfield.
These condensation products {[2-(OH)-5-(R)C6H2-1,3-(CH)2][(O)(2-C6H4N)2]}2 (R = Me L1H2, tBu L2H2, Cl L3H2) can be recrystallized from a variety of solvents; the molecular structures of a number of solvates are described below. Selected bond lengths and angles for each of the solvates are either discussed in the text or, in the case of L2H2, are presented in Table 1, with crystallographic parameters for all structures collated in Table 5. In each case, crystals of LnH2 suitable for an X-ray diffraction study were grown from the respective solvent on prolonged standing at ambient temperature. The molecular structure of L1H2·MeCN is shown in Fig. 1. In the asymmetric unit, there is one macrocycle and one molecule of MeCN. The macrocycle adopts an open, taco-like conformation, and the orientation of the two sides of the macrocycle can be monitored by looking at the cleft angle ϕ (ϕ is defined as the angle subtended between the mean planes of the two phenolate rings (O1 C1–C6, C8, C42, N1, N4 and C21–C27, C29, N2, N3, O3) as illustrated in Fig. 2). Thus, the smaller the cleft angle, the more parallel are the sides and the more taco-like the conformation. In the case of L1H2·MeCN, the open-taco description reflects the approximate cleft angle of 89.2°. A more detailed analysis of the orientation of the rings is presented in Table S1 (see ESI†). The MeCN molecule is encapsulated by the macrocycle between the rings incorporating C19 and C43. The centroid-to-centroid distance is approximately 8.5 Å, whilst the shortest H(MeCN) to centroid distances are 3.76 and 3.66 Å. The closest neighbour of the MeCN methyl group is the phenolic group with O1⋯H52c at 2.51 Å. The compound displays strong intramolecular hydrogen bonds involving the phenolic hydrogen and an imino nitrogen [H1⋯N1 = 1.74(3) Å and H3⋯N3 = 1.59(3) Å; O1–H1⋯N1 = 150(3)° and O3–H3⋯N3 = 152(3)°].
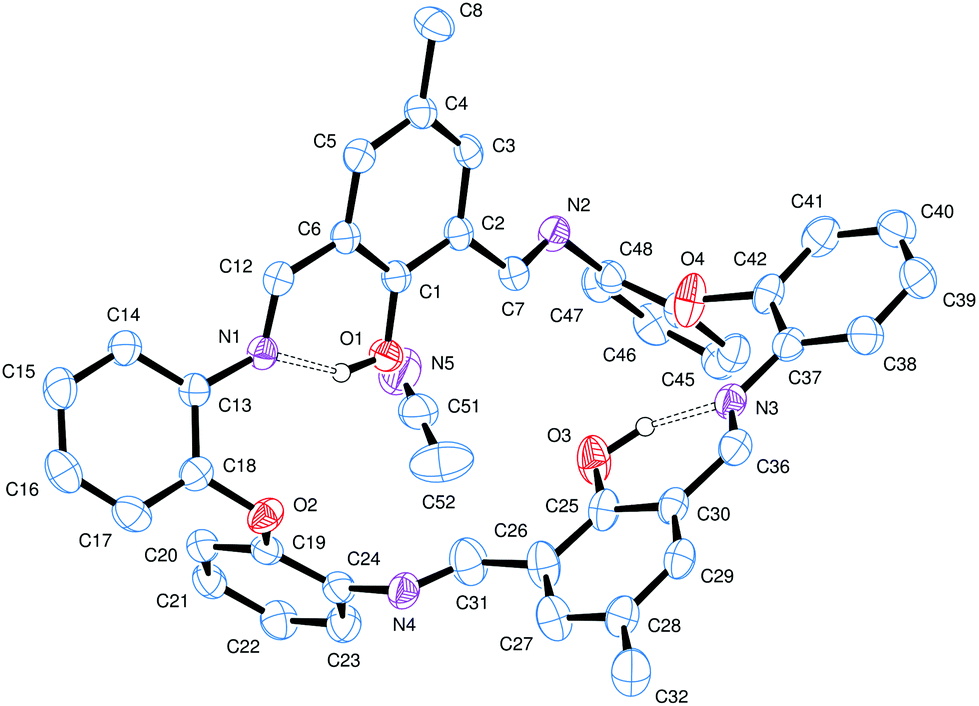 | ||
| Fig. 1 Molecular structure of L1H2·MeCN. Selected bond lengths (Å) and angles (°): N1–C12 1.284(2), N1–C13 1.415(2), N2–C7 1.276(2), N2–C48 1.419(2); C6–C12–N1 121.50(13), C2–C7–N2 121.44(14). H atoms not involved in H-bonding are omitted for clarity. | ||
 | ||
| Fig. 2 The cleft ϕ, defined by the angle subtended by the mean planes of the phenolic rings. | ||
| L 2 H2·MeCN | L 2 H2·EtOAc | L 2 H2·2acetone | L 2 H2·2toluene | |
|---|---|---|---|---|
| N1–C12 | 1.286(3) | 1.288(2) | 1.280(2) | 1.282(3) |
| N1–C13 | 1.412(3) | 1.4188(19) | 1.4112(18) | 1.415(3) |
| N2–C7 | 1.258(3) | 1.2679(19) | 1.2616(19) | 1.276(3) |
| N2–C24′/48 | 1.411(3) | 1.412(2) | 1.417(2) | 1.422(3) |
| C18–O2 | 1.395(3) | 1.3971(19) | 1.385(2) | 1.392(3) |
| O2–C19 | 1.401(3) | 1.4022(19) | 1.398(2) | 1.391(3) |
| C18–O2–C19 | 116.3(2) | 115.40(11) | 117.10(12) | 116.45(19) |
| C12–N1–C13 | 123.2(3) | 119.70(13) | 121.71(15) | 120.45(19) |
| N2–C7–C2 | 122.5(3) | 121.67(14) | 123.80(16) | 122.6(2) |
| N1–C12–C6 | 120.0(3) | 122.75(14) | 121.78(15) | 121.4(2) |
| C14–C13–N1 | 126.1(3) | 123.87(14) | 124.09(15) | 124.1(2) |
| C18–C13–N1 | 116.0(3) | 118.50(14) | 117.58(15) | 117.1(2) |
| C7–N2–C24′/48 | 116.6(3) | 119.29(14) | 116.98(15) | 117.05(19) |
Intermolecular face-to-face interactions give rise to stacks along the c direction (see Fig. S1, ESI†).
In the case of L2H2·MeCN, there are two very similar, independent molecules in the asymmetric unit, together with two molecules of solvent (MeCN), both of which are disordered in several orientations. In this case, the conformation in each macrocyclic molecule is much more closed with ϕ angles of about 13 and 15°, i.e. the two sides of the cleft are almost parallel. The whole molecule shows approximate symmetry about a pseudo two-fold axis (see Fig. S2 and S3†). The pseudo symmetry axes of the two molecules are not parallel. Distinct from L1H2·MeCN, the solvent does not reside in a pocket and has no close interaction with the macrocyclic ring. As expected, the bond lengths in L2H2·MeCN are similar to those observed in L1H2·MeCN, and in each molecule of L2H2·MeCN, the hydroxyl hydrogen atoms of the phenol groups were all located from difference maps and refined well to show clear intramolecular hydrogen bonding with neighbouring imine nitrogen atoms [molecule 1: H1o–N1 = 1.57(3) Å and O1–H1o⋯N1 = 150(3)°, H3o–N3 = 1.79(3) Å and O3–H3o⋯N3 = 148(3)°; molecule 2: H51o–N51 = 1.68(3) Å and O1–H51o⋯N51 = 148(3)°, H53o–N53 = 1.64(3) Å and O3–H53o⋯N53 = 150(3)°].
L 2 H2 can also be readily crystallized from ethyl acetate from which two different solvates were isolated on separate occasions. The molecular structure of one product is shown in Fig. S4 (ESI†), with selected bond lengths and angles given in Table 1. The asymmetric unit contains half a molecule of L2H2 and half a disordered solvent molecule. The second half of the macrocycle molecule is generated by a two-fold symmetry axis. Again, the macrocycle possesses quite a tight cleft angle ϕ at about 17°. As in the previous solvates, there is intramolecular H-bonding involving the phenolic hydrogen and an imino nitrogen [H1o–N1 = 1.75(2) Å and O1–H1o⋯N1 = 153(2)°]. The disordered ethyl acetate solvent molecule resides over an inversion centre, and is located in a pocket between four of the macrocycles.
A separate crystallization afforded a different solvate, namely L2H2·2(ethyl acetate), the asymmetric unit for which (not shown) contains half a molecule of the macrocycle and one solvent molecule. The main difference from the mono-solvate is that there is a pronounced twist about the central bond, resulting in a C12–N1–C13–C14 torsional angle of −33.1(8)° (the same angle in the mono-solvate is −15.8(2)°). The ϕ angle of the V-shaped cleft in L2H2·2(ethyl acetate) is about 7° (i.e. close to parallel), though it should be noted here that the distance between the rings of each side of the cleft (see Fig. S5, ESI†) is larger than in the mono-solvate, with a mean of 3.7 Å (cf. 3.5 Å for the mono-solvate).
In the case of the crystallization from acetone, the asymmetric unit contains half a macrocycle and one molecule of acetone. A similar conformation (Fig. S6, ESI†) to the ethyl acetate solvate is adopted in that the V-shaped cleft has a very tight ϕ angle (ca. 8°). Pairs of acetone molecules, arranged centrosymmetrically, reside in approximately spherically shaped pockets formed between the macrocycle molecules. Again, there is intramolecular H-bonding involving the phenolic hydrogen and an imino nitrogen [H1o–N1 = 1.68(2) Å and O1–H1o⋯N1 = 151(2)°].
The two different ethyl acetate solvates and the acetone solvate all crystallize in similar sized and shaped unit cells in space group C2/c; i.e. they are almost isomorphic (see Table 5 for unit cell geometry).
