
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the importance of tetrel bonding interactions in lead(II) complexes with (iso)nicotinohydrazide based ligands and several anions†
Ghodrat
Mahmoudi
*a,
Antonio
Bauzá
b,
Mojtaba
Amini
a,
Elies
Molins
c,
Joel T.
Mague
d and
Antonio
Frontera
 *b
*b
aDepartment of Chemistry, Faculty of Science, University of Maragheh, P.O. Box 55181-83111, Maragheh, Iran. E-mail: mahmoudi_ghodrat@yahoo.co.uk
bDepartamento de Quimica, Universitat de les Illes Balears, Crta. de Valldemossa km 7.5, 07122 Palma de Mallorca, Baleares, Spain. E-mail: toni.frontera@uib.es
cInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC), Campus de la Universitat Autònoma de Barcelona, 08193 Bellaterra, Spain
dDepartment of Chemistry, Tulane University, New Orleans, LA 70118, USA
First published on 1st June 2016
Abstract
Four Pb(II) complexes of (iso)nicotinohydrazide-based ligands and different anionic coligands (azide, nitrate, acetate and iodide) have been synthesized and characterized by structural, analytical and spectroscopic methods. The mono-deprotonated or neutral forms of the ligands coordinate in a tridentate fashion via two nitrogen and one oxygen donor atoms to the Pb. Single-crystal X-ray crystallography showed that the molecular complexes can aggregate into larger entities depending upon the anion coordinated to the metal centre. In all four complexes the lead center is hemidirectionally coordinated and, consequently, it is sterically ideal for establishing tetrel bonding interactions. In the crystal structures of all complexes, the lead atoms participate in short contacts with nitrogen atoms. These contacts are shorter than the sums of the van der Waals radii and larger than the sums of the covalent radii. The tetrel bonding interactions interconnect the covalently bonded units (monomers or dimers) into supramolecular assemblies (chains and 3D structures). We have analysed the interesting supramolecular assemblies observed in the solid state of all four complexes by means of DFT calculations.
1. Introduction
Metal–organic frameworks (MOFs) are continuously attracting the attention of chemists working in crystal engineering, solid-state and materials chemistry due to their promising applications in multidisciplinary fields like photoluminescence and electronics, magnetism, gas absorption, and many others.1 Nowadays, it is possible to modify and tune the properties of MOFs by using different ligands,2 metal centres with different coordination behaviour,3 and counter ions.4 More importantly, it is also possible to modulate their properties using supramolecular interactions.5Apart from the widely used hydrogen bonding interactions for the construction/modification of MOFS, other specific noncovalent interactions like σ–hole interactions, especially halogen bonds, are becoming prominent players.6 Halogens, however, are not the only atoms susceptible to formation of σ–holes.7 Electron-deficient regions are also present on covalently bonded heavier atoms of groups IV to VII and are located in the extension of the covalent bond. The size and electron deficiency of σ–holes increases with increasing electron-withdrawing nature of the covalently bonded group and with the polarizability of the atom on which the σ–hole is formed. The strongest σ–hole interactions occur in systems where a heavy atom of groups IV to VII is covalently bonded to electronegative ones. Although σ–hole interactions have been widely described for groups VI (chalcogen interaction) and V (pnictogen interaction), those of group IV (tetrel interaction) are scarce,8 However, it has been shown that they can be markedly strong, in particular with Sn and Pb, the metallic members of the group. Previously some of us have reported the design and synthesis of lead(II) metal–organic frameworks (MOFs) based on covalent and noncovalent tetrel bonding.9 In these MOFs, Pb⋯S and Pb⋯N noncovalent tetrel bonds interconnect the covalently bonded units into supramolecular assemblies. A combined DFT study and a statistical survey of the Cambridge Structural Database (CSD) revealed that tetrel bonds with hemidirectionally coordinated lead occur with high probability and with predictable geometries, thus playing an important role in the solid-state chemistry of lead. Moreover, we have recently shown that concurrent noncovalent tetrel bonding, agostic interactions and chelate ring–chelate ring stacking interactions control the supramolecular architectures and organometallic frameworks observed in the solid state architecture of hemidirectionally coordinated PbX2 salts (X = Cl, NO2, I and SCN) to the N′-(phenyl(pyridin-2-yl)methylene)-isonicotinohydrazide ligand.10
Taking advantage of this understanding, herein we report the synthesis and X-ray characterization of four new Pb(II) complexes with (iso)nicotinohydrazide-based ligands and different anions (azide, nitrate, acetate and iodine) as coligands (see Scheme 1). Interestingly, in all complexes the Pb(II) is hemidirectionally coordinated and participates in noncovalent tetrel bonding interactions. These forces along with stacking interactions control the supramolecular architectures and organometallic frameworks observed in their solid state architecture. In line with previous reports, the Pb2+ shows a tendency to establish an unusual coordination mode in which the Pb2+ ion has a clear void in the distribution of bonds to the ligands (hemidirectional coordination).11 Such void in the coordination sphere facilitates the approach of electron donors to the σ–hole of the Pb2+ enabling the formation of a strong tetrel bond with a more predictable geometry. The nature of the tetrel bonds in all four structures was studied by DFT calculations, which showed the presence of the σ–hole at the Pb atom and the considerable strength of these interactions.
 | ||
| Scheme 1 Synthetic route to complexes 1–4. | ||
2. Experimental
2.1. Materials and methods
All reagents used in these studies were obtained from commercial sources and used as received. The ligands HL1, HL2 and HL3 were prepared using the procedure previously described.12 Briefly, they were obtained by the condensation of either 2-acetylpyridine or 2-formylpyridine with (iso)nicotinohydrazide in ethanol solution. Recrystallization from aqueous EtOH afforded pure ligands in >80% yield.Elemental analyses were carried out using an Elemental Vario EL III instrument and FT-IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer with KBr disks in the range 4000–400 cm−1. Caution! Lead and its compounds are toxic.13 Only a small amount of these materials should be prepared and handled with care.
2.2. Synthesis of [Pb2(L1)2(N3)2] (1)
To a magnetically stirred solution of 2-formyl-pyridyl-nicotinoylhydrazone (0.113 g, 0.5 mmol) in ethanol, was added an aqueous solution of PbCl2 (0.138 g, 0.5 mmol) and NaN3 (0.0325 g, 0.5 mmol) in portions. The resulting reaction mixture was left unperturbed for slow evaporation of the solvent. After 7 days, crystals of 1 were obtained. Yield: 70% (0.0166 g). Anal. Calc. for C12H9N7OPb (1): C, 30.4; H, 1.9; N, 20.7. Found: C, 31.0; H, 2.0; N, 21.8%. IR (KBr cm−1) selected bands: ν = CH b (oop): 669 (m) and 775 (m); CH b: 1317 (m); CCst: 1458 (m); C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N st: 1495 and 1584 (m); C–O st (Ligand) 1646; N–NN st: 2012 (s); CH st: 3067 (w), and OH st: 3448 (w) cm−1.
N st: 1495 and 1584 (m); C–O st (Ligand) 1646; N–NN st: 2012 (s); CH st: 3067 (w), and OH st: 3448 (w) cm−1.
2.3. Synthesis of [Pb2(HL2)2(NO3)4] (2) and [Pb(HL2)I2] (3)
Compounds 2–3 were prepared similarly using the same stoichiometry and reaction condition except that 2-acetyl-pyridyl-nicotinoylhydrazone (0.120 g, 0.5 mmol) instead of 2-formyl-pyridyl-nicotinoylhydrazone and Pb(NO3)2 (0.165 g, 0.1 mmol), PbI2 (0.230 g, 0.5 mmol), instead of PbCl2 was used.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 382 (s); CN st: 1467 and 1589 (m); C–O st (Ligand) 1634; CH st: 3073 (w), NH st: 3205 (w) and OH st: 3431 (w) cm−1.
N st: 1501 and 1582 (m); C–O st (Ligand) 1649; CH st: 3043 (w), NH st: 3219 (w) and OH st: 3446 (w) cm−1.
382 (s); CN st: 1467 and 1589 (m); C–O st (Ligand) 1634; CH st: 3073 (w), NH st: 3205 (w) and OH st: 3431 (w) cm−1.
