
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electron poor iridium pincer complex for catalytic alkane dehydrogenation†
Oleksandr O.
Kovalenko
and
Ola F.
Wendt
*
Centre for Analysis and Synthesis, Department of Chemistry, Lund University, P. O. Box 124, S-221 00 Lund, Sweden. E-mail: ola.wendt@chem.lu.se
First published on 4th July 2016
Abstract
A novel electron deficient 4,6-bis(trifluoromethyl)-1,3-phenylene diphosphinite ligand 4 was developed and synthesized. Reaction of Ir precursors with ligand 4 gave chloro(hydride) pincer complex 5, which demonstrated a higher TON in alkane dehydrogenation reactions compared to similar phosphinite based pre-catalysts. The formation of cyclooctene (COE) and tert-butylethylene adducts of the 14e catalysts was also studied and the COE adduct is implicated as the resting state of the catalyst. All compounds were characterized by NMR spectroscopy and, in addition, the molecular structures of key complexes were confirmed by X-ray analysis.
Introduction
The selective functionalization of alkanes is a challenging topic in modern organic chemistry.1 One highly demanded reaction, particularly in industry, is the conversion of inert alkanes into more reactive olefins, which opens up broad possibilities for further transformations and modifications. In fact, olefins are the most important starting material for making industrial organic chemicals. Industrial catalytic alkane dehydrogenations proceed at high temperatures (ca. 400–600 °C),2 and effective homogeneous alkane dehydrogenation catalysts operating under relatively low temperature conditions are therefore highly desirable.The first homogeneous alkane dehydrogenation catalytic systems were described in the 1980s by Crabtree and Felkin.3 The temperature of the catalytic process was decreased down to 150 °C, but the turnover numbers (TONs) were rather low due to catalyst decomposition. The results reported by Kaska and Jensen using a very robust and efficient iridium hydrido complex (I) opened up a new pincer era in catalytic alkane dehydrogenation research (Fig. 1).4
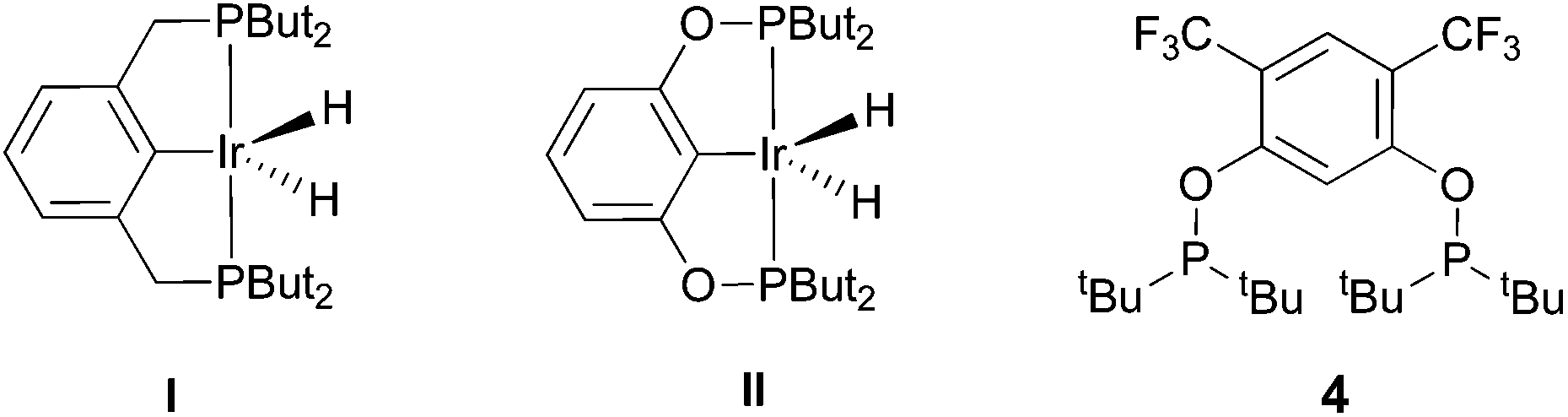 | ||
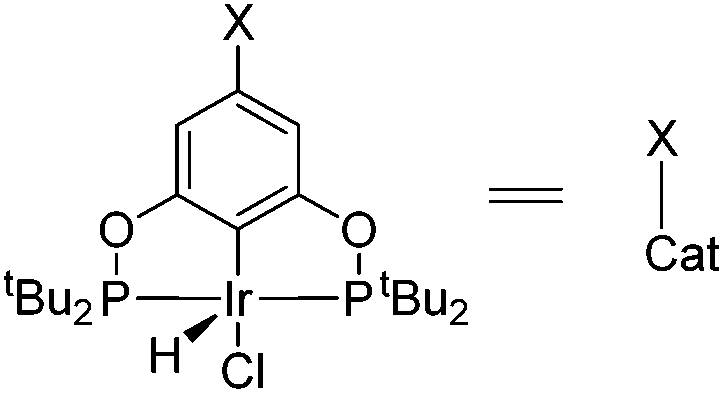
| Fig. 1 Previously reported pincer-iridium catalysts I–II and the herein developed ligand 4 for catalytic alkane dehydrogenation. | ||
Many variations of the pincer framework have been explored including both aromatic and aliphatic backbones and the highest TONs were reported for aromatic backbones with oxygen and/or sulfur linkers on the arm.5–9 Thus, bis-phosphinite complex II demonstrates a significantly higher TON in alkane transfer dehydrogenation compared to complex I (Fig. 1). Furthermore, the electron withdrawing substituents in the aromatic ring increase the catalyst performance.6
Based on these findings, we decided to investigate a bis-phosphinite aryl backbone with two strong electron withdrawing CF3 groups in the meta-position with respect to the metallation site. This should give maximum inductive, electronic communication with the metal and, in addition, undesired orthometallated byproduct formation should be avoided.10 Thus, we herein report the synthesis of electron poor ligand 4 and its metallation to afford the corresponding iridium pincer complex. The reactivity of this complex and its catalytic activity in alkane dehydrogenation are described.