For the toluene solvate (Fig. S7, ESI† and Table 1), the asymmetric unit contains a single macrocycle and two unique solvent molecules. In this case, the conformation adopted by the macrocycle is more open such that the ‘cleft’ has an approximate ϕ angle of 89°. This open conformation allows for the formation of intermolecular π⋯π and CH⋯π interactions. The phenyl rings do not directly overlay, rather they are somewhat slipped such that a C–C bond in one ring is positioned directly below the centroid of an adjacent ring (see Fig. S8, ESI†). The shortest C to centroid distances are 3.38 and 3.42 Å. Intramolecular H-bonding is present involving the phenolic hydrogen and an imino nitrogen [H1o–N1 = 1.74(3) Å and O1–H1o⋯N1 = 150(3)°, H3o–N3 = 1.66(3) Å and O1–H3o⋯N3 = 151(3)°].
In these solvates, the range of CN bond lengths (1.258(3)–1.288(2) Å, see Table 1 and caption for Fig. 1) compares favourably with those reported for the related ethylene bridged phenolic macrocycles [1.2554(17)–1.299(7) Å],4b and those observed in bis(imino)pyridine containing macrocycles [1.246(3)–1.289(3) Å].11
In these L2H2 derived systems, the angular variation in the V-shaped cleft can also be gauged by the gradation of tilting of the t-butyl-phenol groups, from 6.09(8)° in L2H2·MeCOOEt, through L2H2·2(MeCOOEt) at 6.8(2)°, L2H2·2acetone at 7.39(7)°, L2H2·MeCN at 9.49(14) and 12.56(12)° in the two molecules (for further analysis see Table S1, ESI†). By contrast, for the L1H2 system, the structure is more open, for example L1H2·MeCN at 89.03(5)°. L2H2·2toluene is also more open, at 89.88(7), and in L2(tosyl)2, where the two phenolate rings are opposed and related by a centre of symmetry, the angle is 180.0°.
Tosylated macrocycle
The precursor 2,6-dicarboxy-4-R-phenol was prepared via tosylation of the parent tris(hydroxyl) compound 2,6-dimethanol-4-R-phenol, and during these syntheses, we isolated one of the tosylated intermediates, which was subsequently reacted with oxydianiline. The resulting tosylated macrocycle L2(tosyl)2 was crystallized from acetonitrile. The molecular structure is shown in Fig. S9, ESI (and an alternative view is given in Fig. S10 in the ESI†), with selected bond lengths and angles given in the caption. There is half a molecule in the asymmetric unit, and the molecule lies on an inversion centre. In the packing of the molecules, there is off-set π⋯π stacking: C1⋯C2′ = 3.700 Å, C2⋯O1′ = 3.456 Å and C6⋯C7′ = 3.684 Å.Preparation, structure and ROP behaviour of organoaluminium complexes
The reaction of the [2 + 2] macrocyclic Schiff bases {[2-(OH)-5-(R)C6H2-1,3-CH][O(2-C6H4N)2]}2 (R = Me L1H2, tBu L2H2, Cl L3H2) with two equivalents of R′3Al in refluxing hexane afforded, following work-up, cooling and prolonged standing (1–2 days) at ambient temperature, yellow crystals in good yield (ca. 55–67%) of the dinuclear complexes [(AlR′2)2L] (L1, R′ = Me (1), L2, R′ = Me (2), L3, R′ = Me (3), L1, R′ = Et (4), L2, R′ = Et (5), L3, R′ = Et (6)). Unfortunately, we were unable to grow single crystals of 1–6 suitable for X-ray crystallography, and so our attention turned to systems derived from the ethylene-bridged dianiline [(CH2CH2)(2-C6H4NH2)2] prepared under the same conditions. In previous work, we have investigated the reaction of two equivalents of R′3Al with such [2 + 2] Schiff-base macrocycles, but no structural information was reported. Herein, for R′ = Me, we were able to isolate and structurally characterize a secondary product, namely [(AlMe)(AlMe2)L5]·2¼MeCN (7). Small, orange, plate-like crystals were grown from a saturated acetonitrile solution on prolonged standing at ambient temperature. The crystals proved to be weakly diffracting, even when using synchrotron radiation, and so data was only integrated to 2θ = 45°. The asymmetric unit contains two macrocyclic complexes and 4.5 molecules of solvent of crystallization (MeCN). The molecular structure of one of the macrocyclic structures is shown in Fig. 3, with selected bond lengths and angles given in the caption. The interesting features of this complex are (i) the different degree of alkylation of the distorted tetrahedral aluminium centres, with Al1 bearing two methyl groups, whereas Al2 has only one, and (ii) the ‘trans’ positioning of the Al1 centres. Thus for Al1, the macrocycle binds in N,O-bi-dentate fashion, whereas for Al2, the macrocycle coordinates via a tri-dentate N,N,O mode. The conformation of the macrocycle is somewhat twisted to accommodate the tridentate nature of the bonding at Al2.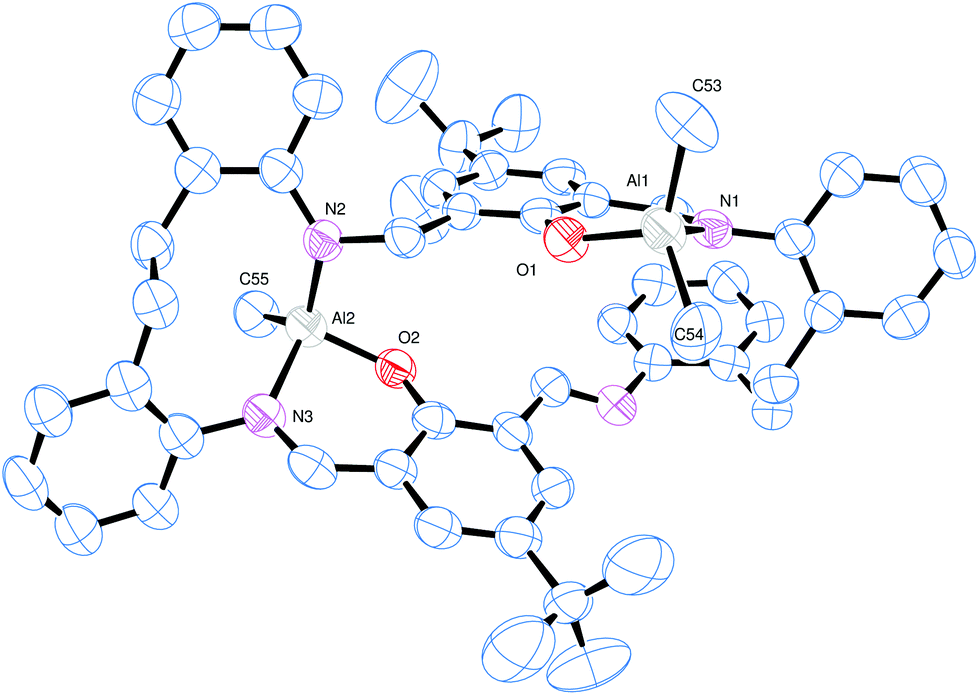 | ||
| Fig. 3 Molecular structure of [(AlMe)(AlMe2)L′]·2¼MeCN (7, R = tBu) (7), with atoms drawn as 50% probability ellipsoids. Hydrogen atoms and MeCN of crystallisation have been omitted for clarity. This is one of two similar macrocyclic complexes in the asymmetric unit. Selected bond lengths (Å) and angles (°): Al1–O1 1.761(4), Al1–N1 1.963(5), Al1–C53 1.977(6), Al1–C54 1.949(5), Al2–O2 1.768(4), Al2–N2 1.860(4), Al2–N3 1.970(4), Al2–C55 1.963(5); O1–Al1–N1 94.49(17), C53–Al1–C54 119.3(3), N2–Al2–N3 110.90(18), O2–Al2–C55 109.2(2). | ||
Given the unexpected nature of complex 7, we re-visited the complex {(Et2Al)[2-(O)-5-(Me)C6H2-1,3-CH][CH2CH2(2-C6H4N)2]}2 (8) and determined the centro-symmetric molecular structure of crystals grown from a saturated acetonitrile solution, see Fig. 4 and Table 6. Interestingly, again the structure reveals a ‘trans’ deposition of the distorted tetrahedral aluminium centres, though in this case there is the anticipated diorganoaluminiums present. Each is bound to the two opposite phenolic oxygen atoms and to a neighbouring imine nitrogen (N1 or N1i). The conformation of the macrocycle is relatively planar. The observed ‘trans’ deposition of the diethylaluminium centres in 8 could be explained in terms of steric effects, but the situation in 7 is less clear.
 | ||
| Fig. 4 Molecular structure of [(AlEt2)2L4] (8), with atoms drawn as 50% probability ellipsoids. Symmetry operator used to generate the second half of the molecule: i = 1 − x, −y, 1 − z. Hydrogen atoms and MeCN of crystallisation have been omitted for clarity. Selected bond lengths (Å) and angles (°): Al1–N1 1.9710(16), Al1–O1i 1.7826(13), N1–C23 1.294(2), N12–C15 1.276(2); N1–Al1–O(1i) 94.67(6), N1–Al1–C26 106.37(10). | ||
Conducting the reaction of L3H2 with limited Et3Al resulted in the isolation of a yellow crystalline material. Crystals grown from a saturated solution of toluene were found to be a bis-chelate structure [Al(L3)(L3H)]·4toluene (9·4toluene) (see Fig. 5, Tables 2 and 6), in which a distorted octahedral aluminium centre is bound to two of the macrocyclic ligands.
