
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBoosting catalyst activity in cis-selective semi-reduction of internal alkynes by tailoring the assembly of all-metal aromatic tri-palladium complexes†
Anna
Monfredini‡
a,
Veronica
Santacroce‡
a,
Pierre-Alexandre
Deyris
b,
Raimondo
Maggi
a,
Franca
Bigi
a,
Giovanni
Maestri
*a and
Max
Malacria
*bc
aDipartimento di Chimica, Università degli Studi di Parma, 17/A Parco Area delle Scienze, 43124 Parma, Italy. E-mail: giovanni.maestri@unipr.it
bICSN CNRS (UPR2301), 1 Av. de la Terrasse, Bat. 27, 91198 Gif s/Yvette, France. E-mail: max.malacria@cnrs.fr
cUPMC Sorbonne Université, IPCM (UMR CNRS 8232), 4 place Jussieu, C. 229, 75005 Paris, France
First published on 22nd June 2016
Abstract
Highly symmetric [Pd3]+ clusters that present delocalized metal–metal bonds can catalyse the selective semi-reduction of internal alkynes to cis-alkenes. Studies on factors governing the formation of all-metal aromatics enabled the design of an optimised catalytic system that delivers cis-alkenes with almost complete selectivity on a gram scale with very low catalyst loadings (0.03 mol%).
All-metal aromatic molecules feature delocalized metal–metal bonds that parallel bonding of their well-known main-group counterparts.1 While they are often synthetically challenging at best,2 we recently developed a simple route3 to a family of bench-stable [Pd3]+ complexes whose core represent a prototypical well-defined sub-nanometric metal surface.4
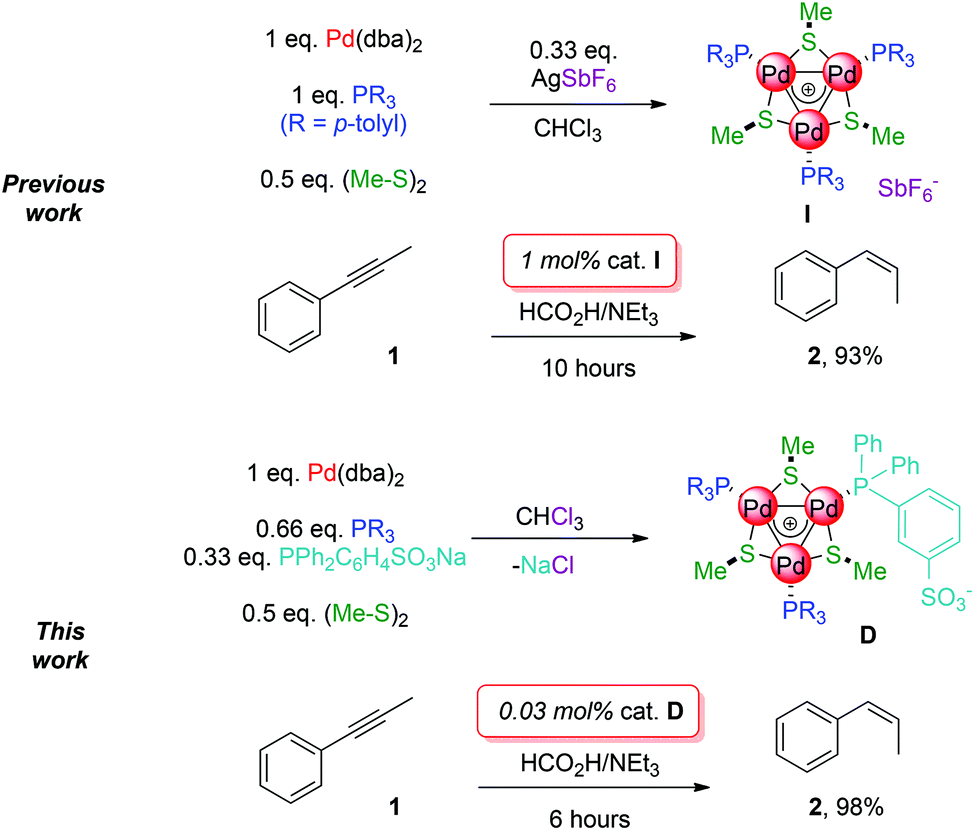
These complexes could be decorated with a variety of organic fragments and represent therefore a valuable asset to assess the consequences of their bonding mode on coordination chemistry and catalysis. Internal alkynes were semi-reduced to alkenes with good cis-selectivity under transfer-hydrogenation conditions,5 which avoids the use of hazardous H2 gas.6 The best first generation catalyst (I, 1 mol%) delivered full conversion of phenylpropyne 1 upon 10 hours in refluxing THF. No alkane was detected and the ratio between semi-reduced alkenes was 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3:4 (cis-, trans- and isomerised-products, respectively, Scheme 1, top).