Experimental section
General information
All manipulations were carried out under a dry Ar or N2 atmosphere using standard Schlenk or glovebox techniques, unless otherwise noted. All catalytic experiments were performed under a dry Ar atmosphere. Hydrocarbon solvents were degassed and distilled from sodium benzophenone ketyl prior to use. Chlorinated solvents were degassed and distilled from CaH2. Silica gel with 60 Å pore size and 230–400 mesh was used for column chromatography. All other solvents, reagents and chemicals were used as received.1H, 19F, 31P, and 13C NMR spectra were recorded at 400, 376, 162 and 100 MHz, respectively, using a Bruker 400 MHz spectrometer. For compounds 1–3 chloroform-d and for compounds 4–9 benzene-d6 were used and chemical shifts were referenced to the residual solvent peaks.11 Elemental analysis was performed by H. Kolbe Microanalytisches Laboratorium, Mülheim an der Ruhr, Germany. High-resolution mass spectra (HRMS) were recorded on a Waters Xevo Q-TOF mass spectrometer using electrospray ionization and lock mass corrections.
Crystallography
The crystallographic measurements were performed at 293 K using an Oxford Diffraction Xcalibur 3 system with Mo-Kα radiation.12 Data collection, cell refinement, and data reduction and analysis were carried out using the Xcalibur PX software, CRYSALIS CCD and CRYSALIS RED.13 Analytical absorption corrections were applied to the data using CRYSALIS RED. All structures were solved by direct methods using SHELXS-2014 and refined by a full-matrix least-squares technique based on F2 using SHELXL-2014 with anisotropic thermal parameters for all non-H atoms.14 Hydrogen atoms were constrained to parent sites, using a riding model.Preparations
1H NMR (CDCl3, 400 MHz): δ 8.12 (s, 1H), 7.95 (s, 1H); 19F NMR (CDCl3, 376 MHz): δ −63.07 (s); 13C NMR (CDCl3, 100 MHz): δ 140.87, 129.84 (q, J = 33.0 Hz), 126.80 (sep, J = 5.5 Hz), 124.76, 122.05 (q, J = 273.0 Hz).
1H NMR (CDCl3, 400 MHz): δ 7.81 (s, 1H), 7.46–7.32 (m, 10H), 6.61 (s, 1H), 5.16 (s, 4H); 19F NMR (CDCl3, 376 MHz): δ −61.55 (s); 13C NMR (CDCl3, 100 MHz): δ 160.38, 135.33, 128.86, 128.39, 126.78, 126.54 (sep, J = 5.5 Hz), 123.27 (q, J = 269.7 Hz), 111.14 (q, J = 32.0 Hz), 99.26, 70.70. HRMS (ESI, m/z) calcd for C22H15O2F6 [M − H+]: 425.0976; found: 425.0976.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM (2:1). Removal of the solvent gave 0.096 g, 65% of a red solid. Deep red crystals suitable for X-ray analysis were obtained by slow evaporation of the hexane solution under an argon atmosphere.
:DCM (2:1). Removal of the solvent gave 0.096 g, 65% of a red solid. Deep red crystals suitable for X-ray analysis were obtained by slow evaporation of the hexane solution under an argon atmosphere.