N st: 1501 and 1582 (m); C–O st (Ligand) 1649; CH st: 3043 (w), NH st: 3219 (w) and OH st: 3446 (w) cm−1.
2.4. Synthesis of [Pb(L3)(OAc)] (4)
Compound 4 was prepared similarly using the same stoichiometry and reaction condition except that 2-acetyl-pyridyl-isonicotinoylhydrazone (0.120 g, 0.5 mmol) instead of 2-formyl-pyridyl-nicotinoylhydrazone and Pb(CH3COO)2·3H2O (0.165 g, 0.1 mmol) instead of PbCl2 was used.N st: 1507 and 1573 (m); C–O st (Ligand) 1648; O–CO st: 1742 (s); CH st: 3096 (w), and OH st: 3446 (w) cm−1.
2.5. X-ray crystallography
A single crystal of compound 1, suitable for X-ray analysis, was used for intensity data collection on a Nonius Kappa CCD FR590 diffractometer with mirror-monochromatized Mo-Kα radiation (λ = 0.71073 Å). The data set for compound 2 was collected on a Bruker Smart APEX CCD diffractometer using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The data set for compound 3 and 4 were collected on a Bruker AXS diffractometer with mirror-monochromatized Mo-Kα radiation (λ = 0.71073 Å). All data collections were performed at different temperatures (100–300 K). The structures of all compounds were determined by direct methods and refined by full-matrix least-squares procedures using the SHELXTL package.14 All the hydrogen atoms were fixed in calculated positions except those that were detected in the Fourier map. The molecular structure plots were prepared with Mercury.15 The details of the crystal parameters, data collection and refinements for all the complexes are summarized in Table 1. Tables S1–S4 (ESI†) contain the coordination bond distances and angles of the structures reported. Further details are available from the Cambridge Crystallographic Centre with quotation numbers CCDC 1474989 and 1480033–1480035.| Compound | 1 | 2 | 3 | 4 |
|---|---|---|---|---|
| R 1 = ∑[|Fo| − |Fc|/∑|Fo|], wR2 = {[∑w(|Fo2 − Fc2|)2]/[∑w(Fo2)2}]1/2. | ||||
| Formula | C24H18N14O2Pb2 | C26H24N12O14Pb2 | C13H12I2N4OPb | C15H14N4O3Pb |
| Fw | 948.92 | 1142.96 | 701.26 | 505.49 |
| Crystal system | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P21/n | P21/n |
| a (Å) | 5.834(7) | 9.0862(4) | 8.0385(12) | 7.4569(15) |
| b (Å) | 8.133(4) | 9.3122(4) | 16.069(2) | 13.319(3) |
| c (Å) | 14.800(4) | 11.5175(5) | 13.0637(19) | 15.894(4) |
| α (°) | 85.65(4) | 84.905(1) | 90 | 90 |
| β (°) | 81.87(10) | 72.809(1) | 97.536(4) | 98.541(7) |
| γ (°) | 70.41(7) | 62.775(1) | 90 | 90 |
| V (Å3) | 654.6(9) | 826.61(6) | 1672.9(4) | 1561.1(6) |
| Temp (K) | 293(2) | 100(2) | 179(2) | 300(2) |
| Z | 1 | 1 | 4 | 4 |
| D c (g cm−3) | 2.412 | 2.063 | 2.784 | 2.151 |
| μ (mm−1) | 12.892 | 10.259 | 13.779 | 10.827 |
| No. reflections | 2859 | 14331 |
3641 | 9580 |
| Reflecs I > 2σ(I) | 2383 | 3973 | 2857 | 2718 |
| R int | 0.0438 | 0.0196 | 0.0332 | 0.0308 |
| F(000) | 442 | 540 | 1256 | 952 |
| R 1 (I > 2σ(I)) | 0.055 | 0.0198 | 0.0507 | 0.0560 |
| wR (all data) | 0.1073 | 0.0484 | 0.0731 | 0.0704 |
| GOF | 1.084 | 1.079 | 1.046 | 1.060 |
2.6. Theoretical methods
The geometries of the complexes included in this study were computed at the BP86-D3/def2-TZVP level of theory using the crystallographic coordinates within the TURBOMOLE program.16 This level of theory that includes the latest available dispersion correction (D3) is adequate for studying non-covalent interactions dominated by dispersion effects like π-stacking. The basis set superposition error for the calculation of interaction energies has been corrected using the counterpoise method.17 The “atoms-in-molecules” (AIM)18 analysis of the electron density has been performed at the same level of theory using the AIMAll program.193. Results and discussion
3.1. Synthesis and spectroscopy
The compounds have been synthesized as described in the Experimental section and the reaction pathways are sketched in Scheme 1.The ligands are expected to exhibit keto–enol tautomerism, and the amide proton can be dissociated through enolization, as observed in compounds 1 and 4. The IR spectrum of the ligands exhibits two bands in the regions 3205 and 1660 cm−1 due to ν(N–H) and ν(CO) stretching, respectively. The free ligand amide N–H and CO stretching vibrations are not observed in the IR spectrum of compounds 1 and 4, but these compounds exhibit a strong absorption band at 1630–1640 cm−1, not observed in the free ligand, which is due to the conjugated CN–NC–O moiety. Therefore in these compounds the deprotonated ligands are in the enolate form. By contrast the IR spectra of 2 and 3, where the amide N-atom is not deprotonated, exhibit both the amide and CO vibrations and there is no peak characteristic of the deprotonated ligand.
3.2. Crystal structures of 1–4
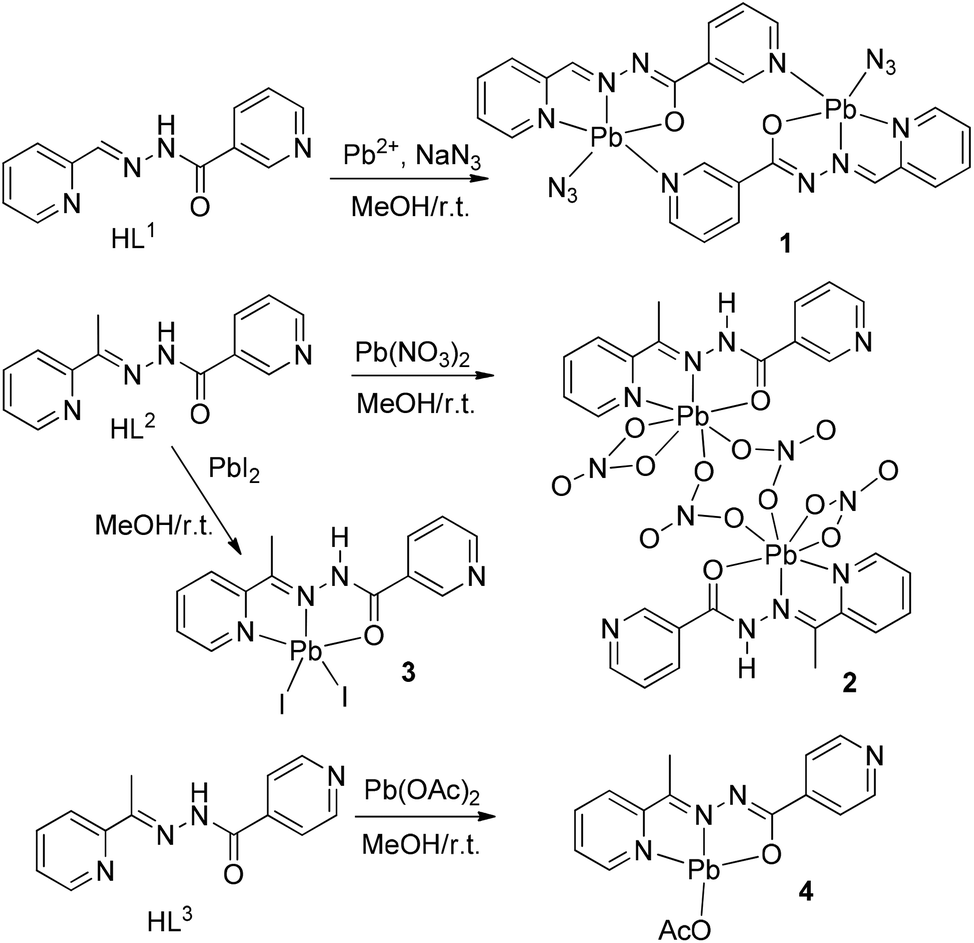
space group and consists of a dinuclear metallacycle complex molecule (Fig. 1) with the asymmetric unit containing one-half of the metallacycle unit. Significant structural parameters are given in Table S1.† The structure of the molecule shows that each mononegative ligand (L1) binds to the Pb(II) center via N,N,O-donor atoms, resulting in two five membered chelate rings. The other two coordination sites around Pb(II) are occupied by the axial azide ligand and the pyridine N atom of the other nicotinohydrazide ligand, thus generating the metallacycle. It is also worth stressing that the bonding parameters are in good agreement with the coordinating pattern of a CN–NC–O moiety via the enolate form: C6–N6 [1.27(1) Å], N6–N7 [1.36(1) Å], N7–C7 [1.32(1) Å] and C7–O7 [1.28(1)] bond distances.