:3:4 (cis-, trans- and isomerised-products, respectively, Scheme 1, top).
 | ||
| Scheme 1 First vs. second generation metal-aromatic transfer hydrogenation catalysts. | ||
We report herein preliminary results on factors triggering the formation of triangular all-metal aromatic [Pd3]+ complexes. Through this approach we were able to decorate their metal core with two different phosphinic ligands, a neutral and an anionic one. This in turn enabled us to develop a catalytic system for the selective semi-reduction of 1 to cis-alkene 2 with unparalleled activity among Pd-based catalysts under transfer hydrogenation conditions. The novel complex delivers the desired cis-alkene almost quantitatively, in shorter times and with a thirty-three-times lower catalytic charge (Scheme 1, bottom).
To gain insights into factors governing the formation of delocalized metal–metal bonds, we tried to monitor these reactions by NMR. In a 50 mL Schlenk tube we mixed 0.2 mmol of Pd(dba)2 (dba = dibenzylideneacetone), 0.2 mmol of PPh3 and 0.1 mmol of diphenyldisulphide in 20 mL of CDCl3, taking 0.5 mL samples at regular intervals to record the 1H and 31P NMR spectra (Fig. 1, spectra in the ESI†).
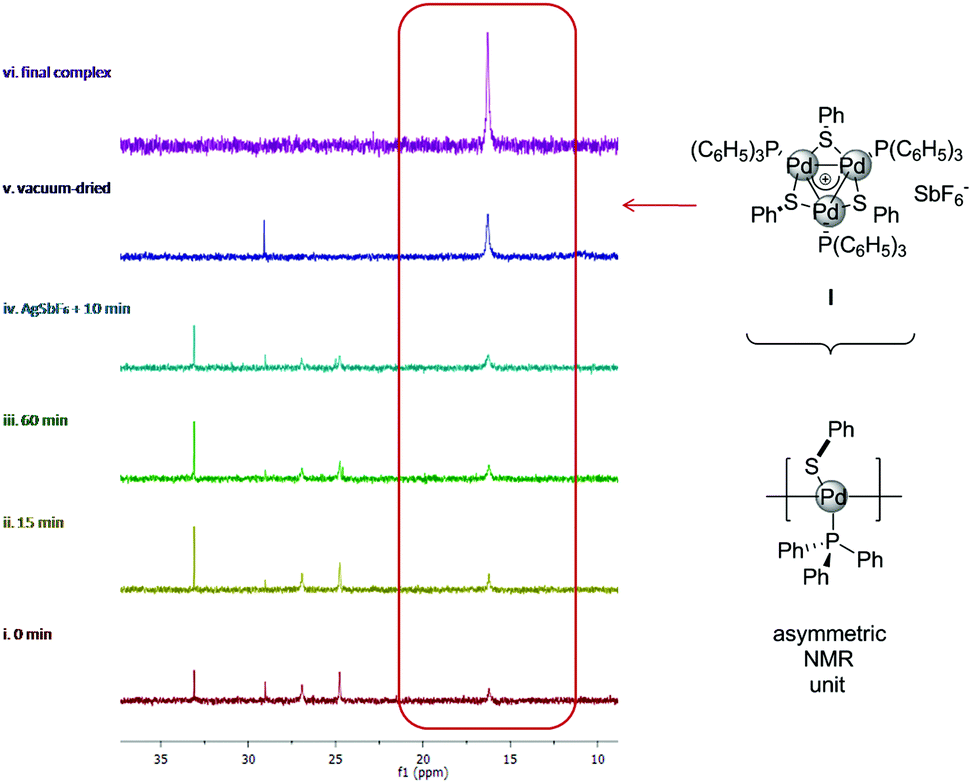 | ||
| Fig. 1 31P spectra of crude samples from the synthesis of [Pd3]SbF6 complex highlighting the diagnostic resonance of its three equivalent PPh3 units. | ||
Surprisingly, the broad and diagnostic 31P resonance of the [Pd3]+ cation at 15.5 ppm was already visible within few minutes (Fig. 1, red line) and free phosphine at −5 ppm was no longer present. From the proton spectrum, we noticed the consumption of Pd(dba)2 and evaluated 18% of the desired [Pd3]+ cation using dba resonances as the internal standard. Two other broad 31P resonances were observed at 24 and 27 ppm respectively. We were so far unable to unambiguously assign these signals, although their intensity slowly fades through time with a concomitant increase of the desired resonance at 15.5 ppm. Upon 60 minutes, the amount of the [Pd3]+ complex is around 40% (green line). Addition of 0.33 equiv. of AgSbF6 resulted in a further increase of the desired resonance at the expense of the two unassigned species (cyan line). Upon celite filtration under nitrogen and concentration in vacuo, the [Pd3]SbF6 complex II became the almost exclusive species present in the crude mixture (Fig. 1, blue line, 86% isolated yield). Taken together, these observations showed that addition of a silver salt is not necessary to trigger the formation of metal-aromatic [Pd3]+ clusters. This in turn suggested that the chlorinated solvent played a key role in the formation of the [Pd3]+ core.
