
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Excited state decay of cyclometalated polypyridine ruthenium complexes: insight from theory and experiment†
Christoph
Kreitner
ab and
Katja
Heinze
*a
aInstitute of Inorganic and Analytical Chemistry, Johannes Gutenberg University, Duesbergweg 10-14, D-55128 Mainz, Germany. E-mail: katja.heinze@uni-mainz.de
bGraduate School Materials Science in Mainz, Staudingerweg 9, D-55128 Mainz, Germany
First published on 17th June 2016
Abstract
Deactivation pathways of the triplet metal-to-ligand charge transfer (3MLCT) excited state of cyclometalated polypyridine ruthenium complexes with [RuN5C]+ coordination are discussed on the basis of the available experimental data and a series of density functional theory calculations. Three different complex classes are considered, namely with [Ru(N^N)2(N^C)]+, [Ru(N^N^N)(N^C^N)]+ and [Ru(N^N^N)(N^N^C)]+ coordination modes. Excited state deactivation in these complex types proceeds via five distinct decay channels. Vibronic coupling of the 3MLCT state to high-energy oscillators of the singlet ground state (1GS) allows tunneling to the ground state followed by vibrational relaxation (path A). A ligand field excited state (3MC) is thermally accessible via a 3MLCT → 3MC transition state with the 3MC state being strongly coupled to the 1GS surface via a low-energy minimum energy crossing point (path B). Furthermore, a 3MLCT → 1GS surface crossing point directly couples the triplet and singlet potential energy surfaces (path C). Charge transfer states either with higher singlet character or with different orbital parentage and intrinsic symmetry restrictions are thermally populated which promote non-radiative decay via tunneling to the 1GS state (path D). Finally, the excited state can decay via phosphorescence (path E). The dominant deactivation pathways differ for the three individual complex classes. The implications of these findings for isoelectronic iridium(III) or iron(II) complexes are discussed. Ultimately, strategies for optimizing the emission efficiencies of cyclometalated polypyridine complexes of d6-metal ions, especially RuII, are suggested.
 Christoph Kreitner | Christoph Kreitner is currently working on his PhD thesis in the research group of Prof. Dr Katja Heinze at the Johannes Gutenberg-University in Mainz, Germany (since 2013). There, his research focusses on the synthesis and understanding of new cyclometalated ruthenium complexes and their application in dye-sensitized solar cells. He studied chemistry at the Johannes Gutenberg-University in Mainz, Germany where he received his diploma degree in 2012. During his studies he spent one semester at the University of Toronto, Toronto, Canada (2010/2011), working in the Department of Inorganic Chemistry in the group of Prof. Douglas W. Stephan on Frustrated Lewis Pairs and their reactions with lactones and lactide. For his PhD thesis, he received a scholarship of the Graduate School of Excellence “Materials Science in Mainz” (2013) and became junior member of the Gutenberg Academy of the Johannes Gutenberg-University Mainz, Germany (2014). His research interests particularly aim at the understanding of electron and energy transfer as well as optoelectronic mechanisms in molecular systems. |
 Katja Heinze | Katja Heinze is professor of organometallic and bioinorganic chemistry at the Johannes Gutenberg-University of Mainz, Germany. After receiving a diploma degree (1995) and a Ph.D. degree (1998) from the Ruprecht Karls University of Heidelberg, Germany (G. Huttner), she went for a postdoctoral stay to the University of Zurich, Zurich, Switzerland (1999). She was appointed Privatdozent in 2004 at the University of Heidelberg, Germany, and Full Professor in 2008 at the Johannes Gutenberg-University of Mainz, Germany. She received the Lieseberg award of the Faculty of Chemistry and Earth Sciences, University of Heidelberg (2002), the Heisenberg fellowship from the Deutsche Forschungsgemeinschaft (2004), the Hengstberger Award of the University of Heidelberg (2007) and the Interregional Research Price of the Greater Region (2014). Currently she serves as chair of the Institute of Inorganic Chemistry and Analytical Chemistry, University of Mainz. From 2011 to 2015 she has served as a member of the International Advisory Board of Organometallics. Her key research interests comprise functional complex systems based on coordination and organometallic compounds with special emphasis on molecular wires, light-harvesting systems, bistable systems, emitters, switches and sensors as well as on (biomimetic) catalysts. She has authored more than 130 international refereed papers. |
Introduction
Polypyridine complexes of a wide range of transition metals have received great interest from coordination chemists and materials scientists over the last few decades due to their versatile applicability. For example, such complexes of copper(I),1–3 iron(II),4–6 osmium(II),7,8 iridium(III)9,10 and, particularly, ruthenium(II)11–19 were successfully applied as sensitizers in dye sensitized solar cells. Additionally, a wide range of complexes of this type have been successfully used as sensitizers in photoredox catalysis.20–31Moreover, the most fascinating and thoroughly studied property observed for a large number of polypyridine transition metal complexes is their luminescence.32,33 The first polypyridine complex reported to exhibit luminescence was [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine).34 Since then, luminescent polypyridine complexes have been described for second and third row transition metal ions with d6, d8 and d10 electron configuration,35–37 namely for rhenium(I),38–41 osmium(II),42–45 rhodium(III),46–48 iridium(III),41,42,49–51 palladium(II),52 platinum(II),52–57 and gold(III).58,59 Even a few luminescent first row transition metal polypyridine complexes are known of chromium(III),60–64 copper(I)65–71 and zinc(II).72
As such luminescence was first observed for ruthenium, most of the effort understanding the underlying electronic processes evolved around this element and in particular around [Ru(bpy)3]2+ as the prototype.73–75 Its key features are an electron-rich low-spin d6 metal center with (t2g)6 electron configuration in idealized Oh symmetry and strongly π-accepting chelate ligands. Upon irradiation with visible light (λmax = 452 nm),73 [Ru(bpy)3]2+ is excited into its lowest excited singlet state which has metal-to-ligand charge transfer (MLCT) character.76–78 As MLCT transitions are not restricted in terms of parity selection rules, the corresponding absorption bands are typically very intense. Due to the spin–orbit coupling induced by the ruthenium atom, the excited 1MLCT state undergoes very efficient intersystem crossing (ϕISC ≈ 1) onto the triplet hypersurface populating a 3MLCT state.79,80 At low temperatures in solid matrix, this state either evolves into the ground state without emission via tunnelling to the singlet energy surface and vibrational cooling or via phosphorescent emission of a photon (at 77 K: λem = 580 nm,77ϕem = 0.38;77 at 298 K: λem = 621 nm,81ϕem = 0.095![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 81).77,82–84 The rate of non-radiative excited state decay is hereby governed by the so-called energy gap law.85–88 In a series of structurally related compounds, the rate increases with decreasing exited state energy. At room temperature however, a third deactivation pathway via metal centered (MC) d–d excited states ((t2g)5(eg)1 electron configuration) is thermally accessible dramatically quenching the emission.84,89–91 The activation barrier for this thermally activated depopulation was determined to be about 45 kJ mol−1 for [Ru(bpy)3]2+.90,91
81).77,82–84 The rate of non-radiative excited state decay is hereby governed by the so-called energy gap law.85–88 In a series of structurally related compounds, the rate increases with decreasing exited state energy. At room temperature however, a third deactivation pathway via metal centered (MC) d–d excited states ((t2g)5(eg)1 electron configuration) is thermally accessible dramatically quenching the emission.84,89–91 The activation barrier for this thermally activated depopulation was determined to be about 45 kJ mol−1 for [Ru(bpy)3]2+.90,91
This general scheme for [Ru(bpy)3]2+ is applicable to other ruthenium polypyridine complexes as well. It enables a fine-tuning of their emissive properties by manipulation of the 3MLCT energies via introduction of functional groups or extension of the aromatic backbone of the pyridine ligands.75,92–96 This allowed the design of specifically tailored complexes for luminescent sensing applications97,98 and for optoelectronics.99–102
The concept was successfully transferred to polypyridine complexes of other transition metals. For example, cyclometalated polypyridine complexes of iridium(III) proved to be exceptionally well suited for PHOLED applications as their room temperature emission is typically very intense and can be tuned throughout the visible range of the electromagnetic spectrum.41,103–106 Cyclometalation hereby refers to the exchange of one or multiple nitrogen atoms of the polypyridine's coordination sphere by isoelectronic carbon anions. This substitution typically yields a reduction of the overall charge of the complex moiety as well as a substantial shift of all redox processes to lower potentials.
Due to the fact that the isoelectronic cyclometalated complexes of ruthenium(II) perform very well as sensitizers in dye sensitized solar cells, they have also received increasing interest in the last years.16,17,19,107–109 Additionally, cyclometalated bridging ligands enhance the electronic coupling between the redox centers in mixed-valent Ru/Ru complexes110–112 and Ru/organic hybrid structures.113–116 Despite the large variety of cyclometalated polypyridine ruthenium complexes synthesized up-to-date, however, no phosphorescence comparable to [Ru(bpy)3]2+ has yet been achieved. Furthermore, a general explanation for the striking difference in the luminescence properties between the isoelectronic cyclometalated complexes of iridium(III) and ruthenium(II) is still missing.117–119
Hence, this issue will be addressed in this perspective. We will discuss the excited state deactivation processes of cyclometalated polypyridine complexes beyond the 3MLCT/3MC and energy gap law schemes by picking illustrative examples from the literature and elaborate why these are weak emitters at room temperature. Several decay pathways of the emissive 3MLCT state are known for polypyridine ruthenium complexes, and their individual contribution to the excited state decay of cyclometalated polypyridine ruthenium complexes will be discussed (Fig. 1):
Decay path A: tunneling into high-lying vibrational levels of the singlet state.86–88 This channel is always available and its efficiency depends on the Franck–Condon overlap of the vibrational wavefunctions of the 3MLCT and singlet ground states (1GS).
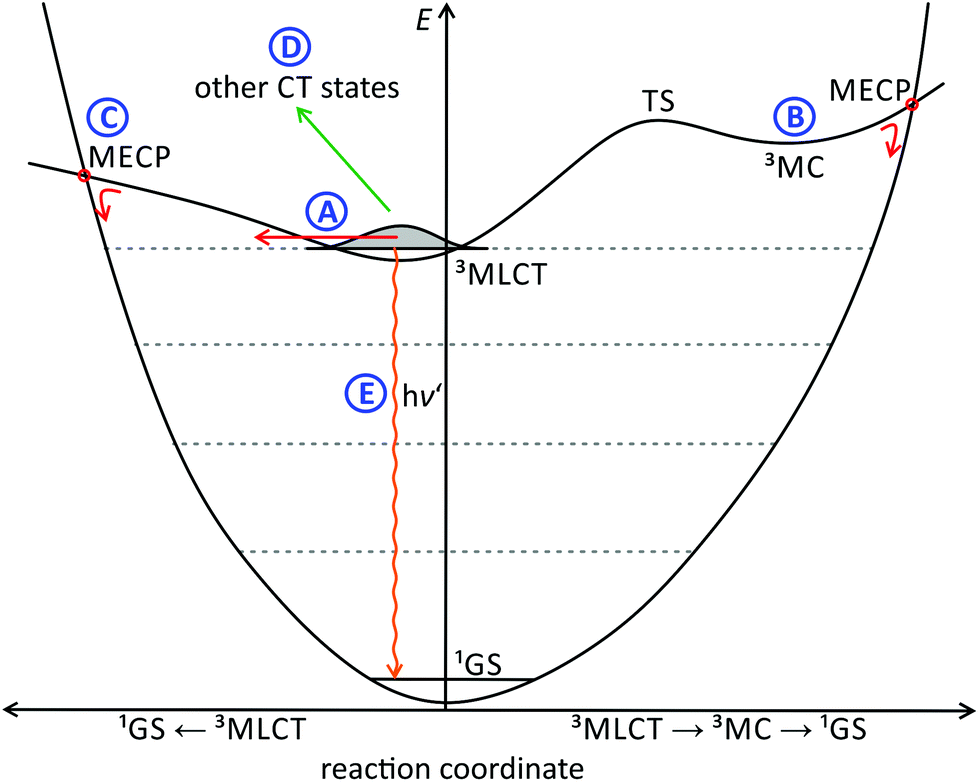 | ||
| Fig. 1 Schematic representation of the energy landscape of a polypyridine ruthenium complex including all relevant deactivation pathways of an emissive 3MLCT state: (A) tunneling into high-energy vibrationally excited singlet states; (B) thermally activated decay into a 3MC state followed by surface crossing at a minimum energy crossing point (MECP), (C) direct thermally activated surface crossing from the 3MLCT state to the singlet ground state, (D) decay via non-emissive charge transfer states, and (E) phosphorescence. | ||
Decay path B: via a thermally accessible 3MC state. Population of this state is followed by rapid surface crossing to the singlet potential energy surface via a close-lying minimum energy crossing point (MECP) and vibrational cooling.56 The energy of the 3MC state mainly depends on the ligand field strength.
Decay path C: direct surface crossing from the 3MLCT state to the singlet potential surface via a low-lying surface crossing point. The energy of this MECP depends on the degree of 3MLCT state distortion and the energy of the 3MLCT state itself. A shallow 3MLCT state potential surface around the minimum favours the occurrence of a surface crossing.
Decay path D: via other non-emissive triplet states or states with higher singlet character as in [Ru(tpy)2]2+ and [Ru(bpy)3]2+ that allow efficient tunnelling into high-lying singlet states.77,83,91,120
Decay path E: phosphoresence.
In the following, we will divide the discussion into tris(bidentate) complexes with [Ru(N^N)2(N^C)]+ coordination sphere and bis(tridentate) complexes with either [Ru(N^N^N)(N^C^N)]+ or [Ru(N^N^N)(N^N^C)]+ motifs to highlight similarities and differences between the different classes. Finally, we will suggest strategies how to improve the room temperature emission of such complexes and how our conclusions might impact the photophysics of cyclometalated polypyridine complexes of still elusive iron(II) emitters6,121–123 and well-known iridium(III) emitters49,104,124 as well.