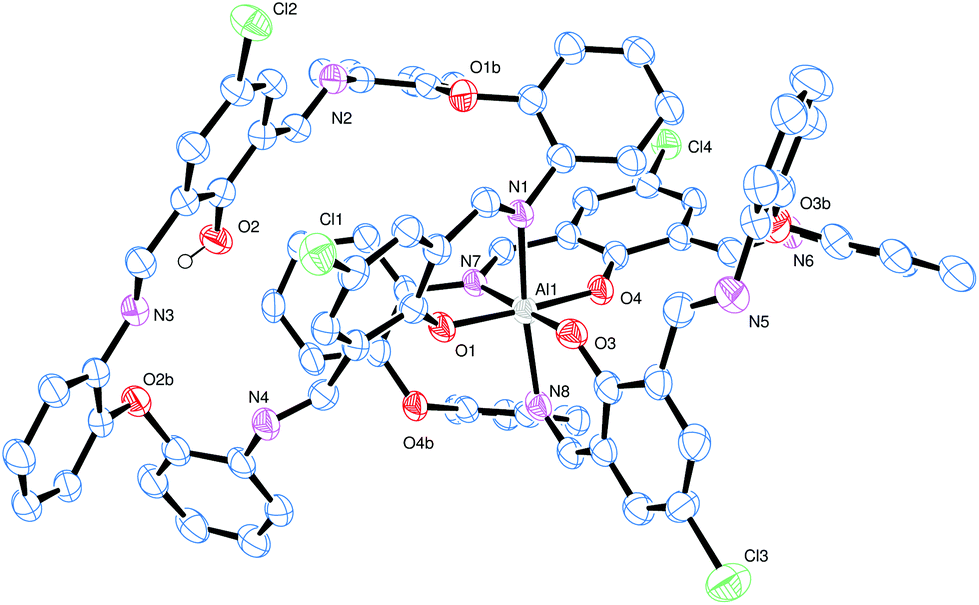 | ||
| Fig. 5 View of the molecular structure of [Al(L3)(L3H)]·4toluene (9·4toluene), with atoms drawn as 50% probability ellipsoids. Hydrogen atoms, except that on O2 which H-bonds to N3, and toluene molecules of crystallisation have been omitted for clarity. | ||
| Bond length (Å)/angle (°) | 9·4toluene | 10·5MeCN |
|---|---|---|
| Al1–O1 | 1.8121(17) | 1.814(3) |
| Al1–O3 | 1.8410(17) | 1.819(3) |
| Al1–O4 | 1.8338(17) | 1.817(3) |
| Al1–N1 | 2.100(2) | 2.090(3) |
| Al1–N7 | 2.079(2) | 2.112(4) |
| Al1–N8 | 2.114(2) | 2.087(3) |
| O1–Al1–O4 | 176.18(8) | 173.51(16) |
| O3–Al1–N7 | 176.96(8) | 178.76(14) |
| N1–Al1–N8 | 168.76(8) | 173.02(15) |
The asymmetric unit contains one complex and four toluene molecules. The central octahedral Al centre is bound by two macrocycles, with one of the macrocycles binding through two atoms [O1 and N1] to form a nearly planar 6-membered chelate ring; the remainder of this macrocycle adopts a taco-like configuration. The remaining coordination sites at aluminium are occupied by two pairs of O/N chelators (both from the other macrocycle), again forming six membered rings that are close to planar. These two chelate rings are linked by a phenyl ring and a single oxo bridge, and are approximately perpendicular at the aluminium. The remainder of this macrocycle adopts a bowl-shaped conformation. There is a single O–H⋯N hydrogen bond formed by the unbound phenol present. Within the solid-state, the crystal packing facilitates a large number of non-classical C–H⋯N and C–H⋯Cl hydrogen bonds. Four unique, crystallographically resolved, toluene molecules lie between the complexes. There is rotational disorder in their positions but no regions of disordered solvent that could be resolved. There is evidence that C–H⋯π interactions help to locate the toluene.
Similar treatment of L5H2 again afforded a bis-chelate structure, namely [Al(L5)(L5H)]·5MeCN (10·5MeCN), for which single crystals suitable for X-ray diffraction were grown from toluene at 0 °C.
The molecular structure of 10·5MeCN is shown in Fig. 6 and S11 and S12 (ESI†) which, along with the geometrical parameters (Table 2), reveals the similarity between complexes 9·4toluene and 10·5MeCN. The asymmetric unit contains one aluminium complex and 5 molecules of acetonitrile. As for 9·4toluene, the coordination at the aluminium is such that one macrocycle is bound only in chelate fashion via N,O-type ligation, whilst the second macrocycle utilizes four atoms to bind in 2× N,O-type fashion. In the bidentate ligand, there is also an intramolecular H-bond involving the phenolic group at O2 and the adjacent imine nitrogen N3. In terms of packing, the aromatic ring at C38 forms a centrosymmetric π⋯π interaction at 3.6 Å.
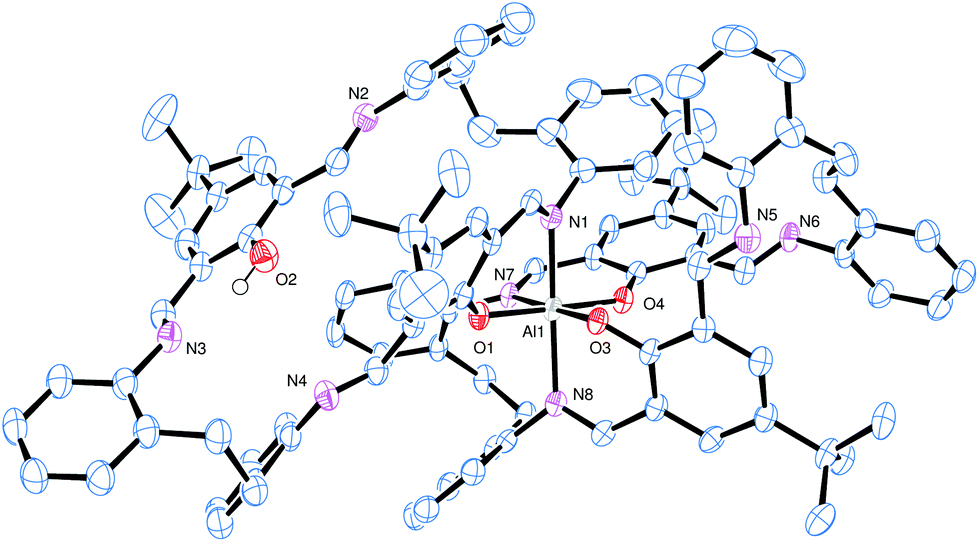 | ||
| Fig. 6 The molecular structure of [Al(L5)(L5H)]·5MeCN (10·5MeCN), with atoms drawn as 50% probability ellipsoids. Hydrogen atoms, except that on O2 which H-bonds to N3, and MeCN solvent molecules of crystallisation have been omitted for clarity. | ||
Treatment of LH2 with excess R′3Al (four equivalents) in refluxing hexane afforded, following work-up (extraction into toluene), cooling and prolonged standing (1–2 days) at ambient temperature, yellow crystals in moderate yield (ca. 30–35%) of the tetra-nuclear complexes [(AlR′2)4L1′–3′] (R = L2′, R′ = Me (11); L3′, R′ = Me (12); L1′, R′ = Et (13); L3′, R′ = Et (14)), where L1′–3′ is the macrocycle resulting from double alkyl transfer to imine, namely {[2-(O)-5-(R)C6H2-1-(CH)-3-C(R′)H][(O)(2-(N)-2′-C6H4N)2]}2. In the case of the reaction involving L1H2 and Me3Al, single crystals of the complex were grown from a saturated hexane/toluene (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) solution at 0 °C. The molecular structure is shown in Fig. 7, with selected bond lengths and angles given in the caption. This reveals the formation of a tetra-nuclear complex (11) akin to that formed form when using the analogous –CH2CH2– bridged Schiff-base macrocycle.12 For a relatively simple compound, the crystal structure displays unwelcome complexity. There are four, symmetry unique, bowl-shaped molecules of 11·1¾toluene·1¼hexane occupying the asymmetric unit. Each of these binds four AlMe2 units; subtle differences in the configuration of the macrocycles render these symmetry independent. Between these macrocycles lie crystallographically resolved and unresolved solvent to give an estimated formula (after Squeeze)13 of 8{(Me2Al)4[2-(O)-5-(tBu)C6H2-1-CH-3C(Me)H][O(2-C6H4N)2]}2·14toluene·9hexane. To simplify the discussion of the four similar units, the orientation of one macrocycle will be discussed. The macrocycle is twisted such that one tert-butyl group is pointing ‘up’ and one ‘down’. At the opposite end of each of the phenyl groups bearing the tert-butyl are bound two AlMe2 units. Each aluminium is coordinated by two methyl groups and one neutral imine and a phenoxide in approximately tetrahedral geometry. The phenoxides bridge between the two aluminium centres (atoms O1 and O3 in Fig. 7). One pair of aluminium atoms reside on one side of the molecule and the others lie on the opposite side. There is evidence for C–H⋯π interactions between adjacent macrocycles but the packing is unremarkable. Between the macrocycles lie ordered and disordered solvent; some hexane and toluene are crystallographically resolved. There are also portions of the structure in which the solvent molecules cannot be located reliably and these regions were modelled using the Squeeze routine.13