The overall process involves the formal oxidation of Pd(0) to Pd(4/3). Considering the stoichiometry of the reaction, the disulphide can oxidise the noble metal on average to Pd(I). Radical palladium species7 might be sufficiently strong reductants to trigger SET (single electron transfer) on a chloroform molecule (−1.09 V vs. SCE).8 This would form the corresponding radical anion, which eventually collapses into a radical and a chloride ion. The formal addition of a cationic Pd(II) fragment on a neutral Pd(I) dimer9 would eventually deliver the [Pd3]+ cation, isolable as a chloride salt, in analogy to the step-wise assembly of [Au3]+ complexes from an Au(I) cation and an Au(0) dimer reported by Bertrand.10
The [Pd3]+ cation barely forms in CH2Cl2 while the reaction is almost instantaneous in CCl4, being limited by the solubilisation of Pd(dba)2 only. These results fit with the trend of C–Cl BDE and reduction potentials among chlorinated solvents.8
Likely, the soluble AgX salt just favours anion metathesis steered by the precipitation of AgCl in chloroform. This suggests that [Pd3]+ complexes can be synthesised in any given solvent if a suitable mild stoichiometric oxidant is added.
This is of particular interest for catalysis as it would allow the in situ preparation of [Pd3]+ complexes in any medium.
We performed our usual reaction in degassed THF without the addition of any additive. Expectedly, no traces of the desired [Pd3]+ cation were observed upon 24 hours at room temperature. On the contrary, mixing reagents for 1 hour and adding then 0.33 equiv. of AgSbF6 allowed the mixture to turn deep red, eventually delivering complex II in 73% yield.
We then performed the same experiment adding a weaker Cu(II) oxidant (+0.28 V vs. SCE, +0.6 V for Ag(I)).
Thus, 0.2 mmol of Pd(dba)2, 0.2 mmol of P(p-tol)3 and 0.1 mmol of dimethyldisulphide were stirred for 1 hour at RT in degassed THF. Two main resonances at 23 and 26 ppm were observed in the corresponding 31P spectra, which likely correspond to the species observed in chloroform presented in Fig. 1 (spectra in the ESI†). Addition of 0.066 mmol of Cu(OTf)2 triggered a slow turning of the solution towards red and, furthermore, the appearance of the diagnostic 31P resonance of the [Pd3]OTf complex III at 15 ppm. The reaction was slower than the previous one but once again a single resonance was eventually detected upon 24 hours stirring at RT. These findings paralleled observations made using chlorinated solvents and provide preliminary insights into the mechanism of formation of all-metal aromatic [Pd3]+ complexes.