Method B. In a thick wall Straus flask, compound 4 (0.214 g, 0.400 mmol) and chlorobis(cyclooctene)iridium dimer [Ir(μ2-Cl)(COE)2]2 (0.179 g, 0.200 mmol) were combined in benzene. The reaction vessel was sealed, placed in an oil bath and stirred overnight at 90 °C. The solvent was removed under vacuum and the dark brown residue was purified by column chromatography gradually changing the eluent from hexane (100%) to hexane:DCM (2:1). Removal of the solvent gave 0.261 g, 86% of a red solid. 1H NMR (C6D6, 400 MHz): δ 7.57 (s, 1H), 1.20 (vt, J = 15.0 Hz, 18H), 1.15 (vt, J = 15.0 Hz, 18H), −40.44 (t, J = 14.0 Hz, 1H); 19F NMR (C6D6, 376 MHz,): δ −61.74 (s); 31P NMR (C6D6, 162 MHz): δ 184.73 (m); 13C NMR (C6D6, 100 MHz): δ 165.88 (vt, J = 12.0 Hz), 124.15 (t, J = 3.0 Hz), 123.63 (q, J = 270.0 Hz), 120.79 (sep, J = 5.0 Hz), 108.35 (qvt, J = 32.3 Hz, J = 9.6 Hz), 43.02 (vt, J = 22.0 Hz), 39.50 (vt, J = 22.0 Hz), 26.90 (vt, J = 6.6 Hz). Anal. Calcd for C24H38O2P2IrClF6: C, 37.82; H, 5.03. Found C, 38.22, H, 5.22.
:DCM (9:1) as the eluent. Yield: 9 mg, 9% of pale red solid. Single crystals suitable for X-ray analysis were obtained by recrystallization from pentane. Spectroscopic data of compound 6 were reported previously by Buono and coworkers.16
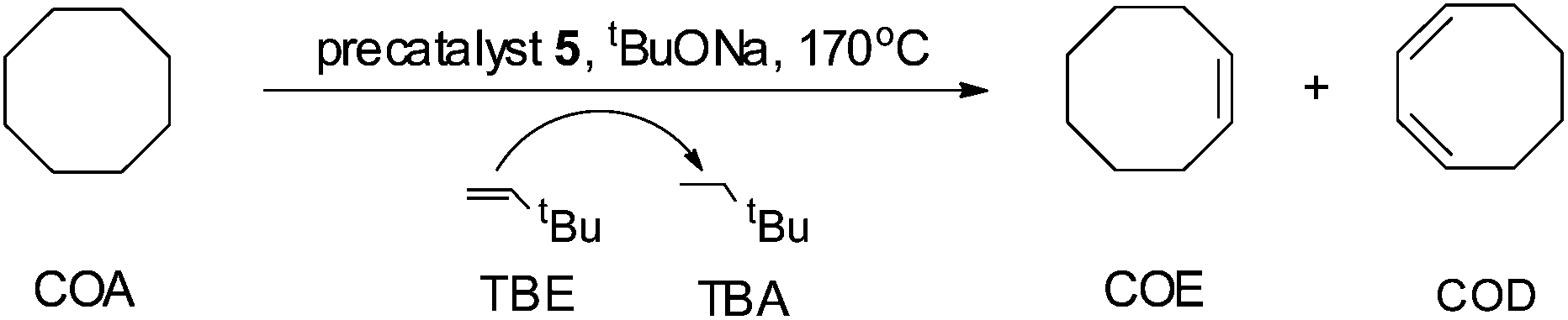
General procedure for the catalytic dehydrogenation of COA with TBE
Complex 5 (0.0030 g, 0.0040 mmol, 1.0 eq.), tBuONa (0.0006 g, 0.0060 mmol, 1.5 eq.), degassed dry cyclooctane (COA) (1.631 mL, 12.12 mmol, 3030 eq.) and tert-butylethylene (TBE) (1.562 mL, 12.12 mmol, 3030 eq.) were placed in a Straus flask. The flask was sealed and fully immersed into a pre-heated oil bath with the specified temperature and time. Thereafter, the flask was cooled down in an ice bath and the sample was analyzed by NMR spectroscopy. Three runs were performed to determine the average TONs. No compounds other than COA, cyclooctene (COE), cyclooctadiene (COD), TBE and tert-butylethane (TBA) could be detected by 1H NMR spectroscopy. This procedure was used also for other iridium complexes.Results and discussion
Synthesis and characterization of ligands and complexes
Ligand 4 was synthesized in five steps following the strategy described in Scheme 1, starting from bis(trifluoromethyl)aniline. Compound 1 was obtained in 32% yield following a previously described procedure of Williams et al.15 All attempts to obtain key resorcinol 3 in one step from 1 using either tetrabutylammonium hydroxide or alkali metal hydroxide media following reported catalytic protocols were unsuccessful, probably due to the electron poor nature of structure 1 and the necessity to substitute two bromine atoms at the same time.17 Instead, the synthesis of benzyl ester 2 was performed using an Ullmann-type reaction with CuBr as the catalyst.18 Thus, the target ligand 4 was obtained in 14% total yield and fully characterized by 31P, 19F, 13C and 1H NMR spectroscopy. Its structure was confirmed with a single crystal X-ray analysis and the molecular structure is given in Fig. 2. This shows that the trifluoromethyl groups cause substantial steric hindrance, obstructing the free rotation of the phosphinite groups across the C–O bond. Hence, the arms of the pincer ligand are oriented in the same direction, possibly providing some assistance in the cyclometallation reactions.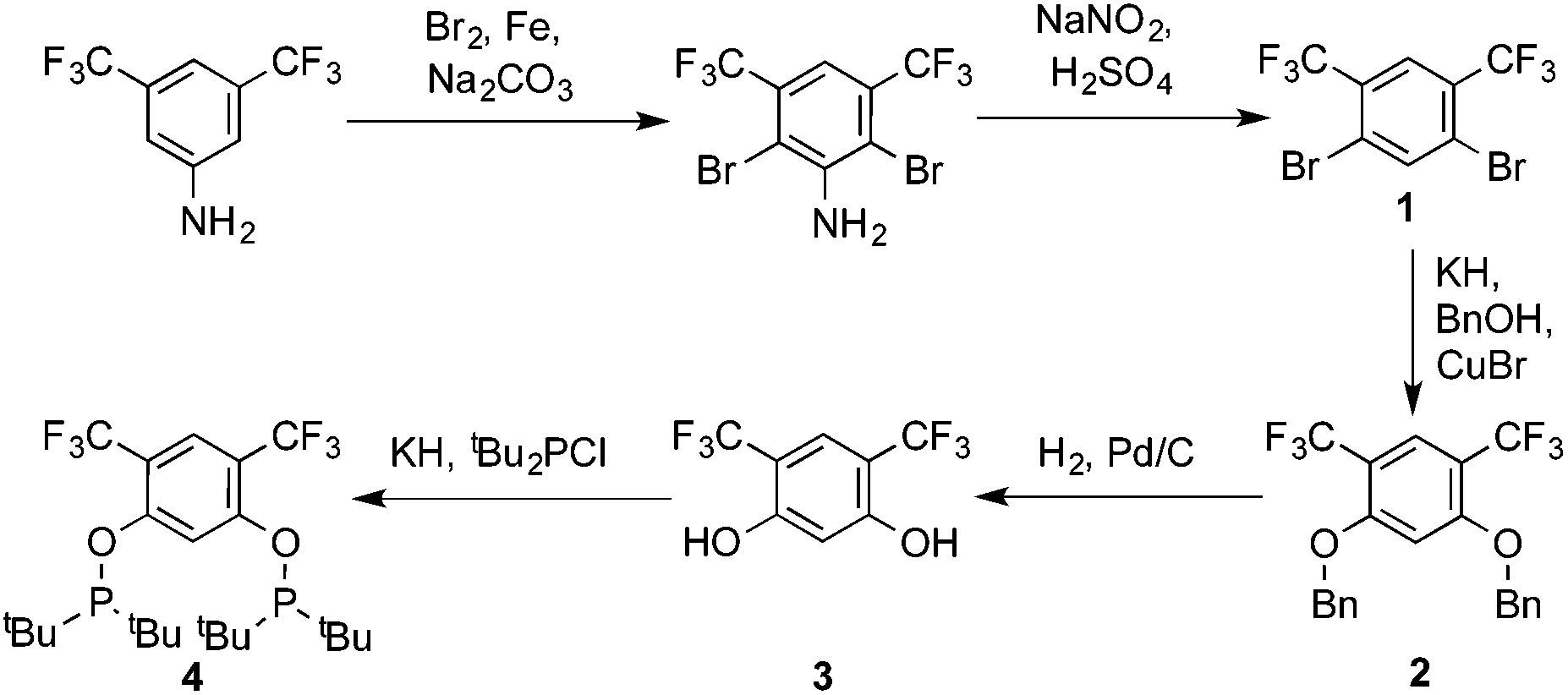 | ||
