
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Five different types of η8-cyclooctatetraenyl-lanthanide half-sandwich complexes from one ligand set, including a “giant neodymium wheel”†
Farid M.
Sroor
a,
Cristian G.
Hrib
b,
Phil
Liebing
b,
Liane
Hilfert
b,
Sabine
Busse
b and
Frank T.
Edelmann
*b
aOrganometallic and Organometalloid Chemistry Department, National Research Centre, 12622 Cairo, Egypt. E-mail: faridsroor@gmx.de
bChemisches Institut der Otto-von-Guericke-Universität Magdeburg, Universitätsplatz 2, D-39106 Magdeburg, Germany
First published on 28th June 2016
Abstract
The lithium-cyclopropylethynylamidinates Li[c-C3H5–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–C(NR)2] (1a: R = iPr, 1b: R = cyclohexyl (Cy)) have been used as precursors for the preparation of five new series of half-sandwich complexes. These complexes contain the large flat cyclooctatetraenyl ligand (C8H82−, commonly abbreviated as COT), and were isolated as solvated, unsolvated and inverse sandwich complexes. Treatment of the halide precursors [(COT)Pr(μ-Cl)(THF)2]2 with 1b and [(COT)Nd(μ-Cl)(THF)2]2 with 1a and 1b in THF in a 1
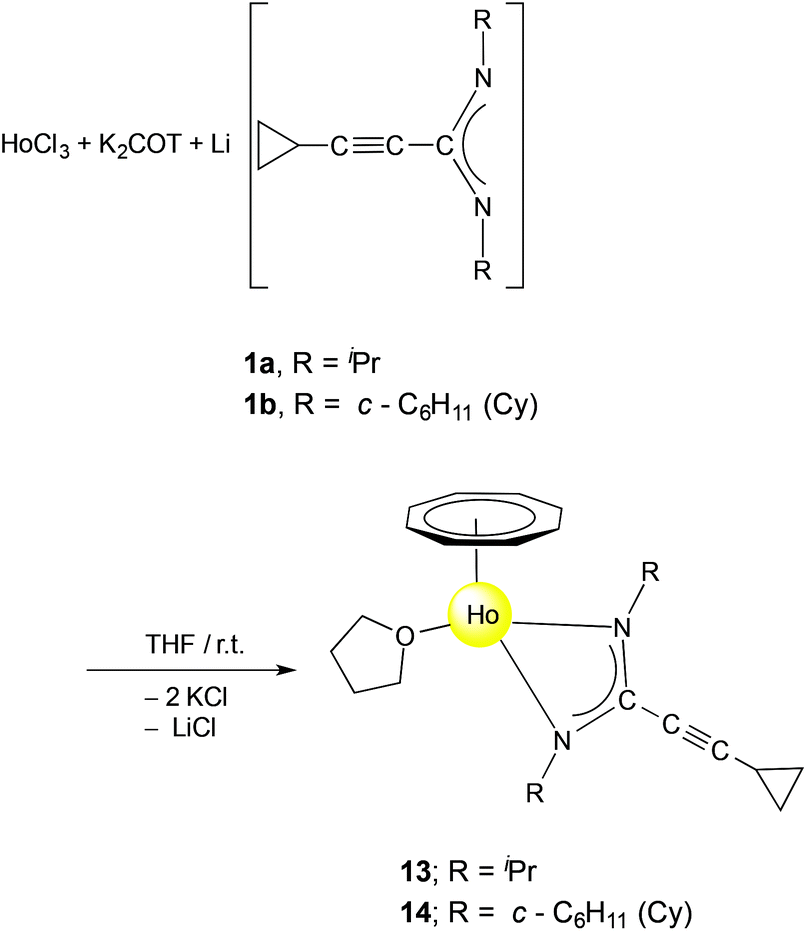
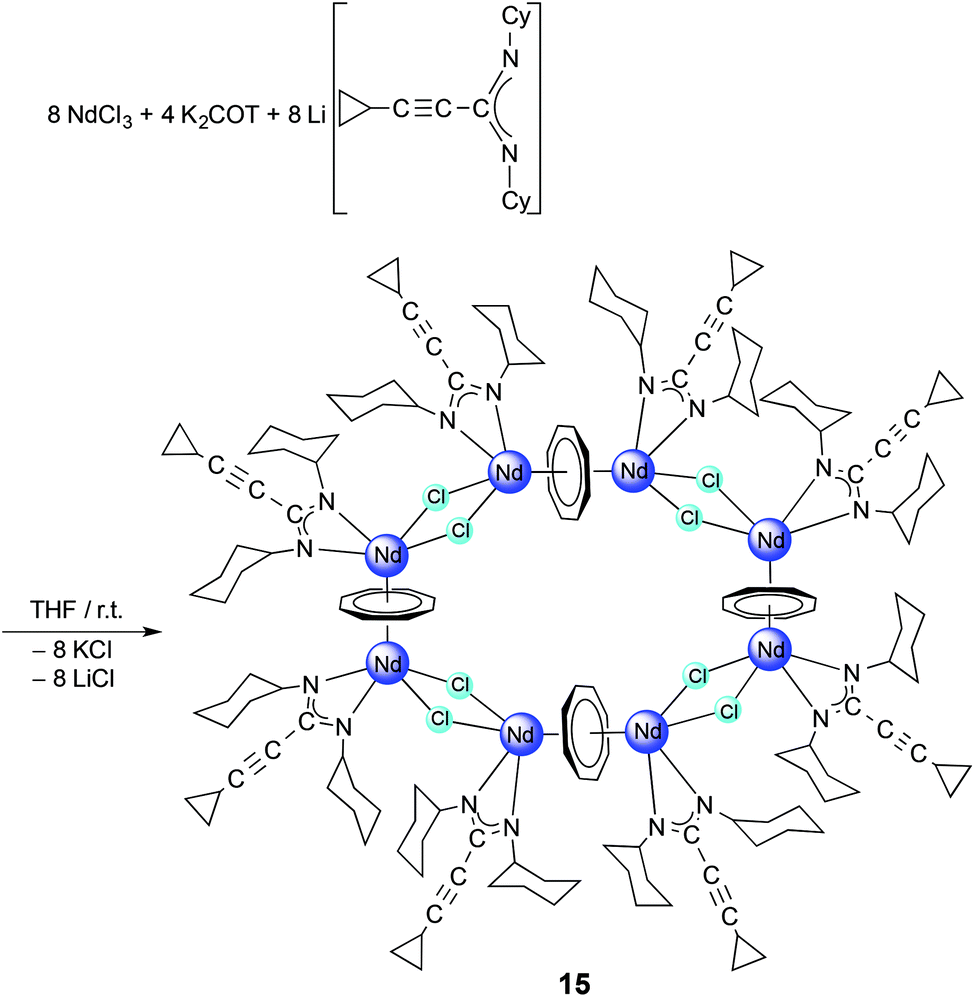
C–C(NR)2] (1a: R = iPr, 1b: R = cyclohexyl (Cy)) have been used as precursors for the preparation of five new series of half-sandwich complexes. These complexes contain the large flat cyclooctatetraenyl ligand (C8H82−, commonly abbreviated as COT), and were isolated as solvated, unsolvated and inverse sandwich complexes. Treatment of the halide precursors [(COT)Pr(μ-Cl)(THF)2]2 with 1b and [(COT)Nd(μ-Cl)(THF)2]2 with 1a and 1b in THF in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, respectively, afforded (COT)Ln[μ-c-C3H5–CC–C(NR)2]2Li(L) (2: Ln = Pr, R = Cy, L = Et2O; 3: Ln = Nd, R = iPr, L = THF; 4: Ln = Nd, R = Cy, L = THF). Treatment of the dimeric cerium(III) bis(cyclopropylethynylamidinate) complexes [{c-C3H5–CC–C(NR)2}2Ce(μ-Cl)(THF)]2 (5: R = iPr; 6: R = Cy) in situ with K2C8H8 in a 1:1 molar ratio in THF at room temperature afforded the inverse-sandwich complexes (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2 (7: R = iPr; 8: R = Cy). This reaction represents a new method for encapsulation of a planar (C8H8)2− ring in lanthanide complexes containing amidinate ligands in the outer decks. Novel unsolvated dinuclear lanthanide half-sandwich complexes were prepared by using the precursors 1a, 1b and COT2−. Unlike the complexes 2–4, the reaction of [(COT)Pr(μ-Cl)(THF)2]2 with 1a afforded the unsolvated centrosymmetric complex [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9). These dimeric structures could be also accessed by reaction of LnCl3 (Ln = Ce or Nd) with 1a or 1b and K2COT in a 1:1:1 molar ratio as a one-pot reaction to give novel [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 complexes (10: Ln = Ce, R = iPr; 11: Ln = Ce, R = Cy; 12: Ln = Nd, R = iPr). Similar treatment of HoCl3 with 1a or 1b and K2COT as three-component reactions in a 1:1:1 molar ratio afforded the solvated half-sandwich complexes (COT)Ho(c-C3H5–CC–C(NR)2)(THF) (13: R = iPr; 14: R = Cy). A unique multidecker sandwich complex [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15) was prepared by reaction of anhydrous NdCl3 with K2COT and 1b in a one-pot reaction. The solid state structure of 15 revealed the presence of an unprecedented macrocyclic sandwich compound (“giant neodymium wheel”) consisting of four COT rings sandwiched between eight Nd3+ ions, and each Nd3+ ion is bonded to one amidinate ligand and bridged by two chlorine atoms with the neighbouring Nd3+ ion.
:2 molar ratio, respectively, afforded (COT)Ln[μ-c-C3H5–CC–C(NR)2]2Li(L) (2: Ln = Pr, R = Cy, L = Et2O; 3: Ln = Nd, R = iPr, L = THF; 4: Ln = Nd, R = Cy, L = THF). Treatment of the dimeric cerium(III) bis(cyclopropylethynylamidinate) complexes [{c-C3H5–CC–C(NR)2}2Ce(μ-Cl)(THF)]2 (5: R = iPr; 6: R = Cy) in situ with K2C8H8 in a 1:1 molar ratio in THF at room temperature afforded the inverse-sandwich complexes (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2 (7: R = iPr; 8: R = Cy). This reaction represents a new method for encapsulation of a planar (C8H8)2− ring in lanthanide complexes containing amidinate ligands in the outer decks. Novel unsolvated dinuclear lanthanide half-sandwich complexes were prepared by using the precursors 1a, 1b and COT2−. Unlike the complexes 2–4, the reaction of [(COT)Pr(μ-Cl)(THF)2]2 with 1a afforded the unsolvated centrosymmetric complex [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9). These dimeric structures could be also accessed by reaction of LnCl3 (Ln = Ce or Nd) with 1a or 1b and K2COT in a 1:1:1 molar ratio as a one-pot reaction to give novel [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 complexes (10: Ln = Ce, R = iPr; 11: Ln = Ce, R = Cy; 12: Ln = Nd, R = iPr). Similar treatment of HoCl3 with 1a or 1b and K2COT as three-component reactions in a 1:1:1 molar ratio afforded the solvated half-sandwich complexes (COT)Ho(c-C3H5–CC–C(NR)2)(THF) (13: R = iPr; 14: R = Cy). A unique multidecker sandwich complex [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15) was prepared by reaction of anhydrous NdCl3 with K2COT and 1b in a one-pot reaction. The solid state structure of 15 revealed the presence of an unprecedented macrocyclic sandwich compound (“giant neodymium wheel”) consisting of four COT rings sandwiched between eight Nd3+ ions, and each Nd3+ ion is bonded to one amidinate ligand and bridged by two chlorine atoms with the neighbouring Nd3+ ion.
Introduction
In organolanthanide chemistry, steric saturation of the coordination sphere of the large rare-earth metal cations is generally more important than the electron count. Thus, the investigation of new spectator ligands which satisfy the coordination requirements of the lanthanides continues to be of significant current interest. Anionic amidinate ligands of the type [RC(NR′)2]− (R = H, alkyl, aryl; R′ = alkyl, cycloalkyl, aryl, SiMe3) are now well-established as highly useful and versatile spectator ligands in that respect. These readily available N-chelating ligands are generally regarded as steric cyclopentadienyl equivalents.1 In the case of rare-earth metals, mono-, di- and trisubstituted lanthanide amidinate and guanidinate complexes are all accessible, just like the mono-, di- and tricyclopentadienyl complexes. Over the past ca. 25 years, lanthanide amidinates have undergone an impressive transformation from laboratory curiosities to highly active homogeneous catalysts as well as valuable precursors in materials science. Numerous rare-earth metal amidinates have been reported to be very efficient homogeneous catalysts e.g. for ring-opening polymerization reactions of lactones, the guanylation of amines or the addition of terminal alkynes to carbodiimides.2 In materials science, certain homoleptic alkyl-substituted lanthanide tris(amidinate) complexes are often highly volatile and can be used as promising precursors for ALD (atomic layer deposition) and MOCVD (metal–organic chemical vapor deposition) processes, e.g. for the deposition of lanthanide oxide (Ln2O3) or lanthanide nitride (LnN) thin films.3The introduction of alkynyl groups to the central carbon atom in amidines leads to alkynylamidines of the type R–CC–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NR′)(NHR′). In organic synthesis, alkynylamidines have been frequently employed in the preparation of various heterocycles.4,5 More recently, alkynylamidines have attracted considerable attention due to their diverse applications in biological and pharmacological systems.6 Moreover, transition metal and lanthanide alkynylamidinate complexes have been shown to be efficient and versatile catalysts e.g. for C–C and C–N bond formation, the addition of C–H, N–H and P–H bonds to carbodiimides as well as ε-caprolactone polymerization.7 Thus far, only few lanthanide complexes containing alkynylamidinate ligands have been described.7,8 Previously used alkynylamidinate ligands include e.g. phenylethynyl derivatives [Ph–CC–C(NR)2]− (R = iPr, tBu)7a,8 and the trimethylsilylacetylene-derived anions [Me3Si–CC–C(NR)2]− (R = cyclohexyl (Cy), iPr).9
NR′)(NHR′). In organic synthesis, alkynylamidines have been frequently employed in the preparation of various heterocycles.4,5 More recently, alkynylamidines have attracted considerable attention due to their diverse applications in biological and pharmacological systems.6 Moreover, transition metal and lanthanide alkynylamidinate complexes have been shown to be efficient and versatile catalysts e.g. for C–C and C–N bond formation, the addition of C–H, N–H and P–H bonds to carbodiimides as well as ε-caprolactone polymerization.7 Thus far, only few lanthanide complexes containing alkynylamidinate ligands have been described.7,8 Previously used alkynylamidinate ligands include e.g. phenylethynyl derivatives [Ph–CC–C(NR)2]− (R = iPr, tBu)7a,8 and the trimethylsilylacetylene-derived anions [Me3Si–CC–C(NR)2]− (R = cyclohexyl (Cy), iPr).9
In the course of our ongoing investigation of lanthanide amidinates we recently initiated a study of alkynylamidinates derived from cyclopropylacetylene. The cyclopropyl group was chosen because of the well-known electron-donating ability of this substituent to an adjacent electron-deficient center.10 This would give us the rare chance to electronically influence the amidinate ligand system rather than altering only its steric demand. The resulting anions, [c-C3H5–CC–C(NR)2]− (R = iPr, Cy), represent a potentially useful addition to the current library of amidinate ligands. In a first contribution, we described the synthesis and full characterization of the lithium-cyclopropylethynylamidinates Li[c-C3H5–CC–C(NR)2] (1a: R = iPr, 1b: R = cyclohexyl (Cy)).11 These precursors are easily available on a large scale and in high yields using commercially available reagents (cyclopropylacetylene, n-butyllithium, N,N′-diorganocarbodiimides). In subsequent contributions we described the first trivalent rare-earth metal complexes comprising the new cyclopropylethynylamidinate ligands and their use as guanylation, hydroacetylenation, and hydroamination catalysts.12–14
On the other hand, the large, flat cyclooctatetraenyl ligand (C8H82−, commonly abbreviated as COT) is one of the carbocyclic ring systems which play an eminent role in organolanthanide chemistry for more than five decades. Streitwieser et al. reported the first anionic sandwich complexes of the type [Ln(COT)2]−,15 as well as the dimeric mono(cyclooctatetraenyl) lanthanide(III) chlorides, [(COT)Ln(μ-Cl)(THF)2]2 which are still important starting materials in the organolanthanide chemistry involving COT ligands.16 In 1995, Schumann and Edelmann et al. described a series of monomeric (cyclooctatetraenyl)lanthanide(III) benzamidinates of the type (C8H8)Ln[4-RC6H4C(NSiMe3)2](THF).17 Until now, these half-sandwich complexes remained the only organolanthanide compounds which combine COT and amidinate ligands in the coordination sphere of Ln3+ ions. We report here that the use of the recently discovered cyclopropylethynylamidinate ligands [c-C3H5–CC–C(NR)2]− (R = iPr, Cy) in combination with COT allows for the synthesis and full characterization of no less than five different types of (COT)Ln half-sandwich complexes.
Results and discussion
Synthesis and structure of (COT)Ln[μ-c-C3H5–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e002.gif) C–C(NR)2]2Li(L) (2–4)
C–C(NR)2]2Li(L) (2–4)
The starting materials for the present study, the lithium-cyclopropylethynylamidinates Li[c-C3H5–CC–C(NR)2] (1a: R = iPr, 1b: R = Cy) were prepared using the published straightforward protocol shown in Scheme 1.11In situ deprotonation of commercially available cyclopropylacetylene with nBuLi followed by treatment with either N,N′-diisopropylcarbodiimide or N,N′-dicyclohexylcarbodiimide afforded solutions of 1a or 1b which were used directly for subsequent reactions with rare-earth metal halide precursors.