We thus attempted the catalytic semi-reduction of phenylpropyne 1 preparing in situ the [Pd3]+ complex. Triethylammonium formate and 1 were added as a THF solution to eventually provide the same concentration previously reported (0.1 M). Sadly, the reaction proved significantly slower, requiring 24 hours to achieve full conversion. The selectivity towards the desired cis-alkene 2 remained similar (91%). Partial decomposition to Pd black was observed through time although no alkane was detected. The reduced activity may be due to dba, which can likely compete with the substrate slowing down its semi-reduction.
We then turned to another strategy to design better catalysts for this reaction. Reasoning that [Pd3]+ is the major species in chloroform even without the addition of any other metal salts, we performed the reaction with 0.33 equiv. of sodium sulfonate phosphine. This should provide a zwitterionic complex by filtering off the coproduced NaCl. We prepared accordingly a small library of complexes (Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | R3 | R′ | Complex | Yield (%) |
| Reaction conditions: on a 0.2 mmol scale, 0.01 M in degassed chloroform. | ||||
| 1 | (4-F-C6H4)3 | 4-Me-C6H4 | A | 71 |
| 2 | (4-F-C6H4)3 | Me | B | 55 |
| 3 | (4-Me-C6H4)3 | Ph | C | 59 |
| 4 | (4-Me-C6H4)3 | Me | D | 76 |
| 5 | (Ph)2C6H4SO3Na | Me | E | 72 |
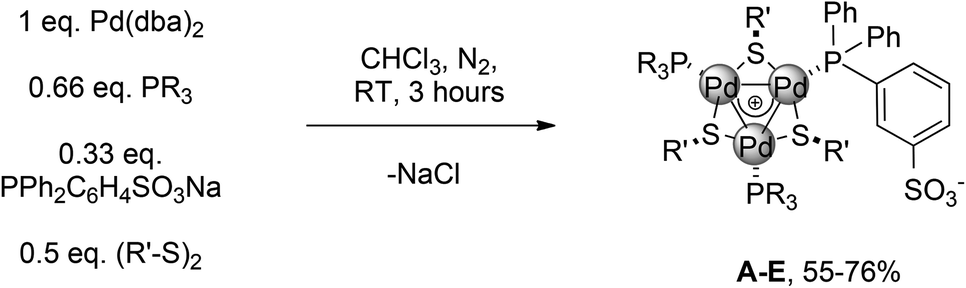
Several organic fragments varying both in electronic and steric features could be easily introduced. Products A–E were purified through a series of solvent washings3 (55–76% yield). They were characterized via HRMS analyses and multinuclear NMR experiments. In contrast to C3-symmetric complexes, as salt I, which showed [Pd3]+ cations in ESI+ experiments, we invariably observed protonated molecular adducts only for A–D. This confirms their zwitterionic nature. Regarding NMR, two patterns of resonances in a 2:1 ratio were observed for bridging the thiolate fragments in the 1H spectra. 31P signals followed suit. For instance, a broad triplet and a broad doublet in a 1:2 ratio were observed for A (at 17 and 13 ppm respectively).
We then tested these new complexes on the catalytic semi-reduction of phenylpropyne 1 in the presence of a five-fold excess of triethylammonium formate as the hydrogen source (Table 2). This excess ensures that the chemoselectivity towards the semi-reduction of alkynes is due to the catalytic system rather than to a mere shortage of a hydrogen donor.11
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | t (h) | Conv. of 1a (%) | Sel. to 2a (%) |
| Conditions: 0.35 mmol of 1, 0.1 M in THF, 1.75 mmol of triethylammonium formate, 1 mol% of complexes A–E.a 0.1 mL samples were periodically taken and analysed by GC and GC-MS using p-xylene (0.3 mmol) as the internal standard. | |||||