 | ||
| Fig. 1 X-ray molecular structure of 1 with the atom numbering scheme. | ||
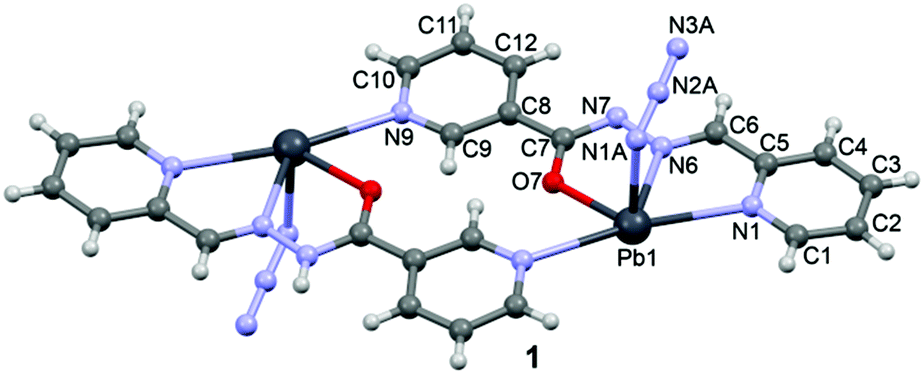
In the crystal structure of 1 the Pb2+ ion is fivefold-coordinated by four atoms of the chelating ligands L1 (N1, N6, N9i, and O1; i = −x, 2 − y, 2 − z), and a nitrogen atom (N1A) of the azido ligand. All bonds are basically concentrated on one hemisphere of the coordination sphere, thus leaving a large gap on the Pb2+ ion, which enables close approach of a second azide nitrogen atom (N3A) from the adjacent complex. These Pb1⋯N3A contacts further interconnect the metallacycles into a polymeric chain in the solid state (Fig. 2).
 | ||
| Fig. 2 Chains in the crystal structure of 1. Tetrel bonds Pb⋯N are shown as dashed lines. Distance in Å. | ||
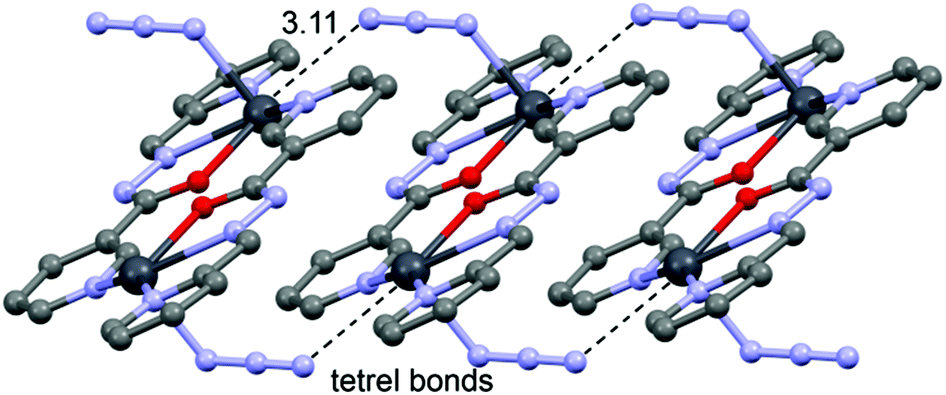
space group and consists of a dinuclear metallacycle complex molecule (Fig. 3) with the asymmetric unit containing one-half of the metallacycle unit. Significant structural parameters are given in Table S2.† The structure of the molecule (Fig. 3) shows that each neutral ligand (HL2) binds to the Pb(II) center via N,N,O-donor atoms resulting in two five membered chelate rings. The other three coordination sites around Pb(II) are occupied by one bidentate and two bridging nitrato ligands forming the metallacycle. The bonding parameters are in good agreement with the coordinating pattern of a CN–NH–CO moiety: C6–N2 [1.288(5) Å], N2–N3 [1.386(4) Å], N3–C8 [1.354(5) Å] and C8–O7 [1.234(3) Å] bond distances.
 | ||
| Fig. 3 Perspective view of the molecular structure of 2 with the atom numbering scheme. | ||
In the crystal structure of 2 the Pb2+ ion is sevenfold-coordinated by three atoms of the chelating ligands HL2 (N1, N2 and O7), and four oxygen atoms of the nitrato ligands (O1, O2, O4ii and O6, ii = 2 − x, 1 − y, −z). Remarkably, all seven coordination bonds are concentrated on one hemisphere of the coordination sphere, thus leaving a large gap on the Pb2+ ion, which enables close approach of the nitrogen atom (N4) of the uncoordinated pyridine ring of the ligand belonging to a neighbouring complex. These Pb1⋯N6 contacts interconnect the metallacycles into a polymeric chain in the solid state (Fig. 4).
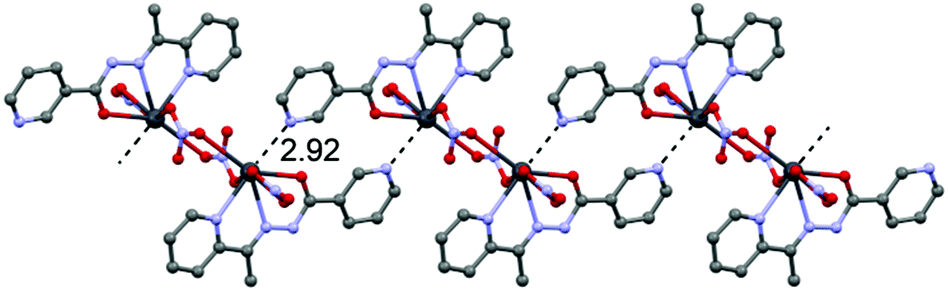 | ||
| Fig. 4 Chains in the crystal structure of 2. Tetrel bonds Pb⋯N are shown as dashed lines. Distance in Å. | ||
N–NH–CO moiety: C2–N2 [1.271(9) Å], N2–N1 [1.389(9) Å], N1–C1 [1.366(9) Å] and C1–O1 [1.212(8) Å] bond distances.
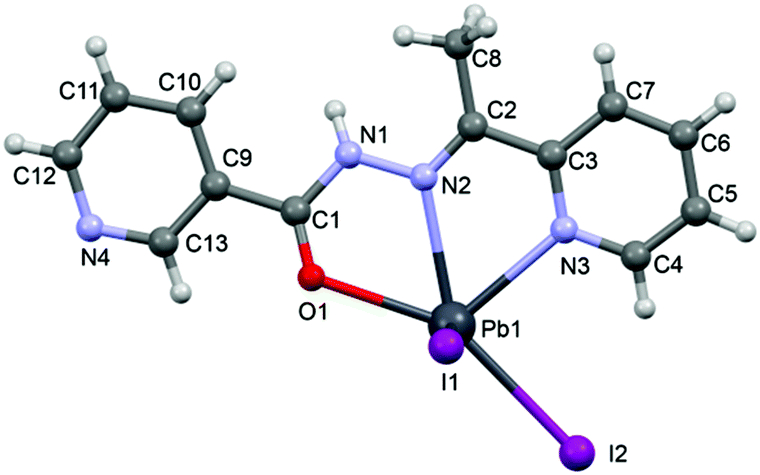 | ||
| Fig. 5 Perspective view of the molecular structure of 3 with the atom numbering scheme. | ||
In the crystal structure of 3 the Pb2+ ion is fivefold-coordinated by three atoms of the chelating ligands HL2 (N1, N3 and O1), and two iodide counterions. Clearly, all five coordination bonds are concentrated on less than one hemisphere of the coordination sphere, thus leaving a large gap on the Pb2+ ion, which enables close approach of the nitrogen atom (N4) of the uncoordinated pyridine ring of the ligand belonging to a neighbouring complex. These Pb1⋯N4 tetrel bonding contacts control the formation of self-assembled dimers (see Fig. 6) in the solid state structure. The coordination bonds and the noncovalent Pb1⋯N4 contact fulfil one half of the coordination sphere. The other hemisphere is used to establish chelate ring⋯π stacking interactions that connect and propagate the self-assembled dimers in the solid state (see Fig. 6) interactions.