Tris(bidentate) ruthenium complexes
The first cyclometalated (polypyridine)ruthenium complex, [Ru(bpy)2(ppy-NO2)]+ [1a]+ (Hppy = 2-phenylpyridine, R2 = NO2), was reported in 1985 by Reveco et al. (Scheme 1).125 Shortly thereafter, the syntheses of unsubstituted [Ru(bpy)2(ppy)]+ [1b]+126 and the bis(tridentate) counterparts [Ru(ttpy)(dpb)]+ (ttpy = 4′-p-tolyl-2,2′;6′,2′′-terpyridine, dpbH = 1,3-di-(2′-pyridyl)benzene)131 and [Ru(tpy-4′-Cl)(pbpy)]+ (tpy = 2,2′;6′,2′′-terpyridine, pbpyH = 6-phenyl-2,2′-bipyridine)132 were presented (vide supra). However, deeper interest in the photophysical properties of cyclometalated (polypyridine)ruthenium(II) complexes did not evolve until the discovery of their excellent performance as solar cell sensitizers in 2007.16 Since then, more effort has been put into the understanding of the photophysical properties of this class of compounds.107,109,117–119,127,133–135
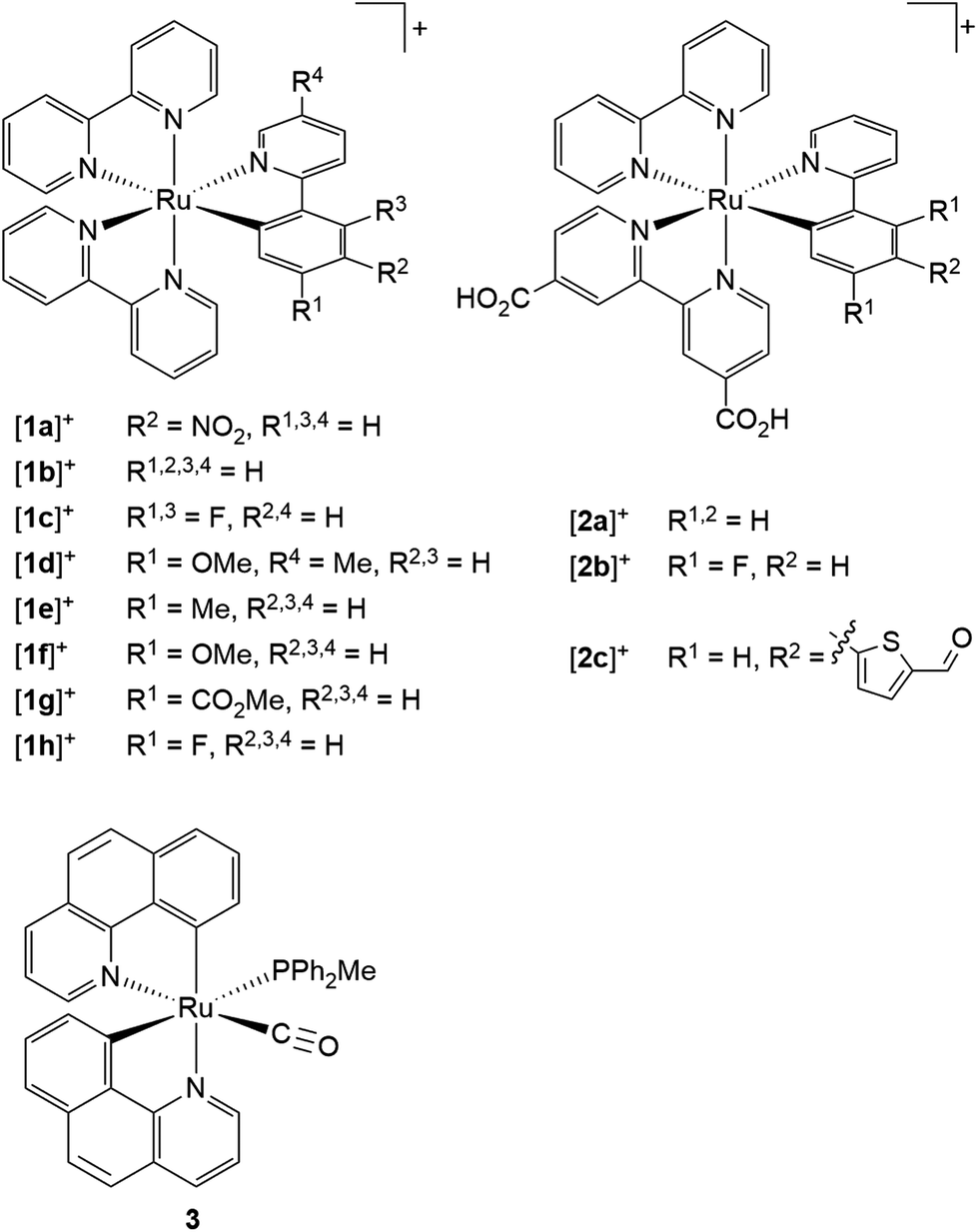 | ||
| Scheme 1 Literature-known tris(bidentate) cyclometalated (polypyridine)ruthenium(II) complexes relevant to this work ([1a]+,125 [1b]+,126–128 [1c]+–[1d]+,127 [1e]+–[1h]+,128 [2a]+–[2c]+,1293130). | ||
The visible range of the absorption spectrum of [Ru(bpy)2(ppy)]+-type complexes typically is dominated by two broad structured absorption bands, one appearing between 320 and 450 nm and the second between 470 and 650 nm.107,127,133–135 The broadness of the visible range absorption features has been attributed to the low symmetry around the metal center which breaks the degeneracy of the metal's d orbitals. Additionally, the lowest unoccupied molecular orbital (LUMO) of the ppy unit (LUMO+2) is substantially higher in energy than the bpy LUMO (Fig. 2). Hence, Berlinguette and coworkers assigned these bands to Ru → ppy (at 400 nm) and Ru → bpy (at 500 nm) MLCT transitions.3,107,127,135 Grätzel, however, suggested that all visible range absorption bands predominantly arise from Ru → bpy MLCT transitions with varying contributions from the highest occupied molecular orbital (HOMO) of π-symmetry at the cyclometalating phenyl ring.17 Indeed, the theoretical data published by Berlinguette107 show that all Ru → ppy transitions are weak in intensity and do not contribute to the absorption spectrum which supports Grätzel's interpretation. Additionally, the shift of the absorption bands induced by the functional groups attached to either the ppy ligand or the bpy units further underlines Grätzel's assignment of all bands as Ru → bpy transitions.3,107,127,135
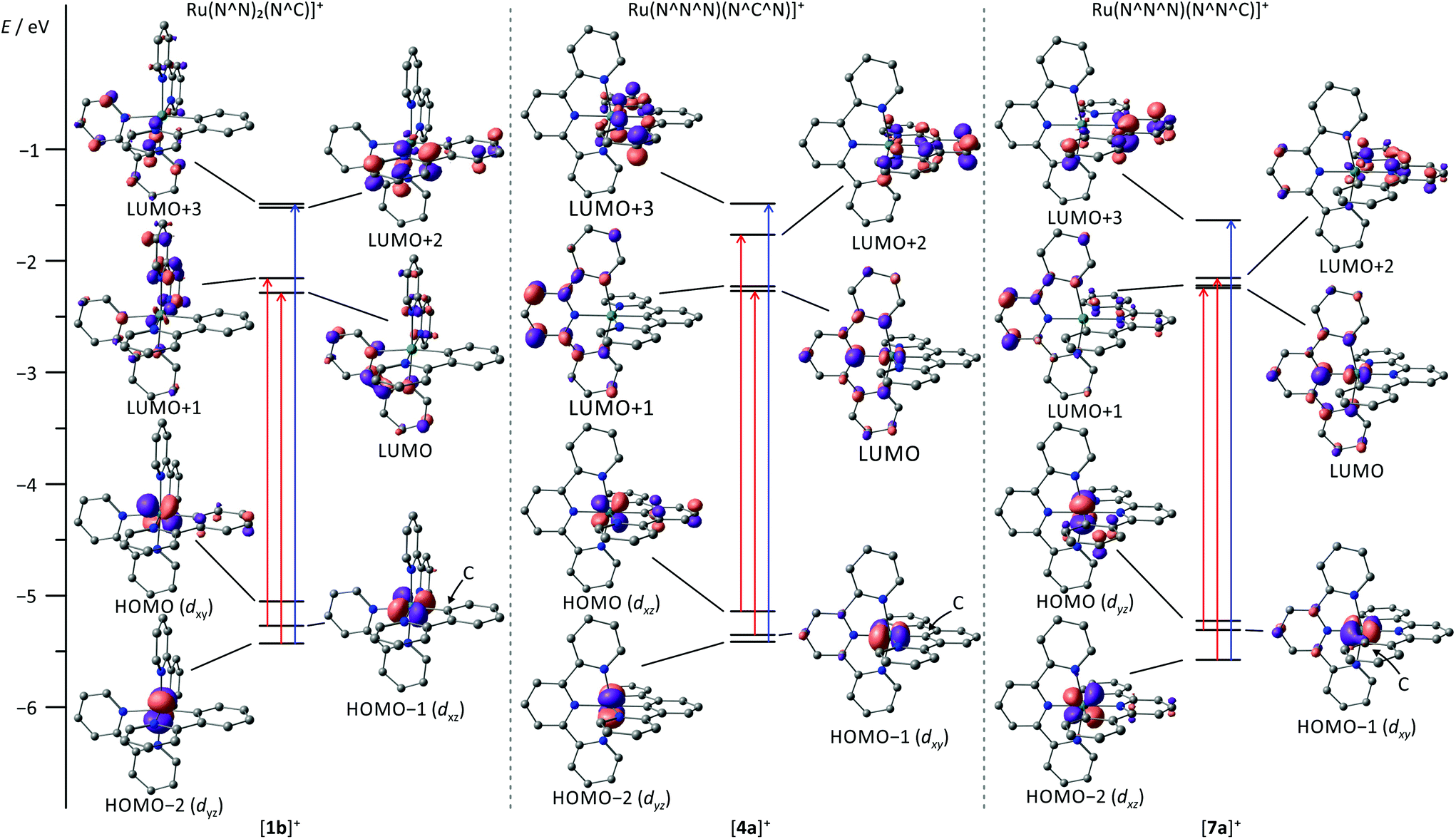 | ||
| Fig. 2 MO diagram of the parent cyclometalated (polypyridine)ruthenium(II) complexes [1b]+, [4a]+ and [7a]+ obtained from DFT calculations (B3LYP, def2-SVP, ZORA, COSMO (acetonitrile) contour value: 0.07). H atoms are omitted for clarity. The most important orbitals involved in the dominant transitions around 500 nm and 400 nm are highlighted in red and blue, respectively. | ||
For most tris(bidentate) complexes of this type, weak room temperature emission in the range between 720 and 820 nm is reported (Table 1).107,127–129,133,135 The emissive state is considered to be a Ru → bpy 3MLCT state.107,133 This is corroborated by the influence of functional groups on the emission energy. In a series of [Ru(bpy)2(ppy-R)]+ complexes ([1e]+–[1h]+, Scheme 1) with functional groups in meta-position to the cyclometalating carbon atom, the emission energy decreases with increasing electron donating strength of the respective substituent as this destabilizes the metal d orbitals (Table 1).128 Similarly, functionalization of the bpy ligands with electron accepting substituents such as COOR groups shifts the emission bathochromically as a consequence of the lowered LUMO energy (Table 1).133 However, missing low-temperature emission data (λem, ϕ, τ) currently impede a quantification of the respective effects.