| 1 | (4-F-C6H4)3 | 4-Me-C6H4 | 8 | 97 | 85 |
| 2 | (4-F-C6H4)3 | Me | 5 | Quant. | 87 |
| 3 | (4-Me-C6H4)3 | Ph | 6 | Quant. | 78 |
| 4 | (4-Me-C6H4)3 | Me | 3 | Quant. | 77 |
| 5 | (Ph)2C6H4SO3Na | Me | 3 | Quant. | 77 |
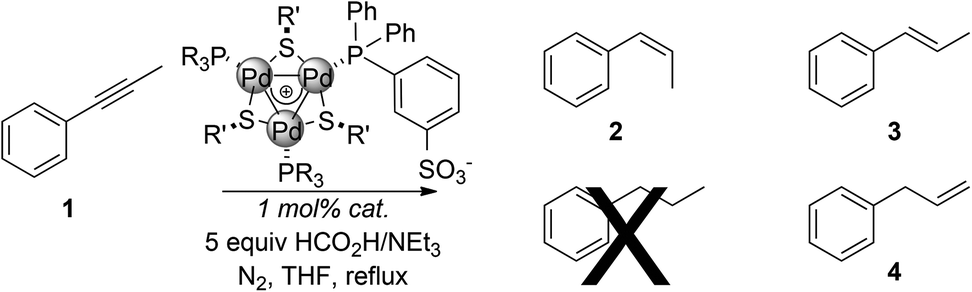
Complexes with fluorinated phosphines proved active with either aromatic or aliphatic bridging thiolates (entries 1 and 2). The latter proved a better substitution pattern providing complete conversion of the substrate upon five hours. In both cases, cis-selectivity resulted to be lower than that previously observed (85% and 87% respectively). No traces of alkanes were detected. Alkenes 3 and 4 formed in a comparable amount (6–8%). Complexes bearing two p-tolylphosphines were even more active (entries 3 and 4). In combination with bridging methylthiolates, complete conversion of 1 was achieved within 3 hours, albeit at the expense of selectivity towards 2 (77%). 3 and 4 formed in 10% and 13% yield respectively. The same outcome was observed using complex E (entry 5, 77%). Interestingly, the catalytic system using any complex of the series A–E proved both more active and less selective than the best one previously developed based on C3-symmetric [Pd3]+ complexes (Scheme 1, top).
We reasoned that this might be a trans-like effect12 of the anionic ligand that would make more labile the phosphine on the opposite edge of the metal kernel. It can correlate with (slight) upfield shifts of the latter observed by 31P NMR and could rationalise the activity of the complexes with p-fluorinated phosphines (A and B, entries 1 and 2). Their corresponding C3-symmetric [Pd3]+ complexes are indeed fairly active only for the semi-reduction of 1, not even attaining full conversion upon 96 hours.5 DFT modeling supports this hypothesis. The Pd–P distance with the anionic phosphine shortens in D compared to I, while those with neutral ones are slightly longer (Fig. 2, details in the ESI†).
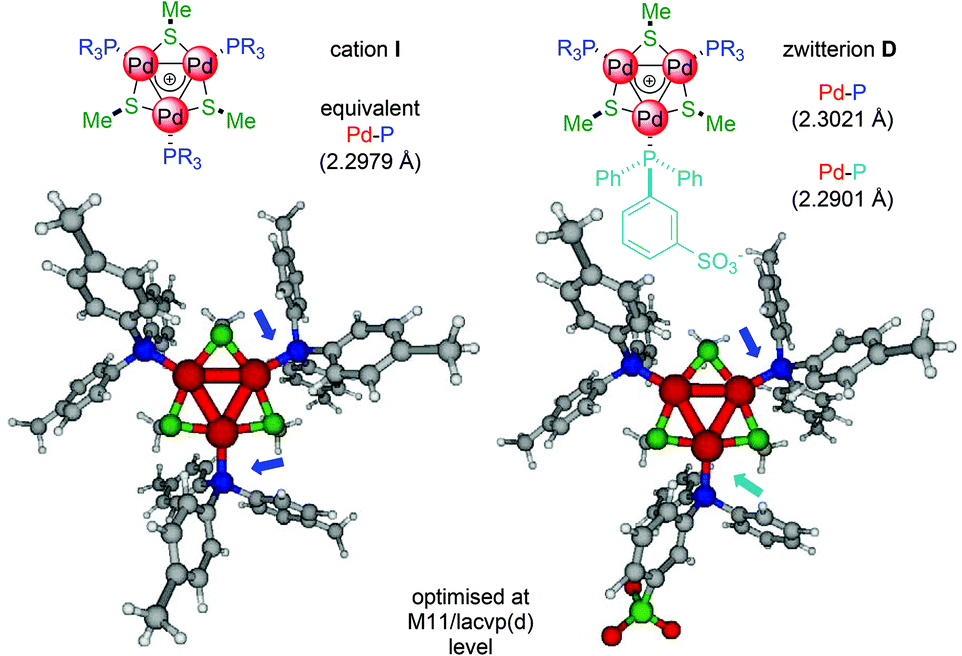 | ||
| Fig. 2 Comparison of optimised structures of cation I and zwitterion D; the Pd–P distances in D at the M11/lacvp(d) level vary by +0.0041 and −0.0078 Å with the neutral and anionic phosphines respectively. | ||
In all cases we noticed the formation of traces of Pd black when the conversion of 1 passed 70%, suggesting that the zwitterionic complexes A–E could be less robust than their cationic analogues. Nonetheless, no trace of over-reduced alkanes has ever been detected, even prolonging warming after full conversion. In these cases, partial isomerization to 3 and 4 was however noticed (below 5% upon 24 hours).