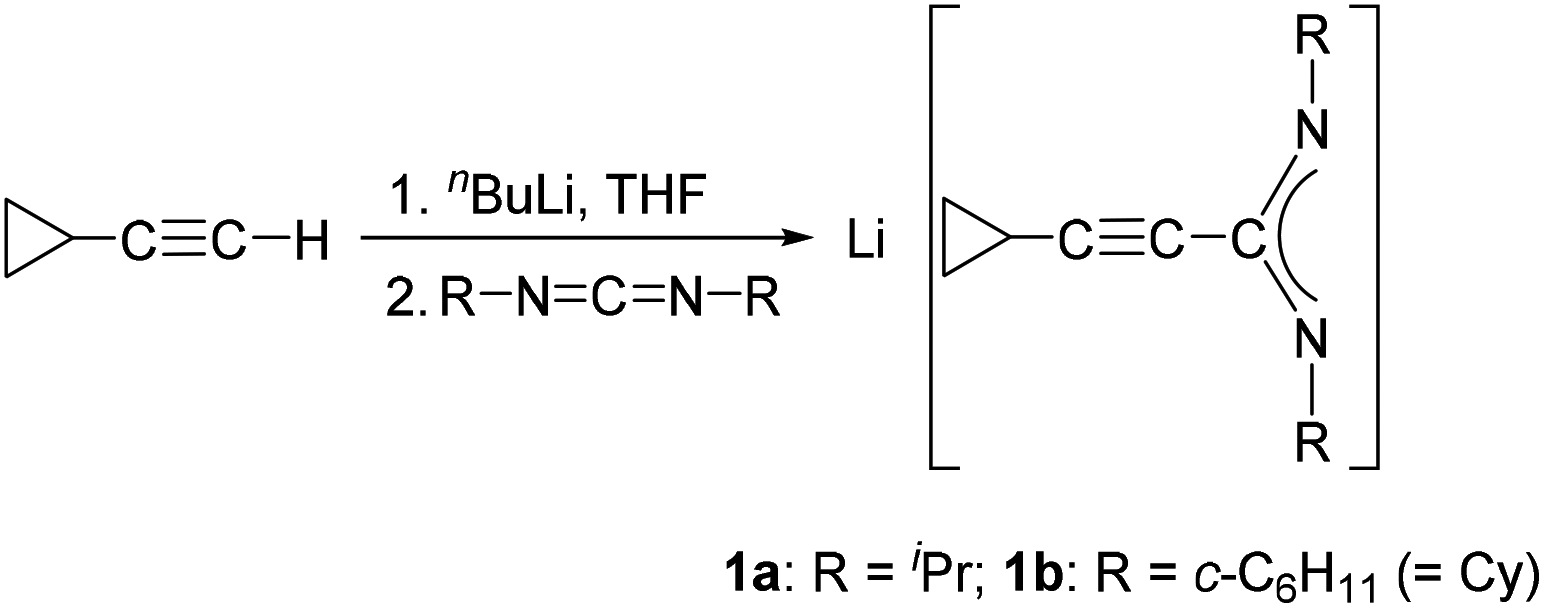 | ||
| Scheme 1 Synthesis of the lithium-cyclopropylethynylamidinates 1a and 1b. | ||
In a first set of experiments, new heterobimetallic (Li/Ln) (COT)Ln–amidinate half-sandwich complexes were accessed by treatment of selected [(COT)Ln(μ-Cl)(THF)2]2 derivatives with 1a and 1b. The starting materials [(COT)Ln(μ-Cl)(THF)2]2 (Ln = Pr, Nd) were prepared from anhydrous LnCl3 and K2COT according to the reported methods.16 Treating a solution of the halide precursor [(COT)Pr(μ-Cl)(THF)2]2 with 1b as well as [(COT)Nd(μ-Cl)(THF)2]2 with 1a and 1b in a 1:2 molar ratio, respectively, at room temperature afforded the new “ate” complexes (COT)Ln[μ-c-C3H5–CC–C(NR)2]2Li(L) (2: Ln = Pr, R = Cy, L = Et2O; 3: Ln = Nd, R = iPr, L = THF; 4: Ln = Nd, R = Cy, L = THF) according to Scheme 2. Compounds 2 and 4 were isolated as pale green crystals by extraction and recrystallization from n-pentane at 5 °C, while 3 was extracted with toluene and recrystallized as bright yellow crystals using diethyl ether (Et2O) at 5 °C. The isolated yields were good (3: 64%) to moderate (2: 53%, 4: 41%).
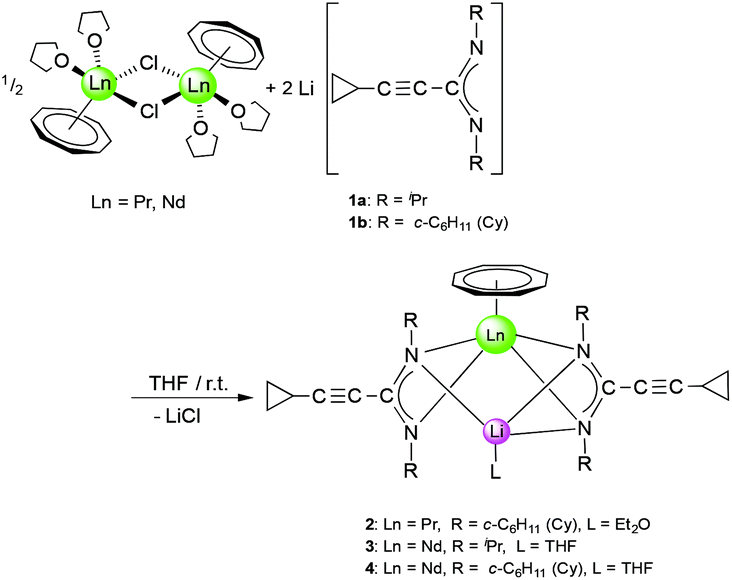 | ||
| Scheme 2 Synthesis of (COT)Ln[μ-c-C3H5–CC–C(NR)2]2Li(L) (2–4). | ||
All three compounds 2–4 were investigated by IR, mass spectra, elemental analysis, and NMR spectra. Crystals of 2 and 3 were found to be suitable for single-crystal X-ray diffraction. In the IR spectra, a strong band in the range of 2217–2226 cm−1 could be assigned to the CC stretching vibration,18 while bands in the range of 1593–1635 cm−1 can be attributed to the CN vibration in the NCN units of the amidinate moieties.19 All protons and carbons in the complexes 2 and 4 could be detected in the NMR spectra. Due to the paramagnetic nature of the Nd3+ ion, the NMR resonances of 3 could not be assigned. 2D experiments of 2 and 4 showed that the protons of η8-C8H8 ligand appear as multiplet in 2 at δ = 5.50–5.90 ppm, while in 4 they appear at high field as singlet at δ = −11.56 ppm.17 The CH protons of the cyclohexyl groups were observed at δ = 3.40 ppm in 2 and at δ = 32.80 ppm in 4. The signals of the cyclopropyl group are shifted to high field in 4 compared to those were observed in the lithium salt of amidinate 1b (δ = 7.56 ppm for CH, δ = 6.15 and 4.55 ppm for the CH2 groups).11 In the 13C NMR spectra, the signals of the COT ligand appear at δ = 128 ppm for 2, while for 4 they appear at δ = 161 ppm. The CH carbon signals of the cyclohexyl groups appear at a similar value at δ = 61 ppm in the spectra of both complexes 2 and 4. The molecular structures of the complexes 2 and 3 were verified by single-crystal X-ray diffraction. The molecular structures of 2 and 3 are shown in Fig. 1 and 2, respectively. Tables 1 and 2 summarize the crystallographic data for all new compounds.
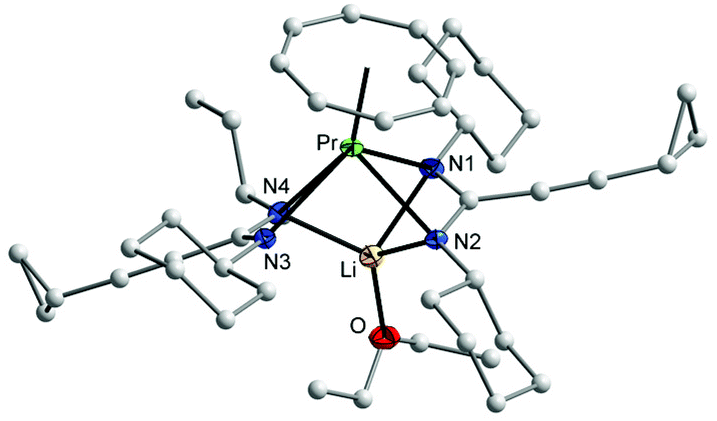 | ||
| Fig. 1 Molecular structure of (COT)Pr[μ-c-C3H5–CC–C(NCy)2]2Li(Et2O) (2) in the crystal. Ellipsoids of the heavier atoms and Li with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Pr–N(1) 2.771(2), Pr–N(2) 2.569(2), Pr–N(3) 2.595(2), Pr–N(4) 2.599(2), N(1)–Pr–N(2) 49.79(7), N(3)–Pr–N(4) 52.39(7), N(1)–Pr–N(3) 116.65(7), N(1)–Pr–N(4) 74.53(7), N(2)–Pr–N(3) 88.49(7), N(2)–Pr–N(4) 83.43(7), Pr–C(COT) 2.697(3)–2.743(3), Pr–centroid(COT) 2.016, Li–N(1) 2.152(6), Li–N(2) 2.130(6), Li–N(4) 2.106(5), N(1)–Li–N(2) 63.56(16), N(1)–Li–N(4) 99.7(2), N(2)–Li–N(4) 108.5(2), N(1)–Li–O 128.1(3), N(2)O–Li–O 114.9(2), N(4)–Li–O 125.5(3), N(1)–C(1) 1.326(3), N(2)–C(1) 1.338(3), N(3)–C(21) 1.305(3), N(4)–C(21) 1.362(4), N(1)–C(1)–N(2) 115.7(2), N(3)–C(21)–N(4) 118.5(2). | ||
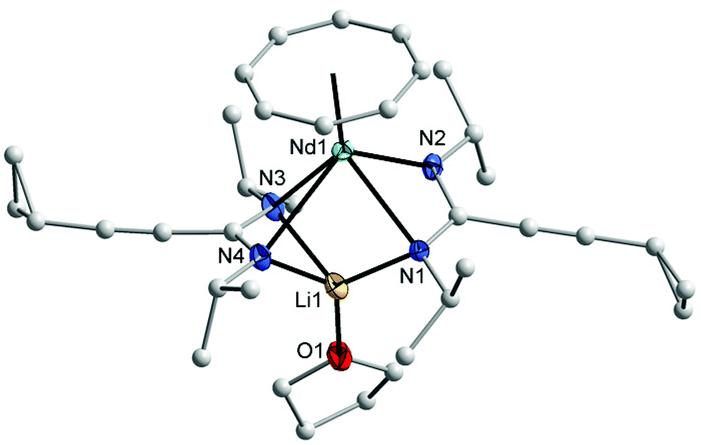 | ||
| Fig. 2 Molecular structure of (COT)Nd[μ-c-C3H5–CC–C(NiPr)2]2Li(THF) (3) in the crystal (molecule 1 of 2 in the asymmetric unit). Ellipsoids of the heavier atoms and Li with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Nd(1)–N(1) 2.603(4), Nd(1)–N(2) 2.555(5), Nd(1)–N(3) 2.549(5), Nd(1)–N(4) 2.771(5), Nd–C(COT) 2.684(7)–2.724(7), Nd–centroid(COT) 2.002, N(1)–Nd(1)–N(2) 52.3(1), N(3)–Nd(1)–N(4) 49.8(2), N(1)–Nd(1)–N(3) 84.6(2), N(1)–Nd(1)–N(4) 75.6(2), N(2)–Nd(1)–N(3) 82.7(2), N(2)–Nd(1)–N(4) 112.8(2), Li–N(1) 2.05(1), Li–N(3) 2.18(1), Li–N(4) 2.15(1), N(1)–Li–N(4) 103.4(6), N(3)–Li–N(4) 62.6(4), N(1)–Li–N(3) 110.4(5), N(1)–Li–O 121.1(7), N(4)–Li–O 130.6(7), N(3)–Li–O 114.0(6), N(1)–C(1) 1.319(8), N(2)–C(1) 1.331(7), N(3)–C(21) 1.315(8), N(4)–C(21) 1.335(8), N(1)–C(1)–N(2) 118.3(6), N(3)–C(21)–N(4) 116.1(6). | ||
| 2 | 3 | 7 | 8 | |
|---|---|---|---|---|
| Molecular formula | C48H72LiN4OPr | C36H54LiN4NdO | C56H84Ce2N8 | C80H116Ce2N8 |
| Formula wt. | 868.95 | 710.01 | 1149.55 | 1470.05 |
| Crystal size/mm3 | 0.46 × 0.35 × 0.14 | 0.36 × 0.31 × 0.16 | 0.57 × 0.18 × 0.17 | 0.48 × 0.34 × 0.33 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic | Monoclinic |
| Space group | Pbca | P212121 | C2/c | Pn |
| a/Å | 21.425(4) | 17.592(3) | 21.938(4) | 21.084(4) |
| b/Å | 20.113(4) | 20.156(4) | 17.320(4) | 13.758(3) |
| c/Å | 21.502(4) | 20.206(4) | 16.644(3) | 26.383(5) |
| α/° | 90 | 90 | 90 | 90 |
| β/° | 90 | 90 | 112.97(3) | 102.71(3) |
| γ/° | 90 | 90 | 90 | 90 |
| Cell volume Vc/Å3 | 9266(3) | 7165(2) | 5823(2) | 7465(3) |
| Molecules per cell z | 8 | 8 | 4 | 4 |
| ρ calc, Mg m−3 | 1.246 | 1.316 | 1.311 | 1.308 |
| μ/mm−1 | 1.089 | 1.481 | 1.584 | 1.251 |
| F 000 | 3664 | 2952 | 2368 | 3072 |
| Index ranges | −26 ≤ h ≤ 26 | −21 ≤ h ≤ 21 | −29 ≤ h ≤ 30 | −25 ≤ h ≤ 25 |
| −25 ≤ k ≤ 21 | −25 ≤ k ≤ 23 | −23 ≤ k ≤ 23 | −16 ≤ k ≤ 16 | |
| −24 ≤ l ≤ 26 | −23 ≤ l ≤ 25 | −21 ≤ l ≤ 22 | −32 ≤ l ≤ 32 | |
| Data/restraints/parameters | 9346/0/496 | 14633/0/775 |
7841/0/299 | 23075/2/1622 |
| GooF (F2) | 1.014 | 0.917 | 1.155 | 0.972 |
| R 1 (all data, I > 2σ(I)) | 0.0515, 0.0355 | 0.0641, 0.0442 | 0.0367, 0.0333 | 0.0324, 0.0268 |
| wR2 (all data, I > 2σ(I)) | 0.0839, 0.0790 | 0.0815, 0.0771 | 0.0821, 0.0806 | 0.0624, 0.0604 |
| Largest diff. peak and hole, e Å−3 | 0.887, −1.490 | 0.834, −1.068 | 1.347, −1.886 | 0.845, −0.748 |
| 10 | 11 | 12 | 14 | 15·6 PhMe | |
|---|---|---|---|---|---|
| Molecular formula | C40H54Ce2N4 | C52H70Ce2N4 | C40H54N4Nd2 | C30H43HoN2O | C190H264ClN16Nd8 (6·C7H8) |
| Formula wt. | 871.11 | 1031.36 | 879.35 | 612.59 | 4209.69 + 552.82 |
| Crystal size/mm3 | 0.28 × 0.08 × 0.06 | 0.14 × 0.11 × 0.03 | 0.12 × 0.10 × 0.06 | 0.44 × 0.38 × 0.22 | 0.18 × 0.12 × 0.11 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/n | P21/c | P21/n | C2/c |
| a/Å | 19.2388(2) | 14.8710(1) | 19.1172(1) | 12.872(3) | 44.794(9) |
| b/Å | 19.8763(2) | 20.0176(1) | 19.8054(1) | 14.491(3) | 21.617(4) |
| c/Å | 9.9758(1) | 15.5860(1) | 9.9578(1) | 15.056(3) | 32.326(7) |
| α/° | 90 | 90 | 90 | 90 | 90 |
| β/° | 99.8124(9) | 93.5556(6) | 99.550(1) | 100.37(3) | 125.03(3) |
| γ/° | 90 | 90 | 90 | 90 | 90 |
| Cell volume Vc/Å3 | 3758.90(6) | 4630.72(5) | 3718.01(5) | 2762(1) | 25631(9) |
| Molecules per cell z | 4 | 4 | 4 | 4 | 4 |
| ρ calc, Mg m−3 | 1.539 | 1.479 | 1.571 | 1.473 | 1.091 |
| μ/mm−1 | 18.699 | 15.273 | 21.293 | 2.888 | 1.712 |
| F 000 | 1752 | 2104 | 1768 | 1248 | 8528 |
| Index ranges | −24 ≤ h ≤ 24 | −18 ≤ h ≤ 18 | −24 ≤ h ≤ 24 | −17 ≤ h ≤ 17 | −55 ≤ h ≤ 56 |
| −24 ≤ k ≤ 24 | −23 ≤ k ≤ 25 | −24 ≤ k ≤ 24 | −19 ≤ k ≤ 19 | −26 ≤ k ≤ 27 | |
| −12 ≤ l ≤ 12 | −19 ≤ l ≤ 19 | −10 ≤ l ≤ 12 | −18 ≤ l ≤ 20 | −40 ≤ l ≤ 40 | |
| Data/restraints/parameters | 7809/54/441 | 9629/38/599 | 7724/48/443 | 7466/48/307 | 26581/0/1001 |
| GooF (F2) | 1.130 | 1.100 | 1.113 | 1.036 | 1.056 |
| R 1 (all data, I > 2σ(I)) | 0.0462, 0.0442 | 0.0407, 0.0379 | 0.0398, 0.0395 | 0.0461, 0.0362 | 0.0554, 0.0478 |
| wR2 (all data, I > 2σ(I)) | 0.1091, 0.1077 | 0.0939, 0.0921 | 0.0962, 0.0960 | 0.0881, 0.0843 | 0.1205 |
| Largest diff. peak and hole, e Å−3 | 1.229, −1.555 | 1.097, −1.122 | 2.307, −2.782 | 1.899, −2.651 | 2.533, −1.664 |
The crystal structures of 2 and 3 confirmed the presence of solvated half-sandwich complexes containing a COT ligand and two amidinate ligands, as well as a lithium ion coordinated by three nitrogen atoms of the amidinate ligands and one neutral ligand (Et2O in 2 and THF in 3). Complex 2 crystallizes from diethyl ether in the orthorhombic space group Pbca with one molecule in the asymmetric unit. The praseodymium ion has a pseudo-tetragonal pyramidal coordination sphere consisting of one η8-coordinated COT ring and four nitrogens of the amidinate ligands. The average Pr–N distance is 2.633(2) Å which is significantly longer than that found in [4-MeOC6H4C(NSiMe3)2]3Pr (2.487(4) Å)20 and (COT)Tm[C6H5C(NSiMe3)2](THF) (2.344(4) Å).17 The Pr–C distances to the η8-coordinated COT are within a range from 2.697(3) to 2.743(3) Å. The Pr–(COT ring-centroid) distance is 2.016 Å.21,22 The lithium ion is coordinated by three nitrogen atoms of the amidinate moieties with an average bond length of 2.129(5) Å, whereas the distance between the lithium atom and the fourth nitrogen atom Li–N3 is 3.435 Å, indicating the absence of a bonding interaction. The Li–O bond length is 1.894(5) Å. The N1–Pr–N2 and N3–Pr–N4 angles are 49.79(7)° and 52.39(7)°, respectively. The (COT ring-centroid)–Pr–Li angle is 161.1°. Despite the fact that complex 2 is insoluble in n-pentane, complex 3 was found to be soluble in n-pentane. Complex 3 crystallizes from n-pentane with two nearly identical crystallographically independent molecules in the orthorhombic space group P212121. Similar to 2, the neodymium ion in 3 has a pseudo-tetragonal-pyramidal coordination sphere consisting of one η8-coordinated COT ring and four nitrogen atoms of the amidinate ligands. The average Nd1–N bond length of 2.619 Å is significantly longer compared to that found in (COT)Nd[Ph2P(NSiMe3)2](THF) (2.473(3) Å).17 The bond length between the neodymium atom and the carbons of the η8-coordinated COT ring are in the range from 2.684(7) to 2.724(7) Å. The distance Nd1–(COT ring-centroid) is 2.002 Å, whereas in the second molecule it is 1.991 Å. The average bond length between lithium and the three nitrogen atoms of the amidinate ligands is 2.125(12) Å, whereas the distance between the lithium ion and the uncoordinated nitrogen atom Li1–N2 (Fig. 2) is 3.247 Å. The bond length Li1–O1 is 1.875(13) Å. The N1–Nd1–N2 and N3–Nd1–N4 angles are 52.32(14)° and 49.78(16)°, respectively, and the angle (COT ring centroid)–Nd–Li is 163.7°.21 In both complexes 2 and 3, the bond length of C1–N1 (1.326(3) Å in 2 and 1.319(8) Å in 3) and C1–N2 (1.338(3) in 2 and 1.331(7) Å in 3) indicate the negative charge delocalization within the NCN fragments. The bond angles of N1–C1–N2 unit are 115.7(2)° in 2 and 118.3(6)° in 3.