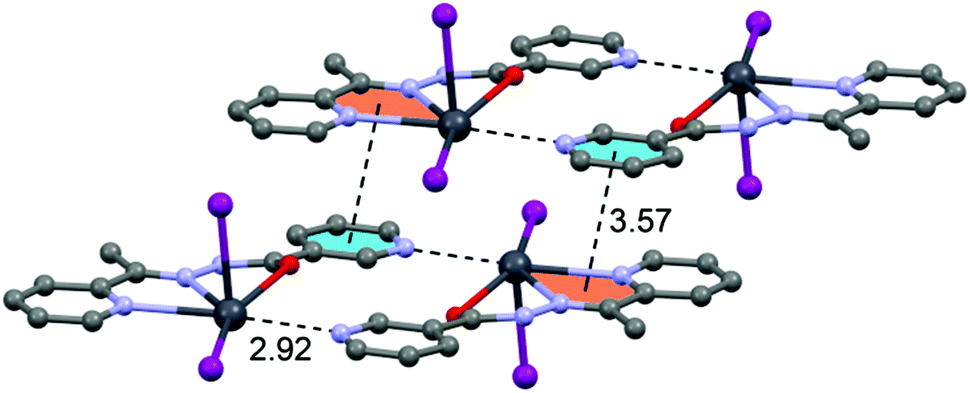 | ||
| Fig. 6 Self-assembled dimers observed in the crystal structure of 3. Tetrel bonds Pb⋯N and chelate ring⋯π interactions are shown as dashed lines. Distance in Å. | ||
N–NC–O moiety via the enolate form: C4–N1 [1.300(10) Å], N2–N4 [1.391(9) Å], N4–C5 [1.320(10) Å] and C5–O3 [1.290(10) Å] bond distances.
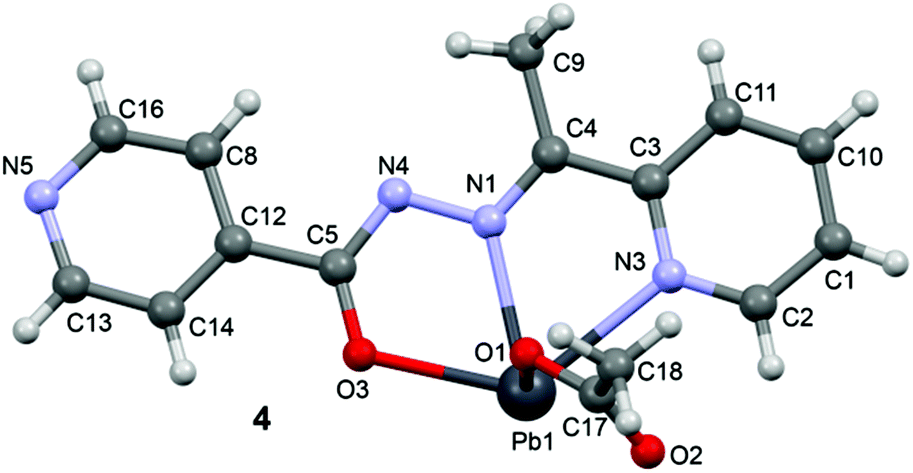 | ||
| Fig. 7 Perspective view of the molecular structure of 4 with the atom numbering scheme. | ||
In the crystal structure of 4 the Pb2+ ion is only fourfold-coordinated by three atoms of the chelating ligands L3 (N1, N3 and O3), and one atom (O1) from the acetate. Clearly, all four coordination bonds are concentrated on less than one hemisphere of the coordination sphere, thus leaving a large gap on the Pb2+ ion, which enables a close interaction with the nitrogen atom (N5) of the uncoordinated pyridine ring of the ligand belonging to another complex. Moreover, opposite to the axial acetate ligand this compound forms a self-assembled π-stacked supramolecular complex. The antiparallel arrangement of the complexes allows the formation of two symmetrically related π–π interactions between the coordinated pyridine ring of one ligand and the uncoordinated pyridine ring of the other ligand. This self-assembled dimer establishes four equivalent Pb1⋯N5 tetrel bonding contacts, two as donor and two as acceptor (see Fig. 8) controlling the final 3D architecture of this compound in solid state structure.
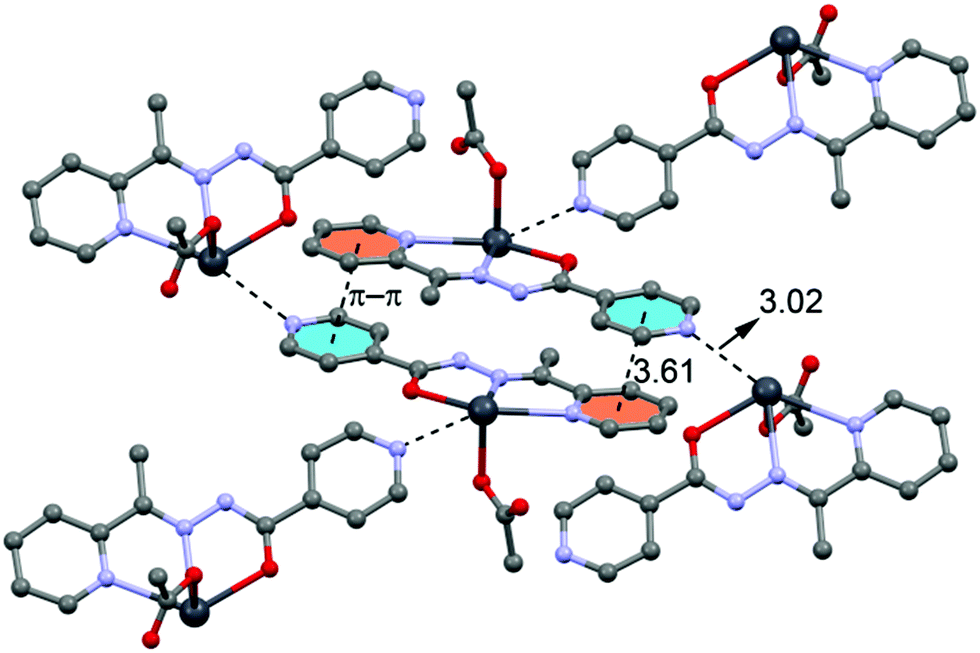 | ||
| Fig. 8 Self-assembled dimer observed in the crystal structure of 4 and its interaction with neighbouring molecules. Tetrel bonds Pb⋯N and π⋯π interactions are shown as dashed lines. Distances in Å. | ||
In addition to the tetrel bonding and stacking interactions described above for compounds 1–4, H-bonding interactions are also important, especially in complexes with neutral ligands (2 and 3). This is due to the presence of the highly acidic CONH amide proton. The coordination of the amide oxygen atom of the ligand to Pb2+ dramatically increases the H-bond donor ability of the amide proton. Consequently both compounds present N–H⋯X (X = O, I) interactions that govern the formation of relevant supramolecular assemblies (1D chains) in their solid state structures, as depicted in Fig. 9.