Taken together, these results confirmed the complete chemoselectivity of the catalytic system towards alkynes and let us speculate that isomerisation of 2 could be due to the Pd species generated from the decomposition of the actual catalyst in the reducing reaction medium.13 We wondered whether addition of a suitable ligand might slow down these undesired processes without quenching activity (Table 3).

We were delighted that addition of 3 mol% of p-tolylphosphine proved decisive (entry 1). Full conversion of 1 was achieved upon 4 hours and the selectivity towards 2 was almost complete (96%). Isomers 3 and 4 formed in 2% yield each. Furthermore, the homogeneous orange solution of complex D did not show any trace of formation of black metal colloids before consumption of 1 (images in the ESI†). We then added 10 mol% of phosphine (entry 2). Conversion of 1 required 24 hours, in agreement with a stiffer competition between substrates and ligands for metal coordination. Selectivity towards 2 remained 96% despite the prolonged warming. As in the previous case, no traces of visible heterogeneous aggregates appeared throughout this (longer) experiment. The use of a coordinating electron-poor olefin as dba confirmed the outcome previously observed generating a [Pd3]+ complex in situ (vide supra), requiring 24 hours to achieve full conversion of 1 (entry 3). In this case, traces of Pd black were observed together with a slightly more pronounced formation of 3 and 4 (4% each). Addition of 3 mol% of the sodium salt of a sulfonated phosphine gave 2 with 96% selectivity within 4 hours (entry 4) and the reaction mixture remained once more a red homogeneous solution.
We performed ESI-MS analyses on samples collected at regular intervals to check the stability of D during the reaction. This method proved useful to detect a fading concentration of cation I throughout the reaction, in agreement with the formation of visible Pd black.5 However, no Pd-containing ion has been detected in this case, likely owing to a difficult ionisation of zwitterion D (spectra in the ESI†). We thus resorted to UV-Vis absorption to monitor the diagnostic bands of D in the region between 300 and 550 nm (Fig. 3, inset), which is not affected by any reagent during the catalytic reaction. Fig. 3 shows the absorption spectra of the reaction samples. Both bands in the near UV (320–380 nm) and the saddle at 450 nm remain clearly visible throughout the reaction, indicating that the complex remains stable in solution until full conversion of 1.
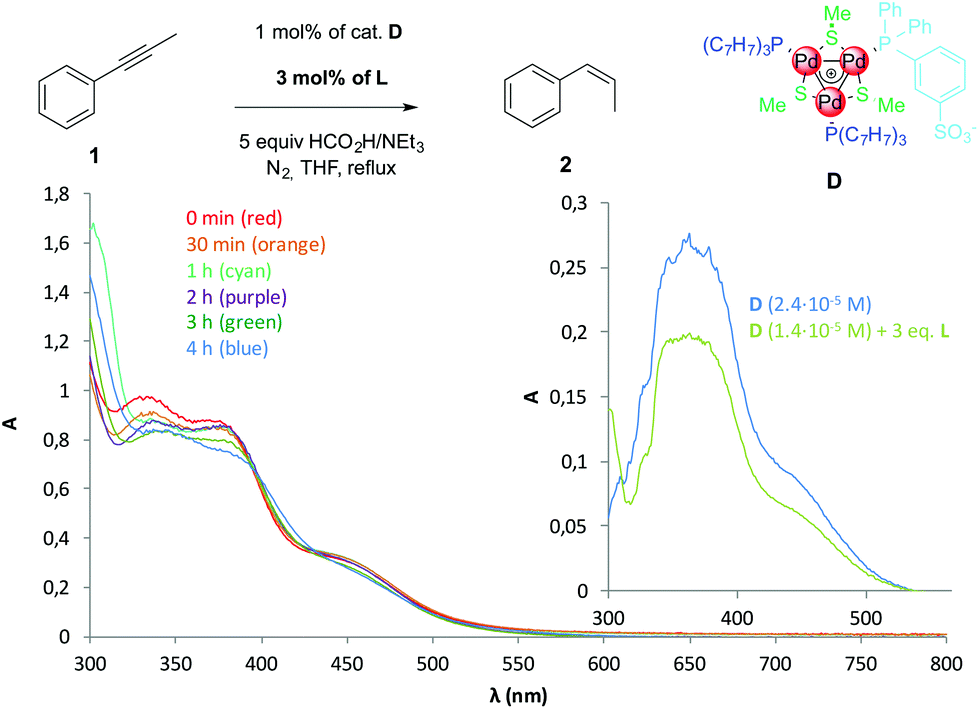 | ||
| Fig. 3 UV-Vis absorption spectra of samples collected from the catalytic reaction of Table 3, entry 1; 100 μl samples were taken via syringe and diluted with 4.9 mL of degassed THF prior to analyses; the inset presents the absorption spectra of pure D (blue) and D with added tritolylphosphine (green) in THF. | ||
These results suggest that addition of a suitable ligand can be a tool to stabilise the active species and to minimise the undesired isomerization of cis-olefins. We thus tried to exploit these hypotheses by reducing the catalyst loading (Table 4).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | X (mol%) | Y (mol%) | t (h) | Conv. of 1a (%) | Sel. to 2a (%) | TOFb (h−1) |