Synthesis and structure of (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2 (7, 8)
In the case of trivalent cerium, the combination of COT with cyclopropylethynylamidinate ligands in the coordination sphere of Ce3+ led to formation of novel inverse sandwich complexes. Inverse sandwich complexes containing a planar (C8H8)2− ring sandwiched between two rare-earth metal atoms are quite rare. A prominent early example was reported by Schumann et al. in 1993. The dinuclear samarium(III) complex (μ–η8:η8-COT)[Sm{N(SiMe3)2}2]2 (Scheme 3A) was prepared by treatment of the dimeric mono(COT) precursor [(COT)Sm(μ-Cl)(THF)2]2 with an excess (molar ratio 3:8) of NaN(SiMe3)2.22 A closely related divalent samarium complex, (μ–η8:η8-COT)[Sm{N(SiMe3)2}(THF)2]2 (Scheme 3B), was prepared by Evans et al. by reaction of [(Me3Si)2N]2Sm(THF)2, SmI2(THF)2 and K2C8H8.23 Encapsulation of a (C8H8)2− ring by twelve-membered Si4O6Li2 inorganic rings was found in the unusual complex (μ–η8:η8-COT)[Nd{(Ph2SiO)2O}2{Li(THF)2}{Li(THF)}]2 (Scheme 3C).24
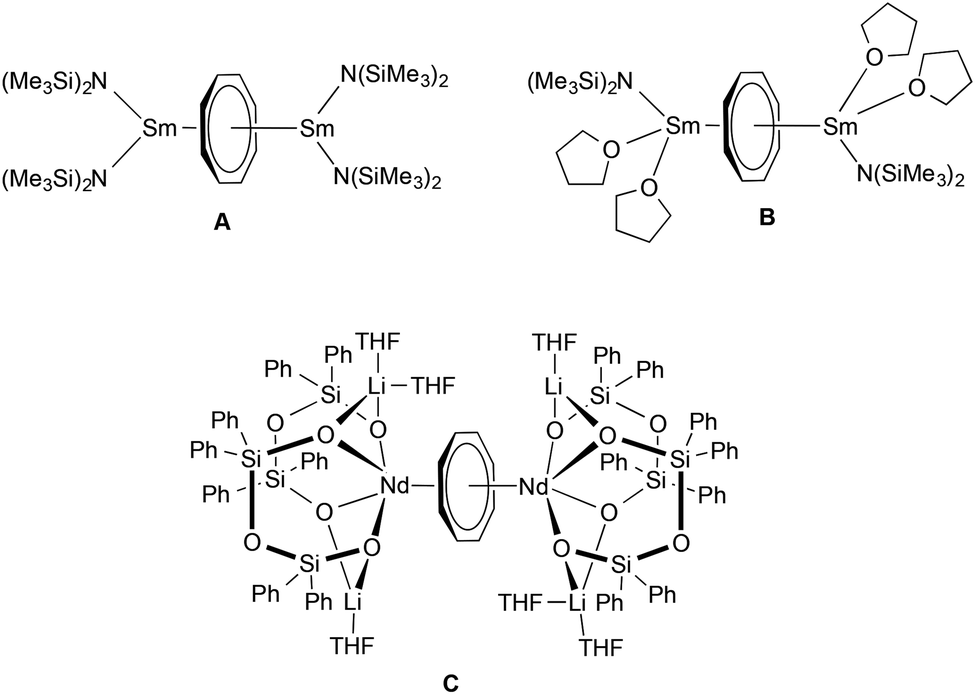 | ||
| Scheme 3 Previously reported inverse sandwich complexes of rare-earth elements. | ||
An unprecedented synthetic route to unsolvated inverse sandwich bimetallic Ln(COT) complexes has now been discovered in the course of the present study. The complexes (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2 (R = iPr or Cy) were prepared by treatment of the dimers 5 and 6, respectively, with K2C8H8 in a 1:1 molar ratio at room temperature to afford (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2 (7: R = iPr; 8: R = Cy) as illustrated in Scheme 4. Both compounds 7 and 8 were extracted using n-pentane affording bright yellow, exceedingly air-sensitive crystals at 5 °C in 45% (7) and 49% (8) yields. The spectroscopic data and elemental analysis were consistent with the structures. Both complexes 7 and 8 were structurally characterized by single-crystal X-ray diffraction.
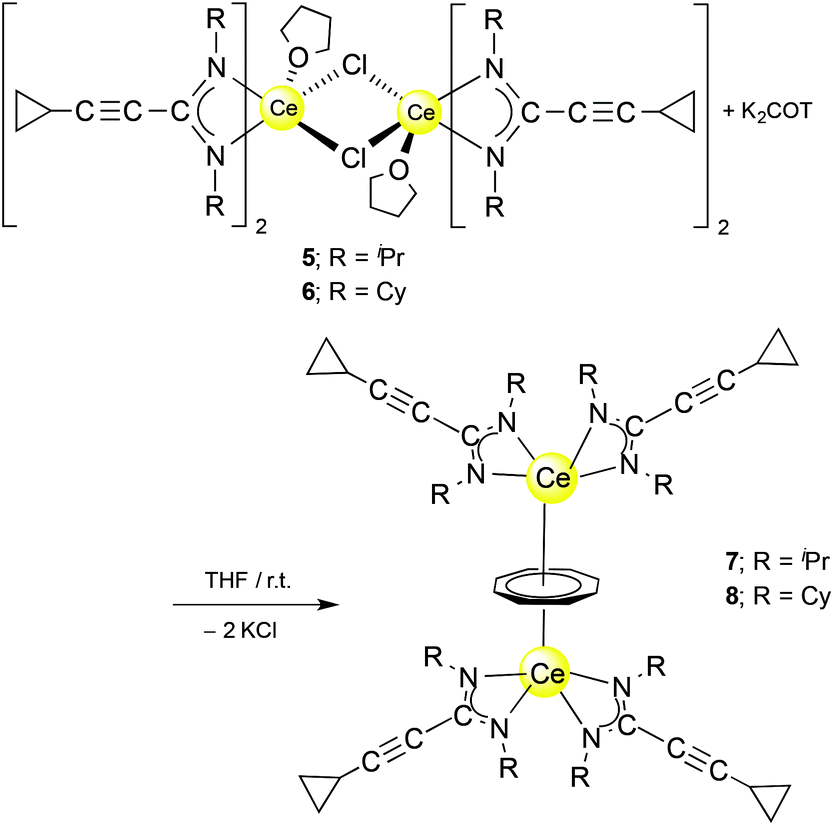 | ||
| Scheme 4 Synthesis of (μ–η8:η8-COT)[Ce(c-C3H5–CC–C(NR)2)2]2 (7, 8). | ||
In the 1H NMR spectra of 7 and 8, the influence of the paramagnetism of the Ce3+ ion on the protons of COT and the amidinate ligands is evident. Thus, the C8H8 protons in THF-d8 solution show a chemical shift of δ = 1.15 ppm in 7 and at δ = 0.91 ppm in 8. The CH protons of isopropyl groups in 7 appear at δ = 10.01 ppm, likewise, the CH protons of cyclohexyl groups in 8 appears at δ = 9.70 ppm. The CH protons of cyclopropyl groups were observed in the range δ = 0.81–1.04 and 1.27–1.35 ppm in 1a and 1b, respectively, and were found to appear at δ = 3.15 and 3.21 ppm in 7 and 8, respectively. The carbon signals of the COT ligand are observed at δ = 107.7 ppm in 7 and at δ = 104.1 ppm in 8. The CH carbons of the isopropyl groups are found at δ = 50 ppm in 1a, whereas they are observed at δ = 58 ppm in 7. Likewise, the CH carbons in cyclohexyl groups found at δ = 59 ppm in 1b, whereas they are observed at δ = 67 ppm in 8. Single-crystals of both 7 and 8 were found to be suitable for single-crystal X-ray diffraction. These were obtained by cooling a saturated n-pentane solution at 5 °C. The compounds 7 and 8 crystallize in the monoclinic space groups C2/c and Pn with one molecule of 7 and two molecules of 8 in the unit cell (cf.Tables 1 and 2). The crystal structure determinations of 7 and 8 confirmed the presence of unsolvated inverse sandwich structures in which a COT ligand is sandwiched between two trivalent cerium ions, and each of the cerium ions is attached to two bidentate amidinate ligands as shown in Fig. 3 and 4. The coordination geometry around the cerium atoms can be described as distorted pseudo-tetragonal pyramidal. The Ce–C(COT) distances range from 2.862(3) to 2.905(3) Å in 7 and from 2.872(4) to 2.908(4) Å in 8. These values are well comparable to those found in (μ–η8:η8-COT)[Sm{N(SiMe3)2}2]2 (2.798(5) to 2.857(5) Å)22 and in (μ–η8:η8-COT)[Sm{N(SiMe3)2}(THF)2]2 (2.863(2) to 2.929(2) Å).23 In 7, the bond lengths Ce1–(COT ring-centroid) and Ce2–(COT ring-centroid) are 2.220 and 2.244 Å, respectively.23,24 Due to the symmetry found in the complex 7, the bond lengths of Ce1–N1 and Ce1–N1A have the same value of 2.521(2) Å, and likewise Ce1–N2 and Ce1–N2A are 2.478(2) Å. Similarly, the distances Ce2–N3, Ce2–N3A are 2.530(2) Å and Ce2–N4, Ce2–N4A are 2.453(2) Å (Fig. 5 (left)). The Ce–N bond lengths are in good agreement with those found in complex 5. As illustrated in Fig. 5 (left), the distance between Ce1 and Ce2 is 4.465 Å. The Ce1–(COT ring centroid)–Ce2 angle is 100.0°. The bond lengths N1–C1 and N2–C1 are 1.327(4) Å and 1.331(4), respectively, indicating negative charge delocalization within the NCN units.
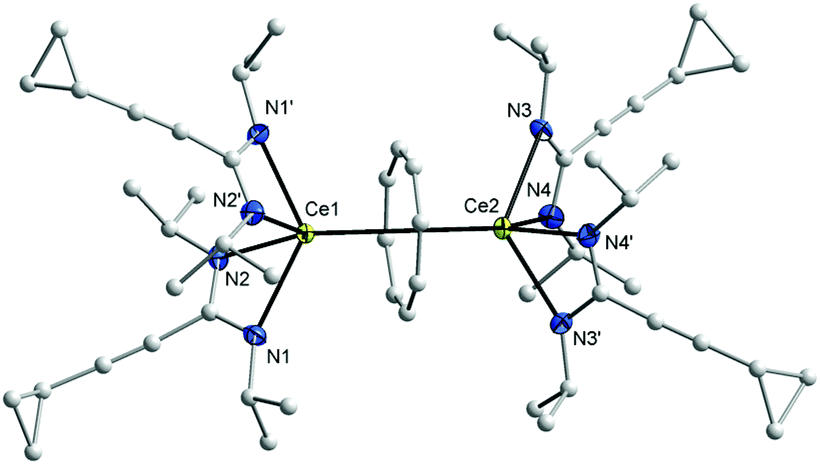 | ||
| Fig. 3 Molecular structure of (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NiPr)2}2]2 (7) in the crystal. Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ce(1)–N(1) 2.521(2), Ce(1)–N(2) 2.478(2), Ce(2)–N(3) 2.530(2), Ce(2)–N(4) 2.453(2), N(1)–Ce(1)–N(2) 54.03(8), N(1)–Ce(1)–N(1)′ 128.3(1), N(1)–Ce(1)–N(2)′ 91.9(1), N(2)–Ce(1)–N(2)′ 101.0(1), N(3)–Ce(2)–N(4) 54.25(8), N(3)–Ce(2)–N(3)′ 128.1(1), N(3)–Ce(2)–N(4)′ 92.2(1), N(4)–Ce(2)–N(4)′ 102.9(1), Ce(1)–C(μ-COT) 2.862(3)–2.888(3), Ce(1)–centroid(μ-COT) 2.220, Ce(2)–C(μ-COT) 2.882(3)–2.905(3), Ce(2)–centroid(μ-COT) 2.244, N(1)–C(1) 1.327(4), N(2)–C(1) 1.331(4), N(3)–C(13) 1.331(4), N(4)–C(13) 1.331(4), N(1)–C(1)–N(2) 117.4(2), N(3)–C(13)–N(4) 117.3(2). Symmetry operator to generate equivalent atoms: ′ 1 − x, y, 1.5 − z. | ||
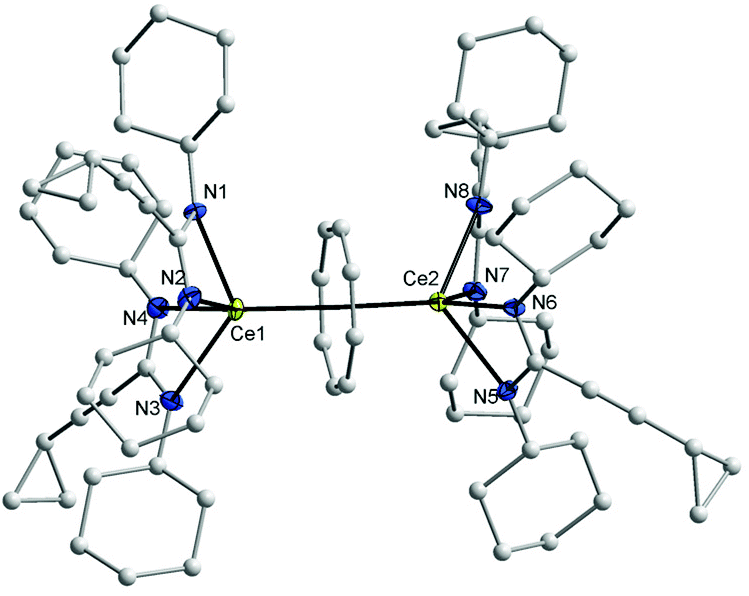 | ||
| Fig. 4 Molecular structure of (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NCy)2}2]2 (8) in the crystal (molecule 1 of 2 in the asymmetric unit). Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ce(1)–N(1) 2.504(4), Ce(1)–N(2) 2.470(4), Ce(1)–N(3) 2.528(4), Ce(1)–N(4) 2.438(4), Ce(2)–N(5) 2.484(3), Ce(2)–N(6) 2.527(4), Ce(2)–N(7) 2.504(4), Ce(2)–N(8) 2.502(4), Ce(1)–C(μ-COT) 2.872(4)–2.950(4), Ce(1)–centroid (μ-COT) 2.247, Ce(2)–C(μ-COT) 2.852(4)–2.905(4) Ce(2)–centroid(μ-COT) 2.229, N(1)–Ce(1)–N(2) 53.9(1), N(3)–Ce(1)–N(4) 54.5(1), N(1)–Ce(1)–N(3) 125.8(1), N(1)–Ce(1)–N(4) 92.2(1), N(2)–Ce(1)–N(3) 96.7(1), N(2)–Ce(1)–N(4) 112.2(1), N(5)–Ce(2)–N(6), 53.9(1), N(7)–Ce(2)–N(8) 54.0(1), N(5)–Ce(1)–N(7) 94.6(1), N(5)–Ce(1)–N(8) 121.3(1), N(6)–Ce(1)–N(7) 121.0(1), N(6)–Ce(1)–N(8) 97.6(1), N(1)–C(1) 1.290(6), N(2)–C(1) 1.336(6), N(3)–C(19) 1.333(6), N(4)–C(19) 1.323(6), N(5)–C(37) 1.329(5), N(6)–C(37) 1.324(5), N(7)–C(55) 1.331(6), N(8)–C(55) 1.334(6), N(1)–C(1)–N(2) 118.3(4), N(3)–C(19)–N(4) 117.8(4), N(5)–C(37)–N(6) 117.8(4), N(7)–C(55)–N(8) 117.0(4). | ||
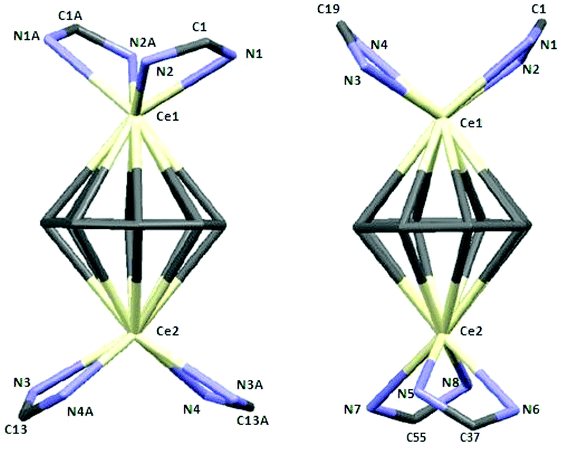 | ||
| Fig. 5 Capped-sticks views of the unit {(μ–η8:η8-COT)[Ce{C(N)2}2]2} of 7 (left) and 8 (right). | ||
In compound 8, the bond lengths of Ce1–(COT ring centroid) and Ce2–(COT ring centroid) are 2.242 and 2.234 Å, respectively.23,24 The Ce–N bond lengths are in the range from 2.438(4) to 2.528(4) Å, which is in good agreement with those found in compound 6. Complex 8 has a somewhat different symmetry than that found in complex 7 as illustrated in Fig. 5 (right). The distance between Ce1 and Ce2 is 4.511 Å. The angle Ce1–(COT ring-centroid)–Ce2 is 177.9°, whereas in the second molecule the Ce3–(COT ring-centroid)–Ce4 angle is 178.7°. In both complexes 7 and 8 no agostic interaction has been observed between Ce3+ and the outer amidinate ligands, although such interaction was found in (μ–η8:η8-COT)[Sm{N(SiMe3)2}2]2.22
Synthesis and structure of [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 complexes
The preparation of unsolvated half-sandwich complexes containing COT ligands is not always straightforward. In the course of the present study we discovered that unsolvated complexes of the type [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 can be accessed via two complementary synthetic routes. Unlike the reaction of [(COT)Pr(μ-Cl)(THF)2]2 with 1b, which afforded a solvated complex as shown in Scheme 2, treatment of [(COT)Pr(μ-Cl)(THF)2]2 with 2 equiv. of 1a in THF at room temperature afforded the unusual complex [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9) in 47% yield as shown in Scheme 5.