![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) em as well as excited state lifetimes τ of selected tris(bidentate) cyclometalated (polypyridine)ruthenium(II) complexes at room temperature in solution
em as well as excited state lifetimes τ of selected tris(bidentate) cyclometalated (polypyridine)ruthenium(II) complexes at room temperature in solution
To the best of our knowledge, no quantum yields have been determined neither at room temperature nor at 77 K for any of the reported tris(bidentate) cyclometalated complexes due to their very weakly emissive character and the associated instrumental limitations.127,128 Castellano and coworkers estimated the phosphorescence quantum yields to be ϕ < 0.005,127 while Housecroft and coworkers reported yields below 0.01 for compounds [1e]+–[1h]+.128 However, Berlinguette129 and Castellano127 provided lifetime data of the emissive excited states for two series of complexes [2a]+–[2c]+ and [1b]+–[1d]+, respectively (Scheme 1, Table 1). The lifetimes are in the nanosecond range for all complexes and correlate nicely with the emission energy: the excited state lifetimes become smaller with decreasing emission energy. In (polypyridine)ruthenium(II) complexes, the emissive 3MLCT state typically is depopulated to some extent via a 3MC state (path B, vide supra).84,89–91 However, cyclometalation substantially increases the 3MC energy, as pointed out by Dixon136 and van Koten,137 efficiently retarding emission quenching via this pathway.118 In fact, we were able to localize the respective 3MLCT and 3MC states of [1b]+via DFT calculations (Fig. 3). The 3MC state (Mulliken spin population at Ru: 1.87) is located 66 kJ mol−1 above the 3MLCT level while for [Ru(bpy)3]2+, the 3MLCT-3MC energy gap was calculated to be −7.7 kJ mol−1 in favour of the 3MC state.138–140 Remarkably, the nitrogen donor atom N1 of the cyclometalating ligand as well as the trans nitrogen atom N4 are essentially decoordinated in the 3MC state of [1b]+ with Ru–N distances of 2.48 and 2.39 Å, respectively (Fig. 4). This tetragonal distortion along the N1–Ru–N4 axis underlines the strongly dissociative character of the 3MC state similar to that described for biscyclometalated tris(bidentate) iridium complexes141–143 and for [Ru(bpy)3]2+.138–140 It resembles the Jahn–Teller mode of d7 low-spin CoII complexes due to the (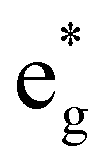 )1 electron configuration.144,145
)1 electron configuration.144,145
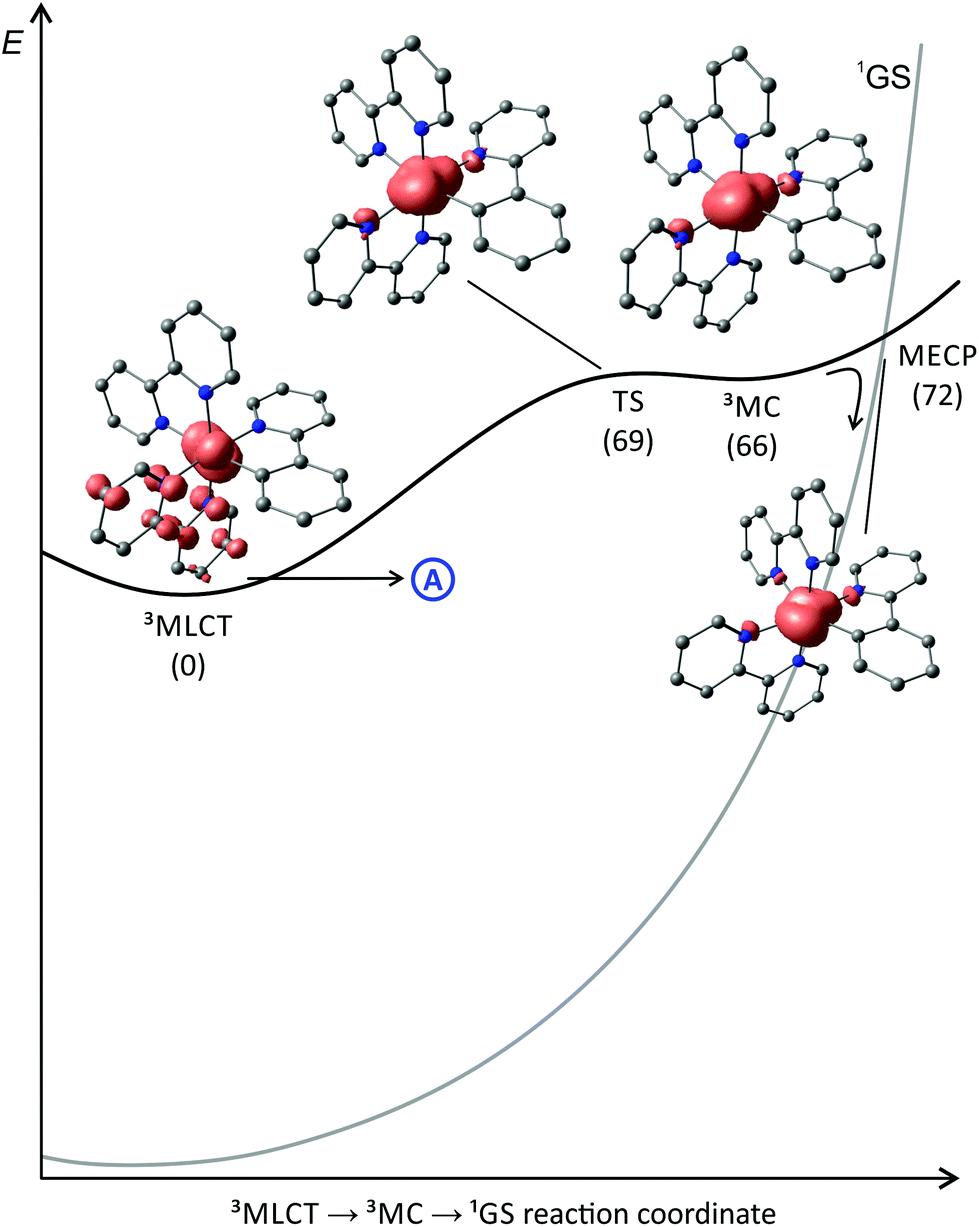 | ||
| Fig. 3 Schematic representation of the energy landscape of [1b]+ including spin density contour plots (contour value: 0.01) of the 3MLCT and the 3MC states as well as the 3MLCT → 3MC transition state and the 3MC → 1GS minimum energy surface crossing point. H atoms are omitted for clarity. Energies (kJ mol−1) are given in parentheses relative to the 3MLCT state (E = 0 kJ mol−1). The shape of the singlet potential energy surface is estimated from the DFT-calculated triplet–singlet energy differences at the various excited state geometries. | ||
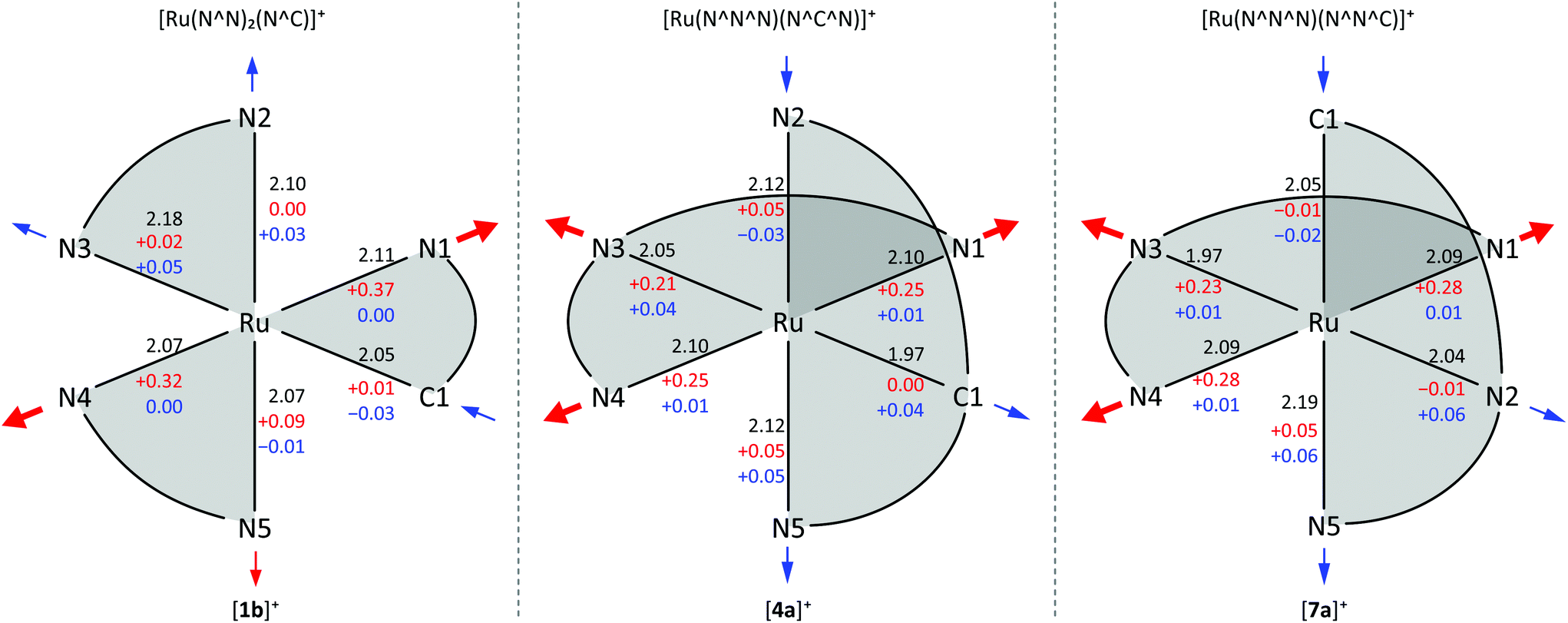 | ||
| Fig. 4 Schematic representation of the 1GS geometries of [1b]+, [4a]+ and [7a]+ including Ru–X bond lengths in Å (black). The bond length changes in Å in the 3MC (red) and 3MLCT states (blue) are given with respect to the 1GS state. Arrows indicate major molecular distortions of the respective states. | ||
The transition state between the 3MC and 3MLCT state was localized on the potential surface with an energy of 69 kJ mol−1 above the 3MLCT state (Fig. 3). Hence, in contrast to the isoelectronic complex [Ru(bpy)3]2+, the 3MC state of [1b]+ is thermally inaccessible. The comparably low quantum yields of [1b]+ are attributed to an increased thermal relaxation into the ground state. Non-emissive excited state decay occurs via vibronic coupling to high-energy oscillators (path A) or via a thermally activated surface crossing to the ground state potential surface (path C).86 The latter requires a low-energy surface crossing point. We attempted to localize such a minimum energy crossing point (MECP) between the 3MLCT and 1GS potential surfaces. However, the lowest 3MLCT → 1GS MECP we could find was localized at 120 kJ mol−1 above the 3MLCT level. An analogous 3MC → 1GS MECP on the other hand is localized at 72 kJ mol−1 merely 6 kJ mol−1 above the 3MC state (path B, Fig. 3). However, since all of these states are thermally inaccessible at room temperature or below, surface crossing to the singlet ground state is irrelevant for the excited state deactivation of [1b]+. As a consequence, emission quenching in [1b]+ appears to occur exclusively via tunnelling into high-energy oscillators of the ground state (path A). According to the energy gap law, the vibronic coupling of the 3MLCT state and 1GS becomes stronger, the smaller the 3MLCT-1GS energy gap is.86–88 Secondly, a more pronounced distortion of the 3MLCT excited state compared to the ground state geometry increases the non-radiative decay rate as it results in a higher Franck–Condon overlap of the vibronic wavefunctions of the ground and excited state.86–88,127,146,147 Indeed, inspection of the DFT-optimized geometries of the 3MLCT and 1GS states reveals a sizable distortion of the former (Fig. 4) allowing for an efficient radiationless deactivation. This distortion is mainly localized at the bpy ligand trans to Ru–C. The Ru–N2 and Ru–N3 bonds are significantly elongated as a consequence of the formal oxidation of ruthenium to +III in the 3MLCT state and the trans influence of the cyclometalating phenyl ring. However, while the Ru–N2 and Ru–N3 bonds are elongated, the formal negative charge on the second bpy ligand compensates the repulsion yielding essentially unaltered Ru–N4 and Ru–N5 bond lengths.
In summary, the high energy of the 3MC state is very favourable for efficient emitters because it eliminates one pathway for exited state deactivation and concomitantly prevents photodecomposition reactions, that typically occur from the dissociative 3MC state.90,148 However, in order to increase phosphorescence quantum yields of cyclometalated (polpyridine)ruthenium, radiationless deactivation via vibronic coupling has to be suppressed. This can be approached in two ways: the distortion of the 3MLCT state compared to the 1GS has to be reduced and the emission energy has to be blueshifted as far as possible. Chou and coworkers provided a beautiful example successfully implementing both approaches.130 Clever molecular design yielded systems with Ru → ppy MLCT states as lowest triplet excited states. This is straight-forwardly accomplished by making the cyclometalating ppy− ligand the strongest π-acceptor in the complex. Chou and coworkers achieved this with carbon monoxide and phosphanes which are rather poor π-acceptors towards RuII in biscyclometalated complexes of the type Ru(bq)2(CO)(PPh2Me) 3 (bqH = benzo-[h]quinoline, Scheme 1).130 As the LUMO of bq− is much higher in energy than that of bpy (Fig. 2), the emission from the corresponding Ru → bq 3MLCT state is blueshifted substantially to 575 nm with a quantum yield of ϕ = 0.24. Additionally, as the excited state involves a cyclometalating ligand, its distortion compared to the ground state geometry should be less pronounced as in complex [1b]+. Similar observations were made for the isoelectronic osmium complexes.149
These findings suggest that with careful choice of suitable, very weakly π-accepting polypyridine ligands, cyclometalated (polypyridine)ruthenium complexes with similar emission behaviour arising from high-energy Ru → ppy 3MLCT states are accessible. Another way of improving the emission behaviour of cyclometalated (polypyridine)ruthenium complexes could be by introducing tridentate chelate ligands as this potentially supresses excited state distortions while maintaining the high energies of parasitic 3MC states. This should yield nested states with poorer Franck–Condon overlap (weakly coupling limit) which reduces tunnelling processes into high-energy singlet states (path A). We will discuss the possibilities and consequences of bis(tridentate) coordination spheres on the phosphorescence properties of cyclometalated ruthenium complexes in the next section.