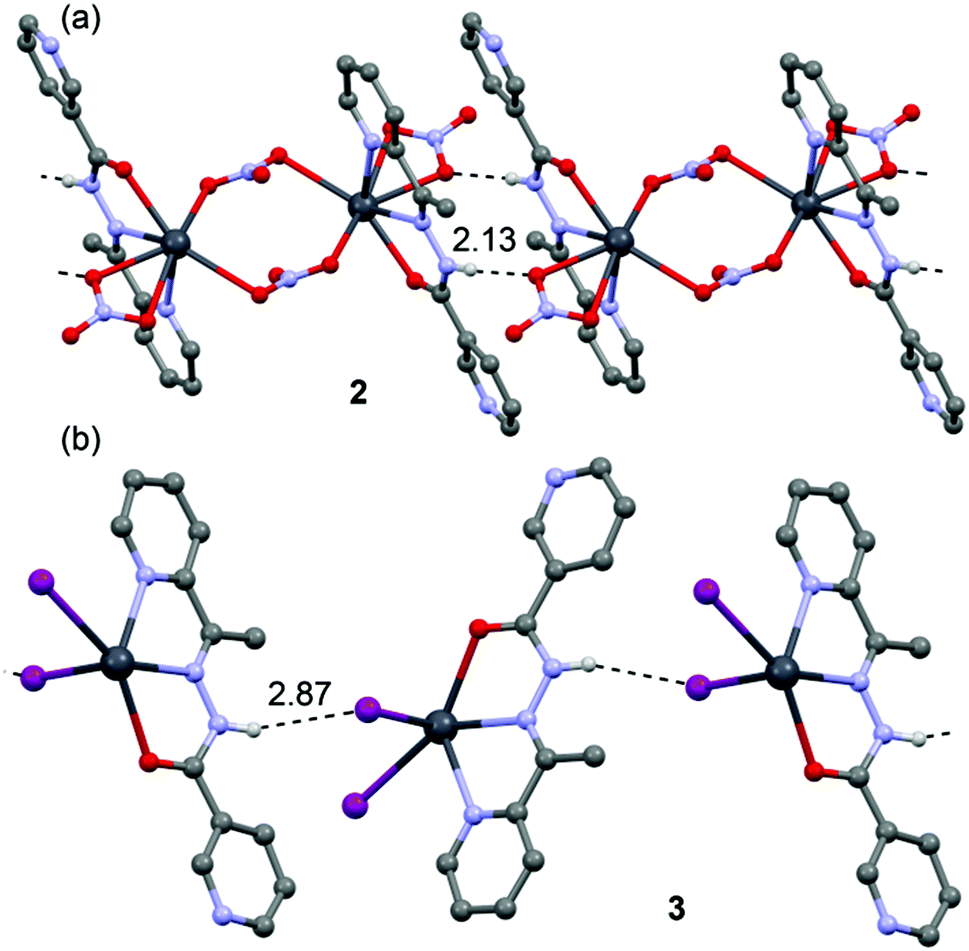 | ||
| Fig. 9 N–H⋯O/I H-bonded chains in the crystal structure of 2 (a) and 3 (b). H-bonds are shown as dashed lines. Distances in Å. | ||
3.3. Theoretical study of the supramolecular assemblies
We have focused the theoretical study on the analysis of the remarkable supramolecular assemblies observed in the solid state of complexes 1–4, directing our attention to the more unconventional, tetrel bonding and stacking interactions. Particularly in compounds 1 and 2 we have analysed the Pb⋯N interactions that have a prominent role in the formation of infinite 1D chains in the solid state. Moreover, in compounds 3 and 4 we have analysed the combination of π-stacking interactions (chelate–π in 3 and π–π in 4) and tetrel bonding interactions that control the formation of self-assembled dimers.In order to analyse the donor–acceptor properties of the Pb(II) complexes ligand (in both enolate and neutral forms), we have computed the molecular electrostatic potential (MEP) surface of compound 1 (enolate) and 3 (neutral), which are represented in Fig. 10 and 11. In the MEP surface of compound 1 the most positive part corresponds to the metal center and, unexpectedly, the most negative part is located at the N atom of the pyridine instead of the N3− ligand, thus explaining the formation of the dinuclear metallacycle in 1. In complex 4, where the ligand is also in the enolate form does not form the metallacycle due to the different directionality of the pyridine N-atom (para). An interesting aspect of the MEP surface is that the MEP values over the pyridine ring centers present opposite signs (see Fig. 10b). Therefore the enolate form of the ligand is well prepared to form strong π–π interactions (electrostatically assisted) between the coordinated and uncoordinated pyridine rings, as observed in 4 (see Fig. 8).
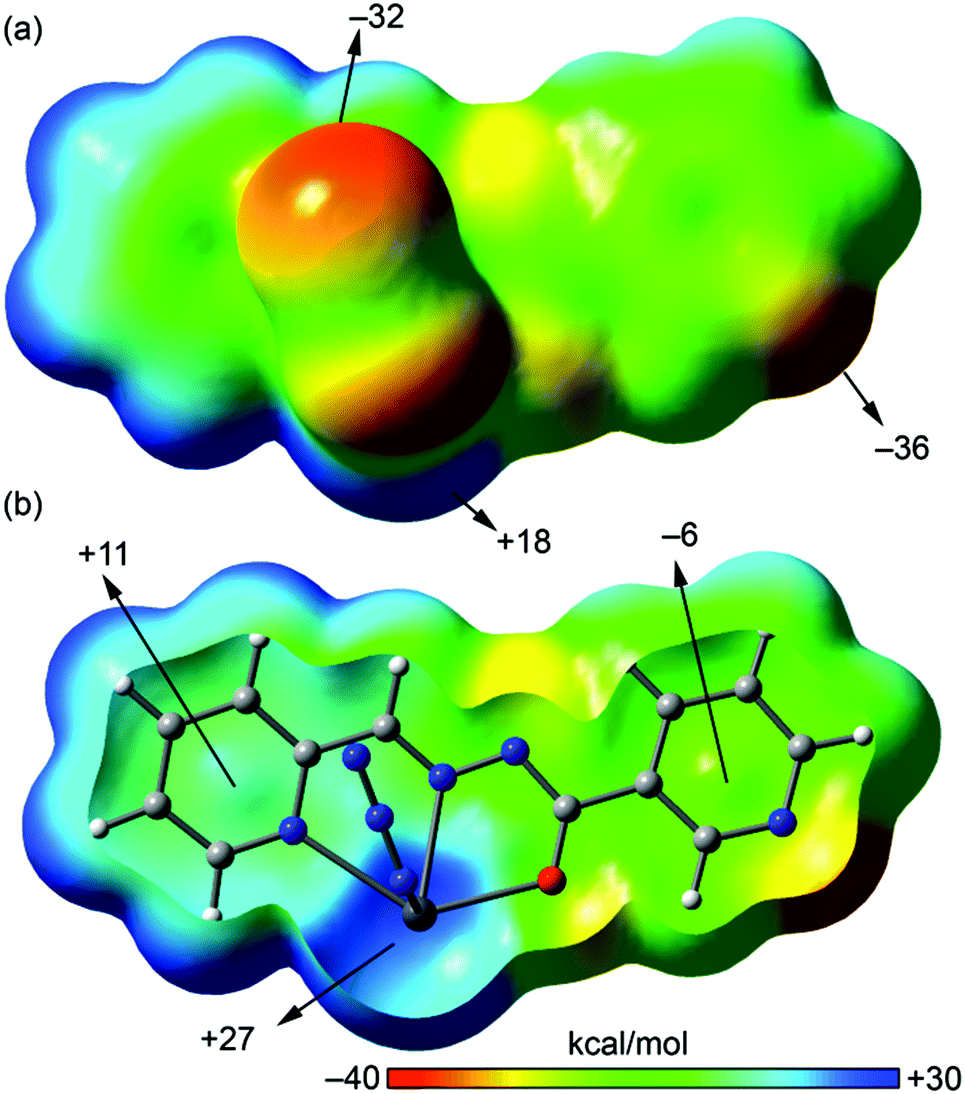 | ||
| Fig. 10 MEP plotted onto the van der Waals surface [close (a) and Z-clipped (b)] of a half of compound 1 computed at the BP86/def2-SVP level of theory. The MEP values at selected points of the surface are indicated. | ||
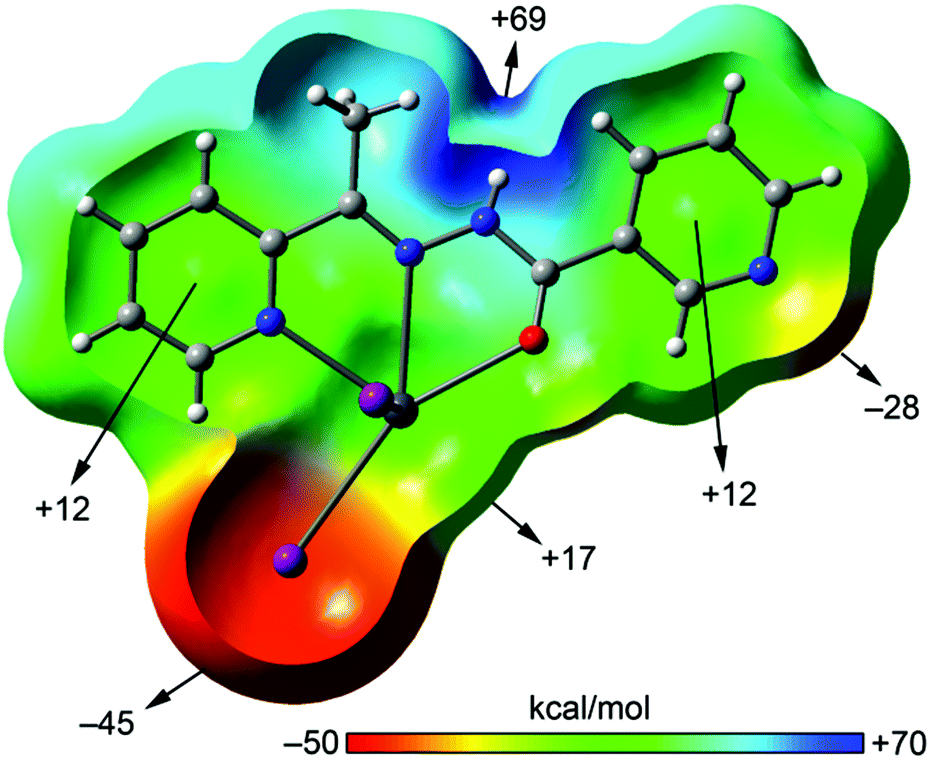 | ||
| Fig. 11 MEP plotted onto the van der Waals surface (Z-clipped) of compound 3 computed at the BP86/def2-SVP level of theory. The MEP values at selected points of the surface are indicated. | ||
The MEP surface of a complex having the neutral form of the ligand is shown in Fig. 11. It exhibits significant differences with respect to the enolate form. First, the most positive value is located on the amide N–H and the most negative in the iodide ligand, thus explaining the formation of H-bonded chains in the solid state. Another significant difference is the behaviour regarding the MEP values over the pyridine rings, since both are positive. Finally, the MEP value on the metal center is positive and that at the uncoordinated pyridine N-atom is negative and smaller in absolute value than the one observed for the enolate form. The directionality of the lone pair of the uncoordinated pyridine and the Pb σ–hole is ideal for the formation of self-assembled dimers as observed in the solid state of 3 (see Fig. 6).