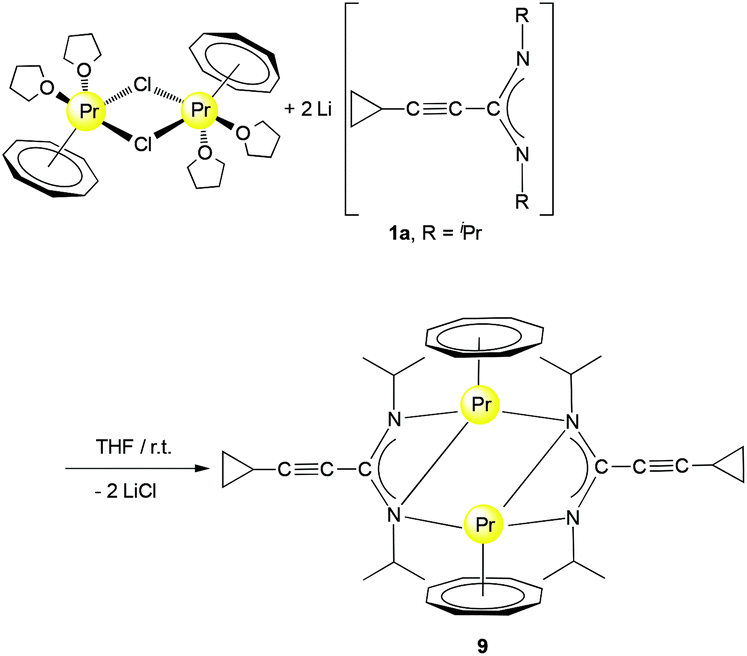 | ||
| Scheme 5 Synthesis of [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9). | ||
The new unsolvated binuclear half-sandwich complex 9 has been fully characterized by spectroscopic and elemental analysis studies. Only on one occasion the well-formed crystals of 9 obtained from a saturated solution in toluene could be successfully subjected to X-ray diffraction, which provided the structure of 9 as a dimer as illustrated in Scheme 5. Unfortunately, the crystal quality was too poor to allow full refinement of the crystal structure. The NMR spectra in toluene-d8 clearly indicated the absence of coordinated THF in the unsolvated half-sandwich complex 9 as shown in Fig. 6. The CH protons of the isopropyl groups are shifted to δ = 10.57 ppm, which can be attributed to the paramagnetic nature of the Pr3+ ion. The singlet of η8-C8H8 is shifted to high magnetic field and is observed at δ = −4.63 ppm. Likewise, the CH3 protons which were observed at δ = 0.65 ppm in 1a, are strongly shifted to higher magnetic field at δ = −10.24 ppm.17,25 All proton signals of the cyclopropyl groups show a marked downfield shift as compared to 1a. The CH protons of the c-C3H5 groups were found at a chemical shift δ = 0.81–1.04 ppm in 1a, while they were observed at δ = 1.94 ppm in 9. Likewise, the CH2 protons were observed at δ = 0.34–0.49 and 0.28–0.32 ppm in 1a and at δ = 1.70 and 1.22 ppm in 9.
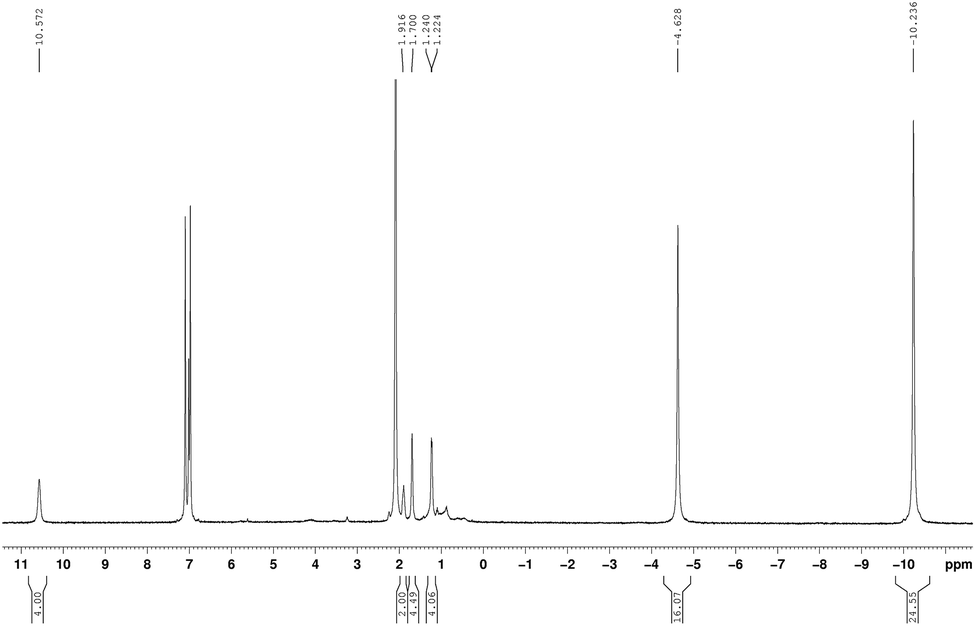 | ||
| Fig. 6
1H NMR spectrum (toluene-d8, 25 °C) of [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9). | ||
The 13C NMR and HSQC spectra of a 9 are shown in Fig. 7. The influence of the paramagnetism of the Pr3+ ion on the carbons of complex 9 is evident. The COT carbons exhibit a chemical shift of δ = 186.1 ppm, and the CH carbons of the isopropyl groups are observed at δ = 33.5 ppm, while the CH3 carbons are observed at δ = 15.6 ppm.
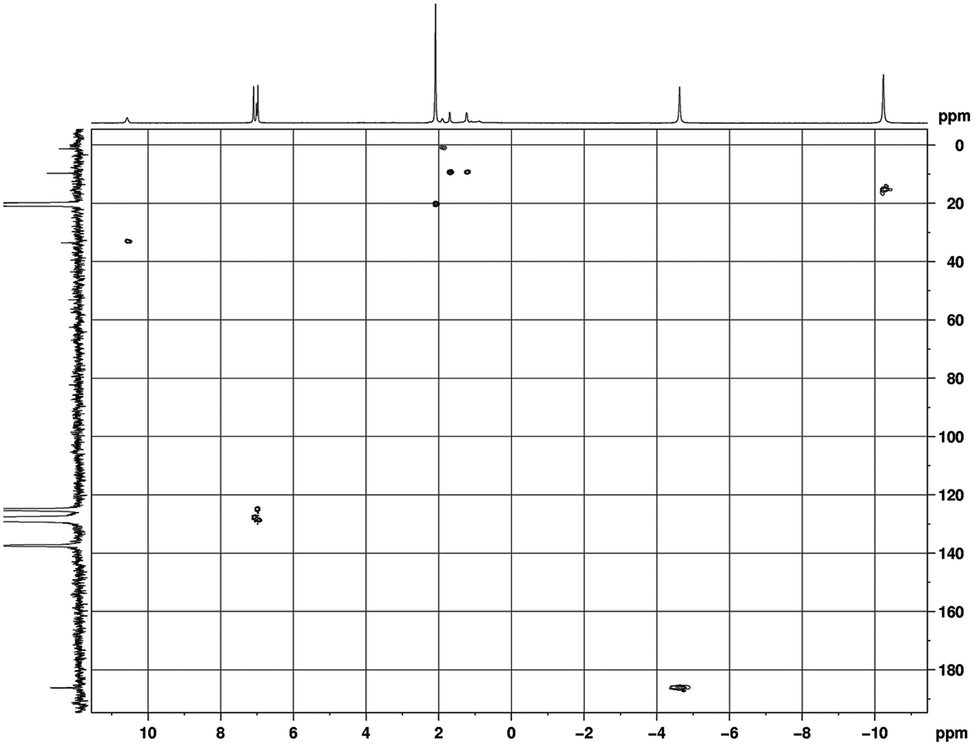 | ||
| Fig. 7 HSQC (H,C-correlation via1J (C, H)) spectrum in (toluene-d8, 25 °C) of [(COT)Pr(μ-c-C3H5–CC–C(NiPr)2)]2 (9). | ||
In the course of this work, novel unsolvated lanthanide half-sandwich complexes like compound 9 have also been prepared by reaction of anhydrous LnCl3 with K2COT and 1a or 1bvia a straightforward one-pot synthetic protocol. A mixture of the cyclopropylethynylamidinates 1a or 1b and K2COT, dissolved in THF, was added to a suspension of LnCl3 (Ln = Ce or Nd) in THF in a 1:1:1 molar ratio as illustrated in Scheme 6. The reaction mixture was stirred for 12 h at room temperature. After evaporation of the solvent, the product was extracted by using toluene to give the novel complexes [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 (10: Ln = Ce, R = iPr; 11: Ln = Ce, R = Cy; 12: L = Nd, R = iPr). Saturated solutions of compounds 10–12 in toluene were kept at 5 °C affording 10 as dark-green crystals (57% yield), 11 as green crystals (17% yield) and 12 as green crystals (43% yield). The unsolvated complexes are readily soluble in THF, Et2O or toluene and insoluble in n-pentane. The new complexes 10–12 have been fully characterized by elemental analysis, spectroscopic methods and single-crystal X-ray diffraction.
 | ||
| Scheme 6 Synthetic route to [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 (10: Ln = Ce, R = iPr; 11: Ln = Ce, R = Cy; 12: L = Nd, R = iPr). | ||
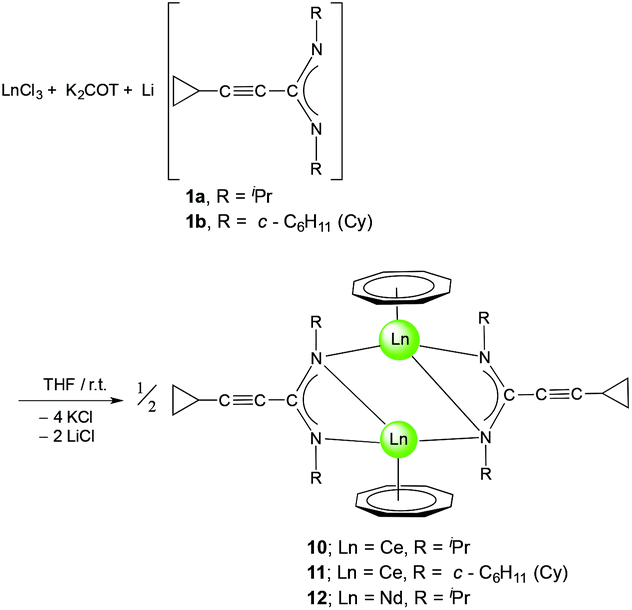
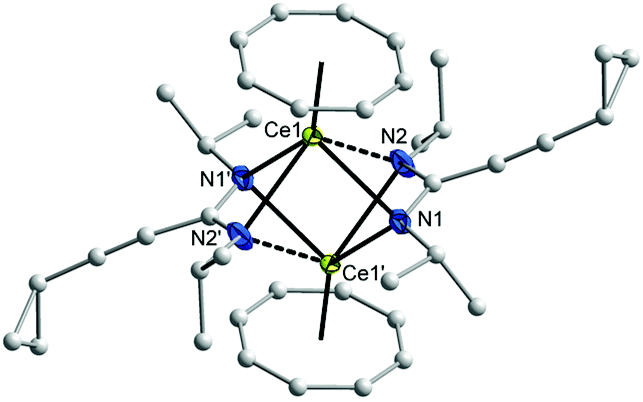
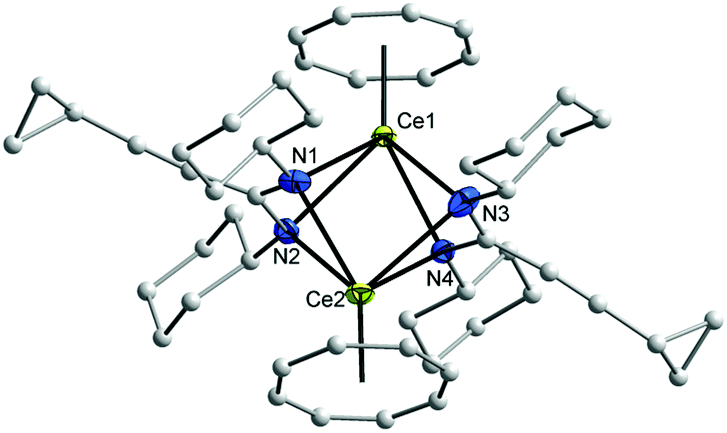
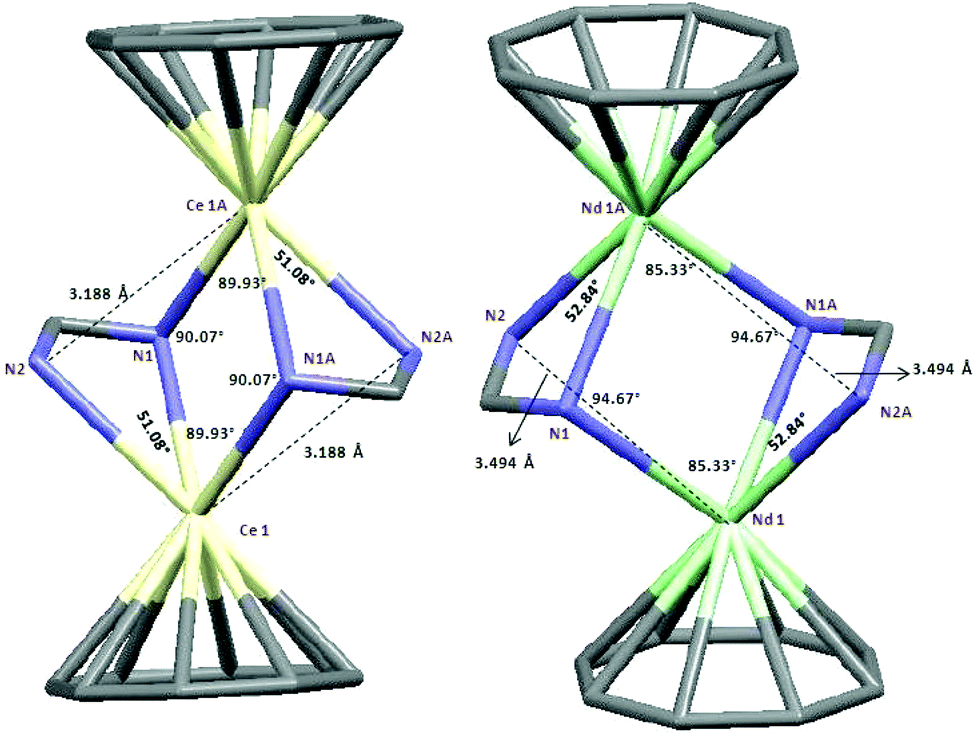
The 1H and 13C NMR spectra of 10–12 indicated the presence of one COT and one cyclopropylethynylamidinate ligand per Ln atom. The protons of η8-C8H8 were observed at δ = 0.91–1.53 and 0.93–1.87 ppm in 10 and 11, respectively,17 whereas the η8-C8H8 protons in 12 appear as singlet at δ = −11.75 ppm.17,26 The COT carbons appear at δ = 108.6 and 115.3 ppm in 10 and 11, respectively, while they appear at δ = 132.7 ppm in 12.27–29 Suitable single-crystals of 10, 11 and 12 for X-ray diffraction studies were obtained from saturated solutions in toluene at 5 °C. The compounds 10 and 12 crystallize in the monoclinic space group P21/c and 11 in the monoclinic space group P21/n (cf.Tables 1 and 2). Compounds 10 and 12 were found have two molecules in the asymmetric unit, whereas 11 was found to crystallize with one molecule in the asymmetric unit. The crystal structures of 10 and 12 showed a centrosymmetric dimeric structure in which each lanthanide ion is coordinated to one η8-COT ring and three nitrogens of the two amidinate ligands. The coordination geometry around the cerium or neodymium atoms in 10 and 12 can be described as distorted pseudo-tetrahedral as shown in Fig. 8 and 10. Unlike the complexes 10 and 12, the X-ray diffraction study of 11 showed that the cerium atom in 11 is coordinated to one η8-COT ring and four nitrogen atoms of the two amidinate ligands. Thus, the coordination geometry around the cerium atom in 11 can be described as distorted pseudo-tetragonal-pyramidal as shown in Fig. 9. In 10, the Ce–C(COT) distances range from 2.694(6) to 2.713(6) Å (average 2.704 Å) in good agreement with (μ–η8:η8-COT)[Sm{N(SiMe3)2}2]2 (2.798(5) to 2.857(5) Å)22 and with (μ–η8:η8-COT)[Sm{N(SiMe3)2}(THF)2]2 (2.863(2) to 2.929(2) Å).23 The distances Ce–(COT ring-centroid) have the same value of 1.992 Å. The Ce–N bond lengths are in the range between 2.625(5) and 2.647(5) Å (average 2.641 Å), whereas the distance of Ce–N2 (N2 is the fourth nitrogen atom which is not attached to the cerium atom) is 3.188 Å as illustrated in Fig. 11 (left). The Ce⋯Ce distance is 4.219 Å. The C1–N1 and C1–N2 bond lengths are 1.350(8) and 1.321(8) Å, respectively, indicating negative charge delocalization within the NCN fragments. The N–Ce–N and Ce–N–Ce bond angles are collected in Fig. 11 (left). Interestingly, compound 10 is a centrosymmetric dimer of the type [(COT)LnL]2 with a planar four-membered Ce1N1Ce1′N1′ ring as the central structural unit with angles of 90.07° (Ce–N–Ce) and 89.93° (N–Ce–N), so that the Ce2N2 moiety has a slightly rhomb-shaped geometry. The Nd–C(COT) distances in 12 are in the range between 2.658(5) to 2.679(5) Å (average 2.670 Å), in good agreement with the 2.852(3) to 2.928(3) Å range found in (μ–η8:η8-COT)[Nd{(Ph2SiO)2O}2{Li(THF)2}{Li(THF)}]2.24 Similar to 10, the Nd–(COT ring-centroid) distances have values of 1.936 Å and 1.949 Å. The Nd–N bond lengths range from 2.570(4) to 2.610(4) Å (average 2.587 Å), while the distance Nd–N2 (N2 is the fourth nitrogen atom which is not attached to the neodymium atom) is 3.494 Å (Fig. 11 (right)). The Nd⋯Nd distance is 3.817 Å. The N–Nd–N and Nd–N–Nd bond angles in 12 are collected in Fig. 11 (right). Similar to the structure of 10, dimeric 12 has a planar four-membered Nd1N1Nd1′N1′ ring as the central structural unit with angles of 94.67° (Nd–N–Nd) and 85.33° (N–Nd–N) and a rhomb-shaped Nd2N2 unit. The torsion angles of (COT ring-centroid)–Ce–Ce–(COT ring-centroid) and (COT ring centroid)–Nd–Nd–(COT ring-centroid) in both complexes 10 and 12 are 180.0°.