Bis(tridentate) ruthenium complexes
Considering cyclometalated bis(tridentate) complexes, a distinction between [Ru(N^N^N)(N^C^N)]+ and [Ru(N^N^N)(N^N^C)]+ coordination environments is reasonable. The next two sections will highlight similarities between the two classes of bis(tridentate) complexes as well as important differences and compare these findings to those concerning tris(bidentate) complexes (vide supra).The [Ru(N^N^N)(N^C^N)]+ coordination sphere
Similar to tris(bidentate) complexes, the visible range of the absorption spectrum of bis(tridentate) complexes with central cyclometalation is dominated by intense and broad absorption bands that have been assigned to MLCT transitions. Again, two bands are observed, one in the range of 350–450 nm and a second between 470 and 650 nm. Van Koten137,150 and Berlinguette107 assigned the high energy MLCT band to Ru → dpb transitions and the low-energy band to Ru → tpy excitations based on relative orbital energies of the lowest π*-orbitals of the respective ligands (Fig. 2). Schubert and coworkers117,151 on the other hand assigned the low-energy band to mixed MLCT and ligand-to-ligand charge transfer (LL′CT) transitions arising from a HOMO–LUMO transition while the blue absorption band was attributed to mixed MLCT/LL′CT and MC transitions. We examined several [Ru(dpb-R1)(tpy-R2)]+ complexes and demonstrated experimentally (resonance Raman spectroscopy) and theoretically that the low energy absorption arises from both Ru → tpy and Ru → dpb excitations (Fig. 2).118,152 Excitation into the tpy-centered LUMO is only possible from the HOMO−1, while the dpb-centered LUMO+2 can be reached from the HOMO as evidenced from time-dependent DFT calculations. Both excitations occur at very similar energy and contribute comparably to the absorption band at 500 nm. The higher energy absorption features result from MLCT transitions targeting the higher π* orbitals of the tpy ligand. In fact, a LL′CT transition as suggested by Schubert117 is symmetry-forbidden as it involves two mutually perpendicular π orbitals. This symmetry argument will become important for the emission properties as well.Weak emission is observed under ambient conditions for most bis(tridentate) complexes with central cyclometalation in the range between 700 and 800 nm while the parent [Ru(tpy)2]2+ complex is virtually non-emissive at room temperature.107,109,118,119,137,151 Van Koten137 and we118,119 determined extremely low quantum yields in the range of 10−5 for complexes of type [Ru(dpb-R1)(tpy-R2)]+. In contrast to the tris(bidentate) series, however, no excited state lifetimes were reported so far (Table 2). Our attempts to obtain lifetimes suggested that they are in the picosecond range.119
em as well as quantum yields ϕ of selected [Ru(N^N^N)(N^C^N)]+ complexes at room temperature
Van Koten137 studied complexes of the type [Ru(dpb-R1)(tpy-R2)]+ bearing carboxy-substituents on either or both ligands (Scheme 2, [4a]+–[4d]+). The effect of the functional groups on the respective emission energy (Table 2) points to a 3MLCT emissive state. In fact, a COOR substituent at the tpy ligand ([4b]+) leads to complexes that are non-emissive at room temperature due to its stabilizing influence on the tpy-centered LUMO. Since the phosphorescence quantum yields decrease with increasing emission wavelength following the energy gap law, van Koten suggested vibrational relaxation as main source of emission quenching.137 We extended this study by also introducing electron-donating substituents such as NH2 and NHCOMe to the dpb ligand (Scheme 2, [5a]+–[5c]+) and the tpy ligand ([5d]+).118,119 Using theoretical methods we showed, that the emissive state is in fact a 3MLCT state. The two singly occupied orbitals (SOMOs) in this state correspond to the dxy (HOMO−1) and the tpy LUMO, respectively (Fig. 2). No dpb ligand participation was observed. However, we localized a second charge transfer state with LL′CT character (dpb → tpy, HOMO → LUMO orbital parentage) on the triplet potential energy surface. The mutually perpendicular SOMOs make this state spectroscopically undetectable and non-emissive (dark state). As third type of triplet states, the 3MC states were found (Fig. 5 and Table 3). While the 3LL′CT state is essentially undistorted compared to the ground state with only a small displacement of ruthenium towards C1 and a concomitant elongation of the Ru–N3 bond, the 3MLCT state exhibits substantial distortions within the tpy ligand. It is inclined with respect to the plane perpendicular to the dpb ligand by 12° (Fig. 5). At the same time the Ru–C and Ru–N5 bonds are slightly elongated while the Ru–N2 bond shortens (Fig. 4). In the 3MC state on the other hand, the peripheral pyridine rings of the tpy ligand are tilted away from the metal center forming dihedral angles of about 11° with the central pyridine ring, respectively. Furthermore, the all Ru–Ntpy bonds Ru–N1, Ru–N3 and Ru–N4 are substantially elongated (Fig. 4) underlining the dissociative character of the 3MC state.
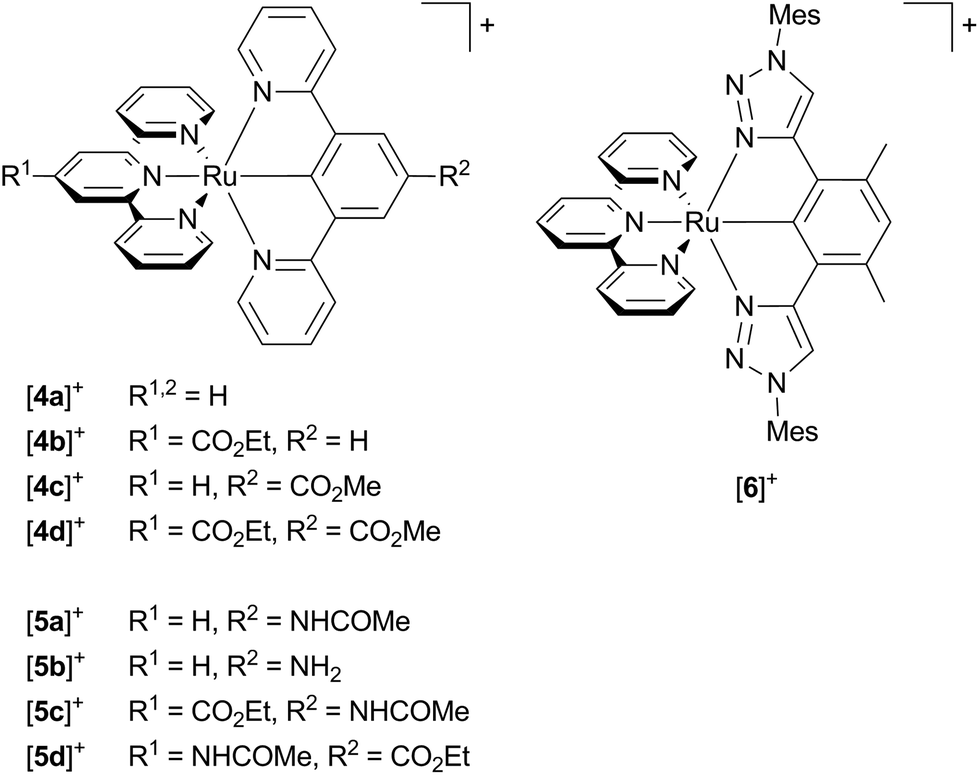 | ||
| Scheme 2 Literature-known [Ru(N^N^N)(N^C^N)]+ complexes relevant to this work ([4a]+,107,137 [4b]+–[4d]+,137 [5a]+ and [5b]+,119 [5c]+ and [5d]+,118 [6]+,117). | ||
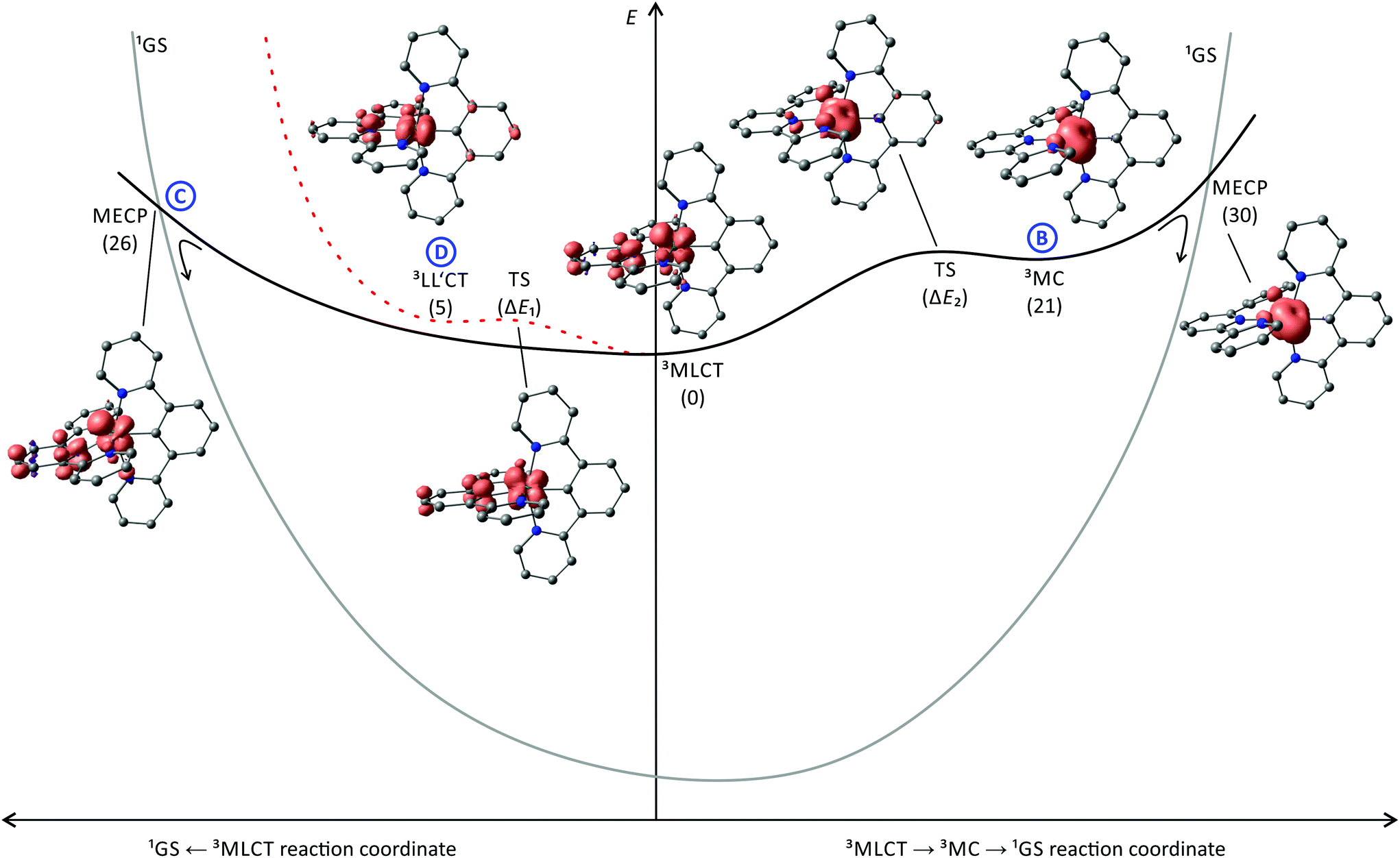 | ||
| Fig. 5 Schematic representation of the energy landscape of [Ru(dpb)(tpy)]+ type of complexes including spin density contour plots (contour value: 0.01) of the 3LL′CT, 3MLCT and 3MC states of [4a]+, the 3MLCT → 3MC and 3MLCT → 3LL′CT transition states as well as the minimum energy points for the 3MLCT → 1GS and the 3MC → 1GS surface crossing. H atoms are omitted for clarity. Energies (kJ mol−1) of the respective states are given in parentheses relative to the 3MLCT state (E = 0 kJ mol−1). The shape of the singlet potential energy surface is estimated from the DFT-calculated triplet–singlet energy differences at the various excited state geometries. | ||
| E(3LL′CT) | ΔE1 | E(3MC) | ΔE2 | |
|---|---|---|---|---|
| [4a]+ | 5 | 5 | 21 | 23 |
| [4c]+ | −3 | 4 (2) | 13 | 21 (22) |
| [5a]+ | 8 | 8 (6) | 13 | 22 (23) |
| [5c]+ | −10 | — | 31 | — |
| [5d]+ | — | — | 9 | (11) |
| [6]+ | — | — (4) | — | — (22) |
The calculated relative energies of the triplet states (Fig. 5) of [5a]+–[5d]+ as well as the transition states connecting them are summarized in Table 3. The emissive 3MLCT state is flanked by two thermally accessible quenching states, namely the 3LL′CT and 3MC states. The lowering of the relative 3MC energy from 66 kJ mol−1 in [1b]+ to 10–30 kJ mol−1 in these bis(tridentate) complexes is attributed to the smaller N–Ru–N bite angles and the weaker overlap of the nitrogen lone pairs with the eg metal orbitals which results in a smaller ligand field splitting. The distortion of the 3MC state allows for tunnelling to the 1GS and for a surface crossing point (MECP) that is just 9 kJ mol−1 above the 3MC level providing an accessible non-emissive deactivation channel for the 3MLCT state (path B, Fig. 5).
Interestingly, a direct 3MLCT → 1GS MECP was found for [4a]+ as well at a moderate energy (26 kJ mol−1 above the 3MLCT state). It is qualitatively similarly distorted as the 3MLCT state but the degree of the distortion is larger. Thus, the geometry of this crossing point can be regarded as a high-amplitude distortional vibration along the 1GS → 3MLCT vibrational mode (3MLCT → 1GS reaction coordinate, Fig. 5). Hence, the 3MLCT distortion of [4a]+ opens up this low-energy deactivation channel (path C), that is absent for [1b]+. However, experimental evidence for such a quenching channel is difficult to obtain as its activation barrier is similar to that of the 3MC deactivation channel and hence a similar temperature-dependent emission behaviour is expected.
The 3LL′CT state is connected to the 3MLCT state via a transition state with a very low activation barrier (Table 3). As the 3LL′CT state is barely distorted compared to the 1GS geometry (vide supra) it is considered a nested state. Indeed, attempts to localize a 3LL′CT → 1GS MECP, that would provide a non-emissive decay channel, failed. Because emission from the 3LL′CT state is symmetry-forbidden, its only decay pathway proceeds via tunnelling into the vibrationally excited singlet state followed by thermal relaxation (path D, Fig. 5).
Hence both, the 3LL′CT and 3MC states (and potentially also the 3MLCT → 1GS MECP) are responsible for the efficient phosphorescence quenching at room temperature. This DFT-based interpretation was evidenced experimentally by recording the temperature dependence of the quantum yield. The respective ln(ϕ) vs. T−1 data of [4c]+ and [5a]+ were reproduced using a fit function accounting for two thermally activated deactivation pathways. The activation barriers obtained from the fit are in excellent agreement with the computed transition state energies (Table 3).