| Conditions: 5.0 mg of complex D, 0.0035 mmol; THF was progressively reduced among the series to keep a fixed concentration of D of 0.001 M.a By GC.b Determined from the linear slope of a time/moles of 2 curve, typically between 0.5 and 2–4 hours.c Without THF.d Without p-xylene as the internal GC standard. | ||||||
| 1 | 1 | — | 3 | Quant. | 77 | 18 |
| 2 | 0.3 | — | 4 | Quant. | 90 | 137 |
| 3 | 0.1 | — | 7 | Quant. | 96 | 329 |
| 4c | 0.05 | — | 6 | 77 | 98 | 582 |
| 5c | 0.05 | 0.15 | 5 | Quant. | 98 | 1026 |
| 6c,d | 0.03 | 0.09 | 6 | Quant. | 98 | 1242 |
Keeping in mind the practical viability of the synthetic method, we decided to fix the concentration of complex D in the mixture, reducing progressively the amount of THF to hold for a higher amount of 1. This is possible thanks to the increased solubility in apolar media of zwitterion D compared to salt I. Reducing the catalyst loading to 0.3 mol% proved beneficial for selectivity (90%, entry 2) and full conversion was attained upon four hours. Further reduction of D to 0.1 mol% provided 96% of 2 upon 7 hours (entry 3). The reaction with 0.05 mol% did not reach full conversion, and the consumption of 1 ended at 77% upon 6 hours (entry 4). In all these cases, the formation of traces of Pd black was observed. For comparison, first generation complexes, such as I, were no longer active before the full conversion of alkynes at catalyst loadings lower than 1 mol%.
We then repeated our last attempt adding 0.15 mmol% of phosphine (entry 5). To our delight, the solution remained homogeneous and 1 was completely converted upon 5 hours, delivering almost exclusively 2 (98%). We further reduced the loading of D to 300 ppm, observing a comparable outcome (entry 6). Full conversion of 1.20 g of 1 was observed upon 6 hours, with 98% selectivity using commercial reagents without any previous purification. Pure 2 has been isolated adsorbing the crude on a 2 cm silica pad and eluting with 5 mL of pentane (1.15 g, 96% yield). This highlights the practical and environmental viability of the present method.14
We are unaware of Pd-based catalytic systems active at so-low loadings under transfer-hydrogenation conditions.6,11,13,14
Conclusions
We reported preliminary studies on the mechanism of formation of triangular Pd complexes with delocalized metal–metal bonds. Through this approach, the panel of readily accessible structures has been further expanded, allowing one to access zwitterionic tripalladium complexes. These clusters formed in one-step from commercial reagents, are definitely stable at the solid state and proved remarkable precatalysts under transfer-hydrogenation conditions, highlighting the great potential of all-metal aromatics in catalysis.Acknowledgements
We thank MIUR, UPCM and CNRS for funding. Research on metal aromaticity is funded through SIR Project “AROMA-TriP” (Grant No.: RBSI14NKFL, GM).Notes and references
- J. M. Mercero, A. I. Boldyrev, G. Merino and J. M. Ugalde, Chem. Soc. Rev., 2015, 44, 6519 RSC; I. Fernández, G. Frenking and G. Merino, Chem. Soc. Rev., 2015, 44, 6452 RSC.