 | ||
| Fig. 8 Molecular structure of [(COT)Ce(μ-c-C3H5–CC–C(NiPr)2)]2 (10) in the crystal (molecule 1 of 2 in the asymmetric unit). Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ce(1)–N(1) 2.664(4), Ce(1)–N(2) 3.187(7), Ce(1)N(1)′ 2.634(4), Ce(1)–N(2)′ 2.625(5), N(1)–Ce(1)–N(2) 44.50(1), N(1)–Ce(1)–N(1)′ 89.9(1), N(1)–Ce(1)–N(2)′ 81.4(2), N(2)–Ce(1)–N(1)′ 71.9(1), N(2)–Ce(1)–N(2)′ 100.3(2), Ce(1)–C(COT) 2.694(7)–2.713(6), Ce(1)–centroid(COT) 1.992, N(1)–C(1) 1.350(8), N(2)–C(1) 1.321(9), N(3)–C(21) 1.342(7), N(4)–C(21) 1.342(6), N(1)–C(1)–N(2) 116.2(5), N(3)–C(21)–N(4) 114.1(4). Symmetry operator to generate equivalent atoms: ′ 1 − x, 1 − y, 2 − z. | ||
 | ||
| Fig. 9 Molecular structure of [(COT)Ce(μ-c-C3H5–CC–C(NCy)2)]2 (11) in the crystal. Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ce(1)–N(1) 2.731(3), Ce(1)–N(2) 2.767(3), Ce(1)–N(3) 2.700(3), Ce(1)–N(4) 2.648(3), Ce(2)–N(1) 2.642(3), Ce(2)–N(2) 2.666(3), Ce(2)–N(3) 2.814(7), Ce(2)–N(4) 2.704(3), N(1)–Ce(1)–N(2) 48.3(1), N(3)–Ce(1)–N(4) 50.0(1), N(1)–Ce(1)–N(3) 79.2(1), N(1)–Ce(1)–N(4) 94.4(1), N(2)–Ce(1)–N(3) 97.2(1), N(2)–Ce(1)–N(4) 72.7(1), N(1)–Ce(2)–N(2) 50.2(1), N(3)–Ce(2)–N(4) 48.3(1), N(1)–Ce(2)–N(3) 78.6(1), N(1)–Ce(2)–N(4) 95.2(1), N(2)–Ce(2)–N(3) 96.8(1), N(2)–Ce(2)–N(4) 73.4(1), Ce(1)–C(COT) 2.693(4)–2.721(4), Ce(1)–centroid(COT) 1.988, Ce(2)–C(COT) 2.686(5)–2.715(5), Ce(2)–centroid(COT) 1.988, N(1)–C(1) 1.349(5), N(2)–C(1) 1.334(5), N(3)–C(21) 1.332(5), N(4)–C(21) 1.348(5), N(1)–C(1)–N(2) 114.1(3), N(3)–C(21)–N(4) 115.1(3). | ||
 | ||
| Fig. 10 Molecular structure of [(COT)Nd(μ-c-C3H5–CC–C(NiPr)2)]2 (12) in the crystal (molecule 1 of 2 in the asymmetric unit). Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Nd(1)–N(1) 2.610(4), Nd(1)–N(1)′ 2.581(4), Nd(1)–N(2)′ 2.570(4), N(1)′–Nd(1)–N(2)′ 51.8(1), N(1)–Nd(1)–N(1)′ 85.3(1) N(1)–Nd(1)–N(2)′ 85.9(1), Nd(1)–C(COT) 2.658(5)–2.679(5), Nd(1)–centroid(COT) 1.936, N(1)–C(1) 1.361(6), N(2)–C(1) 1.329(6), N(1)–C(1)–N(2) 116.9(4). Symmetry operator to generate equivalent atoms: 1 − x, 1 − y, 2 − z. | ||
 | ||
| Fig. 11 Capped-sticks views of the unit [(COT)Ln(μ-C(N)2)]2 of 10 (Ln = Ce; left) and 12 (Ln = Nd; right). | ||
Due to the difference in ionic radii of Ce3+ and Nd3+, slightly shorter bond distances are observed in 12 than in 10. Interestingly, compared to the analogous complexes 10 and 12, complex 11 which has cyclohexyl groups on the nitrogen atoms comprises a different geometry. As shown in Fig. 11, the cerium atoms are coordinated to the COT ring and four nitrogen atoms of amidinate ligands to give a distorted pseudo-tetragonal pyramidal geometry. In 11, the Ce–C(COT) distances range from 2.693(4) to 2.721(4) Å (average 2.704 Å).22–24 The Ce–(COT ring centroid) distance is 1.988 Å. The Ce–N bond lengths are in the range from 2.648(3) to 2.767(3) Å (average 2.711 Å). In comparison with 10, the (COT ring centroid)–Ce–Ce–(COT ring centroid) torsion angle is 166.4°, which is smaller to that found in 10. This can be traced back to the difference in the coordination mode in 11 as compared to that found in 10. Surprisingly, the Ce⋯Ce distance in 11 (3.625 Å) is shorter than that observed in 10 (4.219 Å) with a difference of 0.594 Å. This can attributed to the difference in the substituents on the nitrogen atoms.
Synthesis and structure of (COT)Ho[c-C3H5–CC–C(NR)2](THF) (13, 14)
In contrast to the formation of compounds 10–12, use of the smaller Ho3+ ion gave a different result. Treatment of a mixture of K2COT and 1a or 1b with anhydrous HoCl3 in a 1:1:1 molar ratio in THF in a one-pot reaction afforded the solvated half-sandwich complexes (COT)Ho[c-C3H5–CC–C(NR)2](THF) (13: R = iPr; 14: R = Cy) as shown in Scheme 7. The monomeric complexes 13 and 14 were extracted with n-pentane or toluene and isolated in 48% and 30% yield, respectively.
 | ||
| Scheme 7 Synthesis of (COT)Ho[c-C3H5–CC–C(NR)2](THF) (13: R = iPr; 14: R = Cy). | ||
The new complexes 13 and 14 have been fully characterized by EI/mass, IR, and elemental analyses. In addition, single-crystals of 14 were found to be suitable for an X-ray diffraction study. The effect of the paramagnetism of Ho3+ ion prevented the measurement of NMR data. Both complexes 13 and 14 were characterized by an EI mass spectrum. The EI mass spectrum showed the molecule ion of 13 and its characteristic fragmentation. An EI mass spectrum of 14 showed the molecular ion of 14 without the coordinated THF molecule.17,30 Suitable single-crystals of 14 were obtained by recrystallization from n-pentane. The molecular structure of 14 was established by single-crystal X-ray diffraction as shown in Fig. 12. Compound 14 crystallizes in the monoclinic space group P21/n with one molecule in a symmetric unit. The holmium ion is coordinated to an η8-COT ring and two nitrogen atoms of the amidinate ligand as well as the oxygen atom of a neutral THF ligand. The coordination sphere around the Ho3+ ion can be described as distorted pseudo-tetrahedral.
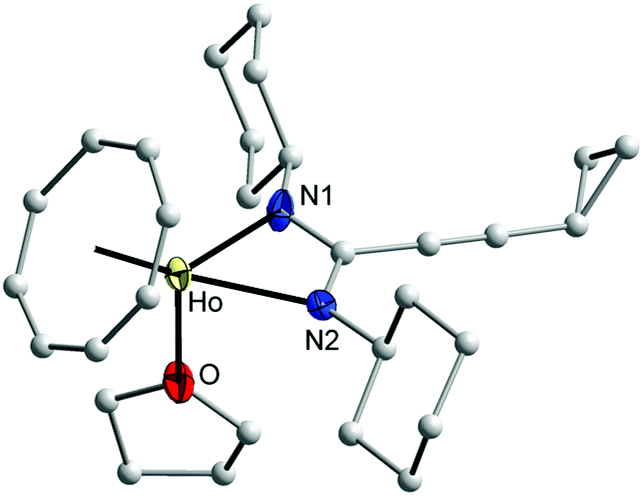 | ||
| Fig. 12 Molecular structure of (COT)Ho[c-C3H5–CC–C(NCy)2](THF) (14) in the crystal. Ellipsoids of the heavier atoms with 50% probability, H atoms omitted for clarity. Selected bond lengths [Å] and angles [°]: Ho–N(1) 2.349(3), Ho–N(2) 2.342(3), N(1)–Ho–N(2) 57.2(1), Ho–O 2.397(2), N(1)–Ho–O 84.6(1), N(2)–Ho–O 84.1(1), Ho–C(COT) 2.552(5)–2.598(4), Ho–centroid(COT) 1.821, N(1)–C(1) 1.324(4), N(2)–C(1) 1.329(4), N(1)–C(1)–N(2) 115.5(3). | ||
The Ho–C(COT) distances, which range from 2.552(5) to 2.598(4) Å (average 2.568 Å) are in good agreement with those reported for (COT)Tm[C6H5C(NSiMe3)2](THF) (average 2.558 Å)17 and [{η8-1,4-(Me3Si)2C8H6}Y{(iPr)2ATI}(THF)] (ATI = N-isopropyl-2-(isopropylamino)troponiminate) (average 2.623 Å).30 The difference in the distances can attributed to the difference in the ionic radii according to Y > Ho > Tm. The Ho–(COT ring-centroid) distance is 1.821 Å.17,30,31 Due to the smaller size of Ho3+, the distance Ho–(COT ring-centroid) is significantly shorter than that observed in the compounds 7–11. The bond lengths Ho–N1, Ho–N2 and Ho–O are 2.349(3), 2.342(3) and 2.397(2) Å, respectively.32 The C1–N1 and C1–N2 distances are 1.324(4) and 1.329(4) Å, respectively, indicating negative charge delocalization in the NCN unit. The N1–Ho–N2 57.15(9)° angle is identical with that found in (COT′′)Yb(DIPPForm)(THF) (DIPPForm = N,N′-bis(2,6-diisopropylphenyl)formamidinate) (57.70(14)°).32 The bond angle (COT ring centroid)–Ho–C1 is 149.3°. The N1–Ho–O and N2–Ho–O angles are similar to each other 84.63(10)° and 84.13(9)°, respectively. The N1–C1–N2 bond angle is 115.5(3)°.
Synthesis and structure of [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15)
Finally, a unique cyclic multidecker sandwich complex was prepared by reaction of anhydrous NdCl3 with K2COT and 1b in a one-pot reaction. According to Scheme 8, treatment of a mixture of K2COT and 2a with anhydrous NdCl3 in THF afforded the unprecedented cyclic sandwich compound [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15) (Scheme 8).
 | ||
| Scheme 8 Synthesis of [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15). | ||
The new compound 15 was extracted using toluene and isolated in the form of blue, needle-like crystals in 20% yield. Complex 15 was fully characterized by elemental analysis, spectroscopic methods and single-crystal X-ray diffraction. In the 1H NMR spectrum, the protons of the η8-C8H8 ligands appear at high field as singlet at δ = −11.34 ppm.17 The CH protons of the cyclohexyl groups include the appearance of two sets of resonances of equal intensity at δ = 3.61 and 3.35 ppm, which can be attributed to the paramagnetic nature of the Nd3+ ion. However, in comparison with the free ligand 1b, the influence of the paramagnetism of the Nd3+ ion on the protons of the cyclopropyl protons is only weak. The CH protons of the c-C3H5 are observed at δ = 1.35 ppm, and the CH2 groups at δ = 0.84 and 0.71 ppm. The 13C NMR spectrum of 15 shows a resonance at δ = 133.7 due to the COT rings. Blue, needle-like single-crystals, grown by slow cooling of a saturated solution in toluene to 5 °C, were found to be suitable for X-ray diffraction study. These crystals were found to contain two molecules of toluene per formula unit. Compound 15 crystallizes in the monoclinic space group C2/c with half a molecule in the asymmetric unit (cf.Tables 1 and 2). The unit cell also contains six molecules of toluene of crystallization. The solid state structure of 15 revealed the presence of an unprecedented macrocyclic sandwich compound of the composition [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4, as shown in Fig. 13.