An energetic DFT study was undertaken to examine more closely the nature of the Pb⋯N interactions observed in the structures of 1–4. In 1 we analysed the Pb1⋯N3A contacts responsible for interconnection of the metallacycles into infinite chains (see Fig. 2). The interaction energy (Fig. 12a) is large and negative (ΔE1 = −40.4 kcal mol−1) and thus indicates a very strong binding motif in the crystal structure. Similarly, in compound 2 we have evaluated the Pb1⋯N6 contacts responsible for interconnection of the metallacycles into infinite chains (Fig. 4). In this complex the interaction energy is considerably less negative (ΔE2 = −19.8 kcal mol−1), indicating a weaker binding. This is likely due to a combination of two factors. First, there is an additional π-stacking interaction in the dimer of 1 that also contributes to the stabilization of the assembly. Secondly, in compound 1 the ligand is in the enolate form, thus favouring the tetrel bonding interaction as anticipated by the MEP analysis. In fact, each Pb2+ metal center is coordinated to three nitrato ligands in 2 that reduce the tetrel bonding donor ability of the Pb atom. Following the same reasoning, the interaction energy of the dimer observed in the solid state of compound 3 (neutral ligand) exhibits an interaction energy (ΔE3 = −26.7 kcal mol−1) that is smaller in absolute value than that of compound 1 (enolate form, ΔE1), but larger than ΔE2 (compound 2). The latter is due to the different coordination environment of the Pb, which is coordinated to two iodido ligands in 3 and to three nitrato ligands in 2, thus decreasing the Pb Lewis acidity in 2 and, consequently, decreasing the Pb⋯N strength.
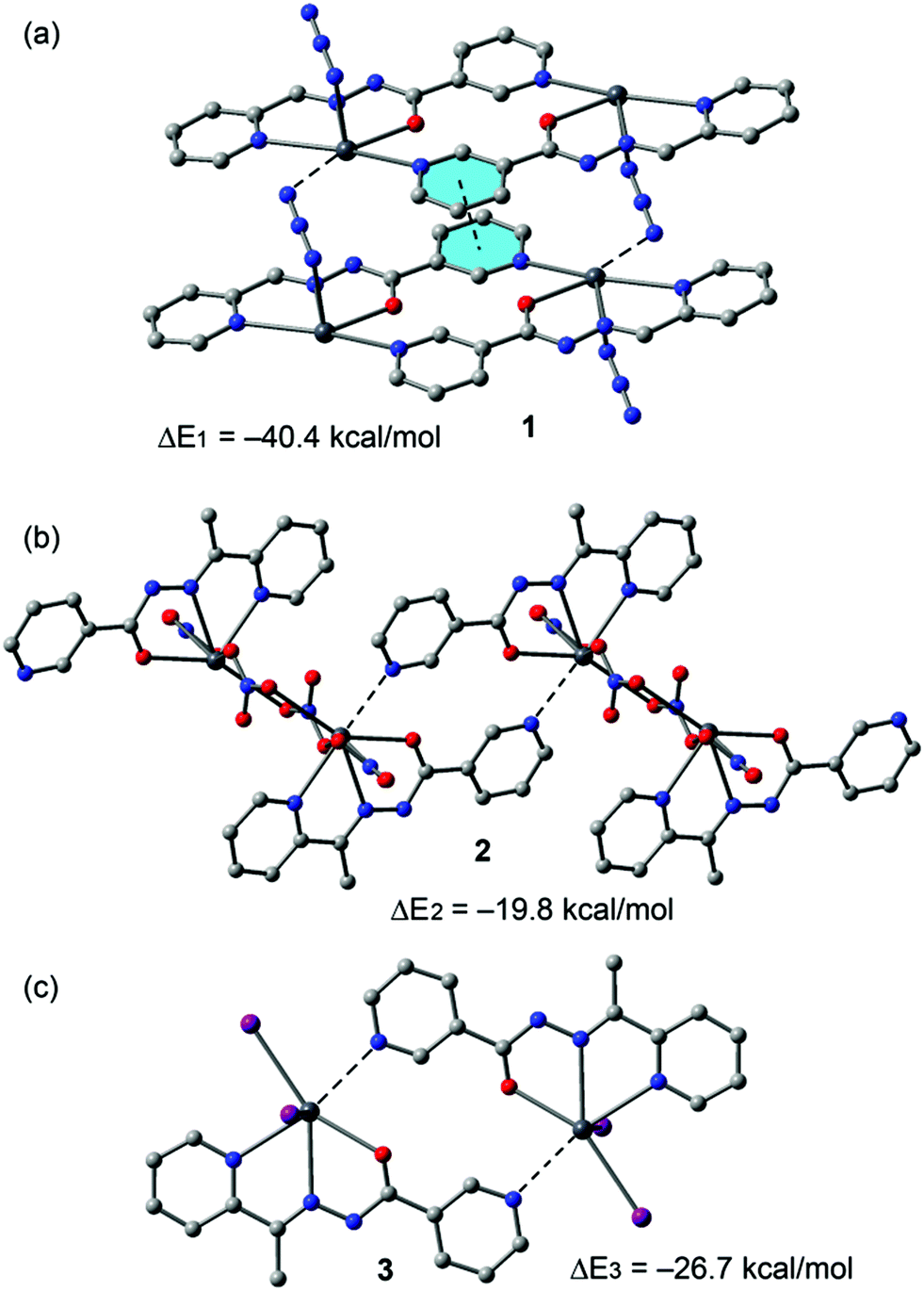 | ||
| Fig. 12 Interaction energies of tetrel bonded dimers of compounds 1 (a), 2 (b) and 3 (c) computed at the BP86-D3/def2-SVP level of theory. | ||
In compound 4 we have focused the theoretical study on the computation of the energy of the interactions responsible for the formation of the supramolecular assembly shown in Fig. 8, which are the antiparallel π–π stacking interaction and the tetrel bonding. The results are gathered in Fig. 13. The interaction energy of the antiparallel p-stacking is very large (ΔE4 = −29.0 kcal mol−1) in agreement with the MEP study that shows opposite signs for the MEP values over the coordinated and uncoordinated pyridine rings. Consequently these electrostatically enhanced π+–π− complexes are very strong binding motifs in the crystal structure. Finally, the interaction energy of the tetrel bond (ΔE5 = −10.5 kcal mol−1) is modest compared to the half of ΔE1 (also enolate form, see Fig. 12a). This is likely due to the additional π–π interaction observed in the solid state of 1 that also contributes to ΔE1.