:50) solution at 0 °C. The molecular structure is shown in Fig. 7, with selected bond lengths and angles given in the caption. This reveals the formation of a tetra-nuclear complex (11) akin to that formed form when using the analogous –CH2CH2– bridged Schiff-base macrocycle.12 For a relatively simple compound, the crystal structure displays unwelcome complexity. There are four, symmetry unique, bowl-shaped molecules of 11·1¾toluene·1¼hexane occupying the asymmetric unit. Each of these binds four AlMe2 units; subtle differences in the configuration of the macrocycles render these symmetry independent. Between these macrocycles lie crystallographically resolved and unresolved solvent to give an estimated formula (after Squeeze)13 of 8{(Me2Al)4[2-(O)-5-(tBu)C6H2-1-CH-3C(Me)H][O(2-C6H4N)2]}2·14toluene·9hexane. To simplify the discussion of the four similar units, the orientation of one macrocycle will be discussed. The macrocycle is twisted such that one tert-butyl group is pointing ‘up’ and one ‘down’. At the opposite end of each of the phenyl groups bearing the tert-butyl are bound two AlMe2 units. Each aluminium is coordinated by two methyl groups and one neutral imine and a phenoxide in approximately tetrahedral geometry. The phenoxides bridge between the two aluminium centres (atoms O1 and O3 in Fig. 7). One pair of aluminium atoms reside on one side of the molecule and the others lie on the opposite side. There is evidence for C–H⋯π interactions between adjacent macrocycles but the packing is unremarkable. Between the macrocycles lie ordered and disordered solvent; some hexane and toluene are crystallographically resolved. There are also portions of the structure in which the solvent molecules cannot be located reliably and these regions were modelled using the Squeeze routine.13
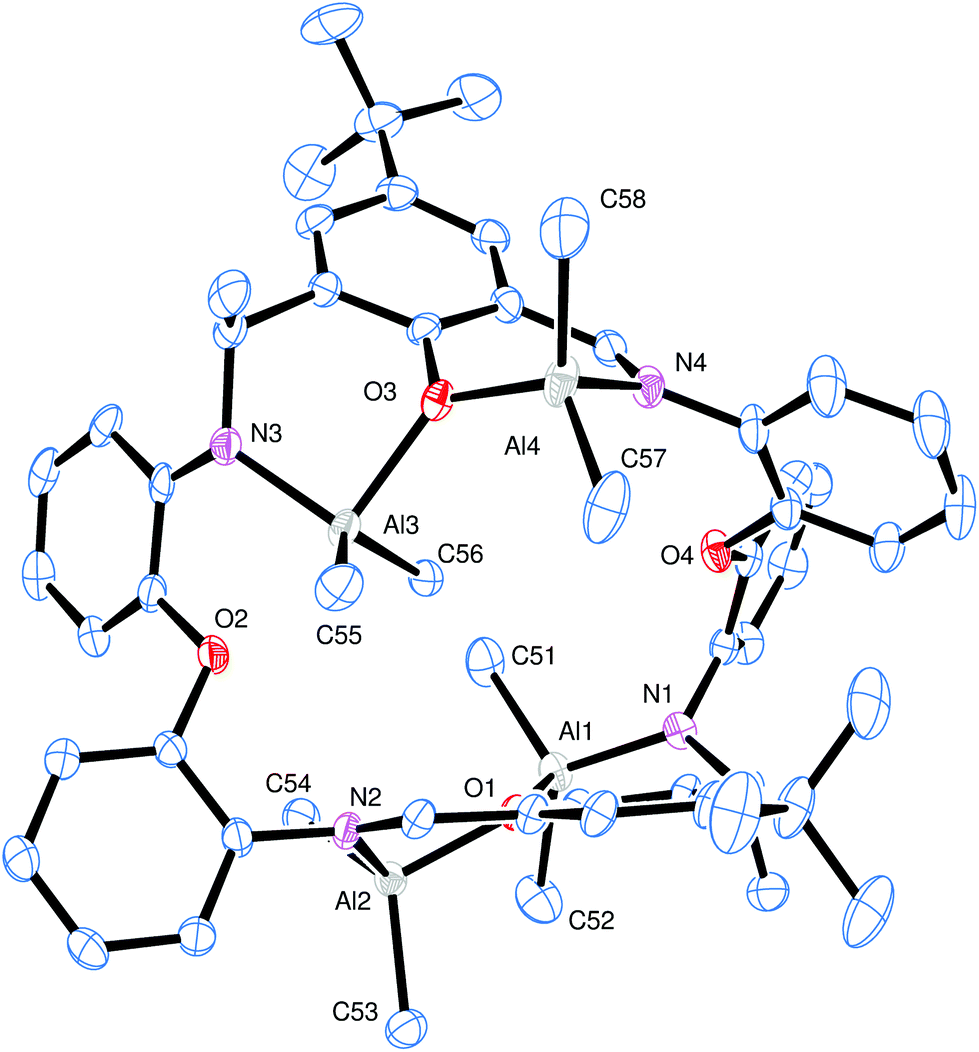 | ||
| Fig. 7 Molecular structure of [(AlMe2)4L1′]·1¾toluene·1¼hexane (11·1¾toluene·1¼hexane), showing the atom numbering scheme. Hydrogen atoms and solvent molecules of crystallisation have been omitted for clarity. This is one of four unique complex molecules in the asymmetric unit. Selected bond lengths (Å) and angles (°): Al1–N1 1.820(3), Al1–O1 1.950(2), Al2–O1 1.857(2), Al2–N2 1.952(2), Al3⋯O2 2.430(2), Al3–O3 1.997(2), N1–C1 1.469(4), N2–C13 1.286(4), N2–C15 1.276(2), N2–C14 1.442(4), N3–C25 1.381(4), N3–C26 1.473(4), N4–C38 1.288(4), N4–C39, Al1⋯Al2 3.1695(12), Al1⋯Al3 5.8984(13), Al1⋯Al4 7.3100(13), Al2⋯Al3 5.0994(13), Al2⋯Al4 7.5339(13), Al3⋯Al4 3.4600(13); Al1–O1–Al2 112.73(10), Al3–O3–Al4 129.79(12), N1–Al1–O1 95.53(10), O1–Al2–N2 94.72(10). | ||
The formation of 11 involves an intramolecular regioselective methyl transfer to two imine moieties of the macrocycle; such methyl transfers are now well established in imine chemistry.14 The methyl transfer occurs at imine groups originating from the same dianiline. In the 1H NMR spectra of 11, the Me–Al resonances occur as eight singlets between −0.52 and −1.39 ppm (and four singlets between −0.49 and −1.01 for 12). In the case of the related ethyl derivatives 13 and 14, two of the Al–Et groups appear to be subject to ring currents which result in unusual low field chemical shifts in the 1H NMR spectra for the CH2 protons (see Experimental section).
Ring opening polymerisation (ROP) of ε-caprolactone and rac-lactide
The dinuclear alkylaluminium complexes 1–6 and the tetranuclear alkylaluminium complexes 11–14 have been screened for their ability to ring open polymerise ε-caprolactone (see Tables 3 and S2†) and rac-lactide (Tables 4 and S4†). Results are compared against the known –CH2CH2– bridged complexes 15 and 16.|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Run | Cat. | CL:Al:BnOH |
T/°C | t/min | Conv.a/% |
M
n × 104b |
M
n,Calcd × 104c |
PDI |
| a By 1H NMR spectroscopic analysis. b Obtained from GPC analysis times 0.56. c (F.W.[M]/[BnOH])(conversion). | ||||||||
| 1 | 5 | 250:1:1 |
20 | 60 | 15.8 | 0.59 | 0.45 | 1.08 |
| 2 | 5 | 250:1:1 |
50 | 30 | 64.4 | 1.57 | 1.82 | 1.15 |
| 3 | 5 | 250:1:1 |
80 | 30 | 98.0 | 3.36 | 2.82 | 1.56 |
| 4 | 5 | 250:1:1 |
110 | 30 | 98.5 | 2.67 | 2.71 | 1.58 |
| 5 | 5 | 250:1:1 |
80 | 10 | 59.0 | 2.98 | 1.68 | 1.29 |
| 6 | 5 | 250:1:1 |
80 | 20 | 92.5 | 3.24 | 2.63 | 1.34 |
| 7 | 5 | 250:1:1 |
80 | 60 | 99.2 | 2.88 | 2.69 | 1.40 |
| 8 | 5 | 100:1:1 |
80 | 30 | 99.1 | 2.16 | 1.12 | 1.13 |
| 9 | 5 | 500:1:1 |
80 | 30 | 86.7 | 4.62 | 4.94 | 4.01 |
| 11 | 5 | 250:1:0 |
80 | 30 | 80.1 | 6.59 | 2.28 | 1.60 |
| 12 | 5 | 250:1:3 |
80 | 30 | 93.1 | 2.02 | 2.65 | 1.26 |
| Run | Lac:M:BnOH |
T/°C | t/h | Conv.a/% |
M
n × 104b |
M
n,Cal × 104c |
PDI |
|---|---|---|---|---|---|---|---|
| a By 1H NMR spectroscopic analysis. b M n values were determined by GPC in THF vs. PS standards and were corrected with a Mark–Houwink factor of 0.58. c Polydispersity index (Mw/Mn) were determined by GPC. | |||||||
| 1 | 100:1:1 |
110 | 1 | 57.8 | 0.42 | 0.83 | 1.02 |
| 2 | 100:1:1 |
110 | 3 | 91.3 | 0.63 | 1.31 | 1.03 |
| 3 | 100:1:1 |
110 | 6 | 95.0 | 1.56 | 1.39 | 1.21 |
| 4 | 100:1:1 |
110 | 12 | 97.7 | 1.60 | 1.40 | 1.19 |
| 5 | 100:1:1 |
110 | 24 | 98.6 | 1.45 | 1.40 | 1.14 |
| 6 | 100:1:1 |
50 | 12 | — | — | — | — |
| 7 | 100:1:1 |
80 | 12 | 66.7 | 0.74 | 0.96 | 1.07 |
| 8 | 50:1:1 |
110 | 12 | 94.3 | 0.80 | 0.67 | 1.41 |
| 9 | 200:1:1 |
110 | 12 | 96.6 | 2.29 | 2.78 | 1.14 |
:1:1 (ε-CL:cat:BnOH) over 30 min (runs 1–4, Table 3), the % conversion dramatically increased, reaching around 98% conversion at 80 °C and then increasing only slightly on further elevating the temperature to 110 °C. Under the same conditions, the molecular weight (Mn) peaked at 80 °C. All the polycaprolactone polymers (PCLs) obtained possessed a narrow distribution/polydispersity index (PDI) with unimodal characteristics [Mw/Mn = 1.12–1.58]. The drop off in molecular weight at 110 °C results in a plot of % conversion versus Mn which is only approximately linear. We have also investigated the effect of the ε-CL/Al molar ratio on the catalytic behaviour (entries 3, 8 and 9, Table 3) in the presence of one equivalent of BnOH. When the molar ratio CL:Al was increased from 100 to 500 over 30 min, the molecular weight increased from 2.16 to 4.62 × 104, whilst the conversion rate exhibited the opposite trend peaking at 99.1% for 100:1:1; the molecular weight distribution increased on increasing the molar ratio CL:Al (from 1.13 to 4.01). On increasing the time from 10 min to 60 min, and using 250:1:1 (CL:Al:BnOH) at 80 °C (runs 3, 5–7, Table 3), the conversion gradually increased with time, whilst the molecular weight (Mn) and polydispersity (PDI) remained relatively constant, except in the case of run 9 where it was, surprisingly, somewhat broader (4.01). Increasing the amount of BnOH (run 12 versus 3, Table 3) was detrimental to the molecular weight (Mn), whilst only slightly narrowing the polydispersity, and lowering the % conversion slightly. Conducting the ROP in the absence of BnOH (run 11 versus 3, Table 3) led to a reduction in the % conversion, but afforded a significant increase in the polymer molecular weight (Mn); there was little change in the PDI.