- R. Hoffmann, Am. Sci., 2015, 103, 18 CrossRef.
- Y. Wang, P.-A. Deyris, T. Caneque, F. Blanchard, Y. Li, F. Bigi, R. Maggi, S. Blanchard, G. Maestri and M. Malacria, Chem. – Eur. J., 2015, 21, 12271 CrossRef CAS PubMed.
- S. Blanchard, L. Fensterbank, G. Gontard, E. Lacôte, G. Maestri and M. Malacria, Angew. Chem., Int. Ed., 2014, 53, 1897 CrossRef PubMed.
- P.-A. Deyris, T. Caneque, Y. Wang, P. Retailleau, F. Bigi, R. Maggi, G. Maestri and M. Malacria, ChemCatChem, 2015, 7, 3266 CrossRef CAS.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed; C. Oger, L. Balas, T. Durand and J.-M. Galano, Chem. Rev., 2013, 113, 1313 CrossRef PubMed; S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226 RSC.
- G. Maestri, M. Malacria and E. Derat, Chem. Commun., 2013, 49, 10424 RSC; T. Troadec, S.-Y. Tan, C. J. Wedge, J. P. Rourke, P. R. Unwin and A. B. Chaplin, Angew. Chem., Int. Ed., 2016, 55, 3754 CrossRef CAS PubMed; G. Manolikades and P. Knochel, Angew. Chem., Int. Ed., 2009, 48, 205 CrossRef PubMed; Q. Liu, X. Dong, J. Li, J. Xiao, Y. Dong and H. Liu, ACS Catal., 2015, 5, 6111 CrossRef.
- A. Studer and D. Curran, Nat. Chem., 2014, 6, 765 CrossRef CAS PubMed; J. Bertrand, I. Gallardo, M. Moreno and J. M. Saveant, J. Am. Chem. Soc., 1992, 114, 9576 CrossRef; X.-Q. Hu, X. Qi, J.-R. Chen, Q.-Q. Zhao, Q. Wei, Y. Lan and W.-J. Xiao, Nat. Commun., 2016, 7, 11188 CrossRef PubMed.
- R. Vilar, D. M. P. Mingos and C. J. Cardin, J. Chem. Soc., Dalton Trans., 1996, 4313 RSC.
- L. Jin, D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 9059 CrossRef CAS PubMed.
- P. Hauwert, R. Boerleider, S. Warsink, J. J. Weigand and C. J. Elsevier, J. Am. Chem. Soc., 2010, 132, 16900 CrossRef CAS PubMed; R. Shen, T. Chen, Y. Zhao, R. Qiu, Y. Zhou, S. Yin, X. Wang, M. Goto and L.-B. Han, J. Am. Chem. Soc., 2011, 133, 17037 CrossRef PubMed; J. Broggi, V. Jurcik, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588 CrossRef PubMed.
- B. J. Coe and S. J. Glenwright, Coord. Chem. Rev., 2000, 203, 5 CrossRef CAS; B. Pinter, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Phys. Chem. Chem. Phys., 2013, 15, 17354 RSC.
- R. M. Drost, V. Rosar, S. Dalla Marta, M. Lutz, N. Demitri, B. Milani, B. de Bruin and C. J. Elsevier, ChemCatChem, 2015, 7, 2095 CrossRef CAS; N. T. S. Phan, M. Van Der Sluys and C. W. Jones, Adv. Synth. Catal., 2006, 348, 609 CrossRef.
- E. D. Slack, C. M. Gabriel and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 14051 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Reaction procedures, characterization of complexes, copies of NMR spectra. See DOI: 10.1039/c6dt01840h |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2016 |