Schubert and coworkers117 published very similar results on structurally related complexes such as [Ru(dtp)(tpy)]+ (dtbH = 1,3-di-(1,2,3-triazol-4-yl)benzene, Scheme 2, [6]+). Two Arrhenius-like activation parameters were required to properly reproduce the temperature-dependent lifetime data yielding very similar activation energies compared to our findings for [4c]+ and [5a]+. In contrast to our assignments, however, they attributed the two deactivation channels of [6]+ to an irreversible 3MLCT → 3MC surface crossing and an internal conversion to a higher-lying MLCT state of increased singlet character. The latter is a common feature of non-cyclometalated (polypyridine)ruthenium(II) complexes.77,83,91,153 In the light of our results and the electronic similarity of the studied structures, it seems plausible that the second deactivation channel in [6]+ actually is via a state of 3LL′CT nature as well. In fact, their DFT-optimization of a triplet state of [6]+ afforded a 3CT state with orthogonal SOMOs.117 Even if the cyclometalating ligand does not contribute significantly to the spin density of this state, its emissive relaxation is symmetry-forbidden.
To summarize, the combination of orthogonal ligands with strongly differing electronic properties, one being an excellent π-acceptor and the second a strong π-donor typically yields a low-lying 3CT state. Even though this state is not directly populated after optical excitation into a 1MLCT state and subsequent intersystem crossing onto the triplet manifold it serves as a further low-barrier channel for radiationless deactivation of the emissive 3MLCT state (path D) besides the 3MC state (path B). Recent reports underline that these results are transferable to structurally similar complexes of other transition metals with orthogonal tridentate ligands. In fact, Williams and coworkers suggested that a 3LL′CT state plays a key role in the excited state deactivation of isostructural and isoelectronic [Ir(N^N^N)(N^C^N)]2+ complexes.154 A beautiful theoretical study on the excited state deactivation pathways in biscyclometalated gold(III) complexes of the general formula [Au(C^N^C)(acetylide)] revealed a 3LL′CT state which efficiently contributed to the radiationless deactivation of the emissive state.56,57,155 Its substantial distortion compared to the ground state increased the rate of non-radiative decay.
In order to optimize the emissive properties of bis(tridentate) cyclometalated ruthenium complexes, circumventing the low-energy non-emissive 3LL′CT state is a key objective, for example by removing the axial symmetry. This can be achieved using a N^N^C coordination mode on the cyclometalating ligand which will be discussed in the next section (vide infra). However, the kinetic parameters extracted by Schubert and coworkers117 from the temperature-dependent lifetime data suggest, that at room temperature the 3LL′CT state (path D) with an activation barrier ΔE2 of just 4.2 kJ mol−1 is only responsible for the quenching of about 25% of the excited molecules of [6]+ and structurally similar complexes. At the same time, the 3MC state with a considerably higher barrier ΔE1 of 21.9 kJ mol−1 is responsible for about 75% of the excited state deactivation (path B and potentially path C), while direct radiationless decay into the ground state only contributes 0.1% (path A). Hence, even avoiding the 3LL′CT state by clever molecular design will not per se yield strong emitters. The strong electronic coupling of the 3MLCT and 3MC states [k0 (3MLCT → 3MC) = 1011–1013 s−1; k0 (3MLCT → 3LL′CT) ≈ 108 s−1]117 renders the former the dominant deactivating state despite the substantially higher activation barrier. The appreciably weaker coupling of the 3MLCT and 3LL′CT states can be traced back to the two states still being electronically nearly orthogonal (vide supra). Concluding, despite the occurrence of a 3LL′CT state in this kind of bis(tridentate) complexes the important states to manipulate for improving emission efficiencies remain the 3MC and 3MLCT states. Some well-thought-out examples have been provided in the recent literature employing ligand bite angle manipulation94,95,156,157 and push–pull concepts93,157,158 to increase the 3MLCT-3MC gap in bis(tridentate) ruthenium complexes which in principle are applicable to cyclometalated complexes as well. However, these conceptual approaches are beyond the scope of this perspective. Recently, Dixon and coworkers159–161 suggested on a computational basis, that two cyclometalating sites in cis-position could be beneficial to increase the ligand field splitting of iron(II) complexes and provide a tool for controlling the relative 3MLCT and 3MC energies. This concept should be transferable to cyclometalated ruthenium complexes as well (cf.3) although it is likely accompanied with synthetic challenges.105,130,162,163 An alternative approach could again involve attaching very weakly π-accepting ligands trans to a N^C^N ligand to yield potentially highly luminescent Ru → N^C^N 3MLCT states.
The [Ru(N^N^N)(N^N^C)]+ coordination sphere
The absorption spectrum of [Ru(tpy)(pbpy)]+ type complexes resembles that of [Ru(bpy)2(ppy)]+ with two absorption bands, one around 400 nm, and a second around 500–600 nm. Again, the low-energy band is composed of MLCT transitions both involving the tpy and the cyclometalating ligand. However, the π* orbital of the coordinating phenyl ring (LUMO+3) is not involved in any of the low-energy transitions, as its energy is substantially higher than the frontier orbitals (Fig. 2).107 Due to the near-degeneracy of the three lowest unoccupied orbitals (LUMO–LUMO+2), the absorption band at 500 nm is markedly sharper than that of [Ru(dpb)(tpy)]+ and [Ru(bpy)2(ppy)]+ complexes. The feature around 400 nm is dominated by an intense Ru → phenyl (LUMO+3) transition (Fig. 2).107Unfortunately, accounts on emission properties of these [Ru(tpy)(pbpy)]+ complexes are very limited.107,134,137,164 A few examples are summarized in Scheme 3 and Table 4. In the series [7a]+–[7d]+ with carboxy substituents at either or both ligands, the quantum yields and trends in the emission energies are similar to those of the isomeric [Ru(tpy)(dpb)]+ complexes [4a]+–[4d]+. Again, carboxy-substitution at the tpy ligand lowers the LUMO energy sufficiently to yield non-emissive ([7b]+) or essentially non-emissive ([7d]+) complexes at room temperature due to the energy gap law.137 However, carboxy-substitution at the pbpy ligand ([7c]+) does not blueshift the emission energy compared to the unsubstituted complex [7a]+ (270 cm−1) as much as in the case of the [Ru(dpb)(tpy)]+ complexes [4c]+ and [4a]+ (660 cm−1).137 This was traced back to a change of the orbital parentage of the emissive 3MLCT state. Instead of being a Ru → tpy state as in [7a]+, emission of [7c]+ arises from a Ru → (pbpy-COOR) 3MLCT state. This is accompanied by an altered energy ordering of the lowest unoccupied orbitals and reflects their energetic similarity (Fig. 2). This example highlights that in [Ru(N^N^N)(N^N^C)]+ type complexes Ru → N^N^C 3MLCT states are obtained by tuning the respective frontier orbital energies of the ligands. However, the cyclometalating phenyl ring does not contribute as π-accepting moiety and the excited electron is entirely localized on the bipyridine fragment of the pbpy ligand. Consequently, the phosphorescence efficiency is not affected significantly (Table 4).
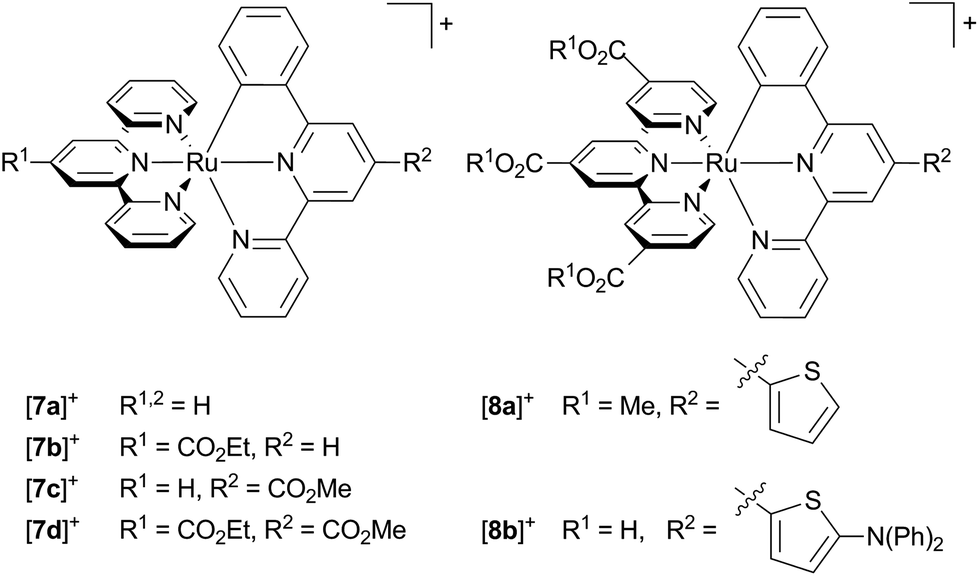 | ||
| Scheme 3 Literature-known [Ru(N^N^N)(N^N^C)]+ complexes relevant to this work ([7a]+–[7d]+,107,137 [8a]+,134 [8b]+164). | ||
em as well as quantum yields ϕ of selected [Ru(N^N^N)(N^N^C)]+ complexes at room temperature in solution
Interestingly, Berlinguette and coworkers reported on a series of [Ru(tpy)(pbpy)]+ based complexes with diarylamine groups appended via a thiophene linker such as [8b]+. These are highly emissive (quantum yields in the range of 0.1–0.3) in some cases, but emit at much higher energy than typically observed for these complexes.164 In fact, the analogous thiophene substituted complex [8a]+ without the diarylamine functionality lacks the strong emission.134 However, Berlinguette showed that the emissive behaviour of [8b]+ actually arises from a singlet intraligand charge transfer state (1ILCT) involving the diarylamine unit as electron donor and the polypyridine moiety as electron acceptor.165,166 An identical emission energy was observed for the free ligand with even higher fluorescence quantum yields (ϕ = 0.91, τ = 3.4 ns) explaining the untypically high emission energy and quantum yield of [8b]+.164–166
To get a better understanding of the states involved in the excited state deactivation of [Ru(tpy)(pbpy)]+ complexes, we studied the triplet potential energy surface of [7a]+ using DFT calculations. Inspecting the 3MLCT state geometry and spin density of [7a]+ reveals a striking similarity to [4a]+ (Fig. 4, 5 and 6). In fact, a similar distortion of the tpy ligand with an offset central pyridine ring is found in both cases (vide supra). Additionally, the bond length changes of the 3MLCT states compared to the respective 1GS geometries of [4a]+ and [7a]+ are very similar (Fig. 4). Given the similar quantum yields of the isoelectronic classes of complexes [Ru(dpb)(tpy)]+ and [Ru(tpy)(pbpy)]+ this suggests excited state deactivation channels with similar barriers are dominant in both cases. However, the 3MC state (path B) is found to be 60 kJ mol−1 (3MLCT → 3MC transition state at 62 kJ mol−1) higher in energy than the 3MLCT state and thus it is thermally inaccessible at room temperature. As a consequence, its contribution to the excited state deactivation of [7a]+ is negligible. The marked increase of the 3MC-3MLCT energy gap by about 30 kJ mol−1 by exchanging N^C^N by N^N^C chelate ligands in bis(tridentate) complexes (Fig. 6) was also found for the isoelectronic iron(II) complexes by Dixon and coworkers.161 They argued that the cyclometalating ligand does not only act as a strong σ-donor but also as a π-donor. In the iron(II) complex [Fe(dpb)(tpy)]+ the π-donor strength is the dominant influence yielding a net reduction of the effective ligand field strength and hence a stabilization of the 3MC state compared to the non-cyclometalated complex [Fe(tpy)2]2+. In [Fe(tpy)(pbpy)]+ on the other hand, the π-overlap between the peripheral cyclometalating phenyl ring and the metal d orbitals is not as pronounced. As a consequence of the σ-overlap an increased ligand field splitting and a higher 3MC energy compared to [Fe(dpb)(tpy)]+ are calculated. At the same time, the 3MLCT energies of [Fe(tpy)(pbpy)]+ and [Fe(dpb)(tpy)]+ are essentially identical yielding an overall higher 3MLCT-3MC energy gap by about 30 kJ mol−1 for [Fe(tpy)(pbpy)]+. As for ruthenium, the d orbitals are more diffuse than for iron, the destabilization of the t2g orbitals via π-donor interactions is much less pronounced yielding 3MC states well above the 3MLCT level in all cyclometalating complexes, but the same principles apply explaining the trends we observe for the isoelectronic ruthenium complexes.161
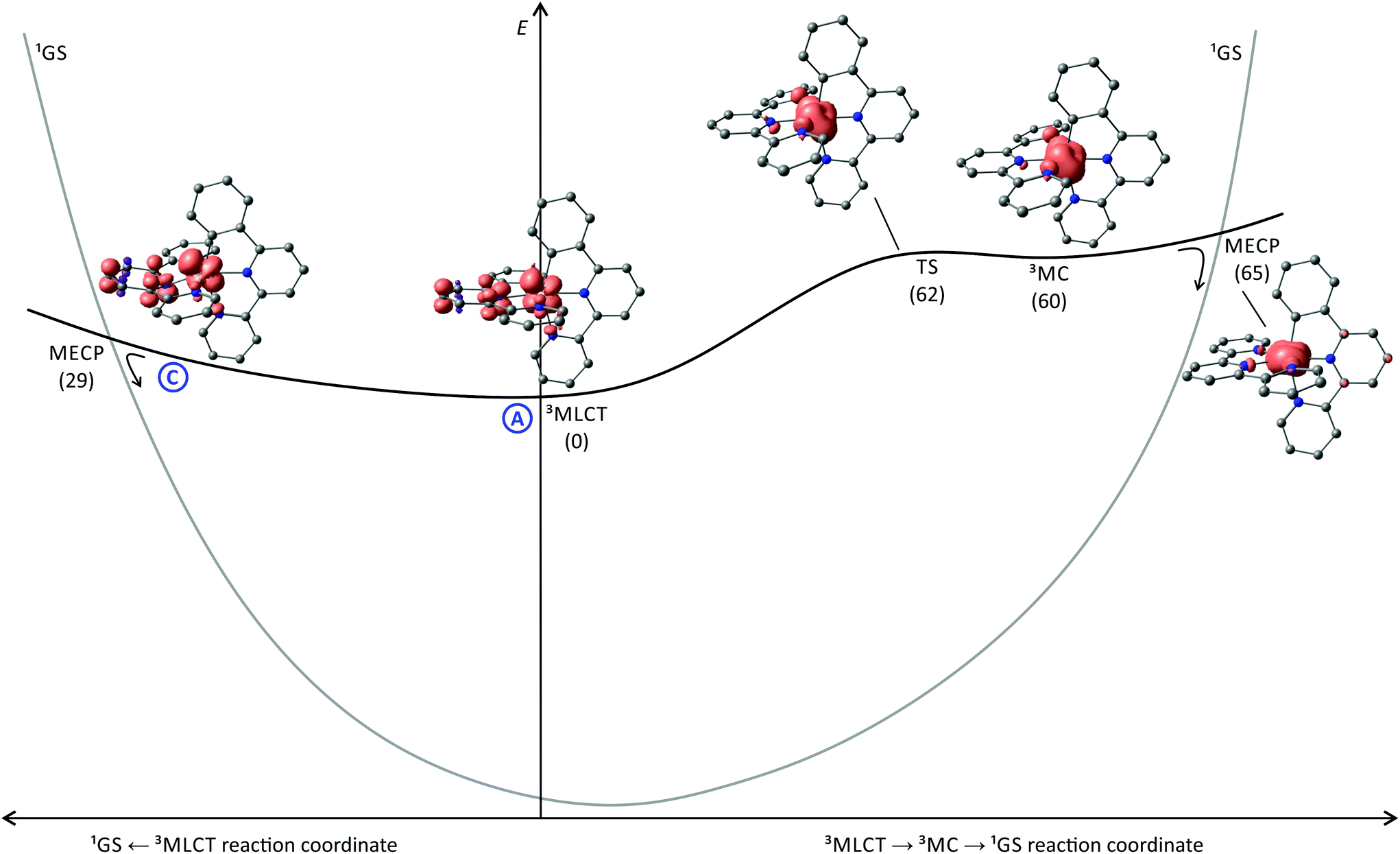 | ||
| Fig. 6 Schematic representation of the energy landscape of [Ru(tpy)(pbpy)]+ type of complexes including spin density contour plots (contour value: 0.01) of the 3MLCT and 3MC states of [7a]+, the 3MLCT → 3MC transition state as well as the minimum energy points for the 3MLCT → 1GS and the 3MC → 1GS surface crossing. H atoms are omitted for clarity. Energies (kJ mol−1) of the respective states are given in parentheses relative to the 3MLCT state (E = 0 kJ mol−1). The shape of the singlet potential energy surface is estimated from the DFT-calculated triplet–singlet energy differences at the various excited state geometries. | ||
In principle, a 3LL′CT state similar to that described for [Ru(dpb)(tpy)]+ complexes (vide supra) is also conceivable for complexes of the [Ru(N^N^N)(N^N^C)]+ class. However, an analogous symmetry restriction as discussed above for the former does not apply in this case due to the lowered molecular symmetry. Although we tried to localize such a 3LL′CT state it remained elusive. Whether such a state actually contributes to the excited state deactivation of [Ru(N^N^N)(N^N^C)]+ complexes has to be evaluated based on experimental emission data at variable temperatures. This exceeds the scope of this article.