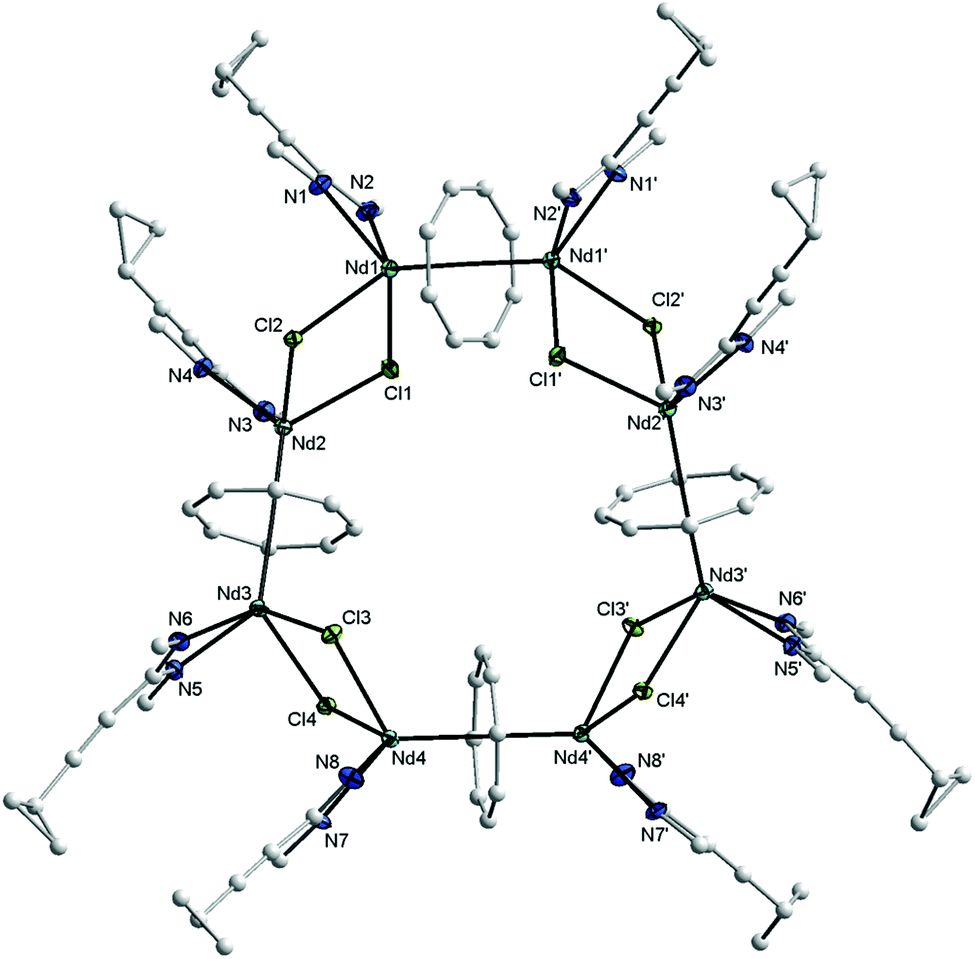 | ||
| Fig. 13 Molecular structure of [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4 (15) in the crystal. Ellipsoids of the heavier atoms with 50% probability, H atoms and peripheral C atoms of the cyclohexyl groups omitted for clarity. Selected bond lengths [Å] and angles [°]: Nd(1)–N(1) 2.424(4), Nd(1)–N(2) 2.403(4), Nd(2)–N(3) 2.434(4), Nd(2)–N(4) 2.380(4), Nd(3)–N(5) 2.439(4), Nd(3)–N(6) 2.404(3), Nd(4)–N(7) 2.395(3), Nd(4)–N(8) 2.418(4), Nd(1)–Cl(1) 2.829(1), Nd(1)–Cl(2) 2.790(1), Nd(2)–Cl(1) 2.768(1), Nd(2)–Cl(2) 2.832(1), Nd(3)–Cl(3) 2.808(2), Nd(3)–Cl(4) 2.777(1), Nd(4)–Cl(3) 2.766(1), Nd(4)–Cl(4) 2.838(2), N(1)–Nd(1)–N(2) 56.0(1), N(3)–Nd(2)–N(4) 56.2(1), N(5)–Nd(3)–N(6) 56.0(1), N(7)–Nd(4)–N(8) 55.6(1), N(1)–Nd(1)–Cl(1) 126.4(1), N(1)–Nd(1)–Cl(2) 86.9(1), N(2)–Nd(1)–Cl(1) 85.0(1), N(2)–Nd(1)–Cl(2) 112.9(1), N(3)–Nd(2)–Cl(1) 83.0(1), N(3)–Nd(2)–Cl(2) 127.3(1), N(4)–Nd(2)–Cl(1) 108.0(1), N(4)–Nd(2)–Cl(2) 85.5(1), N(5)–Nd(3)–Cl(3) 124.6(1), N(5)–Nd(3)–Cl(4) 83.2(1), N(6)–Nd(3)–Cl(3) 83.5(1), N(6)–Nd(3)–Cl(4) 110.4(1), N(7)–Nd(4)–Cl(3) 108.7(1), N(7)–Nd(4)–Cl(4) 86.5(1), N(8)–Nd(4)–Cl(3) 84.5(1), N(8)–Nd(4)–Cl(4) 129.3(1), Cl(1)–Nd(1)–Cl(2) 75.44(4), Cl(1)–Nd(1)–Cl(2) 75.72(4), Cl(3)–Nd(3)–Cl(4) 77.36(3), Cl(3)–Nd(4)–Cl(4) 77.05(3), Nd(1)–C(μ-COT) 2.815(5)–2.844(6), Nd(1)–centroid(COT) 2.162, Nd(2)–C(μ-COT) 2.819(5)–2.851(5), Nd(2)–centroid(COT) 2.164, Nd(3)–C(μ-COT) 2.804(5)–2.875(5), Nd(3)–centroid(COT) 2.171, Nd(4)–C(μ-COT) 2.812(5)–2.858(4), Nd(4)–centroid(COT) 2.161, N(1)–C(1) 1.331(6), N(2)–C(1) 1.341(6), N(3)–C(23) 1.327(6), N(4)–C(23) 1.343(6), N(5)–C(49) 1.339(6), N(6)–C(49) 1.335(6), N(7)–C(67) 1.328(6), N(8)–C(67) 1.331(6), N(1)–C(1)–N(2) 116.2(4), N(3)–C(23)–N(4) 116.3(4), N(5)–C(49)–N(6) 116.4(4), N(7)–C(67)–N(8) 115.0(4). Symmetry operator to generate equivalent atoms: ′ −x, y, 0.5 − z. | ||
The molecule consists of four COT rings sandwiched between eight Nd3+ ions, and each Nd3+ ion is bonded to one amidinate ligand and bridged by two chlorine atoms with the neighbouring Nd3+ atom (Fig. 13). All four COT rings are μ–η8:η8-coordinated to neodymium. The coordination sphere around the Nd3+ ion can be described as distorted pseudo-tetragonal-pyramidal. The average Nd–C(COT) distances range from 2.826 Å to 2.835 Å, similar to those found in (μ–η8:η8-COT)[Sm{N(SiMe3)2}2]2 with a range from 2.798(5) to 2.857(5) Å (ref. 22) and in the triple-decker sandwich-complex (η8-COT′′)Nd(μ–η8:η8-COT′′)Nd(η8-COT′′) with a range from 2.815(3) to 2.922(3) Å.33 The Nd–(COT ring-centroid) distances are ranging from 2.162 to 2.171 Å in good agreement with those found in (η8-COT′′)Nd(μ–η8:η8-COT′′)Nd(η8-COT′′) (2.126 to 2.156 Å).33 The Nd–Cl bond lengths are between 2.7655(11) and 2.8377(15) Å, similar to Nd–Cl (from 2.822(1) to 2.8463(12) Å) in [(COT′′)Nd(μ-Cl)(THF)]2.32 The Nd–N bond lengths are ranging from 2.395(3) to 2.439(4) Å [147]. The Nd–(COT ring-centroid)–Nd (178.9°, 178.6° and 179.8°) angles are almost linear. The N–Nd–N angles range from 55.57(13)° to 56.19(12)° [147]. The unit NCN angles are between 115.0(4)° and 116.4(4)°. The Cl–Nd–Cl angles are ranging from 75.44(3)° to 77.36(3)°, and the Nd–Cl–Nd from 101.96(3)° to 104.02(4)°.32 Although compound 15 is quite unique, it should be noted that a few wheel-shaped organolanthanide complexes of comparable size have previously been reported by Roesky et al. However, these compounds differ from 15 in that lanthanide and potassium ions are bridged by cyclopentadienyl rings or η6-coordinated phenyl substituents.34
Conclusions
The results reported here further underline the utility and versatility of cyclopropylethynylamidinate ligands, [c-C3H5–CC–C(NR)2]− (R = iPr, Cy), in organolanthanide chemistry. Although these amidinate ligands my seem exotic at the first glance, they offer significant advantages. First of all, the precursors Li[c-C3H5–CC–C(NR)2] (1a: R = iPr, 1b: R = cyclohexyl (Cy)) are readily available in large quantities and in high yields using commercially available starting materials (cyclopropyl-acetylene, n-butyllithium, N,N′-diorganocarbodiimides). A second important aspect is the well-known electron-donating ability of the cyclopropyl substituent to an adjacent electron-deficient center. This offers the rare chance to electronically influence the amidinate ligand system rather than just altering its steric demand. The exceptional position of the cyclopropylethynylamidinate ligands was manifested by the results of the present study. In combination with COT in the ligand sphere of Ln3+ ions, these ligands allowed for the successful synthesis and full characterization of no less than five different type of (COT)Ln half-sandwich complexes, namely:
1. heterometallic complexes of the type (COT)Ln[μ-c-C3H5–CC–C(NR)2]2Li(L) (L = Et2O, THF),
2. inverse sandwich complexes of the type (μ–η8:η8-COT)[Ce{c-C3H5–CC–C(NR)2}2]2,
3. binuclear complexes of the type [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2,
4. the mononuclear solvated complexes (COT)Ho[c-C3H5–CC–C(NR)2](THF), and
5. the unique “giant neodymium wheel” [(μ–η8:η8-COT){Nd(c-C3H5–CC–C(NCy)2)(μ-Cl)}2]4.
Together with the previously reported synthetic and catalytic studies,11–14 these results clearly demonstrate the synthetic value of the cyclopropylethynylamidinate ligands [c-C3H5–CC–C(NR)2]− (R = iPr, Cy). Thus, further investigation of the use of these ligands in organolanthanide (and perhaps organoactinide) chemistry appears highly desirable. Future studies in this area should also address the question if the same variety of products can also be achieved when the cyclopropyl substituents are replaced by more common groups such as phenyl.
Experimental section
Materials and methods
All manipulations were performed using glovebox (<1 ppm O2, <1 ppm H2O) and standard Schlenk line techniques under an inert atmosphere of dry argon. THF, Et2O, n-pentane and toluene were distilled from sodium/benzophenone under nitrogen atmosphere prior to use. All glassware was oven-dried at 120 °C for at least 24 h, assembled while hot, and cooled under vacuum prior to use. Lithium-cyclopropylethynylamidinates 1a and 1b were prepared according to the literature method.11 The starting materials LnCl3,35 K2COT,36 [(COT)Nd(THF)2(μ-Cl)]2, [(COT)Pr(THF)2(μ-Cl)]216a and compounds 5 and 612 were also prepared according to known literature procedures. 1H NMR (400 MHz) and 13C NMR (100.6 MHz) were recorded in C6D6, THF-d8 or toluene-d8 solutions on a Bruker DPX 400 spectrometer at 25 °C. Chemical shifts were referenced to TMS. Assignment of signals was made from 1H–13C HSQC NMR experiments. IR spectra were recorded using KBr pellets on a Perkin Elmer FT-IR spectrometer system 2000 between 4000 cm−1 and 400 cm−1. Microanalyses of the compounds were performed using a Leco CHNS 932 apparatus.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C–C(NCy)2]2Li(Et2O) (2).
A solution of [(COT)Pr(μ-Cl)(THF)2]2 (1.0 g, 1.15 mmol) in 20 mL THF was added to a solution of 1b (0.8 g, 2.3 mmol) in 50 mL THF. The resulting orange reaction mixture was stirred over night at room temperature. After evaporation to dryness, the residue was extracted with 30 mL of toluene. After filtration, the toluene was replaced by 10 ml of Et2O to give a bright yellow solution. Crystallization at 5 °C afforded 2 as bright yellow crystals in 53% yield (0.85 g). 1H NMR (THF-d8): δ (ppm) 5.50–5.90 (m br, 8H, C8H8), 3.45 (s br, Et2O), 3.36 (m br, 4H, CH, Cy), 1.39 (m, 2H, CH, c-C3H5), 1.05–1.61 (m br, 40H, CH2, Cy), 0.84 (s br, 4H, CH2, c-C3H5), 0.69 (s br, 4H, CH2, c-C3H5), 0.89 (s br, Et2O). 13C{1H} NMR (THF-d8): δ (ppm) 141.3 (NCN), 127.8 (C8H8), 94.9 (CC–C), 61.9 (CH2, Et2O), 60.5 (CH, Cy), 35.5 (CH2, Cy), 27.1 (CH2, Cy), 25.5 (CH2, Cy), 14.4 (CH3, Et2O), 8.6 (CH2, c-C3H5), −0.3 (CH, c-C3H5). IR (KBr): 3677w, 3440w, 3096w, 3015w, 2961s, 2865s, 2697m, 2217s (CC), 1593vs (NCN), 1495m, 1384m, 1333w, 1263w, 1169m, 1090w, 965w, 918w, 870w, 812w, 715w, 687w, 529w, 436w cm−1. Anal. Calcd for C48H72LiN4OPr (868.95): C, 66.24; H, 8.28; N, 6.42. Found: C, 65.93; H, 8.14; N, 6.34%. EI-MS: m/z (%) 515.5 (10) [Pr(COT)(c-C3H5–CC–C(NCy)2)], 378.4 (83) [(COT)(c-C3H5–CC–C(NCy)2)]+, 272.2 (87) [c-C3H5–CC–C(NCy)2]+, 243.2 (36) [Pr(COT)]+.
C–C(NiPr)2]2Li(THF) (3).
A solution of [(COT)Nd(μ-Cl)(THF)2]2 (1.0 g, 1.16 mmol) in 20 mL THF was added to a solution of 1a (0.62 g, 2.3 mmol) in 50 mL THF. The resulting blue solution was evaporated to dryness under vacuum, followed by extraction with n-pentane (30 mL) to give a clear pale blue solution. The filtrate was concentrated in vacuum to ca. 10 mL. Crystallization at 5 °C afforded 3 in the form of pale blue crystals in 64% yield (0.4 g). IR (KBr): 3678w, 3439w, 3011w, 2932w, 2850s, 2664w, 2592w, 2219s (CC), 2074w, 1890m, 1818w, 1598s (NCN), 1447m, 1390m, 1361w, 1309w, 1255m, 1159m, 1116m, 1067w, 1027w, 971s, 858m, 728m, 593w, 499w, 439w cm−1. Anal. Calcd for C36H54LiN4NdO (710.01): C, 60.90; H, 7.67; N, 7.89. Found C, 60.84; H, 7.10; N, 7.75%. EI-MS: m/z (%) 701.5 (28) [M − Li]+, 677.4 (17) [M − 2CH3]+, 524.3 (100) [Nd(c-C3H5–CC–C(NiPr)2)2]+ or [(COT)Nd(c-C3H5–CC–C(NiPr)) + 2iPr]+, 482.2 (15) [M − (c-C3H5–CC–C(NiPr)2Li(THF))]+, 398.1 (34) [(COT)Nd(CC–C(NiPr)2)]+.
C–C(NCy)2]2Li(THF) (4).
A solution of [(COT)Nd(μ-Cl)(THF)2]2 (1.0 g, 1.16 mmol) in 20 mL THF was added to a solution of 1b (0.8 g, 2.3 mmol) in 50 mL THF, following the procedure for 3. Compound 4 was isolated as a pale blue solid in 41% yield (0.65 g). 1H NMR (THF-d8): δ (ppm) 32.78 (s br, CH, 4H, Cy), 7.56 (s, 2H, CH, c-C3H5), 6.15 (s, 4H, CH2, c-C3H5), 4.55 (s, 4H, CH2, c-C3H5), 3.32–3.64 (m br, 4H, CH2, Cy), 1.30–1.40 (m br, 16H, CH2, Cy), −1.33 (s br, 4H, CH2, Cy), −4.63 (s br, 16H, CH2, Cy), −11.56 (s br, 8H, C8H8). 13C NMR (THF-d8): δ (ppm) 183.5 (NCN), 160.8 (C8H8), 115.5 (CC–C), 92.2 (H–C–CC), 60.6 (CH, Cy), 40.7 (CH2, Cy), 27.2 (CH2, Cy), 26.0 (CH2, Cy), 14.9 (CH2, c-C3H5), 6.6 (CH, c-C3H5). IR (KBr): 3437w, 3225w, 3091w, 2928s, 2853m, 2536w, 2226vs (CC), 1635s (NCN), 1607m, 1479w, 1449w, 1366m, 1313m, 1254w, 1180w, 1157w, 1106m, 1030m, 975s, 890m, 841w, 700w, 566w, 465w cm−1. Anal. Calcd for C48H70LiN4NdO (870.30): C, 66.20; H, 8.04; N, 6.43. Found: C, 66.08; H, 7.98; N, 6.10%. EI-MS: m/z (%) 517.3 (98) [M − (c-C3H5–CC–C(NCy)2Li(THF))], 476.2 (37) [Nd(COT)(CC–C(NCy)2)], 270.2 (43) [(c-C3H5–CC–C(NCy)2)]+, 248.0 (59) [Nd(COT)]2+.
C–C(NiPr)2)2]2 (7).
A solution of [{c-C3H5–CC–C(NiPr)2}2Ce(μ-Cl)(THF)]2 (0.4 g, 0.33 mmol) in 50 mL THF was injected with K2COT (0.6 mL of a 0.6 M solution in THF). The reaction mixture was stirred for 12 h at room temperature. THF was removed under vacuum and the residue was extracted with 30 mL of n-pentane. The filtered solution was concentrated to 10 mL and then kept at 5 °C to afford 7 as yellow, needle-like crystals in 45% yield (0.17 g). 1H NMR (THF-d8): δ (ppm) 10.01 (s br, 8H, CH, iPr), 3.15 (s br, 4H, CH, c-C3H5), 2.22 (s, 8H, CH2, c-C3H5), 1.87 (s, 8H, CH2, c-C3H5), 1.15 (s br, C8H8), −0.32 (s br, 48H, CH3, iPr). 13C{1H} NMR (THF-d8): δ (ppm) 161.2 (NCN), 108.1 (CC–C), 107.7 (C8H8), 77.1 (H–C–CC), 58.7 (CH, iPr), 25.9 (CH3), 10.4 (CH2, c-C3H5), −0.4 (CH, c-C3H5). Anal. Calcd for C56H84Ce2N8 (1149.58): C, 58.51; H, 7.37; N, 9.75. Found: C, 58.59; H, 7.94; N, 9.72.
C–C(NCy)2)2]2 (8).