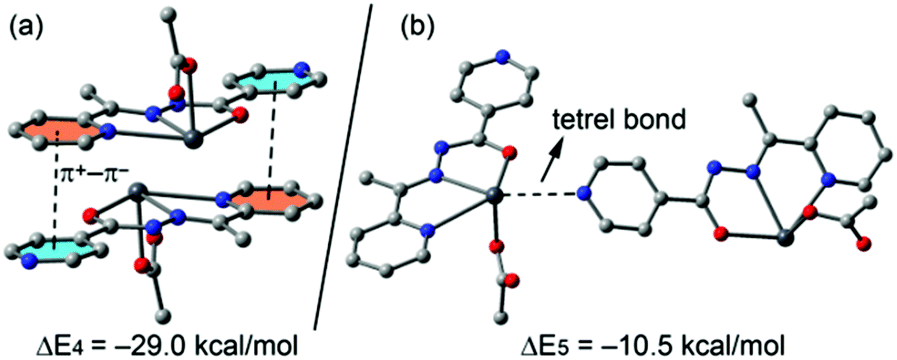 | ||
| Fig. 13 Interaction energies of π-stacked (a) and tetrel bonded (b) dimers of compound 4 (computed at the BP86-D3/def2-TZVP level of theory). | ||
Finally, we have used the Bader's theory of “atoms in molecules”, which provides an unambiguous definition of chemical bonding, to further describe the noncovalent tetrel bonding interactions described above. The AIM theory has been effectively utilized to characterize a great variety of interactions.20
In Fig. 14 we show the AIM analysis of the tetrel bonding complex (a) and the self-assembled π-stacked dimer of compound 4 (the AIM analyses of compounds 1–3 are included in the ESI, Fig. S1–S3†). The Pb⋯N interaction is confirmed by the presence of a bond CP and a bond path connecting the N atom of pyridine to the Pb atom. It can be also observed two intermolecular H bonds involving two aromatic C–H bonds as acceptors. The density at the bond CP that characterizes the tetrel bond is larger than those observed for the H-bonds, indicating that it is the strongest interaction.
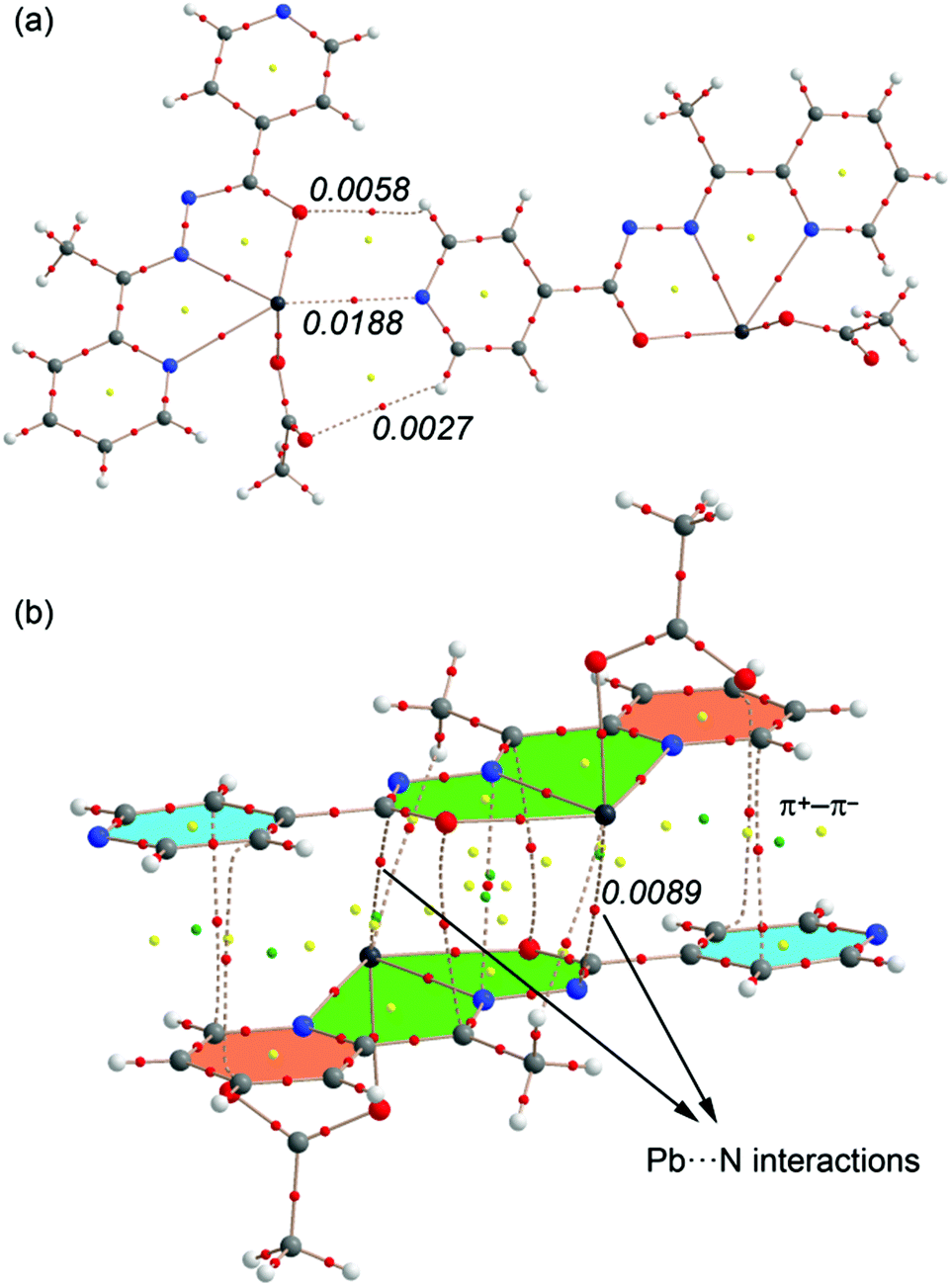 | ||
| Fig. 14 AIM analyses of two dimers of complex 4. Bond, ring and cage critical points are represented by red, yellow and green spheres, respectively. The bond paths connecting bond critical points are also represented by dashed lines. The value of ρ(r) at the bond CPs is given for some interactions in a.u. | ||
Regarding the π-stacked dimer (Fig. 14b) It can be observed that the π+–π− interactions are characterized by the presence of several bond (red spheres), ring (red spheres) and cage (green spheres) critical points (CPs). Basically, two bond CPs connect the carbon atoms of one pyridine to two carbon atoms of the other pyridine. The π–π complex is further characterized by additional CPs that connect the chelate rings. As a consequence of this complicated distribution of bond CPs, several ring and cage CPs are also generated upon complexation due to the formation of supramolecular ring and cages. Remarkably, the AIM reveals the existence of two Pb⋯N contacts that explain the large interaction energy obtained for this dimer (in addition to the electrostatically enhanced π+–π− interactions, see 13a). The value of the Laplacian of the charge density computed at the bond critical points in both assemblies is positive, as is common in closed-shell interactions.