Complexes 1–14 (not 8–10) were then screened using the ratio 250:1:1 (ε-CL:cat:BnOH) over 30 min at 80 °C, and for comparison, the known complexes 15 and 16 were screened under the conditions employed herein. For the di-nuclear complexes 1–6 (runs 1–6, Table S2†), in terms of the % conversion, these complexes behave similarly, which does not allow for the observation of any significant structure/activity relationships. Given this, we provide only a brief discussion here and the tabulated data can be found in the ESI (Table S2,† runs 1–13). For 1–6, the highest conversion was observed for 5 (R = tBu, R′ = Et: 98.0%) and the lowest for 1 (R = R′ = Me: 93.2%). For pairs of complexes where R is constant, the ethyl derivatives were more active than the methyl derivatives and the molecular weights (Mn) were higher; such trends have been noted previously;15 the opposite trends in activity have also been noted.16 The spread of molecular weights (Mn) [5.14–10.12 × 104] also followed no obvious trend, whilst in all cases, the PDI remained relatively constant [1.22–1.49]. However, in all cases, the performance of the oxy bridged systems was superior to that of the di-nuclear –CH2CH2– bridged complexes 7 and 15, for which the % conversion was only 25.6% and 38.5%, respectively under the conditions employed herein.
In the case of the tetra-nuclear complexes 11–14 (runs 8–11, Table S2†), the complexes bearing methyl at the para position of the phenolic group afforded high conversions of about 99%, whilst the systems (12 and 14), employing a para Cl, gave lower conversions of 80.9 and 94.3%, respectively. This may be attributed to observed solubility issues rather than electronic effects. The polymer molecular weight (Mn) associated with 12 and 14 was also somewhat lower than that observed for the other tetra-nuclear systems. Again, the performance of the related –CH2CH2– bridged complex, namely 16 was inferior under the conditions employed herein affording a % conversion of 29.1% and a much lower molecular weight (Mn). This enhanced activity is tentatively ascribed to the ability of the oxygen bridge to stabilize the catalytically active species, akin to the situation observed in dimethyleneoxa-bridged calixarene systems during ethylene polymerization.17 As for the di-nuclear systems, the tetra-nuclear ethylaluminium derivatives (13 and 14) were more active than the methylaluminium counterparts (11 and 12).
In general, the resulting PCL polymer molecular weights were in reasonable agreement with the calculated values, which indicates that there are, in most cases, little in the way of trans-esterification reactions occurring. However, in the MALDI-ToF mass spectra, as well as the population of peaks separated by 114.14 mass units (see Fig. S13 and S14†), there was evidence of a second, albeit minor, population which is more pronounced at 25 °C. A plot of average molecular weight (Mn) versus conversion (Fig. S15†) exhibited a near linear relationship. In the 1H NMR spectra of the PCL (Fig. S16 and S17†), signals at around 7.34 and 5.15 ppm (C6H5CH2–) and 3.62 (CH2CH2OH), with an integral ratio 5:2:2, indicated that the polymer chains are capped by a benzyl group and a hydroxy end group.
:1:1 for rac-Lac:Al:BnOH, whilst prolonging the screening time from 6 to 24 h only afforded a slight increase in the % conversion. In all cases, the system was relatively well controlled with polydispersities in the range 1.03–1.41.
Complexes 1–14 (not 8–10) were then screened using the ratio 100:1:1 (rac-LA:cat:BnOH) over 12 h at 110 °C (Table S2,† runs 14–23). The ROP appeared to be well controlled in terms of PDI with values in the range 1.07–1.38. There was no obvious advantage in the use of di- versus tetra-nuclear systems under the conditions employed. For the di-nuclear systems, the ethylaluminium derivatives were slightly more active than their methylaluminium counterparts and the polymers possessed slightly higher molecular weight (Mn), however this trend was not evident for the tetra-nuclear systems. 1H NMR spectroscopic investigations were conducted in order to verify the polymer molecular weights and to identify the end groups present. The results were similar (e.g. see Fig. S18†) to the results obtained for the PCL runs, i.e. insertion of a benzyloxy group during polymerization. Again, there was reasonable agreement between observed and calculated molecular weights (Mn), whilst MALDI-ToF spectra (e.g. Fig. S19†) revealed a number of minor populations. To assign the stereochemistry of the PLA polymers, we employed 2D J-resolved 1H NMR (e.g. see Fig. S20 and S21†) and assigned the peaks by reference to the literature.18 These systems gave moderately isotactic PLA with Pr values in the range 0.64–0.67.
In conclusion, [2 + 2] Schiff base macrocycles of the type {[2-(OH)-5-(R)C6H2-1,3-(CH)2][O(2-C6H4N)2]}2 (R = Me L1H2, tBu L2H2, Cl L3H2) are readily accessible by reacting 2,6-dicarboxy-4-R-phenol with the diamine 2,2′-oxydianiline, (2-NH2C6H4)2O. The molecular structures of a number of solvates have been determined. The molecular structures of the various solvates reveal a tendency to form a taco-shaped conformation, the cleft angle ϕ associated with the latter varies greatly with that of L1H2·MeCN and L2H2·2toluene being very open at about 89°, whilst the other solvates (MeCN, acetone and ethyl acetate) of L2H2 were more closed with cleft angles ϕ in the range 8–17°. The solvent is only encapsulated by the macrocycle in L1H2·MeCN. Ethyl acetate and acetone reside in similar locations exo to the macrocycle in a series of three pseudo-isomorphic structures. Furthermore, we have found that the interaction of alkylaluminium reagents can be more complicated than originally thought (from studies of the –CH2CH2– bridged systems) and a number of unexpected products can be formed. In particular, we have found that for the di-nuclear species, ‘trans’ as well as the previous ‘cis’ structures can readily be isolated, as can complexes in which one of the methylaluminium centres is bound in tridentate fashion by the macrocycle. Moreover, species in which there are no alkyl groups at aluminium, but where two macrocycles bind such that the Al centre is near octahedral, are readily formed in the presence of limited organoaluminium reagent. Tetra-nuclear complexes can be accessed which have undergone alkyl transfer (×2) to one side of the macrocycle by employing excess organoaluminium reagent. These organoaluminium species are capable of the ROP of ε-caprolactone and rac-lactide and can out-perform the related systems bearing –CH2CH2– bridged Schiff-base macrocycles under similar conditions. However, there appears to be little benefit in the use of di- versus tetra-nuclear species under the ROP conditions employed herein.
Experimental
General
Methanol was dried over magnesium. Hexane was refluxed over sodium and benzophenone. Toluene was refluxed over sodium. Acetonitrile was refluxed over calcium hydride. IR spectra (nujol mulls, KBr windows) were recorded on a Nicolet Avatar 360 FT IR spectrometer; 1H NMR and 13C NMR spectra were recorded at room temperature on a Varian VXR 400 S spectrometer at 400 MHz or a Gemini 300 NMR spectrometer or a Bruker Advance DPX-300 spectrometer. The 1H NMR spectra were calibrated against the residual protio impurity of the deuterated solvent. Elemental analyses were performed by the elemental analysis service at the London Metropolitan University, the Chemistry Department at the University of Hull or at Sichuan University, Chengdu. The precursors 2,6-(CHO)2-4-R-C6H2OH and (2-NH2C6H4)2O and 2,2′-ethylenedianiline and the complexes 15 and 16 were prepared by the literature methods.12,19,20 The Schiff-base ligands were prepared as outlined below, and the respective solvates were crystallized by taking about 100 mg of the macrocycle and dissolving in the appropriate solvent. In the case of acetonitrile and toluene, the solvates crystallized out at ambient temperature, whereas for acetone and ethyl acetate, cooling to −20 °C was required. For the organoaluminium complexes, all manipulations were carried out under an atmosphere of dry nitrogen using conventional Schlenk and cannula techniques or in a conventional nitrogen-filled glove box. All solvents were distilled and degassed prior to use.N), 7.54 (s, 4H, Ar–H), 7.12–7.24 (m, 16H, Ar–H), 2.27 (s, 3H, –CH3), 2.24 (s, 3H, –CH3). 13C NMR (100 MHz, DMSO-d6) δ: 20.4, 116.0, 116.6, 117.7, 120.1, 124.2, 127.7, 140.1, 143.6, 149.7, 160.4.
N), 7.25 (s, 4H, Ar–H), 7.06–7.25 (m, 16H, Ar–H), (s, 18H, C(CH3)3). 13C NMR (100 MHz, DMSO-d6) δ: 31.7, 34.3, 116.0, 116.8, 118.2, 120.6, 124.2, 125.1, 140.1, 140.3, 143.6, 160.9.
N), 7.58 (s, 4H, Ar–H), 7.22–7.34 (m, 12H, Ar–H), 7.07 (d, J = 11.2 Hz, 4H, Ar–H). This compound proved to be too insoluble to obtain meaningful 13C NMR spectra, even upon heating in DMSO-d6.