| Scheme 1 Synthesis of ligand 4. | ||
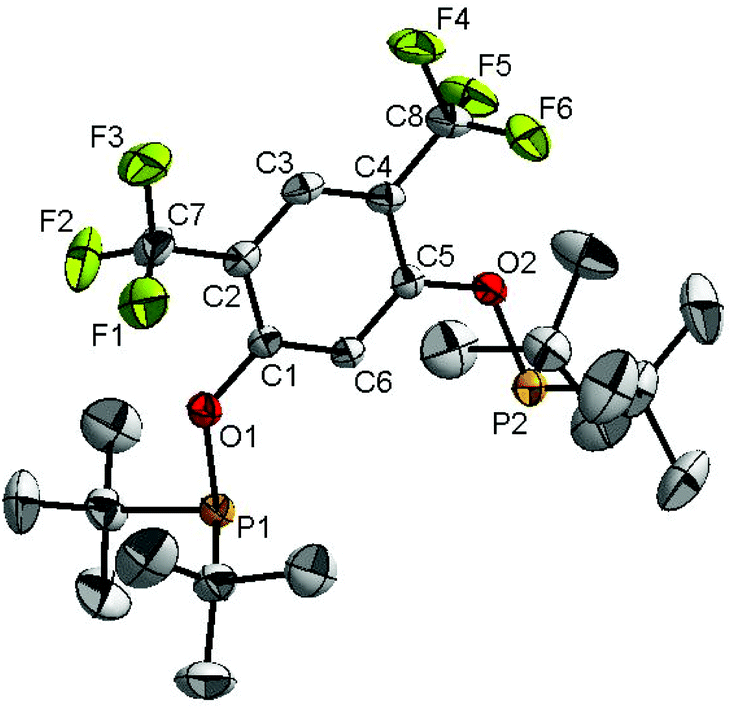 | ||
| Fig. 2 The molecular structure of compound 4. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): C1–O1 1.359(3), O1–P1 1.6915(18), C5–O2 1.360(3), O2–P2 1.6937(18), C1–O1–P1 122.83(15), C5–O2–P2 122.80(15). | ||
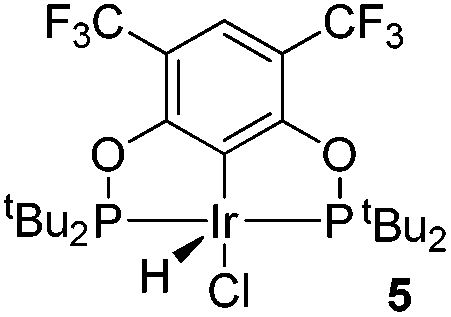
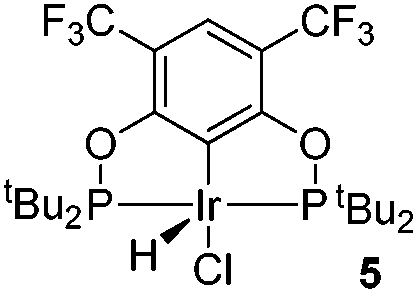
The reaction progress of the cyclometallation of ligand 4 with iridium precursors could be conveniently monitored by 31P{1H} and/or 19F{1H} NMR spectroscopy. After stirring 4 with the cyclooctadiene iridium chloride dimer [Ir(μ2-Cl)(COD)]2 in toluene at room temperature for one hour, an orange precipitate was formed and no signal could be observed in the 31P{1H} NMR spectrum. This behaviour is typical in the formation of non-cyclometallated dimers, which usually have very low solubility.19 However, in this case the dimer could be transformed further; heating the reaction mixture at 120 °C dissolved the precipitate, forming a deep red solution. There was a concomitant appearance of new signals at 184.73 ppm and −61.74 ppm in the 31P{1H} and 19F{1H} NMR spectra, correspondingly. From this reaction the iridium hydrido chloride complex, 5, was obtained in 65% yield (cf. Scheme 2). The 1H NMR spectrum of 5 displays the characteristic hydride triplet at −40.44 ppm,6b and the molecular structure was also confirmed using X-ray crystallography (Fig. 3). As seen from the figure, the iridium atom displays a distorted square-pyramidal geometry assuming that the hydride is in an axial position. The complex has the usual P–Ir–P angle of less than 180° and the Ir–P bond lengths are very similar. The electron withdrawing nature of the ligand is displayed in the unusually short Ir–Cl distance of 2.384(2) Å, which is ca. 0.01 Å shorter than the corresponding unsubstituted one (Table 1).19b
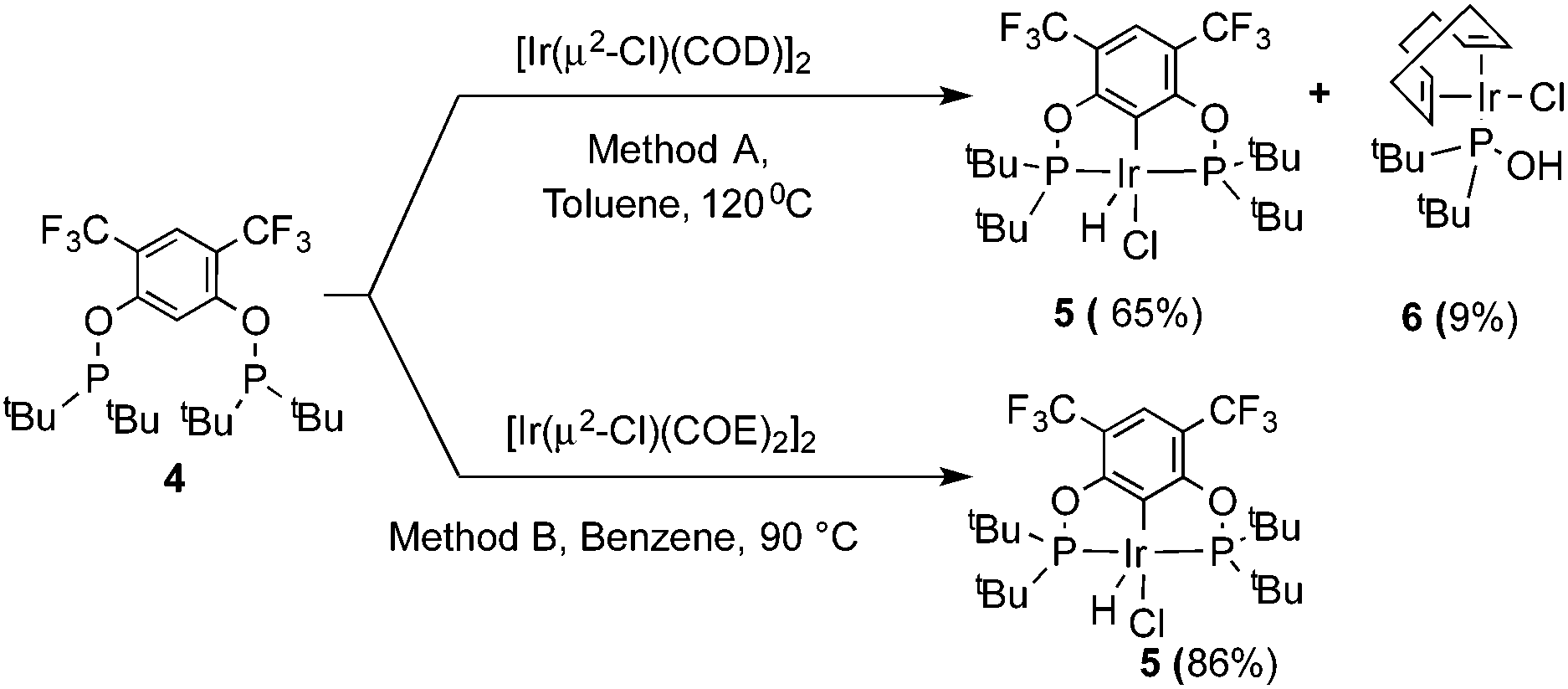 | ||
| Scheme 2 Synthesis of complex 5. | ||
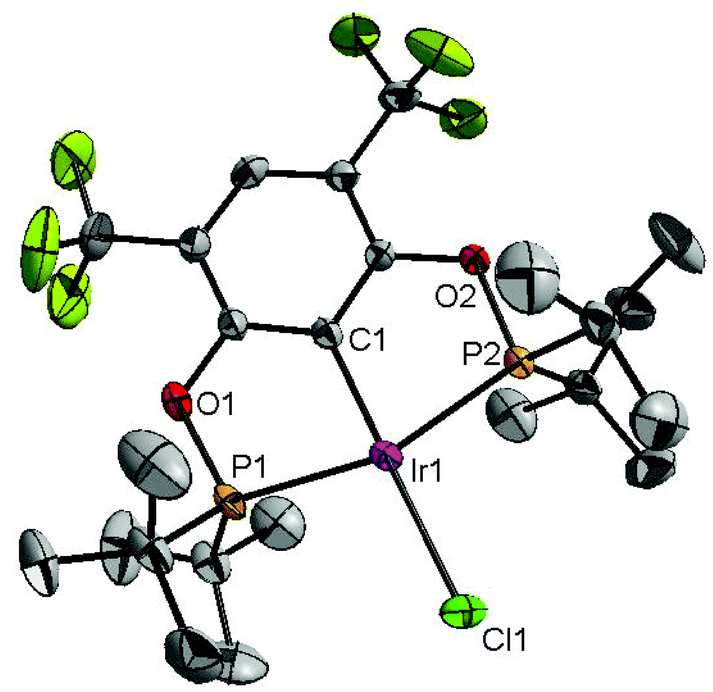 | ||
| Fig. 3 The molecular structure of complex 5. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1–C1 1.997(5), Ir1–P1 2.2946(15), Ir1–P2 2.2919(15), Ir1–Cl1 2.3840(16), P1–Ir1–P2 160.59(5), C1–Ir1–Cl1 174.67(15). | ||
|
|
|
|
|
|
|
|
|---|---|---|---|---|---|---|
| Ir–Cl | 2.3840(16) | 2.3933(13) | 2.3947(12) | 2.3954(8) | 2.4044(14) | 2.4115(12) |
| Ir–C(1) | 1.997(5) | 1.992(4) | 2.000(3) | 1.996(3) | 2.004(5) | 2.014(4) |
| Ir–P(1) | 2.2946(15) | 2.2871(13) | 2.2897(10) | 2.2890(8) | 2.2949(16) | 2.2944(12) |
| Ir–P(2) | 2.2919(15) | 2.2864(13) | 2.2925(11) | 2.2940(8) | 2.2889(15) | 2.2926(11) |
In addition to the main product 5, complex 6 was isolated as a byproduct in 9% yield (Scheme 2). We could establish the molecular structure of 6 using single crystal X-ray analysis; the molecular structure is given in Fig. 4. This compound was previously synthesized and characterized by Buono and coworkers,16 but in our hands it crystallizes in a higher symmetry space group, P21/n. From the structure it is evident that iridium cleaved the C–O bond in one of the pincer phosphinite arms, forming a di(tert-butyl)phosphinous acid iridium complex with a distorted square planar geometry. Such a C–O bond cleavage in pincer complexes has been observed previously.20