4. Conclusions
In conclusion, we report the syntheses and structural characterization of four new Pb2+ complexes with (iso)nicotinohydrazide-based ligands All compounds 1–4 exhibit relevant tetrel bonding interactions in the solid state that have been described and characterized using DFT calculations. The energies associated with the interactions have been computed using DFT calculations. In general, these noncovalent Pb⋯N interactions are strong due to electrostatic effects, as shown by the MEP analysis. Our results might be important to understand the solid state architecture of organic–inorganic materials systems that contain hemidirectionally coordinated Pb2+ metal centers and organic aromatic molecules.Acknowledgements
We are grateful to the University of Maragheh for the generous financial support of this research. A. B. and A. F. thank DGICYT of Spain (projects CTQ2014-57393-C2-1-P and CONSOLIDER INGENIO CSD2010-00065, FEDER funds) for funding. We thank the CTI (UIB) for free allocation of computer time. E. M. is grateful to Generalitat de Catalunya (2014SGR1643), Ministerio de Economia y Competividad (projects ENE2012-36368 and SEV2015-0496), and EU's Seventh Framework Programme (IRSES EULANETCERMAT). JTM thanks Tulane University for support of the Tulane X-ray Crystallography Laboratory.Notes and references
- Y. Wang, L. J. Zhou, M. L. Feng and X. Y. Huang, Dalton Trans., 2012, 41, 6689 RSC; C. S. Liu, X. S. Shi, J. R. Li, J. J. Wang and X. H. Bu, Cryst. Growth Des., 2006, 6, 656 Search PubMed; S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed; S. G. Telfer and R. Kuroda, Coord. Chem. Rev., 2003, 242, 33 CrossRef; S. A. Barnett and N. R. Champness, Coord. Chem. Rev., 2003, 246, 145 CrossRef.
- Y.-F. Hsu, H.-L. Hu, C.-J. Wu, C.-W. Yeh, D. M. Proserpio and J.-D. Chen, CrystEngComm, 2009, 11, 168 RSC.
- J. Yang, B. Wu, F. Zhuge, J. Liang, C. Jia, Y.-Y. Wang, N. Tang, X.-J. Yang and Q.-Z. Shi, Cryst. Growth Des., 2010, 10, 2331 CAS.
- F. F. B. J. Janssen, L. P. J. Veraart, J. M. M. Smits, R. de Gelder and A. E. Rowan, Cryst. Growth Des., 2011, 11, 4313 CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, Oxford, 1997 RSC; Y. Xia, S. Li, B. Wu, Y. Liua and X.-J. Yanga, CrystEngComm, 2011, 13, 5763 RSC; P. Dechambenoit, S. Ferlay, M. W. Hosseini and N. Kyritsakas, Chem. Commun., 2007, 4626 RSC; W. Li, A. Thirumurugan, P. T. Barton, Z. Lin, S. Henke, H. H.-M. Yeung, M. T. Wharmby, E. G. Bithell, C. J. Howard and A. K. Cheetham, J. Am. Chem. Soc., 2014, 136, 7801 CrossRef CAS PubMed; J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334 CrossRef PubMed; R. S. Forgan, R. J. Marshall, M. Struckmann, A. B. Bleine, D.-L. Long, M. C. Bernini and D. Fairen-Jimenez, CrystEngComm, 2015, 17, 299 RSC; M. Servati-Gargari, G. Mahmoudi, S. R. Batten, V. Stilinović, D. Butler, L. Beauvais, W. S. Kassel, W. G. Dougherty and D. VanDerveer, Cryst. Growth Des., 2015, 15, 1336 Search PubMed.
- P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC; A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496 CrossRef CAS PubMed; A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340 CrossRef PubMed.
- J. S. Murray, K. E. Riley, P. Politzer and T. Clark, Aust. J. Chem., 2010, 63, 1598 CrossRef CAS; P. Politzer and J. S. Murray, ChemPhysChem, 2013, 14, 278 CrossRef PubMed; P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2010, 12, 7748 RSC; P. Politzer, K. E. Riley, F. A. Bulat and J. S. Murray, Comput. Theor. Chem., 2012, 998, 2 CrossRef; A. Bauzá, I. Alkorta, A. Frontera and J. Elguero, J. Chem. Theory Comput., 2013, 9, 5201 CrossRef PubMed; G. Sánchez-Sanz, C. Trujillo, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2014, 16, 15900–15909 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317 CrossRef CAS PubMed; S. J. Grabowski, Phys. Chem. Chem. Phys., 2014, 16, 1824 RSC; A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. – Eur. J., 2014, 20, 10245 CrossRef PubMed; E. C. Escudero-Adán, A. Bauzá, A. Frontera and P. Ballester, ChemPhysChem, 2015, 16, 2530 CrossRef PubMed; A. Bauzá, T. J. Mooibroek and A. Frontera, Phys. Chem. Chem. Phys., 2014, 16, 19192 RSC; A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Commun., 2014, 50, 12626 RSC; A. Bauzá, T. J. Mooibroek and A. Frontera, Chem. Rec., 2016, 16, 473 CrossRef PubMed; J. S. Murray, P. Lane and P. Politzer, J. Mol. Model, 2009, 15, 7239 Search PubMed; A. Bundhun, P. Ramasami, J. S. Murray and P. Politzer, J. Mol. Model, 2013, 19, 2739 CrossRef PubMed; R. S. Ruoff, T. Emilsson, A. I. Jaman, T. C. Germann and H. S. Gutowsky, J. Chem. Phys., 1992, 96, 3441 CrossRef; R.-D. Urban, G. Rouillé and M. Takami, J. Mol. Struct., 1997, 413–414, 511 CrossRef; I. Alkorta, I. Rozas and J. Elguero, J. Phys. Chem. A, 2001, 105, 743 CrossRef.
- M. Servati Gargari, V. Stilinović, A. Bauzá, A. Frontera, P. McArdle, D. Van Derveer, S. W. Ng and G. Mahmoudi, Chem. – Eur. J., 2015, 21, 17951–17958 CrossRef CAS PubMed.
- G. Mahmoudi, A. Bauzá and A. Frontera, Dalton Trans., 2016, 45, 4965–4969 RSC.
- L. Shimoni-Livny, J. P. Glusker and C. W. Bock, Inorg. Chem., 1998, 37, 1853 CrossRef CAS; R. L. Davidovich, V. Stavila, D. V. Marinin, E. I. Voit and K. H. Whitmire, Coord. Chem. Rev., 2009, 253, 1316 CrossRef.
- P. V. Bernhardt, G. J. Wilson, P. C. Sharpe, D. S. Kalinowski and D. R. Richardson, J. Biol. Inorg. Chem., 2008, 13, 107 CrossRef CAS PubMed.
- E.-I. Ochiai, J. Chem. Educ., 1995, 72, 479 CrossRef CAS.
- G. M. Sheldrick, SHELXTL: Structure Determination Software Suite, Version 6.14, Bruker AXS, Madison, WI, USA, 2003 Search PubMed.
- Mercury 1.4.1, Copyright Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge, CB2 1EZ, UK, 2001–2005 Search PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park KS, USA, 2013 Search PubMed.
- (a) A. Bauzá and A. Frontera, ChemPhysChem, 2015, 16, 3108–3113 CrossRef PubMed; (b) A. Bauzá and A. Frontera, ChemPhysChem, 2015, 16, 3625–3630 CrossRef PubMed; (c) A. Bauzá, T. J. Mooibroek and A. Frontera, ChemPhysChem, 2015, 16, 2496–2517 CrossRef PubMed; (d) E. C. Escudero-Adán, A. Bauzá, A. Frontera and P. Ballester, ChemPhysChem, 2015, 16, 2530–2533 CrossRef PubMed; (e) M. Mirzaei, M. Nikpour, A. Bauza and A. Frontera, ChemPhysChem, 2015, 16, 2260–2266 CrossRef CAS PubMed; (f) A. Bauzá and A. Frontera, Chem. Phys. Lett., 2015, 633, 282–286 CrossRef; (g) A. Bauzá and A. Frontera, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Tables S1–S4 and Fig. S1–S3. CCDC 1474989 and 1480033–1480035. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01947a |
| This journal is © The Royal Society of Chemistry 2016 |