N), 7.56 (d, J = 8.0 Hz, 2H, C6H2), 7.43 (m, 4H, arylH), 7.31 (d, 4H, arylH), 7.10 (m, H, arylH), 6.99 (d, J = 8.4 Hz, 2H, arylH), 6.35 (s, 2H, CHN), 2.20 (s, 6H, CH3), −0.74 (s, 6H, Al–CH3), −0.75 (s, 6H, Al–CH3).
N), 7.62 (s, 2H, CHN), 7.50 (d, 2H, J = 8.4 Hz, arylH), 7.41–7.46 (m, 2H, arylH), 7.26–7.30 (t, 4H, arylH), 7.11–7.16 (m, 2H, arylH), 7.04–7.08 (m, arylH), 6.99 (dd, 2H, J1 = 7.6 Hz, J2 = 1.6 Hz, arylH), 6.92 (dd, 2H, J1 = 8.0 Hz, J2 = 1.2 Hz, arylH), 6.70–6.71 (d, 2H, J = 2.8 Hz, C6H2), 1.26 (s, 18H, (CH3)3), −0.83 (s, 6H, Al–CH3), −0.84 (s, 6H, Al–CH3).
N), 6.97–7.58 (m, 18H, arylH), 6.59 (d, J = 2.8, 2H, CHN), −0.67 (s, 6H, Al–CH3), −0.73 (s, 6H, Al–CH3).
N), 7.57 (d, J = 8.0 Hz, 2H, arylH), 7.52 (s, 2H, CHN), 7.43–7.48 (m, 2H, arylH), 7.34 (m, 4H, arylH), 7.06–7.13 (m, 6H, arylH), 6.39 (dd, J = 8.0 Hz, J = 1.6 Hz, 2H, arylH), 6.39 (d, 2H, J = 2.4 Hz, C6H2), 2.19 (s, 6H, CH3), 0.94 (t, J = 8.0 Hz, 6H, Al–CH2CH3), 0.74 (t, J = 8.4 Hz, 6H, Al–CH2CH3), −0.07 to −0.09 (overlapping m, 8H, Al–CH2CH3).
N), 7.73 (s, 2H, CHN), 6.97–7.46 (m, 16H, arylH), 6.06 (d, J = 2.8 Hz, C6H2), 0.94 (t, J = 8.0 Hz, 6H, Al–CH2CH3), 0.63 (t, J = 8.0 Hz, 6H, Al–CH2CH3), −0.06 to −0.22 (overlapping m, 8H, Al–CH2CH3).
N), 7.02–7.73 (m, 18H, arylH), 6.06 (d, J = 4.2 Hz, C6H2), 0.94 (t, J = 8.0 Hz, 6H, Al–CH2CH3), 0.73 (t, J = 8.0 Hz, 6H, Al–CH2CH3), −0.05 to −0.11 (overlapping m, 8H, Al–CH2CH3).
N), 8.17 (d, 2H, J = 2.0 Hz, C6H2), 7.60 (d, J = 7.60 Hz, 2H, arylH), 7.40 (t, 2H, J = 7.2 Hz, arylH), 7.26 (t, 4H, J = 6.0 Hz, arylH), 6.99 (d, 2H, J = 5.6 Hz, arylH), 6.90 (t, 2H, J = 7.2 Hz, arylH), 6.81 (d, 2H, J = 7.6 Hz, arylH), 6.68 (d, 2H, J = 2.4 Hz, C6H2), 6.62 (d, 2H, J = 6.8 Hz, arylH), 6.42 (s, 2H, CHN), 3.81 (dt, J1 = 12.8 Hz, J2 = 4.0 Hz, 2H, CH2), 3.69 (td, J1 = 13.2 Hz, J2 = 4.0 Hz, 2H, CH2), 3.01 (dt, J1 = 14.0 Hz, J2 = 4.8 Hz, 2H, CH2), 2.68 (td, J1 = 12.8 Hz, J2 = 4.4 Hz, 2H, CH2), 2.41 (s, 6H, CH3), 0.94 (t, 6H, J = 8.4 Hz, Al–CH2CH3), 0.72 (t, 6H, J = 8.0 Hz, Al–CH2CH3), 0.05 (m, 4H, Al–CH2), 0.32 (m, 4H, Al–CH2).
N), 8.50 (s, 2H, CHN), 8.32 (s, 2H, CHN), 7.61 (s, 4H, Ar–H), 7.25–7.12 (m, 28H, Ar–H), 7.02 (overlapping m, 10H, Ar–H).
N), 8.71 (bs, 2H, CHN), 8.35 (bs, 4H, CHN), 8.29 (m, 2H, arylH), 7.91–6.18 (overlapping m, 32 H, arylH), 5.88 (d, 2H, arylH), 5.86 (d, 2H, J = 18.0 Hz, arylH), 5.62 (d, 2H, J = 14.4 Hz, arylH), 5.34 (bm, 2H, CH2), 4.56 (bm, 2H, CH2), 3.86 (bm, 2H, CH2), 3.74 (bm, 2H, CH2), 3.30 (bm, 2H, CH2), 3.13 (overlapping m, 2H, CH2), 3.07 (bm, 2H, CH2), 2.91 (bm, 2H, CH2), 2.44 (s, 3H, MeCN), 2.01 (s, 3H, MeCN), 0.92 (s, 6H, MeCN), 1.56 (s, 9H, C(CH3)3), 1.41 (s, 9H, C(CH3)3), 1.29 (s, 9H, C(CH3)3), 1.19 (s, 9H, C(CH3)3).
:50) solution at 0 °C afforded 11·1.75toluene·1.25hexane as a red crystalline solid on prolonged standing at 0 °C (1–2 days). Yield 0.25 g, 36.9%. Elemental analysis calculated for C58H72N4O4Al4: C 69.87, H 7.28, N 5.62%; found (sample dried in vacuo for 12 h): C 69.52, H 6.93, N 5.22%. IR (cm−1): 3413 (s), 3064 (m), 2929 (m), 2857 (m) 1624 (s), 1608 (s), 1551 (m), 1508 (s), 1486 (s), 1456 (s), 1377 (w), 1329 (m), 1261 (s), 1233 (m), 1192 (m), 1157 (w), 1101 (s), 1024 (s), 863 (m), 801 (w), 741 (m), 660 (w). MS (E.I.): 1017.43 [M + Na]+. 1H NMR (CDCl3, 400 MHz): δ 8.29 (d, J = 2.4 Hz, 2H, C6H2), 8.02 (s, 2H, CHN), 7.02–7.73 (m, 16H, arylH), 6.06 (d, 2H, J = 4.2 Hz, C6H2), 4.55 (m, 1H, CHCH3), 4.28 (m, 1H, CHCH3), 1.66 (d, 3H, CH3CH), 1.53 (d, 3H, CH3CH), 1.25 (s, 9H, C(CH3)3), 0.89 (s, 9H, C(CH3)3), −0.52 (2 × s, 6H, Al–CH3), −0.77 (s, 3H, Al–CH3), −0.87 (s, 3H, Al–CH3), −0.89 (s, 3H, Al–CH3), −1.14 (s, 3H, Al–CH3), −1.37 (s, 3H, Al–CH3), −1.39 (s, 3H, Al–CH3).
N), 7.43 (td, 2H, J1 = 8.4 Hz, J2 = 1.6 Hz, arylH), 7.36 (m, 2H, arylH), 7.32 (dd, 2H, J1 = 7.2 Hz, J2 = 1.6 Hz, arylH), 7.27 (d, 2H, J = 2.8 Hz, C6H2), 7.18 (m, 2H, arylH), 7.08 (td, 2H, J1 = 8.4 Hz, J2 = 1.6 Hz, arylH), 6.99 (d, 2H, J = 7.6 Hz, arylH), 6.71 (d, 2H, J = 2.4 Hz, C6H2), 6.52 (m, 4H, arylH), 4.47 (q, 2H, J = 7.2 Hz, CHCH3), 1.59 (d, 6H, J = 7.2 Hz, CHCH3), −0.49 (s, 6H, Al–CH3), −0.73 (s, 6H, Al–CH3), −0.83 (s, 6H, Al–CH3), −1.01 (s, 6H, Al–CH3).
N), 4.55 (m, 2H, NCHEt), 2.26 (m, 2H, CHCH2CH3), 2.17 (m, 2H, CH2CH3), 1.91 (s, 6H, CH3 toluene), 1.84 (s, 6H, CH3 toluene), 1.63 (m, 6H, CH3), 1.49 (m, 2H, AlCH2CH3), 1.42 (m, 2H, AlCH2CH3), 0.94 (overlapping m, 12H, CHCH2CH3 + Al–CH2CH3), 0.78 (t, J = 8.4 Hz, 6H, Al–CH2CH3), 0.53 (t, J = 7.2 Hz, 6H, Al–CH2CH3), 0.42 (t, J = 8.2 Hz, 6H, Al–CH2CH3), −0.05 (m, 4H, Al–CH2CH3), −0.26 (m, 4H, Al–CH2CH3), −1.21 (m, 2H, Al–CH2CH3) and −1.50 (m, 2H, Al–CH2CH3).
N), 4.60 (m, 2H, J1 = 9.6, J2 = 1.4 Hz, CHEt), 2.36 (m, 2H, CHCH2CH3), 2.20 (m, 2H, CHCH2CH3), 2.10 (s, 8.4H, CH3 of 2.8toluene), 1.77 (m, 2H, Al–CH2CH3), 1.65 (m, 2H, Al–CH2CH3), 1.02 (overlapping m, J = 8.0 Hz, 12H, CHCH2CH3 + Al–CH2CH3), 0.86 (t, J = 7.2 Hz, 6H, Al–CH2CH3), 0.74 (t, J = 8.2 Hz, 6H, Al–CH2CH3), 0.52 (t, J = 8.2 Hz, 6H, Al–CH2CH3), 0.04 (m, 4H, Al–CH2CH3), −0.14 (m, 4H, Al–CH2CH3), −1.13 (m, 2H, Al–CH2CH3), −1.41 (m, 2H, Al–CH2CH3).