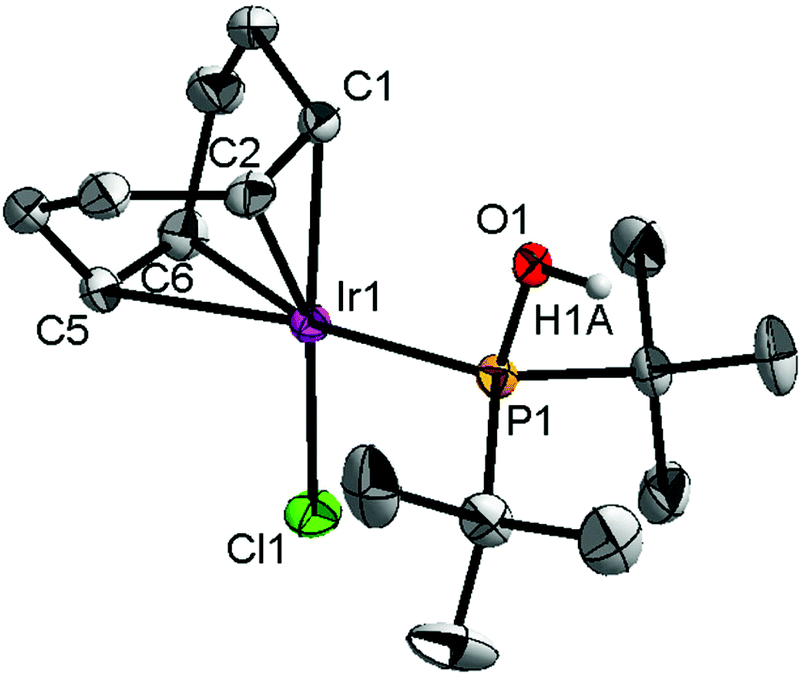 | ||
| Fig. 4 The molecular structure of complex 6. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1–C1 2.125(5), Ir1–C2 2.117(5), Ir1–C5 2.228(5), Ir1–C6 2.217(5), Ir1–Cl1 2.3573(13), Ir1–P1 2.2728(13), P1–O1 1.593(3), P1–Ir1–Cl1 94.74(5), Ir1–P1–O1 110.76(13). | ||
Dehydrogenation catalysis
In order to investigate the catalytic activity in alkane transfer dehydrogenation reactions, we chose a common benchmark reaction – transfer dehydrogenation of cyclooctane in the presence of tert-butylethylene (TBE). The standard conditions and COA/TBE/catalyst ratio (3030/3030/1) were used in the initial catalytic studies, which gave us the possibility to compare catalyst 5 with previously reported ones.6,7 To generate an active catalyst, tBuONa (1.5 eq.) was added in order to remove HCl from 5. This formed a compound, 7, which could be isolated as an orange solid and characterized by 31P, 19F, 13C and 1H NMR spectroscopy. The 31P and 19F{1H} NMR spectra contain signals at 188.64 ppm and a sharp singlet at −61.45 ppm, respectively. Furthermore, the 1H NMR spectrum only contains signals from the ligand with no indication of any dynamic behaviour as previously observed for more electron rich pincer systems.22 It seems unlikely that 7 is a 14e complex and we tentatively describe it as the benzene adduct. The electron poor ligand in 7 probably prohibits dynamic oxidative addition/reductive elimination of benzene (see the ESI†). However, it is likely that there is a fast benzene exchange at the metal centre (without oxidative addition). Attempts to characterize 7 with elemental analysis failed since it quickly forms a dinitrogen complex.After heating the reaction vessel for 24 h at 200 °C, complex 7 demonstrated a turnover number (TON) of 2086. This value is higher than the original Brookhart's catalyst with an unsubstituted benzene ring and also slightly higher than values obtained using other electron withdrawing substituents (Table 3).6,8 A temperature screening was performed and an improved performance of 7 was detected at 170 °C (Table 2); thus, this temperature was used for the following experiments. A kinetic study showed that around 80% of the turnovers were observed within the first 8 hours of the reaction and ≈90% after 24 h. Prolonged heating did not improve turnovers substantially (Table 4 and Fig. 5). An initial TOF could be calculated and it was around 0.8 s−1, which is lower than that reported for {p-C6F5-C6H2(OCH2PtBu2)2}Ir, but on the other hand our experiment was performed at a 30 °C lower temperature. For a direct comparison the same experiment was performed with the Brookhart catalyst (II), reproducing the literature results fairly well (Table S2† and Fig. 5). It again shows that the initial TOF of 5 is lower (at a lower temperature) but the TON is higher.