The reaction of [{c-C3H5–CC–C(NCy)2}2Ce(μ-Cl)(THF)]2 (0.5 g, 0.32 mmol) with K2COT (0.6 mL of a 0.6 M solution in THF) was carried out as described for 7 and afforded 8 as yellow crystals in 49% yield (0.23 g). 1H NMR (THF-d8): δ (ppm) 9.67 (s br, 8H, CH, Cy), 3.22 (s br, 4H, CH, c-C3H5), 2.28 (s br, 8H, CH2, c-C3H5), 1.92 (s br, 8H, CH2, c-C3H5), 0.97–1.60 (m br, 82H, CH2, Cy, C8H8), −0.45 (s br, 6H, CH2, Cy). 13C{1H} NMR (THF-d8): δ (ppm) 163.2 (NCN), 114.5 (CC–C), 106.9 (C8H8), 94.8 (CC–C), 67.1 (CH, Cy), 36.0 (CH2, Cy), 33.5 (CH2, Cy), 26.0 (CH2, Cy), 10.6 (CH2, c-C3H5), 2.4 (CH, c-C3H5). Anal. Calcd for C80H116Ce2N8 (1470.10): C, 65.36; H, 7.95; N, 7.62. Found: C, 62.61; H, 7.81; N, 7.91.
C–C(NiPr)2)]2 (9).
A solution of [(COT)Pr(μ-Cl)(THF)2]2 (1.0 g, 1.15 mmol) in 20 mL of THF was added to a solution of 2a (0.31 g, 1.15 mmol) in 50 mL of THF. The resulting orange reaction mixture was stirred for 12 h at room temperature. Work-up as described for 8 using toluene (30 mL) for extraction gave 9 as yellow solid in 47% yield (0.3 g). 1H NMR (toluene-d8): δ (ppm) 10.57 (s, 4H, CH, iPr), 1.94 (m, 2H, CH, c-C3H5), 1.70 (s br, 4H, CH2, c-C3H5), 1.22 (s br, 4H, CH2, c-C3H5), −4.63 (s br, 16H, C8H8), −10.24 (s br, 24H, CH3, iPr). 13C{1H} NMR (toluene-d8): δ (ppm) 186.1 (C8H8), 33.5 (CH, iPr), 15.6 (CH3, iPr), 9.7 (CH2, c-C3H5), 0.9 (CH, c-C3H5). IR (KBr): 3833w, 3621w, 3221w, 3013w, 2964s, 2930m, 2537w, 2215 vs (CC), 1836w, 1701w, 1612s (NCN), 1466w, 1381w, 1244m, 1179m, 1133m, 1080w, 1052w, 1032w, 983s, 966m, 946m, 841w, 732w, 702w, 646w, 587w, 442w cm−1. Anal. Calcd for C40H54N4Pr2 (872.14): C, 55.04; H, 6.19; N, 6.42. Found: C, 55.14; H, 6.24; N, 6.29%.
C–C(NCy)2]2Li(Et2O) (2).
A solution of [(COT)Pr(μ-Cl)(THF)2]2 (1.0 g, 1.15 mmol) in 20 mL THF was added to a solution of 1b (0.8 g, 2.3 mmol) in 50 mL THF. The resulting orange reaction mixture was stirred over night at room temperature. After evaporation to dryness, the residue was extracted with 30 mL of toluene. After filtration, the toluene was replaced by 10 ml of Et2O to give a bright yellow solution. Crystallization at 5 °C afforded 2 as bright yellow crystals in 53% yield (0.85 g). 1H NMR (THF-d8): δ (ppm) 5.50–5.90 (m br, 8H, C8H8), 3.45 (s br, Et2O), 3.36 (m br, 4H, CH, Cy), 1.39 (m, 2H, CH, c-C3H5), 1.05–1.61 (m br, 40H, CH2, Cy), 0.84 (s br, 4H, CH2, c-C3H5), 0.69 (s br, 4H, CH2, c-C3H5), 0.89 (s br, Et2O). 13C{1H} NMR (THF-d8): δ (ppm) 141.3 (NCN), 127.8 (C8H8), 94.9 (CC–C), 61.9 (CH2, Et2O), 60.5 (CH, Cy), 35.5 (CH2, Cy), 27.1 (CH2, Cy), 25.5 (CH2, Cy), 14.4 (CH3, Et2O), 8.6 (CH2, c-C3H5), −0.3 (CH, c-C3H5). IR (KBr): 3677w, 3440w, 3096w, 3015w, 2961s, 2865s, 2697m, 2217s (CC), 1593vs (NCN), 1495m, 1384m, 1333w, 1263w, 1169m, 1090w, 965w, 918w, 870w, 812w, 715w, 687w, 529w, 436w cm−1. Anal. Calcd for C48H72LiN4OPr (868.95): C, 66.24; H, 8.28; N, 6.42. Found: C, 65.93; H, 8.14; N, 6.34%. EI-MS: m/z (%) 515.5 (10) [Pr(COT)(c-C3H5–CC–C(NCy)2)], 378.4 (83) [(COT)(c-C3H5–CC–C(NCy)2)]+, 272.2 (87) [c-C3H5–CC–C(NCy)2]+, 243.2 (36) [Pr(COT)]+.
C–C(NiPr)2]2Li(THF) (3).
A solution of [(COT)Nd(μ-Cl)(THF)2]2 (1.0 g, 1.16 mmol) in 20 mL THF was added to a solution of 1a (0.62 g, 2.3 mmol) in 50 mL THF. The resulting blue solution was evaporated to dryness under vacuum, followed by extraction with n-pentane (30 mL) to give a clear pale blue solution. The filtrate was concentrated in vacuum to ca. 10 mL. Crystallization at 5 °C afforded 3 in the form of pale blue crystals in 64% yield (0.4 g). IR (KBr): 3678w, 3439w, 3011w, 2932w, 2850s, 2664w, 2592w, 2219s (CC), 2074w, 1890m, 1818w, 1598s (NCN), 1447m, 1390m, 1361w, 1309w, 1255m, 1159m, 1116m, 1067w, 1027w, 971s, 858m, 728m, 593w, 499w, 439w cm−1. Anal. Calcd for C36H54LiN4NdO (710.01): C, 60.90; H, 7.67; N, 7.89. Found C, 60.84; H, 7.10; N, 7.75%. EI-MS: m/z (%) 701.5 (28) [M − Li]+, 677.4 (17) [M − 2CH3]+, 524.3 (100) [Nd(c-C3H5–CC–C(NiPr)2)2]+ or [(COT)Nd(c-C3H5–CC–C(NiPr)) + 2iPr]+, 482.2 (15) [M − (c-C3H5–CC–C(NiPr)2Li(THF))]+, 398.1 (34) [(COT)Nd(CC–C(NiPr)2)]+.
C–C(NCy)2]2Li(THF) (4).
A solution of [(COT)Nd(μ-Cl)(THF)2]2 (1.0 g, 1.16 mmol) in 20 mL THF was added to a solution of 1b (0.8 g, 2.3 mmol) in 50 mL THF, following the procedure for 3. Compound 4 was isolated as a pale blue solid in 41% yield (0.65 g). 1H NMR (THF-d8): δ (ppm) 32.78 (s br, CH, 4H, Cy), 7.56 (s, 2H, CH, c-C3H5), 6.15 (s, 4H, CH2, c-C3H5), 4.55 (s, 4H, CH2, c-C3H5), 3.32–3.64 (m br, 4H, CH2, Cy), 1.30–1.40 (m br, 16H, CH2, Cy), −1.33 (s br, 4H, CH2, Cy), −4.63 (s br, 16H, CH2, Cy), −11.56 (s br, 8H, C8H8). 13C NMR (THF-d8): δ (ppm) 183.5 (NCN), 160.8 (C8H8), 115.5 (CC–C), 92.2 (H–C–CC), 60.6 (CH, Cy), 40.7 (CH2, Cy), 27.2 (CH2, Cy), 26.0 (CH2, Cy), 14.9 (CH2, c-C3H5), 6.6 (CH, c-C3H5). IR (KBr): 3437w, 3225w, 3091w, 2928s, 2853m, 2536w, 2226vs (CC), 1635s (NCN), 1607m, 1479w, 1449w, 1366m, 1313m, 1254w, 1180w, 1157w, 1106m, 1030m, 975s, 890m, 841w, 700w, 566w, 465w cm−1. Anal. Calcd for C48H70LiN4NdO (870.30): C, 66.20; H, 8.04; N, 6.43. Found: C, 66.08; H, 7.98; N, 6.10%. EI-MS: m/z (%) 517.3 (98) [M − (c-C3H5–CC–C(NCy)2Li(THF))], 476.2 (37) [Nd(COT)(CC–C(NCy)2)], 270.2 (43) [(c-C3H5–CC–C(NCy)2)]+, 248.0 (59) [Nd(COT)]2+.
C–C(NiPr)2)2]2 (7).
A solution of [{c-C3H5–CC–C(NiPr)2}2Ce(μ-Cl)(THF)]2 (0.4 g, 0.33 mmol) in 50 mL THF was injected with K2COT (0.6 mL of a 0.6 M solution in THF). The reaction mixture was stirred for 12 h at room temperature. THF was removed under vacuum and the residue was extracted with 30 mL of n-pentane. The filtered solution was concentrated to 10 mL and then kept at 5 °C to afford 7 as yellow, needle-like crystals in 45% yield (0.17 g). 1H NMR (THF-d8): δ (ppm) 10.01 (s br, 8H, CH, iPr), 3.15 (s br, 4H, CH, c-C3H5), 2.22 (s, 8H, CH2, c-C3H5), 1.87 (s, 8H, CH2, c-C3H5), 1.15 (s br, C8H8), −0.32 (s br, 48H, CH3, iPr). 13C{1H} NMR (THF-d8): δ (ppm) 161.2 (NCN), 108.1 (CC–C), 107.7 (C8H8), 77.1 (H–C–CC), 58.7 (CH, iPr), 25.9 (CH3), 10.4 (CH2, c-C3H5), −0.4 (CH, c-C3H5). Anal. Calcd for C56H84Ce2N8 (1149.58): C, 58.51; H, 7.37; N, 9.75. Found: C, 58.59; H, 7.94; N, 9.72.
C–C(NCy)2)2]2 (8).
The reaction of [{c-C3H5–CC–C(NCy)2}2Ce(μ-Cl)(THF)]2 (0.5 g, 0.32 mmol) with K2COT (0.6 mL of a 0.6 M solution in THF) was carried out as described for 7 and afforded 8 as yellow crystals in 49% yield (0.23 g). 1H NMR (THF-d8): δ (ppm) 9.67 (s br, 8H, CH, Cy), 3.22 (s br, 4H, CH, c-C3H5), 2.28 (s br, 8H, CH2, c-C3H5), 1.92 (s br, 8H, CH2, c-C3H5), 0.97–1.60 (m br, 82H, CH2, Cy, C8H8), −0.45 (s br, 6H, CH2, Cy). 13C{1H} NMR (THF-d8): δ (ppm) 163.2 (NCN), 114.5 (CC–C), 106.9 (C8H8), 94.8 (CC–C), 67.1 (CH, Cy), 36.0 (CH2, Cy), 33.5 (CH2, Cy), 26.0 (CH2, Cy), 10.6 (CH2, c-C3H5), 2.4 (CH, c-C3H5). Anal. Calcd for C80H116Ce2N8 (1470.10): C, 65.36; H, 7.95; N, 7.62. Found: C, 62.61; H, 7.81; N, 7.91.
C–C(NiPr)2)]2 (9).
A solution of [(COT)Pr(μ-Cl)(THF)2]2 (1.0 g, 1.15 mmol) in 20 mL of THF was added to a solution of 2a (0.31 g, 1.15 mmol) in 50 mL of THF. The resulting orange reaction mixture was stirred for 12 h at room temperature. Work-up as described for 8 using toluene (30 mL) for extraction gave 9 as yellow solid in 47% yield (0.3 g). 1H NMR (toluene-d8): δ (ppm) 10.57 (s, 4H, CH, iPr), 1.94 (m, 2H, CH, c-C3H5), 1.70 (s br, 4H, CH2, c-C3H5), 1.22 (s br, 4H, CH2, c-C3H5), −4.63 (s br, 16H, C8H8), −10.24 (s br, 24H, CH3, iPr). 13C{1H} NMR (toluene-d8): δ (ppm) 186.1 (C8H8), 33.5 (CH, iPr), 15.6 (CH3, iPr), 9.7 (CH2, c-C3H5), 0.9 (CH, c-C3H5). IR (KBr): 3833w, 3621w, 3221w, 3013w, 2964s, 2930m, 2537w, 2215 vs (CC), 1836w, 1701w, 1612s (NCN), 1466w, 1381w, 1244m, 1179m, 1133m, 1080w, 1052w, 1032w, 983s, 966m, 946m, 841w, 732w, 702w, 646w, 587w, 442w cm−1. Anal. Calcd for C40H54N4Pr2 (872.14): C, 55.04; H, 6.19; N, 6.42. Found: C, 55.14; H, 6.24; N, 6.29%.
General procedure for the synthesis of the complexes [(COT)Ln(μ-c-C3H5–CC–C(NR)2)]2 (10–12) or (COT)Ho(c-C3H5–CC–C(NR)2)(THF) (13, 14)
Anhydrous LnCl3 (2 mmol) (Ln = Ce, Nd or Ho) in 40 mL THF was added to a mixture of the relevant cyclopropylethynyl-amidinate (1a or 1b) (2 mmol) and K2COT (3.3 mL, 0.6 M solution in THF), dissolved in 50 mL of THF. The reaction mixture was stirred over night at room temperature. The solvent was removed under vacuum followed by extraction of the residue with 40 mL toluene (n-pentane in the cases of 13 and 14), the solution was concentrated to 20 mL and then kept at 5 °C to afford 10–14.
C–C(NiPr)2)]2 (10).
Compound 10 was isolated as deep green, needle-like single-crystals suitable for X-ray diffraction in 57% yield (0.96 g). 1H NMR (THF-d8): δ (ppm) 12.32 (s br, 4H, CH, iPr), 3.43 (s br, 2H, CH, c-C3H5), 2.60 (s br, 4H, CH2, c-C3H5), 2.11 (s br, 4H, CH2, c-C3H5), 0.91–1.53 (m, 40H, CH3, iPr, C8H8). 13C{1H} NMR (THF-d8): δ (ppm) 183.2 (NCN), 109.9 (CC–C), 108.6 (C8H8), 81.1 (H–C–CC), 60.8 (CH, iPr), 27.3 (CH3, iPr), 10.6 (CH2, c-C3H5), 3.4 (CH, c-C3H5). IR (KBr): 3852w, 3743w, 3436w, 3224w, 3091w, 2965m, 2930m, 2870w, 2609w, 2533w, 2328w, 2318w, 2226s (CC), 2029m, 1976w, 1959w, 1634s (NCN), 1613w, 1560w, 1504w, 1449m, 1375w, 1307m, 1244m, 1180w, 1157w, 1029w, 985s, 945w, 895w, 878m, 844w, 813w, 747w, 700w, 667w, 615w, 588m, 555w, 504w, 466w, 458w cm−1. Anal. Calcd for C40H54Ce2N4 (871.13): C, 55.15; H, 6.25; N, 6.43. Found: C, 54.73; H, 6.25; N, 6.62.
C–C(NCy)2)]2 (11).
Compound 11 was obtained in the form of green, needle-like single-crystals suitable for X-ray diffraction in 17% yield (0.35 g). 1H NMR (THF-d8): δ (ppm) 12.22 (s br, 4H, CH, Cy), 0.93–1.87 (m br, 58H, CH2, Cy, C8H8, CH, c-C3H5), 0.73 (m br, 8H, CH2, c-C3H5). 13C{1H} NMR (THF-d8): δ (ppm) 140.7 (NCN), 115.3 (C8H8), 94.7 (CC–C), 61.5 (CH, Cy), 35.8 (CH2, Cy), 33.9 (CH2, Cy), 26.8 (CH2, Cy), 8.7 (CH2, c-C3H5), 1.37 (CH, c-C3H5). IR (KBr): 3833w, 3747w, 3435w, 3247w, 3090w, 2926s, 2853m, 2530w, 2356w, 2318w, 2225s (CC), 1959w, 1633vs (NCN), 1448m, 1310m, 1238w, 1180w, 1154w, 984s, 890w, 864w, 809w, 745w, 717w, 667w, 638m, 626w, 554w, 505w, 466w, 450w cm−1. Anal. Calcd for C52H70Ce2N4 (1031.36): C, 60.50; H, 6.78; N, 5.42. Found: C, 59.82; H, 6.63; N, 5.38.
C–C(NiPr)2)]2 (12).
Compound 12 was isolated as purple, needle-like single-crystals suitable for X-ray diffraction in 43% yield (0.74 g). 1H NMR (THF-d8): δ (ppm) 3.96 (s br, 2H, CH, iPr), 3.71 (s br, 2H, CH, iPr), 1.40 (s br, 2H, CH, c-C3H5), 0.72–0.81 (s br, 8H, CH2, c-C3H5), 1.08 (s br, 12H, CH3, iPr), 1.00 (s br, 12H, CH3, iPr), −11.75 (s br, 16H, C8H8). 13C{1H} NMR (THF-d8): δ (ppm) 158.0 (NCN), 132.7 (C8H8), 52.2 (CH, iPr), 42.3 (CH, iPr), 22.7 (CH3, iPr), 8.8 (CH2, c-C3H5), 0.4 (CH, c-C3H5). IR (KBr): 3852w, 3438w, 3282w, 3222w, 3093w, 3012w, 2964m, 2929s, 2868s, 2610w, 2350w, 2350w, 2227s (CC), 1614vs (NCN), 1466w, 1375m, 1361m, 1315w, 1259w, 1179w, 1168m, 1133m, 1053w, 1031w, 984s, 966s, 944w, 880w, 845w, 812w, 774w, 745w, 701w, 668m, 607m, 506w, 467w, 450w cm−1. Anal. Calcd for C40H54N4Nd2 (879.38): C, 54.63; H, 6.19; N, 6.37. Found: C, 54.39; H, 6.26; N, 6.49. EI-MS: m/z (%) 435.5 (20) [(COT)Nd(c-C3H5–CC–C(NiPr)2)]+.
C–C(NiPr)(THF) (13).