Since the 3MLCT state of [7a]+ is electronically very similar to the 3MLCT state of the tris(bidentate) complex [1b]+, an argumentation based on emission quenching via vibronic coupling to the ground state (path A) is insufficient to account for the substantially lower emission quantum yields of the former (10−6–10−5 as compared to 10−4–10−3). Additionally, deactivation channels via low-lying 3MC (path B) or 3LL′CT states (path D) as found for [Ru(dpb)(tpy)]+ complexes do not contribute to the efficient non-emissive excited state decay of [Ru(tpy)(pbpy)]+ complexes. A surface crossing point between the 3MLCT and 1GS potential energy surfaces (path C), however, similar to that found for [4a]+ (Fig. 5), would provide a concise explanation for the marked difference between the tris(bidentate) and bis(tridentate) complexes. Indeed, we localized a thermally accessible 3MLCT → 1GS surface crossing point that is only 29 kJ mol−1 higher in energy than the 3MLCT state (Fig. 6). Remarkably, the geometry and energy of this crossing point is similar to that of the 3MLCT → 1GS MECP of [4a]+. Again, the distortion of the 3MLCT state provides an excited state deactivation pathway for polypyridine ruthenium complexes. This finding has some predictive value as well. The 3MC state is thermally inaccessible at temperatures below 298 K in [7a]+ and does not contribute to the excited state decay. Hence, the temperature dependence of the emission of [7a]+ can provide information on the contribution of a minimum energy surface crossing point in proximity to the relaxed 3MLCT geometry to the emission quenching. An increasing emission intensity upon cooling would support this hypothesis. Additionally, as the dissociative anti-bonding 3MC state is out of reach at room temperature, no photosubstitution reactions should occur for [7a]+ in contrast to [Ru(bpy)3]2+ which is very prone to such reactivity.90 We will devote future work into elucidating these predictions.
In conclusion, it does not suffice to reduce the molecular symmetry and circumvent the parasitic 3LL′CT state to increase phosphorescence quantum yields in bis(tridentate) cyclometalated complexes. Bis(tridentate) [Ru(N^N^N)(N^N^C)]+ complexes with peripheral cyclometalation suffer from the same distortion and low energy of the 3MLCT state as the analogous tris(bidentate) complexes. As the relative 3MC state energy of [Ru(tpy)(pbpy)]+ complexes is substantially higher than for comparable [Ru(N^N^N)(N^C^N)]+ complexes, its contribution to the excited state decay can be neglected (path B). Hence, similar strategies are applicable for increasing the luminescence quantum yields as suggested before for tris(bidentate) complexes. These should focus on reducing the excited state distortion yielding a nested emissive state and shutting down the deactivation via direct 3MLCT → 1GS surface crossing (path C). This might be achieved by shifting the LUMO to higher energies and making the π* orbital of the cyclometalating moiety the acceptor site of the lowest 3MLCT state. At the same time, a lower excited state distortion would shift the energy of the 3MLCT → 1GS MECP to higher energies as well. This could potentially be accomplished by a combination of a very weakly π-accepting spectator ligand with a cyclometalating ligand that also contains a weakly π-accepting site such as an N-heterocyclic carbene.167,168
Experimental section
Density functional theory calculations
DFT calculations were carried out using the ORCA program package (version 3.0.2).169 Tight convergence criteria were chosen for all calculations (Keywords TightSCF and TightOpt). All calculations were performed using the hybrid functional B3LYP170 and employ the RIJCOSX approximation.171,172 Relativistic effects were calculated at the zeroth order regular approximation (ZORA) level. The ZORA keyword automatically invokes relativistically adjusted basis sets.173 To account for solvent effects, a conductor-like screening model (COSMO) modelling acetonitrile was used in all calculations.174 Geometry, transition state and minimum energy crossing point optimizations were performed using Ahlrichs’ split-valence double-ζ basis set def2-SV(P) which comprises polarization functions for all non-hydrogen atoms.175,176 Optimized geometries were confirmed to be mimina or first-order saddle points by subsequent frequency analysis (nimag = 0 or 1, respectively). Surface crossing geometries were subjected to SurfCrossNumFreq calculations to confirm that they are minima in the 3N–7 dimensional subspace excluding the surface crossing reaction coordinate. Computed free Gibbs enthalpies were used to compare the relative energies of all structures. Explicit counterions and/or solvent molecules were not taken into account in any case.The 3MLCT and 3LL′CT states were localized by triplet geometry optimizations from the optimized 1GS geometry. 3MC states were found by elongating two opposite Ru–N bonds to 2.40 Å and subsequent geometry optimizations. Transition state optimizations were started from geometries obtained by averaging all coordinates of the starting and final state geometries using the exact Hessian matrix of the initial geometry. MECP geometries were obtained by starting at the respective optimized triplet state geometry (3MLCT geometry for 3MLCT-1GS MECP, 3MC geometry for 3MC-1GS MECP).
Conclusions
Cyclometalated polypyridineruthenium(II) complexes with N5C coordination sphere typically exhibit very weak room temperature emission in the near infrared range (700–800 nm) of the electromagnetic spectrum. The reasons for the weak emission are various and depend on the chelate coordination sphere around the metal center. In tris(bidentate) [Ru(N^N)2(N^C)]+ complexes, the 3MC state (path B) that is typically a major channel for excited state decay in polypyridineruthenium(II) complexes,84,89,90 does not contribute as it is thermally inaccessible (ΔE(3MLCT → 3MC) = 60–70 kJ mol−1) at room temperature. Furthermore, no low energy 3MLCT → 1GS surface crossing point was found (path C) which suggests that tunnelling into high-energy vibrationally excited singlet states is the main channel of excited state deactivation (path A).In contrast, the emission quenching of [Ru(N^N^N)(N^C^N)]+ complexes is dominated by two thermally accessible triplet states, that flank the emissive 3MLCT state, namely the 3MC state (path B, ΔE(3MLCT → 3MC) = 10–30 kJ mol−1) and a 3LL′CT state (path D, ΔE(3MLCT → 3LL′CT) < 10 kJ mol−1).117–119 The 3LL′CT state is a peculiarity of C2-symmetric cyclometalated complexes and provides a second, unprecedented non-emissive deactivation channel. Additionally, a 3MLCT → 1GS surface crossing point provides another decay channel (path C) whose contribution yet needs to be quantified. These three channels B, C and D are responsible for almost 100% of the emission quenching. Although significantly faster than the emission process itself, direct non-emissive decay via3MLCT → 1GS tunnelling (path A) only plays a subordinate role simply because excited state decay via paths B, C and D is so efficient. This is reflected by the very low excited state lifetimes below the nanosecond range.
In [Ru(N^N^N)(N^N^C)]+ complexes, the 3MC states are thermally inaccessible (ΔE(3MLCT → 3MC) ≈ 60 kJ mol−1) and no quenching 3LL′CT states are relevant. However, the triplet and singlet potential surfaces intersect close to the relaxed 3MLCT state providing an efficient deactivation channel (path C) with an activation barrier of only about 30 kJ mol−1. This explains the similarly low emission quantum yields of bis(tridentate) complexes with central and peripheral cyclometalating site despite the markedly different triplet states relevant to the two systems. However, the amount of emission quenching via tunneling (path A) in [Ru(N^N^N)(N^N^C)]+ complexes remains unclear until temperature-dependent lifetime data are acquired.
Strategies for increasing the phosphorescence quantum yields are proposed. In [Ru(N^N)2(N^C)]+ complexes emission quenching is dominated the very low emission energies and the efficient tunnelling into high-energy singlet states following the energy gap law (path A). Hence, improving the emission efficiency is very challenging and only achieved by structurally restraining the already small excited state distortions or increasing the emission energy drastically. The latter is possible by making the cyclometalating ligand the π-accepting site of the 3MLCT state as shown by Chou and coworkers.130
In bis(tridentate) cyclometalated ruthenium complexes, emission quenching predominantly arises from the distortion of the 3MLCT state compared to the ground state. The triplet potential energy surface is relatively flat around the 3MLCT geometry leading to a 3MLCT/1GS surface intersection less than 30 kJ mol−1 above the emissive 3MLCT state giving rise to a deactivation channel via a direct 3MLCT → 1GS surface crossing. Minimizing the excited state distortion via structural constraints could circumvent this channel. Additionally, by making the cyclometalating ligand the π-accepting site within the 3MLCT state via tuning the energy levels of the lowest unoccupied molecular orbitals, a substantial increase of the 3MLCT state energy can be achieved which would be beneficial for suppressing the 3MLCT → 1GS surface crossing as well. This will, however, also shift the emission well into the visible range of the electronic spectrum.
Furthermore, it is crucial to avoid orthogonal π-donor and π-acceptor sites in trans position across the metal center as in [Ru(N^N^N)(N^C^N)]+ because this inherently invokes orthogonal, non-emissive 3LL′CT states as quenching channels and also lowers the energy of the 3MC states into a thermally accessible region due to a lower ligand field splitting.
In order to elucidate the excited state properties of cyclometalated complexes, temperature-dependent excited state lifetime or emission quantum yield measurements provide an invaluable tool.71,117–119 Additionally, quantum chemical approaches can deliver lots of information about energies and geometries of relevant excited states. Tong, Che and coworkers (vide supra) demonstrated this on luminescent cyclometalated gold(III) complexes.155 They assessed the radiative and non-radiative decay rates from a computational standpoint and quantified key processes that yield or prevent efficient emission in these species. Similarly, Dixon and coworkers studied mono- and bis(cyclometalated) iron(II) complexes using DFT calculations.159–161 The 3MC state in [Fe(pbpy)(tpy)]+ with peripheral cyclometalation is substantially higher in energy than in [Fe(dpb)(tpy)]+ with central cyclometalation, very similar to the results presented here for the ruthenium homologues. Furthermore, they highlighted, that bis(cyclometalated) iron(II) complexes such as [Fe(dpb)(pbpy)] and [Fe(pbpy)2] have very low-lying 3MLCT states that are, in the case of [Fe(dpb)(pbpy)], only marginally distorted compared to the ground state geometry. We suggest that these findings apply to the analogous ruthenium complexes potentially opening a route to highly luminescent near-IR emitters. However, since only very few bis(cyclometalated) polypyridine ruthenium complexes are known so far130,162,163 and none of them contain tridentate ligands, the synthesis of Ru(dpb)(pbpy) and Ru(pbpy)2 complexes might be challenging. We will devote future work to the design and synthesis of ruthenium-based emitters with cyclometalating ligands to improve and exploit their excited state properties.