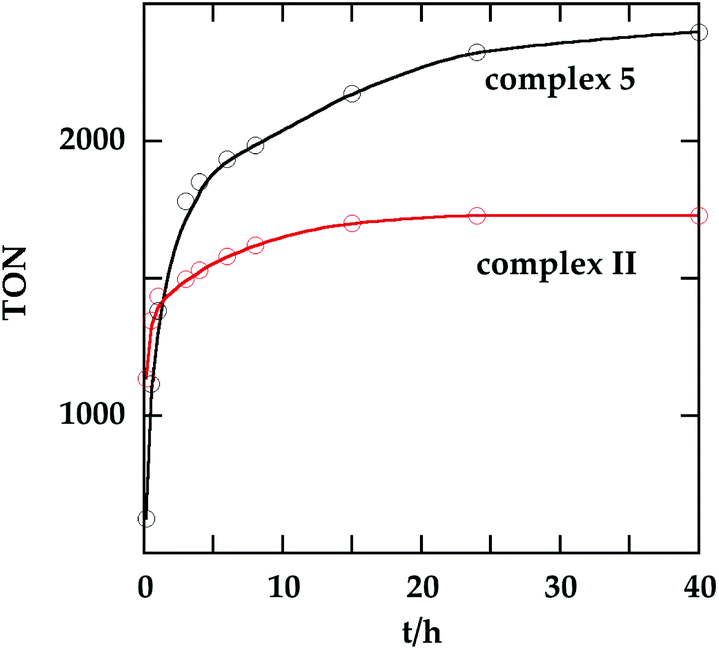 | ||
| Fig. 5 Turnover number as a function of time for 5 (at 170 °C) and II (at 200 °C). The final TON of II is 1730. See Tables 4 and S2† for details. | ||
|
|
|
|---|---|
| Complex | TONsa |
|
a Results were obtained using a 3030:3030:1 ratio of COA/TBE/precatalyst and tBuON under an argon atmosphere. TONs were determined by 1H NMR.
b Obtained at 170 °C for 40 h.
c Obtained at 200 °C for 24 h.
d Obtained at 200 °C for 40 h.
|
|
|
2398b |
| 2086c | |
|
2070d |
| C6F5-Cat | 2041d |
| F-Cat | 1530d |
| H-Cat | 1583d |
|
|
|||||
|---|---|---|---|---|---|
| Entrya | t [h] | TONsb | Entrya | t [h] | TONsb |
|
a Average of three runs, using a 3030:3030:1 ratio of COA/TBE/precatalyst 5 and 1.5 equiv. of tBuONa, unless otherwise noted. All reactions were performed under an argon atmosphere.
b Determined by 1H NMR, the sum of COE and COD double bonds equals the TON of TBE within 2% difference.
c Using a 3030:1 ratio of COA/precatalyst 5 and 1.5 equiv. of tBuONa under argon under refluxing conditions.
|
|||||
| 1 | 0.17 | 625 | 7 | 8 | 1985 |
| 2 | 0.5 | 1117 | 8 | 15 | 2174 |
| 3 | 1 | 1382 | 9 | 24 | 2324 |
| 4 | 3 | 1781 | 10 | 40 | 2398 |
| 5 | 4 | 1851 | 11 | 170 | 2537 |
| 6 | 6 | 1935 | 12c | 24 | 10 |
Also, catalysis without a hydrogen acceptor (TBE) was investigated. This was conducted in an open vessel protected with an argon backstop. This approach provides unimpeded hydrogen elimination from the reaction flask, but puts limits on the maximum reaction temperature because of the boiling point of COA and COE (max. t ≈ 150 °C). Under these conditions we only observed ca. 10 turnovers after 24 h (Table 4).