Compound 13 was obtained as a pale yellow solid in 48% yield (0.84 g). IR (KBr): 3800w, 3571w, 3436w, 3317w, 3222w, 3091w, 3015w, 2960w, 2928s, 2865m, 2605w, 2221s (CC), 2108w, 1959w, 1843w, 1741w, 1718m, 1611s (NCN), 1464w, 1402m, 1373m, 1356w, 1330w, 1262w, 1220m, 1186m, 1140w, 1121w, 1079w, 1053w, 1029w, 968s, 891s, 843w, 811w, 788w, 745w, 712w, 702w, 644w, 595w, 530w, 472w, 453w, 440w cm−1. Anal. Calcd for C24H35HoN2O (532.49): C, 54.14; H, 6.63; N, 5.26. Found: C, 56.02; H, 6.00; N, 5.46. EI-MS: m/z (%) 460.35 (8) [M − THF], 531.46 (30) [M]+, 547.51 (80) [M + CH3)]+.
C–C(NCy)(THF) (14).
Compound 14 was isolated as bright yellow, needle-like single-crystals suitable for X-ray diffraction in 30% yield (0.65 g). IR (KBr): 3436w, 3224w, 3092w, 3011w, 2927s, 2852s, 2666s, 2225vs (CC), 1959w, 1821w, 1603s (NCN), 1476w, 1449s, 1402w, 1363w, 1310w, 1253w, 1209m, 1180m, 1156m, 1123w, 1075w, 1053w, 1029w, 974s, 922s, 890m, 858w, 810w, 775w, 701w, 680w, 642w, 612w, 589w, 504w, 465w cm−1. Anal. Calcd for C30H43HoN2O (612.62): C, 58.82; H, 7.08, N; 4.57. Found: C, 58.87; H, 6.53; N, 6.21. EI-MS: m/z (%) 269.12 (86) [M − (THF + (c-C3H5–CC–C(NCy)2))]+ or [c-C3H5–CC–C(NCy)2)]2+, 433.28 (10) [M − (THF + COT)]2+, 501.26 (33) [M − (THF + c-C3H5)]2+, 540.42 (31) [M − THF].
C–C(NCy)2)2]4 (15).
Anhydrous NdCl3 (1.0 g, 4 mmol) in 30 mL of THF was added to a mixture of 1b (1.10 g, 4 mmol) and K2COT (3.3 mL, 0.6 M in THF) in 50 mL of THF. The reaction mixture was stirred over night at room temperature. THF was removed under vacuum, followed by extraction the residue with 40 mL of toluene, the solution concentrated to 20 mL and then kept at 5 °C to afford 15 as blue crystals in 20% yield (1.9 g). 1H NMR (THF-d8): δ (ppm) 3.61 (s br, 8H, CH, Cy), 3.35 (s br, 8H, CH, Cy), 1.35 (s br, 18H, CH, c-C3H5), 0.84 (s br, 16H, CH2, c-C3H5), 0.71 (s br, 16H, CH2, c-C3H5), 1.01–2.01 (m br, 160H, CH2, Cy), −11.34 (s br, 32H, C8H8). 13C{1H} NMR (THF-d8): δ (ppm) 140.8 (NCN), 133.7 (C8H8), 61.1 (CH, Cy), 50.3 (CH, Cy), 36.1 (CH2, Cy), 33.7 (CH2, Cy), 26.7 (CH2, Cy), 8.9 (CH2, c-C3H5), 0.1 (CH, c-C3H5). IR (KBr): 3221w, 3091w, 3009w, 2929s, 2854s, 2668m, 2230s (CC), 1959w, 1627s (NCN), 1478w, 1450w, 1405(w), 1365w, 1345m, 1310m, 1247w, 1190w, 1151m, 1001m, 1075m, 1030w, 974s, 959s, 926w, 891w, 862w, 842w, 811w, 793w, 754w, 697w, 668w, 628w, 588w, 502w, 466w cm−1. Anal. Calcd for C190H264Cl8N16Nd8 (15·6C7H8) (4209.69 + 552.82): C, 58.45; H, 6.55; N, 4.70. Found: C, 57.28; H, 6.45; N, 5.20. EI-MS: m/z (%) 229.3 (100) [CC–C(c-C6H11N)2]+, 272.4 (83) [c-C3H5–CC–C(c-C6H11N)2]+, 363.5 (75) [(COT) Nd (μ-Cl)(c-C6H11)]+, 446.6 (20) [Nd(μ-Cl)(c-C3H5–CC–C(c-C6H11N)2)]2+.
Crystallographic details
The crystallographic data of compounds 2, 3, 7, 8 and 14 were collected on a STOE IPDS 2T diffractometer at −140 to −120 °C using graphite monochromated Mo-Kα radiation. Data collection, data reduction, space group determination and spherical absorption correction were performed with the STOE software X-AREA and X-RED32.37 The intensity data of compounds 10, 11, 12 and 15 were registered on Xcalibur Atlas Nova diffractometer using mirror-focussed Cu-Kα radiation. Data collection, data reduction and space group determination were performed with the Agilent software CrysAlisPro.38 Absorption corrections were applied using the multi-scan method. The structures were solved by direct methods (SHELXS-97)39 and refined by full matrix least-squares methods on F2 using SHELXL-97.40 Data collection parameters are given in Tables 1 and 2.Acknowledgements
Generous support of this work through a research grant by the Deutsche Forschungsgemeinschaft DFG (Grant No. AN 238/16 “Organocer(IV)-Chemie”) is gratefully acknowledged. This work was also financially supported by the Otto-von-Guericke-Universität Magdeburg. Farid M. Sroor is grateful to the Ministry of Higher Educational Scientific Research (MHESR), Egypt, and the Germany Academic Exchange Service (DAAD), Germany, for a Ph.D. scholarship within the German Egyptian Research Long-Term Scholarship (GERLS) program.Notes and references
- Recent review articles: (a) F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS; (b) M. P. Coles, Chem. Commun., 2009, 3659–3676 RSC; (c) C. Jones, Coord. Chem. Rev., 2010, 254, 1273–1289 CrossRef CAS; (d) A. A. Trifonov, Coord. Chem. Rev., 2010, 254, 1327–1347 CrossRef CAS; (e) A. A. Mohamed, H. E. Abdou and J. P. Fackler Jr., Coord. Chem. Rev., 2010, 254, 1253–1259 CrossRef CAS; (f) S. Collins, Coord. Chem. Rev., 2011, 255, 118–138 CrossRef CAS; (g) F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- (a) F. T. Edelmann, Chem. Soc. Rev., 2009, 38, 2253–2268 RSC; (b) F. T. Edelmann, Chem. Soc. Rev., 2012, 41, 7657–7672 RSC.
- A. Devi, Coord. Chem. Rev., 2013, 257, 3332–3384 CrossRef CAS , and references cited therein.
- (a) H. Fujita, R. Endo, A. Aoyama and T. Ichii, Bull. Chem. Soc. Jpn., 1972, 45, 1846–1852 CrossRef CAS; (b) G. Himbert, M. Feustel and M. Jung, Liebigs Ann. Chem., 1981, 1907–1927 CrossRef CAS; (c) G. Himbert and W. Schwickerath, Liebigs Ann. Chem., 1984, 85–97 CrossRef CAS; (d) G. F. Schmidt and G. Süss-Fink, J. Organomet. Chem., 1988, 356, 207–211 CrossRef CAS; (e) T.-G. Ong, J. S. O'Brien, I. Korobkov and D. S. Richeson, Organometallics, 2006, 25, 4728 CrossRef CAS; (f) X. Xu, J. Gao, D. Cheng, J. Li, G. Qiang and H. Guo, Adv. Synth. Catal., 2008, 350, 61–64 CrossRef CAS; (g) W. Weingärtner, W. Kantlehner and G. Maas, Synthesis, 2011, 265–272 Search PubMed; (h) W. Weingärtner and G. Maas, Eur. J. Org. Chem., 2012, 6372–6382 CrossRef.
- (a) H. Fujita, R. Endo, K. Murayama and T. Ichii, Bull. Chem. Soc. Jpn., 1972, 45, 1581 CrossRef CAS; (b) W. Ried and M. Wegwitz, Liebigs Ann. Chem., 1975, 89–94 CrossRef CAS; (c) W. Ried and R. Schweitzer, Chem. Ber., 1976, 109, 1643–1649 CrossRef CAS; (d) W. Ried and H. Winkler, Chem. Ber., 1979, 112, 384–388 CrossRef CAS.
- (a) P. Sienkiewich, K. Bielawski, A. Bielawska and J. Palka, Environ. Toxicol. Pharmacol., 2005, 20, 118–124 CrossRef PubMed; (b) T. M. Sielecki, J. Liu, S. A. Mousa, A. L. Racanelli, E. A. Hausner, R. R. Wexler and R. E. Olson, Bioorg. Med. Chem. Lett., 2001, 11, 2201–2204 CrossRef CAS PubMed; (c) C. E. Stephens, E. Tanious, S. Kim, D. W. Wilson, W. A. Schell, J. R. Perfect, S. G. Franzblau and D. W. Boykin, J. Med. Chem., 2001, 44, 1741–1748 CrossRef CAS PubMed; (d) C. N. Rowley, G. A. DiLabio and S. T. Barry, Inorg. Chem., 2005, 44, 1983–1991 CrossRef CAS PubMed.
- (a) W.-X. Zhang, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2005, 127, 16788–16789 CrossRef CAS PubMed; (b) S. Zhou, S. Wang, G. Yang, Q. Li, L. Zhang, Z. Yao, Z. Zhou and H.-B. Song, Organometallics, 2007, 26, 3755–3761 CrossRef CAS; (c) W.-X. Zhang and Z. Hou, Org. Biomol. Chem., 2008, 6, 1720–1730 RSC; (d) Z. Du, W. Li, X. Zhu, F. Xu and Q. Shen, J. Org. Chem., 2008, 73, 8966–8972 CrossRef CAS PubMed; (e) C. N. Rowley, T.-G. Ong, J. Priem, D. S. Richeson and T. K. Woo, Inorg. Chem., 2008, 47, 12024–12031 CrossRef CAS PubMed; (f) Y. Wu, S. Wang, L. Zhang, G. Yang, X. Zhu, Z. Zhou, H. Zhu and S. Wu, Eur. J. Org. Chem., 2010, 326–332 CrossRef; (g) F. Zhang, J. Zhang, Y. Zhang, J. Hong and X. Zhou, Organometallics, 2014, 33, 6186–6192 CrossRef CAS.
- (a) P. Dröse, C. G. Hrib and F. T. Edelmann, J. Organomet. Chem., 2010, 695, 1953–1956 CrossRef; (b) L. Xu, Y.-C. Wang, W.-X. Zhang and Z. Xi, Dalton Trans., 2013, 42, 16466–16469 RSC.
- W. W. Seidel, W. Dachtler and T. Pape, Z. Anorg. Allg. Chem., 2012, 638, 116–121 CrossRef CAS.
- Selected references: (a) P. Fischer, W. Kurtz and F. Effenberger, Chem. Ber., 1973, 106, 549–559 CrossRef CAS; (b) C. F. Wilcox, L. M. Loew and R. Hoffman, J. Am. Chem. Soc., 1973, 95, 8192–8193 CrossRef CAS; (c) A. de Meijere, Angew. Chem., Int. Ed. Engl., 1979, 18, 809–826 CrossRef; (d) H. C. Brown, M. Periasamy and P. T. Perumal, J. Org. Chem., 1984, 49, 2754–2757 CrossRef CAS; (e) D. D. Roberts, J. Org. Chem., 1991, 56, 5661–5665 CrossRef CAS; (f) R. R. Sauers, Tetrahedron, 1998, 54, 337–348 CrossRef CAS; (g) A. de Meijere and S. I. Kozhushkov, Mendeleev Commun., 2010, 20, 301–311 CrossRef CAS.
- F. M. A. Sroor, C. G. Hrib, L. Hilfert and F. T. Edelmann, Z. Anorg. Allg. Chem., 2013, 639, 2390–2394 CrossRef CAS.
- F. M. Sroor, C. G. Hrib, L. Hilfert, P. G. Jones and F. T. Edelmann, J. Organomet. Chem., 2015, 785, 1–10 CrossRef CAS.
- F. M. Sroor, C. G. Hrib, L. Hilfert, S. Busse and F. T. Edelmann, New J. Chem., 2015, 39, 7595–7601 RSC.
- F. M. Sroor, C. G. Hrib, L. Hilfert, L. Hartenstein, P. W. Roesky and F. T. Edelmann, J. Organomet. Chem., 2015, 799–800, 160–165 CrossRef CAS.
- (a) F. Mares, K. O. Hodgson and A. Streitwieser, J. Organomet. Chem., 1970, 24, C68–C70 CrossRef CAS; (b) K. O. Hodgson and K. N. Raymond, Inorg. Chem., 1972, 11, 3030–3035 CrossRef CAS; (c) K. O. Hodgson, F. Mares, D. Starks and A. Streitwieser, J. Am. Chem. Soc., 1973, 95, 8650–8658 CrossRef CAS; (d) S. A. Kinsley, A. Streitwieser and A. Zalkin, Organometallics, 1985, 4, 52–57 CrossRef CAS; (e) T. R. Boussie, D. C. Eisenberg, J. Rigsbee, A. Streitwieser and A. Zalkin, Organometallics, 1991, 10, 1922–1928 CrossRef CAS; (f) P. Poremba, U. Reißmann, M. Noltemeyer, H.-G. Schmidt, W. Brüser and F. T. Edelmann, J. Organomet. Chem., 1997, 544, 1–6 CrossRef.
- (a) F. Mares, K. O. Hodgson and A. Streitwieser, J. Organomet. Chem., 1971, 28, C24–C26 CrossRef CAS; (b) K. O. Hodgson and K. N. Raymond, Inorg. Chem., 1972, 11, 171–175 CrossRef CAS; (c) A. Streitwieser Jr. and T. R. Boussie, Eur. J. Solid State Inorg. Chem., 1991, 28, 399–405 Search PubMed; (d) W. E. Evans, R. D. Clarck, M. A. Ansari and J. W. Ziller, J. Am. Chem. Soc., 1998, 120, 9555–9563 CrossRef CAS; (e) G. W. Rabe, M. Zhang-Presse, J. A. Golen and A.-L. Rheingold, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m255–m256 CAS.
- H. Schumann, J. Winterfeld, H. Hemling, F. E. Hahn, P. Reich, K.-W. Brezezinka, F. T. Edelmann, U. Kilimann, M. Schäfer and R. Herbst-Irmer, Chem. Ber., 1995, 128, 395–404 CrossRef CAS.
- P. Dröse, C. G. Hrib and F. T. Edelmann, J. Organomet. Chem., 2010, 695, 1953–1956 CrossRef.
- J. Richter, J. Feiling, H.-G. Schmidt, M. Noltemeyer, W. Brüser and F. T. Edelmann, Z. Anorg. Allg. Chem., 2004, 630, 1269–1275 CrossRef CAS.
- M. Wedler, F. Knösel, U. Pieper, D. Stalke, F. T. Edelmann and H.-D. Amberger, Chem. Ber., 1992, 125, 2171–2181 CrossRef CAS.
- V. Lorenz, S. Blaurock, C. G. Hrib and F. T. Edelmann, Dalton Trans., 2010, 39, 6629–6631 RSC.
- H. Schumann, J. Winterfeld, L. Esser and G. Kociok-Köhn, Angew. Chem., Int. Ed. Engl., 1993, 32, 1208–1210 CrossRef.
- W. J. Evans, M. A. Johnston, R. D. Clark, R. Anwander and J. W. Ziller, Polyhedron, 2001, 20, 2483–2490 CrossRef CAS.
- V. Lorenz, S. Blaurock, C. G. Hrib and F. T. Edelmann, Dalton Trans., 2010, 39, 6629–6631 RSC.
- U. Reißmann, P. Poremba, M. Noltemeyer, H.-G. Schmidt and F. T. Edelmann, Inorg. Chim. Acta, 2000, 303, 156–162 CrossRef.
- S. M. Cendrowski-Guillaume, G. Le Gland, M. Nierlich and M. Ephritikhine, Organometallics, 2000, 19, 5654–5660 CrossRef CAS.
- T. M. Gilbert, R. R. Ryan and A. P. Sattelberger, Organometallics, 1988, 7, 2514–2518 CrossRef CAS.
- W. J. Evans, M. K. Takase, J. W. Ziller and A. L. Rheingold, Organometallics, 2009, 28, 5802–5808 CrossRef CAS.
- M. K. Takase, N. A. Siladke, J. W. Ziller and W. J. Evans, Organometallics, 2011, 30, 458–465 CrossRef CAS.
- P. W. Roesky, J. Organomet. Chem., 2001, 621, 277–283 CrossRef CAS.
- G. W. Rabe, M. Zhang-Preße and G. P. A. Yap, Inorg. Chim. Acta, 2003, 348, 245–248 CrossRef CAS.
- A. Edelmann, V. Lorenz, C. G. Hrib, L. Hilfert, S. Blaurock and F. T. Edelmann, Organometallics, 2013, 32, 1435–1444 CrossRef CAS.
- V. Lorenz, S. Blaurock, C. G. Hrib and F. T. Edelmann, Organometallics, 2010, 29, 4787–4789 CrossRef CAS.
- J. H. Freeman and M. L. Smith, J. Inorg. Nucl. Chem., 1958, 7, 224–227 CrossRef CAS.
- T. J. Katz, J. Am. Chem. Soc., 1960, 82, 3784–3785 CrossRef CAS.
- M. T. Gamer and P. W. Roesky, Inorg. Chem., 2005, 44, 5963–5965 CrossRef CAS PubMed.
- Stoe & Cie, X-AREA Program for X-ray Crystal Data collection (X-RED32 included in X-AREA), 2002 Search PubMed.
- Agilent Technologies, CrysAlisPro Program for X-ray Crystal Data collection, Vers. 1.171.36.28, 2013 Search PubMed.
- G. M. Sheldrick, SHELXS-97 Program for Crystal Structure Solution, Universität Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97 Program for Crystal Structure Refinement, Universität Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full crystallographic data for 2, 3, 4, 7, 8, 10, 11, 12 and 15. CCDC 1057405–1057413. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01974a |
| This journal is © The Royal Society of Chemistry 2016 |