ROP procedure
Experimental crystallography
Diffraction data for L1H2·MeCN and L2(tosyl)2 were measured on Bruker SMART 1000 CCD and APEX 2 CCD diffractometers respectively, with Mo-Kα radiation, at 150(2) K using 0.3° ω-scans.21 Corrections were made for absorption and for Lorentz and Lp effects.21 The structures were solved by direct methods and refined on F2 by full-matrix-least squares.22For the L2H2.solv. samples, diffraction intensities were measured on Oxford Diffraction Xcalibur-3 or New Gemini CCD diffractometers equipped with Mo-Kα radiation and graphite monochromator. The data for L2H2·2(acetone) were recorded at room temperature but the other samples were measured at temperatures between 120 and 140 K. Intensity data were measured by thin-slice ω- and ϕ-scans. Data were processed using the CrysAlis-CCD and -RED23 programs. The structures were determined by the direct methods routines in the SHELXS program22 and refined by full-matrix least-squares methods, on F2, in SHELXL.22
For 7·2¼MeCN, data collected at Daresbury Laboratory Station 9.8.21 The crystal was weakly diffracting, so data were only integrated to 2θ = 45°. The tBu group at C89 was modeled as two-fold disordered with a major component of 72.8(9)%, whilst the MeCN containing N12 was refined at half weight. For 8, data were collected using an Agilent Xcalibur diffractometer with an Eos detector. Single crystal diffraction data for 9·4toluene and 10·5MeCN were collected by the UK National Crystallography Service using a Rigaku FR-E+ diffractometer. This operates with a SuperBright rotating anode X-ray generator and high flux optics. For 10·5MeCN, one MeCN was refined as point atoms, the other four as regions of diffuse electron density using the Platon Squeeze procedure.13 Squeeze identifies 2 voids per unit cell, each containing 207 electrons. Inspection of the residual electron density prior to squeeze strongly suggested 4 MeCNs. Each MeCN contains 22 electrons so, although 207 electrons indicates ca. 9.4 MeCNs, only 8 were added per void, or 4 per metal complex. For 11·1¾toluene·1¼hexane, data were collected with an Agilent Gemini diffractometer using molybdenum radiation and an Eos S2 detector. Disordered solvent was modelled using the Squeeze routine, which identified two voids per unit cell containing a total of 1210 electrons. This was modelled using 9 toluene and 4 hexane molecules (the ratio of disordered toluene to hexane cannot be estimated by this technique).
Structures of the complexes 7–11 were solved using Direct Methods implemented within SHELXS-2013 and refined within SHELXL-2014.24 Further details are provided in Tables 5 and 6.
| Compound | L 1 H2·MeCN | L 2 H2·MeCN | L 2 H2·MeCOOEt | L 2 H2·2(MeCOOEt) | L 2 H2·2(Me2CO) | L 2 H2·2(PhMe) | L 2 (tosyl)2 |
|---|---|---|---|---|---|---|---|
| Formula | C42H32N4O4·C2H3N | C48H44N4O4·C2H3N | C48H44N4O4·C4H8O2 | C48H44N4O4·2(C4H8O2) | C48H44N4O4·2(C3H6O) | C48H44N4O4·2(C7H8) | C62H56N4O8S2 |
| Formula weight | 697.77 | 781.92 | 828.97 | 917.08 | 857.02 | 925.14 | 1049.23 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
C2/c | C2/c | C2/c | P21/n | P21/n |
| Unit cell dimensions | |||||||
| a (Å) | 11.0841(6) | 15.1737(5) | 24.8335(10) | 24.9034(15) | 24.5582(10) | 13.8127(5) | 13.201(3) |
| b (Å) | 12.2117(6) | 15.3473(6) | 11.2046(4) | 11.5371(6) | 12.1677(7) | 16.8060(6) | 13.348(3) |
| c (Å) | 13.8841(7) | 19.2180(7) | 15.9714(11) | 16.9261(12) | 16.0892(7) | 22.5196(9) | 14.966(3) |
| α (°) | 86.1299(8) | 98.169(13) | 90 | 90 | 90 | 90 | 90 |
| β (°) | 74.9778(8) | 109.862(3) | 101.497(6) | 96.003(6) | 98.942(4) | 105.428(4) | 94.913(3) |
| γ (°) | 89.6361(8) | 91.656(3) | 90 | 90 | 90 | 90 | 90 |
| V (Å3) | 1810.81(16) | 4152.1(3) | 4354.9(4) | 4836.4(5) | 4749.3(4) | 5039.2(3) | 2627.4(10) |
| Z | 2 | 4 | 4 | 4 | 4 | 4 | 2 |
| Temperature (K) | 150(2) | 140(2) | 120.0(2) | 120.0(2) | 293(2) | 130.0(1) | 150(2) |
| Wavelength (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Calculated density (g cm−3) | 1.280 | 1.251 | 1.264 | 1.259 | 1.199 | 1.219 | 1.326 |
| Absorption coefficient (mm−1) | 0.08 | 0.08 | 0.083 | 0.084 | 0.078 | 0.076 | 0.164 |
| Transmission factors (min./max.) | 0.947, 0.979 | 0.942, 1.062 | 0.784, 1.000 | 0.799, 1.000 | 0.952, 1.000 | 0.709, 1.000 | 0.960, 0.985 |
| Crystal size (mm3) | 0.66 × 0.45 × 0.25 | 0.38 × 0.29 × 0.10 | 0.49 × 0.40 × 0.38 | 0.48 × 0.42 × 0.27 | 0.20 × 0.20 × 0.30 | 0.50 × 0.40 × 0.30 | 0.25 × 0.18 × 0.09 |
| θ(max) (°) | 29.0 | 22.5 | 27.5 | 25.0 | 25.0 | 25.0 | 25.0 |
| Reflections measured | 16012 |
33814 |
12474 |
12476 |
9158 | 27782 |
19214 |
| Unique reflections | 8329 | 10758 |
4880 | 4267 | 4173 | 8856 | 4626 |
| R int | 0.013 | 0.086 | 0.031 | 0.032 | 0.018 | 0.055 | 0.051 |
| Reflections with F2 > 2σ(F2) | 6933 | 5230 | 3517 | 3777 | 3045 | 6118 | 3019 |
| Number of parameters | 487 | 1093 | 303 | 365 | 323 | 654 | 360 |
| R 1 [F2 > 2σ(F2)] | 0.050 | 0.043 | 0.049 | 0.117 | 0.047 | 0.059 | 0.051 |
| wR2 (all data) | 0.141 | 0.083 | 0.130 | 0.253 | 0.133 | 0.154 | 0.163 |
| GOOF, S | 1.023 | 0.788 | 1.058 | 1.222 | 1.049 | 1.048 | 1.070 |
| Largest difference peak and hole (e Å−3) | 1.30 and −0.53 | 0.32 and −0.28 | 0.25 and −0.31 | 0.37 and −0.39 | 0.14 and −0.16 | 0.68 and −0.36 | 0.33 and −0.54 |
| Compound | 7·2¼MeCN | 8 | 9·4toluene | 10·5MeCN | 11·1.75toluene·1.25hexane |
|---|---|---|---|---|---|
| Formula | C59.50H66.75Al2N6.25O2 | C54H58Al2N4O2 | C108H81AlCl4N8O8 | C114H116AlN13O4 | C264.50H342Al16N16O16 |
| Formula weight | 955.40 | 849.00 | 1787.58 | 1759.17 | 4433.20 |
| Crystal system | Triclinic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group |
P |
P |
P |
P21/c |
P |
| Unit cell dimensions | |||||
| a (Å) | 15.2938(19) | 9.7916(5) | 13.8593(10) | 16.2328(2) | 13.1640(3) |
| b (Å) | 15.671(2) | 11.2215(4) | 14.7463(10) | 27.3761(3) | 31.8640(5) |
| c (Å) | 25.086(3) | 11.7840(6) | 23.7238(17) | 23.7006(3) | 36.2145(5) |
| α (°) | 93.9493(17) | 84.624(4) | 95.508(7) | 90 | 113.2940(10) |
| β (°) | 97.1008(16) | 66.196(5) | 101.879(7) | 107.9523(6) | 94.715(2) |
| γ (°) | 112.5747(16) | 84.347(4) | 109.459(7) | 90 | 95.712(2) |
| V (Å3) | 5464.4(12) | 1176.81(10) | 4401.9(6) | 10019.5(2) |
13759.6(4) |
| Z | 4 | 1 | 2 | 4 | 4 |
| Temperature (K) | 150(2) | 143(2) | 143(2) | 120.0(2) | 120(2) |
| Wavelength (Å) | 0.6884 | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Calculated density (g cm−3) | 1.161 | 1.198 | 1.343 | 1.166 | 1.072 |
| Absorption coefficient (mm−1) | 0.100 | 0.107 | 0.209 | 0.080 | 0.113 |
| Transmission factors (min./max.) | 0.987, 0.997 | 0.906, 1.000 | 0.514, 1.000 | 0.973, 0.990 | 0.564, 1.000 |
| Crystal size (mm3) | 0.14 × 0.10 × 0.03 | 0.80 × 0.50 × 0.40 | 0.35 × 0.30 × 0.20 | 0.35 × 0.25 × 0.12 | 0.80 × 0.50 × 0.40 |
| θ(max) (°) | 22.6 | 26.4 | 27.4 | 25.0 | 29.5 |
| Reflections measured | 36298 |
9795 | 67195 |
191662 |
155744 |
| Unique reflections | 15657 |
4806 | 20011 |
17619 |
64526 |
| R int | 0.065 | 0.023 | 0.067 | 0.105 | 0.051 |
| Reflections with F2 > 2σ(F2) | 9183 | 3428 | 12308 |
13161 |
43448 |
| Number of parameters | 1319 | 283 | 1054 | 1095 | 2792 |
| R 1 [F2 > 2σ(F2)] | 0.082 | 0.047 | 0.099 | 0.066 | 0.085 |
| wR2 (all data) | 0.263 | 0.127 | 0.291 | 0.153 | 0.255 |
| GOOF, S | 1.030 | 1.030 | 1.021 | 1.026 | 1.029 |
| Largest difference peak and hole (e Å−3) | 0.76 and −0.32 | 0.45 and −0.35 | 0.90 and −0.51 | 0.28 and −0.29 | 1.44 and −0.59 |
Acknowledgements
Sichuan Normal University and the National Natural Science Foundation of China (grants 51273133 and 51443004) are thanked for financial support. The Special Funds for sharing large precision equipment (no. DJ2014-22) at Sichuan Normal University is also thanked. CR thanks the EPSRC for a travel grant (EP/L012804/1) and the National Crystallographic Service at the University of Southampton (for data collection of 9·4toluene and 10·5MeCN). The CCLRC is thanked for the award of beamtime (complex 7·2¼MeCN) at SRS Daresbury Laboratory (Station 9.8), and the EPSRC Mass Spectrometry Service at Swansea is thanked for data.References
- See for example: (a) E. M. Hodnet and J. W. Dunn, J. Med. Chem., 1970, 13, 768 CrossRef; (b) S. N. Pandeya, D. Sriram, G. Nath and E. De Clercq, Il Farmaco, 1999, 54, 624 CrossRef CAS PubMed; (c) A. H. El-Masry, H. H. Fahmy and S. H. Abdelwahed, Molecules, 2000, 5, 1429 CrossRef CAS; (d) A. A. Jarrahpour and M. Zarei, Molbank, 2004, M377 CrossRef CAS; (e) H. L. Siddiqui, A. Iqbal, S. Ahmad and G. W. Weaver, Molecules, 2006, 11, 206 CrossRef CAS PubMed.