Isolation of intermediates
To probe the resting state of the catalyst the reaction mixture after catalysis was analyzed by 31P{1H} NMR spectroscopy. The presence of a dominant broad signal at 185.48 ppm was observed, which triggered us to examine the nature of the contributing species. Previous mechanistic studies23 have shown that the resting state varies with the electronic nature of the catalyst, with the lower oxidation states being favoured for more electron poor systems.24Compound 7 reacts with COE and TBE in benzene solution at room temperature forming deep red solutions of complexes 8 and 9, correspondingly (Scheme 3). 31P and 19F{1H} NMR spectra demonstrate chemical shifts at 185.48 ppm, 185.45 ppm, −61.04 ppm and −61.47 ppm, respectively. The signal from 8 agrees very well with the signal of the reaction mixture after catalysis. Evaporation of volatile TBE and benzene from the solution of 9 led to the formation of compound 7 again, and 9 could not be isolated in a pure form. Contrary to 9, complex 8 did not lose coordinated COE readily, and based on 31P, 19F, 13C and 1H NMR studies of the deuterated benzene solutions an equilibrium was established between complex 8, precursor 7 and free COE in a ratio of 2:1:1. Compound 8 is poorly soluble in benzene, and X-ray quality crystals were obtained and an X-ray analysis was performed to confirm the molecular structure of 8, which is given in Fig. 6. The structure exhibits a distorted square planar geometry with the olefin bonded perpendicular to the coordination plane. Bond lengths and angles are in good agreement with similar, previously reported complexes with coordinated ethylene and COE. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is 1.380(18) indicating a fairly weak back-donation from the Ir(I) metal centre.25,26
C bond is 1.380(18) indicating a fairly weak back-donation from the Ir(I) metal centre.25,26
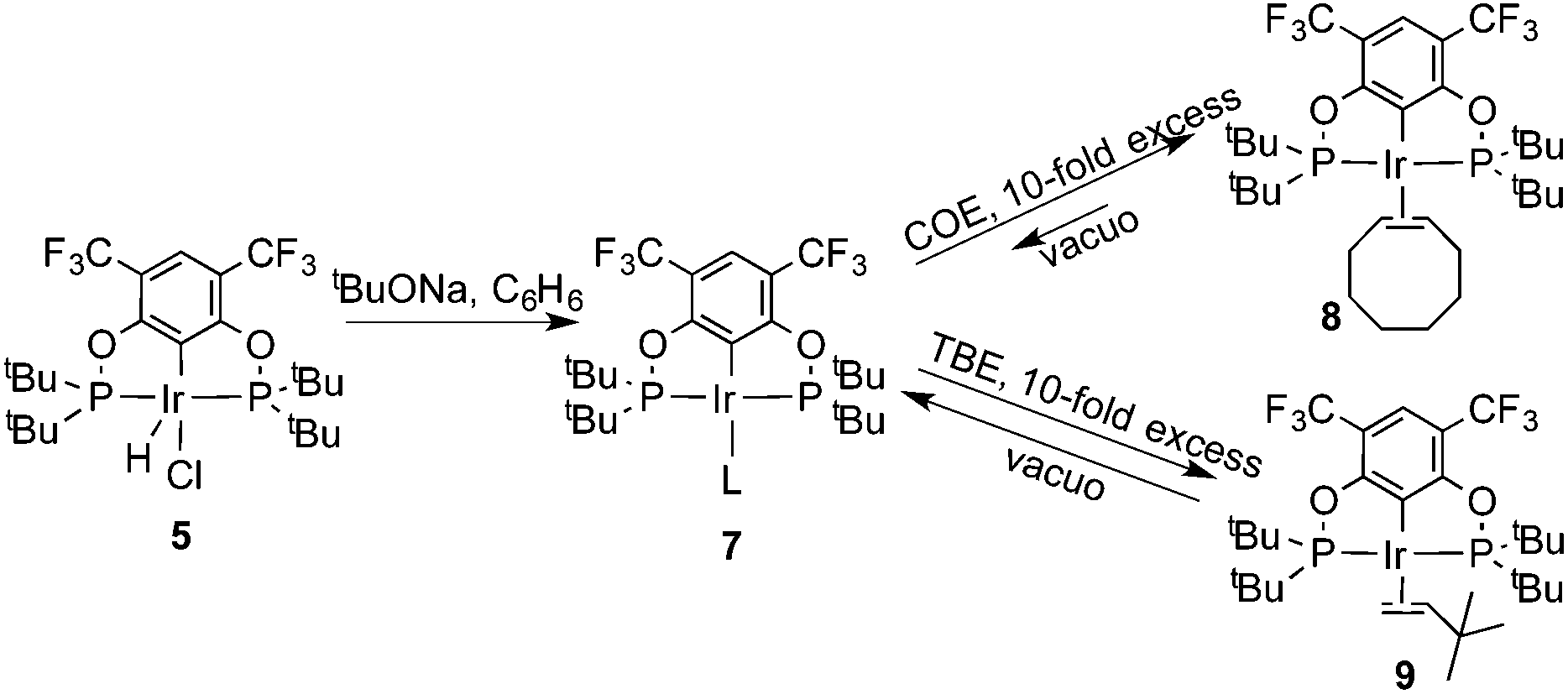 | ||
| Scheme 3 Reactions of complex 5 with TBE and COE. | ||
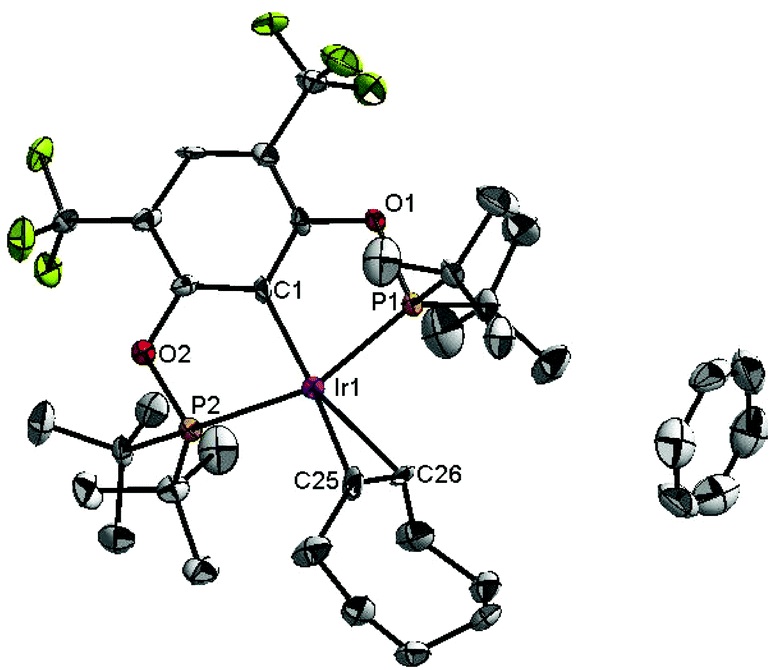 | ||
| Fig. 6 The molecular structure of complex 8. Thermal ellipsoids are drawn at the 30% probability level. Hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ir1–C1 2.032(9), Ir1–P1 2.285(5), Ir1–P2 2.315(5), Ir1–C25 2.289(12), Ir1–C26 2.230(12), C25–C26 1.380(18), P1–Ir1–P2 157.15(10), C1–Ir1–C25 159.4(5), C1–Ir1–C26 160.2(7). | ||
These experiments show that COE binds much more strongly than TBE to compound 7. As the COE concentration rises, the activity consequently decreases and already at moderate COE concentrations, 8 is probably the resting state of the catalyst. In keeping with the electron poorness of the ligand system there is no oxidative addition observed. As mentioned previously, 7 does not seem to undergo oxidative addition of benzene and the olefin substrates show no tendency for any vinylic C–H oxidative addition; the oxidative addition of COA is therefore probably the rate determining step in the catalytic dehydrogenation.