Acknowledgements
Parts of this research were conducted using the supercomputer MOGON and advisory services offered by Johannes Gutenberg Univ. Mainz (http://www.hpc.uni-mainz.de), which is a member of the AHRP and the Gauss Alliance e.V. This work was financially supported by the Deutsche Forschungsgemeinschaft (GSC 266, Materials Science in Mainz, scholarship for C. K.).References
- N. Alonso-Vante, J.-F. Nierengarten and J.-P. Sauvage, J. Chem. Soc., Dalton Trans., 1994, 1649–1654 RSC.
- T. Bessho, E. C. Constable, M. Graetzel, A. Hernandez Redondo, C. E. Housecroft, W. Kylberg, M. K. Nazeeruddin, M. Neuburger and S. Schaffner, Chem. Commun., 2008, 3717–3719 RSC.
- C. E. Housecroft and E. C. Constable, Chem. Soc. Rev., 2015, 44, 8386–8398 RSC.
- S. Ferrere and B. A. Gregg, J. Am. Chem. Soc., 1998, 120, 843–844 CrossRef CAS.
- S. Ferrere, Chem. Mater., 2000, 12, 1083–1089 CrossRef CAS.
- T. C. B. Harlang, Y. Liu, O. Gordivska, L. A. Fredin, C. S. Ponseca, P. Huang, P. Chábera, K. S. Kjaer, H. Mateos, J. Uhlig, R. Lomoth, R. Wallenberg, S. Styring, P. Persson, V. Sundström and K. Wärnmark, Nat. Chem., 2015, 7, 883–889 CrossRef CAS PubMed.
- G. Sauvé, M. E. Cass, S. J. Doig, I. Lauermann, K. Pomykal and N. S. Lewis, J. Phys. Chem. B, 2000, 104, 3488–3491 CrossRef.
- T. Kinoshita, J.-i. Fujisawa, J. Nakazaki, S. Uchida, T. Kubo and H. Segawa, J. Phys. Chem. Lett., 2012, 394–398 CrossRef CAS PubMed.
- E. I. Mayo, K. Kilså, T. Tirrell, P. I. Djurovich, A. Tamayo, M. E. Thompson, N. S. Lewis and H. B. Gray, Photochem. Photobiol. Sci., 2006, 5, 871–873 Search PubMed.
- E. Baranoff, J. Yum, I. Jung, R. Vulcano, M. Grätzel and M. K. Nazeeruddin, Chem. – Asian J., 2010, 5, 496–499 CrossRef CAS PubMed.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Graetzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. K. Nazeeruddin, S. M. Zakeeruddin, R. Humphry-Baker, M. Jirousek, P. Liska, N. Vlachopoulos, V. Shklover, C.-H. Fischer and M. Grätzel, Inorg. Chem., 1999, 38, 6298–6305 CrossRef CAS PubMed.
- M. K. Nazeeruddin, P. Péchy, T. Renouard, S. M. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. B. Deacon, C. A. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS PubMed.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef.
- S. H. Wadman, J. M. Kroon, K. Bakker, M. Lutz, A. L. Spek, G. P. M. van Klink and G. van Koten, Chem. Commun., 2007, 1907–1909 RSC.
- T. Bessho, E. Yoneda, J.-H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930–5934 CrossRef CAS PubMed.
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed.
- P. G. Bomben, K. C. Robson, B. D. Koivisto and C. P. Berlinguette, Coord. Chem. Rev., 2012, 256, 1438–1450 CrossRef CAS.
- H. D. Abruna, A. Y. Teng, G. J. Samuels and T. J. Meyer, J. Am. Chem. Soc., 1979, 101, 6745–6746 CrossRef CAS.
- I. Okura and N. Kim-Thuan, J. Mol. Catal., 1979, 5, 311–314 CrossRef CAS.
- J.-M. Lehn and R. Ziessel, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 701–704 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1983, 536 RSC.
- H. Ozawa, M. Haga and K. Sakai, J. Am. Chem. Soc., 2006, 128, 4926–4927 CrossRef CAS PubMed.
- S. Rau, B. Schäfer, D. Gleich, E. Anders, M. Rudolph, M. Friedrich, H. Görls, W. Henry and J. G. Vos, Angew. Chem., 2006, 118, 6361–6364 ( Angew. Chem., Int. Ed. , 2006 , 45 , 6215–6218 ) CrossRef.
- A. Fihri, V. Artero, M. Razavet, C. Baffert, W. Leibl and M. Fontecave, Angew. Chem., 2008, 120, 574–577 ( Angew. Chem., Int. Ed. , 2008 , 47 , 564–567 ) CrossRef.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed.
- J. W. Tucker, Y. Zhang, T. F. Jamison and C. R. J. Stephenson, Angew. Chem., 2012, 124, 4220–4223 ( Angew. Chem., Int. Ed. , 2012 , 51 , 4144–4147 ) CrossRef.
- M. Majek and A. Jacobi von Wangelin, Angew. Chem., 2013, 125, 6033–6035 ( Angew. Chem., Int. Ed. , 2013 , 52 , 5919–5921 ) CrossRef.
- S. Paria and O. Reiser, ChemCatChem, 2014, 6, 2477–2483 CrossRef CAS.
- S. M. Stevenson, M. P. Shores and E. M. Ferreira, Angew. Chem., 2015, 127, 6606–6610 ( Angew. Chem., Int. Ed. , 2015 , 54 , 6506–6510 ) CrossRef.
- V. Balzani and S. Campagna, Top. Curr. Chem., 2007, 280, 1–273 CrossRef CAS.
- V. Balzani and S. Campagna, Top. Curr. Chem., 2007, 281, 1–327 CrossRef.
- J. P. Paris and W. W. Brandt, J. Am. Chem. Soc., 1959, 81, 5001–5002 CrossRef CAS.
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579–11592 RSC.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128–6185 RSC.
- H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259–3302 RSC.
- S. M. Fredericks, J. C. Luong and M. S. Wrighton, J. Am. Chem. Soc., 1979, 101, 7415–7417 CrossRef CAS.
- A. Juris, S. Campagna, I. Bidd, J. M. Lehn and R. Ziessel, Inorg. Chem., 1988, 27, 4007–4011 CrossRef CAS.
- R. A. Kirgan, B. P. Sullivan and D. P. Rillema, Top. Curr. Chem., 2007, 281, 45–100 CrossRef CAS.
- K. K.-W. Lo, M.-W. Louie and K. Y. Zhang, Coord. Chem. Rev., 2010, 254, 2603–2622 CrossRef CAS.
- J. N. Demas, E. W. Harris, C. M. Flynn and D. Diemente, J. Am. Chem. Soc., 1975, 97, 3838–3839 CrossRef CAS.
- C. Creutz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309–1319 CrossRef CAS.
- J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. de Cola and L. Flamigni, Chem. Rev., 1994, 94, 993–1019 CrossRef CAS.
- D. Kumaresan, K. Shankar, S. Vaidya and R. H. Schmehl, Top. Curr. Chem., 2007, 281, 101–142 CrossRef CAS.
- D. Carstens and G. Crosby, J. Mol. Spectrosc., 1970, 34, 113–135 CrossRef CAS.
- A. Zilian, U. Maeder, A. von Zelewski and H. U. Guedel, J. Am. Chem. Soc., 1989, 111, 3855–3859 CrossRef CAS.
- M. T. Indelli, C. Chiorboli and F. Scandola, Top. Curr. Chem., 2007, 280, 215–255 CrossRef CAS.
- K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1985, 107, 1431–1432 CrossRef CAS.
- M. G. Colombo, A. Hauser and H. U. Guedel, Inorg. Chem., 1993, 32, 3088–3092 CrossRef CAS.
- L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143–203 CrossRef CAS.
- F. Barigelletti, D. Sandrini, M. Maestri, V. Balzani, A. von Zelewsky, L. Chassot, P. Jolliet and U. Maeder, Inorg. Chem., 1988, 27, 3644–3647 CrossRef CAS.
- H.-K. Yip, L.-K. Cheng, K.-K. Cheung and C.-M. Che, J. Chem. Soc., Dalton Trans., 1993, 2933–2938 RSC.
- D. R. McMillin and J. J. Moore, Coord. Chem. Rev., 2002, 229, 113–121 CrossRef CAS.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2013, 135, 15135–15143 CrossRef CAS PubMed.
- E. S.-H. Lam, W. H. Lam and V. W.-W. Yam, Inorg. Chem., 2015, 54, 3624–3630 CrossRef CAS PubMed.
- V. W.-W. Yam and E. C.-C. Cheng, Top. Curr. Chem., 2007, 281, 269–309 CrossRef CAS.
- C. Bronner and O. S. Wenger, Dalton Trans., 2011, 40, 12409–12420 RSC.
- N. A. P. Kane-Maguire and C. H. Langford, J. Chem. Soc. D, 1971, 895–896 RSC.
- M. Maestri, F. Bolletta, L. Moggi, V. Balzani, M. S. Henry and M. Z. Hoffman, J. Am. Chem. Soc., 1978, 100, 2694–2701 CrossRef CAS.
- A. D. Kirk and G. B. Porter, J. Phys. Chem., 1980, 84, 887–891 CrossRef CAS.
- N. A. P. Kane-Maguire, Top. Curr. Chem., 2007, 280, 37–67 CrossRef CAS.
- S. Otto, M. Grabolle, C. Förster, C. Kreitner, U. Resch-Genger and K. Heinze, Angew. Chem., 2015, 127, 11735–11739 ( Angew. Chem., Int. Ed. , 2015 , 54 , 11572–11576 ) CrossRef.
- A. Juris and R. Ziessel, Inorg. Chim. Acta, 1994, 225, 251–254 CrossRef CAS.
- N. Armaroli, Chem. Soc. Rev., 2001, 30, 113–124 RSC.
- N. Armaroli, G. Accorsi, F. Cardinali and A. Listorti, Top. Curr. Chem., 2007, 280, 69–115 CrossRef CAS.
- M. Iwamura, S. Takeuchi and T. Tahara, Acc. Chem. Res., 2015, 48, 782–791 CrossRef CAS PubMed.
- J. Nitsch, C. Kleeberg, R. Fröhlich and A. Steffen, Dalton Trans., 2015, 44, 6944–6960 RSC.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- G. Capano, U. Rothlisberger, I. Tavernelli and T. J. Penfold, J. Phys. Chem. A, 2015, 119, 7026–7037 CrossRef CAS PubMed.
- L.-Y. Zhang, G.-F. Liu, S.-L. Zheng, B.-H. Ye, X.-M. Zhang and X.-M. Chen, Eur. J. Inorg. Chem., 2003, 2003, 2965–2971 CrossRef.
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- V. Balzani and A. Juris, Coord. Chem. Rev., 2001, 211, 97–115 CrossRef CAS.
- J. R. Winkler, T. L. Netzel, C. Creutz and N. Sutin, J. Am. Chem. Soc., 1987, 109, 2381–2392 CrossRef CAS.
- G. D. Hager and G. A. Crosby, J. Am. Chem. Soc., 1975, 97, 7031–7037 CrossRef CAS.
- T. J. Meyer, Pure Appl. Chem., 1986, 58, 1193–1206 CrossRef CAS.
- J. N. Demas and D. G. Taylor, Inorg. Chem., 1979, 18, 3177–3179 CrossRef CAS.
- S. Yoon, P. Kukura, C. M. Stuart and R. A. Mathies, Mol. Phys., 2006, 104, 1275–1282 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- G. A. Crosby, Acc. Chem. Res., 1975, 8, 231–238 CrossRef CAS.
- G. D. Hager, R. J. Watts and G. A. Crosby, J. Am. Chem. Soc., 1975, 97, 7037–7042 CrossRef CAS.
- J. van Houten and R. J. Watts, J. Am. Chem. Soc., 1976, 98, 4853–4858 CrossRef CAS.
- M. Kasha, Discuss. Faraday Soc., 1950, 9, 14–19 RSC.
- R. Englman and J. Jortner, Mol. Phys., 1970, 18, 145–164 CrossRef CAS.
- J. V. Caspar, E. M. Kober, B. P. Sullivan and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 630–632 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Phys. Chem., 1983, 87, 952–957 CrossRef CAS.
- J. van Houten and R. J. Watts, Inorg. Chem., 1978, 17, 3381–3385 CrossRef CAS.
- B. Durham, J. V. Caspar, J. K. Nagle and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4803–4810 CrossRef CAS.
- F. Barigelletti, A. Juris, V. Balzani, P. Belser and A. von Zelewsky, Inorg. Chem., 1983, 22, 3335–3339 CrossRef CAS.
- G. H. Allen, R. P. White, D. P. Rillema and T. J. Meyer, J. Am. Chem. Soc., 1984, 106, 2613–2620 CrossRef CAS.
- M. Maestri, N. Armaroli, V. Balzani, E. C. Constable and A. M. W. C. Thompson, Inorg. Chem., 1995, 34, 2759–2767 CrossRef CAS.
- M. Abrahamsson, M. Jäger, T. Österman, L. Eriksson, P. Persson, H.-C. Becker, O. Johansson and L. Hammarström, J. Am. Chem. Soc., 2006, 128, 12616–12617 CrossRef CAS PubMed.
- F. Schramm, V. Meded, H. Fliegl, K. Fink, O. Fuhr, Z. Qu, W. Klopper, S. Finn, T. E. Keyes and M. Ruben, Inorg. Chem., 2009, 48, 5677–5684 CrossRef CAS PubMed.
- A. Breivogel, C. Kreitner and K. Heinze, Eur. J. Inorg. Chem., 2014, 2014, 5468–5490 CrossRef CAS.
- R. D. Gerardi, N. W. Barnett and S. W. Lewis, Anal. Chim. Acta, 1999, 378, 1–41 CrossRef.
- S. Ji, W. Wu, W. Wu, P. Song, K. Han, Z. Wang, S. Liu, H. Guo and J. Zhao, J. Mater. Chem., 2010, 20, 1953–1963 RSC.
- F. G. Gao and A. J. Bard, J. Am. Chem. Soc., 2000, 122, 7426–7427 CrossRef CAS.