- (a) S. Brooker, Coord. Chem. Rev., 2001, 222, 33 CrossRef CAS; (b) W. Radecka-Paryzek, V. Patroniak and J. Lisowski, Coord. Chem. Rev., 2005, 249, 2156 CrossRef CAS.
- (a) F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef; (b) W. Yang, K.-Q. Zhao, B.-Q. Wang, C. Redshaw, M. R. J. Elsegood, J.-L. Zhao and T. Yamato, Dalton Trans., 2016, 45, 226–236 RSC.
- (a) A. Arbaoui, C. Redshaw and D. L. Hughes, Chem. Commun., 2008, 4717 RSC; (b) A. Arbaoui, C. Redshaw and D. L. Hughes, Supramol. Chem., 2009, 21, 35 CrossRef CAS.
- F. Majoumo-Mbe, E. Smolensky, P. Lőnnecke, D. Shpasser, M. S. Eisen and E. Hey-Hawkins, J. Mol. Catal. A: Chem., 2005, 240, 91 CAS.
- R. Mazarro, I. Gracia, J. F. Rodriquez, G. Storti and M. Morbidelli, Polym. Int., 2012, 61, 265 CrossRef CAS.
- P. Chakraborty, J. Adhikary, S. Samanta, I. Majumder, C. Massera, D. Escudero, S. Ghosh, A. Bauza, A. Frontera and D. Das, Dalton Trans., 2015, 44, 20032 RSC.
- Y. Wei, S. Wang and S. Zhou, Dalton Trans., 2016, 45, 4471 RSC.
- (a) S. R. Korupoju and P. S. Zacharias, Chem. Commun., 1998, 1267 RSC; (b) S. J. Na, D. J. Joe, S. Sujith, W.-S. Han, S. O. Kang and B. Y. Lee, J. Organomet. Chem., 2006, 691, 611 CrossRef CAS.
- (a) S. Brooker, G. S. Dunbar and T. Weyhermuller, Supramol. Chem., 2001, 13, 601 CrossRef CAS; (b) J. Gao, J. H. Reibenspies, R. A. Zingaro, R. Woolley, A. E. Martell and A. Clearfield, Inorg. Chem., 2005, 44, 232 CrossRef CAS PubMed; (c) S. J. Na, D. J. Joe, S. Sujith, W.-S. Han, O. S. Kang and B. Y. Lee, J. Organomet. Chem., 2006, 691, 611 CrossRef CAS; (d) M. Paluch, J. Lisowski and T. Lis, Dalton Trans., 2006, 381 RSC.
- (a) R. W. Stotz and R. C. Stoufer, J. Chem. Soc., Chem. Commun., 1970, 1682 RSC; (b) J. D. O. Cabral, M. F. Cabral, M. G. B. Drew, F. S. Esho, O. Haas and S. M. Nelson, J. Chem. Soc., Chem. Commun., 1982, 1066 RSC; (c) T. W. Bell and F. Guzzo, J. Chem. Soc., Chem. Commun., 1986, 769 RSC; (d) T. Sato, K. Sakai and T. Tsubomura, Chem. Lett., 1993, 859 CrossRef CAS; (e) S. W. A. Bligh, N. Cho, V. W. J. Cummins, E. G. Evagoroula, D. J. Kelly and M. McPartlin, J. Chem. Soc., Dalton Trans., 1994, 3369 RSC; (f) D. A. Plattner, A. K. Beck and M. Neuburger, Helv. Chim. Acta, 2002, 85, 4000 CrossRef CAS; (g) M. Allmendinger, P. Zell, A. Amin, U. Thewalt, M. Klinga and B. Rieger, Heterocycles, 2003, 60, 1065 CrossRef CAS; (h) J. Gregolinski, J. Lisowski and T. Lis, Org. Biomol. Chem., 2005, 3, 3161 RSC.
- A. Arbaoui, C. Redshaw and D. L. Hughes, Chem. Commun., 2008, 4717 RSC.
- A. L. Spek, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 1990, 46, C34 Search PubMed.
- (a) J. M. Klerks, D. J. Stufkens, G. van koten and K. Vrieze, J. Organomet. Chem., 1979, 181, 271 CrossRef CAS; (b) V. C. Gibson, C. Redshaw, A. J. P. White and D. J. Williams, J. Organomet. Chem., 1998, 550, 453 CrossRef CAS; (c) M. Bruce, V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Chem. Commun., 1998, 2523 RSC; (d) V. C. Gibson, D. Nienhuis, C. Redshaw, A. J. P. White and D. J. Williams, Dalton Trans., 2004, 1761 RSC; (e) V. C. Gibson, C. Redshaw, G. A. Solan, A. J. P. White and D. J. Williams, Organometallics, 2007, 26, 5119 CrossRef CAS; (f) A. Arbaoui, C. Redshaw and D. L. Hughes, Supramol. Chem., 2009, 21, 35 CrossRef CAS; (g) W. Alkarekshi, A. P. Armitage, O. Boyron, C. J. Davies, M. Govere, A. Gregory, K. Singh and G. A. Solan, Organometallics, 2013, 32, 249–259 CrossRef CAS.
- (a) M. Shen, W. Zhang, K. Nomura and W.-H. Sun, Dalton Trans., 2009, 9000 RSC; (b) M. Shen, W. Huang, W. Zhang, X. Hao, W.-H. Sun and C. Redshaw, Dalton Trans., 2010, 39, 9912 RSC; (c) M.-C. Chang, W.-Y. Lu, H.-Y. Chang, Y.-C. Lai, M. Y. Chiang, H.-Y. Chen and H.-Y. Chen, Inorg. Chem., 2015, 54, 11292 CrossRef CAS PubMed.
- (a) N. Iwasa, S. Katao, J. Liu, M. Fujiki, Y. Furukawa and K. Nomura, Organometallics, 2009, 28, 2179 CrossRef CAS; (b) D. Li, Y. Peng, C. Geng, K. Liu and D. Kong, Dalton Trans., 2013, 42, 11295 RSC; (c) W.-L. Kong, Z.-Y. Chai and Z.-X. Wang, Dalton Trans., 2014, 43, 14470 RSC; (d) W. Zhang, Y. Wang, L. Wang, C. Redshaw and W.-H. Sun, J. Organomet. Chem., 2014, 750, 65 CrossRef CAS.
- C. Redshaw, M. A. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. Pérez-Casas, S. Matsui and S. Matsuura, Chem. – Eur. J., 2007, 13, 1090–1107 CrossRef CAS PubMed.
- T. K. Sen, A. Mukherjee, A. Modak, S. K. Mandel and D. Koley, Dalton Trans., 2013, 42, 1893 RSC.
- (a) R. S. Drago, M. J. Desmond, B. B. Corden and K. A. Miller, J. Am. Chem. Soc., 1983, 105, 2295 Search PubMed; (b) J. J. Randell, C. E. Lewis and P. M. Slagan, J. Org. Chem., 1962, 27, 4098 CrossRef.
- V. C. Gibson, C. Redshaw, W. Clegg, M. R. J. Elsegood, U. Siemeling and T. Türk, Polyhedron, 2004, 23, 189 CrossRef CAS.
- SMART and SAINT software for CCD diffractometers, Bruker AXS Inc., Madison, USA, 2001 and 2007 Search PubMed.
- G. M. Sheldrick, SHELX-97 – Programs for crystal structure determination (SHELXS) and refinement (SHELXL), Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- Programs CrysAlis-CCD and -RED, Oxford Diffraction Ltd., Abingdon, UK, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic files CIF format for the structure determinations of compound L1H2·MeCN, L2H2·MeCN, L2H2·2(Me2CO) and L2H2·n(MeCOOEt), n = 1 and 2, L2H2·2(PhMe), L2(tosyl)2, 7·2¼MeCN, 8, 9·4toluene, 10·5MeCN and 11·1¾toluene·1¼hexane. Alternative views of structures and further polymerisation data. CCDC 1442772–1442778 (Schiff-base pro-ligands) and 1463685–1463689 (organoaluminium complexes). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01997h |
| This journal is © The Royal Society of Chemistry 2016 |