- H. J. Bolink, L. Cappelli, E. Coronado and P. Gaviña, Inorg. Chem., 2005, 44, 5966–5968 CrossRef CAS PubMed.
- H. J. Bolink, L. Cappelli, E. Coronado, M. Grätzel and M. K. Nazeeruddin, J. Am. Chem. Soc., 2006, 128, 46–47 CrossRef CAS PubMed.
- H. J. Bolink, L. Cappelli, E. Coronado, M. Grätzel, E. Ortí, R. D. Costa, P. M. Viruela and M. K. Nazeeruddin, J. Am. Chem. Soc., 2006, 128, 14786–14787 CrossRef CAS PubMed.
- X. Yang, D. Neher, D. Hertel and T. K. Däubler, Adv. Mater., 2004, 16, 161–166 CrossRef CAS.
- M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970–7977 CrossRef CAS PubMed.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638–655 RSC.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. de Cola, Chem. Mater., 2012, 24, 3684–3695 CrossRef CAS.
- P. G. Bomben, K. C. D. Robson, P. A. Sedach and C. P. Berlinguette, Inorg. Chem., 2009, 48, 9631–9643 CrossRef CAS PubMed.
- T. Funaki, H. Funakoshi, O. Kitao, N. Onozawa-Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Angew. Chem., 2012, 124, 7646–7649 ( Angew. Chem., Int. Ed. , 2012 , 51 , 7528–7531 ) CrossRef.
- C. Kreitner, A. K. Mengel, T. K. Lee, W. Cho, K. Char, Y. S. Kang and K. Heinze, Chem. – Eur. J., 2016 Search PubMed.
- M. Beley, J. P. Collin and J. P. Sauvage, Inorg. Chem., 1993, 32, 4539–4543 CrossRef CAS.
- C. Patoux, J.-P. Launay, M. Beley, S. Chodorowski-Kimmes, J.-P. Collin, S. James and J.-P. Sauvage, J. Am. Chem. Soc., 1998, 120, 3717–3725 CrossRef CAS.
- W.-W. Yang, J. Yao and Y.-W. Zhong, Organometallics, 2012, 31, 1035–1041 CrossRef CAS.
- C.-J. Yao, R.-H. Zheng, Q. Shi, Y.-W. Zhong and J. Yao, Chem. Commun., 2012, 48, 5680–5682 RSC.
- C.-J. Yao, H.-J. Nie, W.-W. Yang, J.-Y. Shao, J. Yao and Y.-W. Zhong, Chem. – Eur. J., 2014, 20, 17466–17477 CrossRef CAS PubMed.
- J.-J. Shen and Y.-W. Zhong, Sci. Rep., 2015, 5, 13835 CrossRef PubMed.
- Y.-W. Zhong, Z.-L. Gong, J.-Y. Shao and J. Yao, Coord. Chem. Rev., 2016, 312, 22–40 CrossRef CAS.
- B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Görls, S. Sinn, M. Thomas, S. Mai, J. Popp, B. Dietzek, L. González and U. S. Schubert, Chem. – Eur. J., 2012, 18, 4010–4025 CrossRef CAS PubMed.
- C. Kreitner, E. Erdmann, W. W. Seidel and K. Heinze, Inorg. Chem., 2015, 54, 11088–11104 CrossRef CAS PubMed.
- C. Kreitner and K. Heinze, Dalton Trans., 2016, 45, 5640–5658 RSC.
- A. C. Benniston, G. Chapman, A. Harriman, M. Mehrabi and C. A. Sams, Inorg. Chem., 2004, 43, 4227–4233 CrossRef CAS PubMed.
- L. A. Fredin, M. Pápai, E. Rozsályi, G. Vankó, K. Wärnmark, V. Sundström and P. Persson, J. Phys. Chem. Lett., 2014, 5, 2066–2071 CrossRef CAS PubMed.
- L. L. Jamula, A. M. Brown, D. Guo and J. K. McCusker, Inorg. Chem., 2014, 53, 15–17 CrossRef CAS PubMed.
- A. K. C. Mengel, C. Förster, A. Breivogel, K. Mack, J. R. Ochsmann, F. Laquai, V. Ksenofontov and K. Heinze, Chem. – Eur. J., 2015, 21, 704–714 CrossRef CAS PubMed.
- K. A. King and R. J. Watts, J. Am. Chem. Soc., 1987, 109, 1589–1590 CrossRef CAS.
- P. Reveco, J. H. Medley, A. R. Garber, N. S. Bhacca and J. Selbin, Inorg. Chem., 1985, 24, 1096–1099 CrossRef CAS.
- E. C. Constable and J. M. Holmes, J. Organomet. Chem., 1986, 301, 203–208 CrossRef CAS.
- M. L. Muro-Small, J. E. Yarnell, C. E. McCusker and F. N. Castellano, Eur. J. Inorg. Chem., 2012, 2012, 4004–4011 CrossRef CAS.
- C. D. Ertl, D. P. Ris, S. C. Meier, E. C. Constable, C. E. Housecroft, M. Neuburger and J. A. Zampese, Dalton Trans., 2015, 44, 1557–1570 RSC.
- P. G. Bomben, K. D. Thériault and C. P. Berlinguette, Eur. J. Inorg. Chem., 2011, 2011, 1806–1814 CrossRef.
- E. Y. Li, Y.-M. Cheng, C.-C. Hsu, P.-T. Chou, G.-H. Lee, I.-H. Lin, Y. Chi and C.-S. Liu, Inorg. Chem., 2006, 45, 8041–8051 CrossRef CAS PubMed.
- M. Beley, J. P. Collin, R. Louis, B. Metz and J. P. Sauvage, J. Am. Chem. Soc., 1991, 113, 8521–8522 CrossRef CAS.
- E. C. Constable, A. M. W. C. Thompson and S. Greulich, J. Chem. Soc., Chem. Commun., 1993, 1444–1446 RSC.
- P. G. Bomben, B. D. Koivisto and C. P. Berlinguette, Inorg. Chem., 2010, 49, 4960–4971 CrossRef CAS PubMed.
- H. Kisserwan, A. Kamar, T. Shoker and T. H. Ghaddar, Dalton Trans., 2012, 41, 10643–10651 RSC.
- Z. Ji, G. Natu, Z. Huang, O. Kokhan, X. Zhang and Y. Wu, J. Phys. Chem. C, 2012, 116, 16854–16863 CrossRef CAS.
- I. M. Dixon, F. Alary and J.-L. Heully, Dalton Trans., 2010, 39, 10959–10966 RSC.
- S. H. Wadman, M. Lutz, D. M. Tooke, A. L. Spek, F. Hartl, R. W. A. Havenith, G. P. M. van Klink and G. van Koten, Inorg. Chem., 2009, 48, 1887–1900 CrossRef CAS PubMed.
- F. Alary, J.-L. Heully, L. Bijeire and P. Vicendo, Inorg. Chem., 2007, 46, 3154–3165 CrossRef CAS PubMed.
- J.-L. Heully, F. Alary and M. Boggio-Pasqua, J. Chem. Phys., 2009, 131, 184308 CrossRef PubMed.
- T. Guillon, M. Boggio-Pasqua, F. Alary, J.-L. Heully, E. Lebon, P. Sutra and A. Igau, Inorg. Chem., 2010, 49, 8862–8872 CrossRef PubMed.
- R. D. Costa, E. Ortí, D. Tordera, A. Pertegás, H. J. Bolink, S. Graber, C. E. Housecroft, L. Sachno, M. Neuburger and E. C. Constable, Adv. Energy Mater., 2011, 1, 282–290 CrossRef CAS.
- F. Monti, A. Baschieri, I. Gualandi, J. J. Serrano-Pérez, J. M. Junquera-Hernández, D. Tonelli, A. Mazzanti, S. Muzzioli, S. Stagni, C. Roldan-Carmona, A. Pertegás, H. J. Bolink, E. Ortí, L. Sambri and N. Armaroli, Inorg. Chem., 2014, 53, 7709–7721 CrossRef CAS PubMed.
- S. Yi, J.-H. Kim, Y.-J. Cho, J. Lee, T.-S. Choi, D. W. Cho, C. Pac, W.-S. Han, H.-J. Son and S. O. Kang, Inorg. Chem., 2016, 55, 3324–3331 CrossRef CAS PubMed.
- I. Krivokapic, M. Zerara, M. L. Daku, A. Vargas, C. Enachescu, C. Ambrus, P. Tregenna-Piggott, N. Amstutz, E. Krausz and A. Hauser, Coord. Chem. Rev., 2007, 251, 364–378 CrossRef CAS.
- R. G. Miller, S. Narayanaswamy, J. L. Tallon and S. Brooker, New J. Chem., 2014, 38, 1932–1941 RSC.
- J. P. Claude and T. J. Meyer, J. Phys. Chem., 1995, 99, 51–54 CrossRef CAS.
- N. H. Damrauer, T. R. Boussie, M. Devenney and J. K. McCusker, J. Am. Chem. Soc., 1997, 119, 8253–8268 CrossRef CAS.
- J. V. Caspar and T. J. Meyer, J. Am. Chem. Soc., 1983, 105, 5583–5590 CrossRef CAS.
- K.-C. Hwang, J.-L. Chen, Y. Chi, C.-W. Lin, Y.-M. Cheng, G.-H. Lee, P.-T. Chou, S.-Y. Lin and C.-F. Shu, Inorg. Chem., 2008, 47, 3307–3317 CrossRef CAS PubMed.
- S. H. Wadman, J. M. Kroon, K. Bakker, R. W. A. Havenith, G. P. M. van Klink and G. van Koten, Organometallics, 2010, 29, 1569–1579 CrossRef CAS.
- B. Schulze, D. G. Brown, K. C. D. Robson, C. Friebe, M. Jäger, E. Birckner, C. P. Berlinguette and U. S. Schubert, Chem. – Eur. J., 2013, 19, 14171–14180 CrossRef CAS PubMed.
- C. Kreitner, M. Grabolle, U. Resch-Genger and K. Heinze, Inorg. Chem., 2014, 53, 12947–12961 CrossRef CAS PubMed.
- F. Barigelletti, P. Belser, A. von Zelewsky, A. Juris and V. Balzani, J. Phys. Chem., 1985, 89, 3680–3684 CrossRef CAS.
- A. J. Wilkinson, H. Puschmann, J. A. K. Howard, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2006, 45, 8685–8699 CrossRef CAS PubMed.
- G. S. M. Tong, K. T. Chan, X. Chang and C.-M. Che, Chem. Sci., 2015, 6, 3026–3037 RSC.
- A. Breivogel, C. Förster and K. Heinze, Inorg. Chem., 2010, 49, 7052–7056 CrossRef CAS PubMed.
- A. Breivogel, M. Meister, C. Förster, F. Laquai and K. Heinze, Chem. – Eur. J., 2013, 19, 13745–13760 CrossRef CAS PubMed.
- K. Heinze, K. Hempel and M. Beckmann, Eur. J. Inorg. Chem., 2006, 2006, 2040–2050 CrossRef.
- I. M. Dixon, F. Alary, M. Boggio-Pasqua and J.-L. Heully, Inorg. Chem., 2013, 52, 13369–13374 CrossRef CAS PubMed.
- I. M. Dixon, S. Khan, F. Alary, M. Boggio-Pasqua and J.-L. Heully, Dalton Trans., 2014, 43, 15898–15905 RSC.
- I. M. Dixon, F. Alary, M. Boggio-Pasqua and J.-L. Heully, Dalton Trans., 2015, 44, 13498–13503 RSC.
- J. M. Patrick, A. H. White, M. I. Bruce, M. J. Beatson, D. C. St. Black, G. B. Deacon and N. C. Thomas, J. Chem. Soc., Dalton Trans., 1983, 2121–2123 RSC.
- M. I. Bruce, M. J. Liddell, G. N. Pain, M. A. Bennett and H. Neumann, Inorg. Synth., 1989, 26, 171–180 CAS.
- K. C. D. Robson, B. D. Koivisto, A. Yella, B. Sporinova, M. K. Nazeeruddin, T. Baumgartner, M. Grätzel and C. P. Berlinguette, Inorg. Chem., 2011, 50, 5494–5508 CrossRef CAS PubMed.
- K. C. D. Robson, B. D. Koivisto, T. J. Gordon, T. Baumgartner and C. P. Berlinguette, Inorg. Chem., 2010, 49, 5335–5337 CrossRef CAS PubMed.
- K. C. D. Robson, B. Sporinova, B. D. Koivisto, E. Schott, D. G. Brown and C. P. Berlinguette, Inorg. Chem., 2011, 50, 6019–6028 CrossRef CAS PubMed.
- W. A. Herrmann and C. Köcher, Angew. Chem., 1997, 109, 2256–2282 ( Angew. Chem., Int. Ed. Engl. , 1997 , 36 , 2162–2187 ) CrossRef.
- Y.-M. Zhang, J.-Y. Shao, C.-J. Yao and Y.-W. Zhong, Dalton Trans., 2012, 41, 9280–9282 RSC.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- F. Neese, F. Wennmohs, A. Hansen and U. Becker, Chem. Phys., 2009, 356, 98–109 CrossRef CAS.
- R. Izsák and F. Neese, J. Chem. Phys., 2011, 135, 144105 CrossRef PubMed.
- D. A. Pantazis, X.-Y. Chen, C. R. Landis and F. Neese, J. Chem. Theory Comput., 2008, 4, 908–919 CrossRef CAS PubMed.
- S. Sinnecker, A. Rajendran, A. Klamt, M. Diedenhofen and F. Neese, J. Phys. Chem. A, 2006, 110, 2235–2245 CrossRef CAS PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: DFT optimized Cartesian coordinates of the 1GS and all relevant triplet states of complexes [1b]+, [4a]+ and [7a]+. See DOI: 10.1039/c6dt01989g |
| This journal is © The Royal Society of Chemistry 2016 |