Conclusions
A new electron deficient ligand 4 was synthesized and the iridium precatalyst 5 was successfully obtained and characterized. With a base, this gives the active species 7, which could be isolated probably as the benzene adduct. The catalyst demonstrated an increase in the TON compared to other iridium phosphinite systems. Also, a relatively stable COE complex 8 was obtained and characterized. This compound is probably the resting state of the catalyst already at low conversion.Acknowledgements
Financial support from the Swedish Research Council, the Knut and Alice Wallenberg Foundation and the Royal Physiographic Society in Lund is gratefully acknowledged.References
- (a) J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 2–24 CrossRef CAS PubMed; (b) J. A. Labinger and J. E. Bercaw, Nature, 2002, 417, 507–514 CrossRef CAS PubMed; (c) K. M. Waltz and J. F. Hartwig, Science, 1997, 277, 211–213 CrossRef CAS.
- K. Weissermel and H.-J. Arpel, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 2003, pp. 59–89 Search PubMed.
- (a) M. J. Burk and R. H. Crabtree, J. Am. Chem. Soc., 1987, 109, 8025–8032 CrossRef CAS; (b) H. Felkin, T. Fillebeen-Khan, R. Holmes-Smith and Y. Lin, Tetrahedron Lett., 1985, 26, 1999–2000 CrossRef CAS.
- (a) M. Gupta, C. Hagen, R. J. Flesher, W. C. Kaska and C. M. Jensen, Chem. Commun., 1996, 2083–2084 RSC; (b) M. Gupta, C. Hagen, W. C. Kaska, R. E. Cramer and C. M. Jensen, J. Am. Chem. Soc., 1997, 119, 840–841 CrossRef CAS; (c) C. M. Jensen, Chem. Commun., 1999, 2443 RSC. For reviews, see: (d) D. Morales-Morales and C. M. Jensen, The Chemistry of Pincer Compounds, Elsevier, Amsterdam, 2007 Search PubMed; (e) Activation and functionalization of C–H bonds, ed. K. I. Goldberg and A. S. Goldman, ACS symposium series, American Chemical Society, Washington, DC, 2004, vol. 885 Search PubMed; (f) D. Morales-Morales, Iridium-mediated alkane dehydrogenation, in Iridium complexes in organic synthesis, ed. L. A. Oro and C. Claver, Wiley-VCH, Weinheim, 1st edn, 2009 Search PubMed; (g) D. Bézier and M. Brookhart, Transfer Dehydrogenations of Alkanes and Related Reactions Using Iridium Pincer Complexes, in C–H Bond Activation and Catalytic Functionalization II, ed. P. H. Dixneuf and H. Doucet, Topics in organometallic chemistry, Springer, Heidelberg, 2016, vol. 56, p. 189 Search PubMed.
- (a) K. Krogh-Jespersen, M. Czerw, K. Zhu, B. Singh, M. Kanzelberger, N. Darji, P. D. Achord, K. B. Renkema and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 10797–10809 CrossRef CAS PubMed; (b) K. Zhu, P. D. Achord, X. Zhang, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2004, 126, 13044–13053 CrossRef CAS PubMed; (c) S. Kundu, Y. Choliy, G. Zhuo, R. Ahuja, T. J. Emge, R. Warmuth, M. Brookhart, K. Krogh-Jespersen and A. S. Goldman, Organometallics, 2009, 28, 5432–5444 CrossRef CAS; (d) Z. Huang, M. Brookhart, A. S. Goldman, S. Kundu, A. Ray, S. L. Scott and B. C. Vicente, Adv. Synth. Catal., 2009, 351, 188–206 CrossRef CAS.
- (a) I. Göttker-Schnetmann, P. S. White and M. Brookhart, Organometallics, 2004, 23, 1766–1776 CrossRef; (b) I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed.
- W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS PubMed.
- A. V. Polukeev, R. Gritcenko, K. J. Jonasson and O. F. Wendt, Polyhedron, 2014, 84, 63–66 CrossRef CAS.
- J. J. Adams, N. Arulsamy and D. M. Roddick, Organometallics, 2012, 31, 1439–1447 CrossRef CAS.
- (a) X. Zhang, T. J. Emge and A. S. Goldman, Inorg. Chim. Acta, 2004, 357, 3014–3018 CrossRef CAS; (b) R. B. Bedford, M. E. Blake, S. J. Coles, M. B. Hursthouse and P. N. Scully, Dalton Trans., 2003, 2805–2807 RSC.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- Crysalis CCD, Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- Crysalis RED, Oxford Diffraction Ltd, Abingdon, Oxfordshire, UK, 2005 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813–3826 CrossRef CAS PubMed.
- D. Martin, D. Moraleda, T. Achard, L. Giordano and G. Buono, Chem. – Eur. J., 2011, 17, 12729–12740 CrossRef CAS PubMed.
- (a) Y. Xiao, Y. Xu, H.-S. Cheon and J. Chae, J. Org. Chem., 2013, 78, 5804–5809 CrossRef CAS PubMed; (b) J. Chen, T. Yuan, W. Hao and M. Cai, Catal. Commun., 2011, 12, 1463–1465 CrossRef CAS; (c) R. Paul, M. A. Ali and T. Punniyamurthy, Synthesis, 2010, 4268–4272 CAS.
- K. Kunz, U. Scholz and D. Ganzer, Synlett, 2003, 2428–2439 CrossRef CAS.
- (a) D. Olsson, A. Arunachalampillai and O. F. Wendt, Dalton Trans., 2007, 5427–5433 RSC; (b) A. Arunachalampillai, D. Olsson and O. F. Wendt, Dalton Trans., 2009, 8626–8630 RSC; (c) M. T. Johnson and O. F. Wendt, Inorg. Chim. Acta, 2011, 367, 222–224 CrossRef CAS.
- K. J. Jonasson, N. Ahlsten and O. F. Wendt, Inorg. Chim. Acta, 2011, 379, 76–80 CrossRef CAS.
- N. T. Mucha and R. Waterman, Organometallics, 2015, 34, 3865–3872 CrossRef CAS.
- (a) M. Kanzelberger, B. Singh, M. Czerw, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2000, 122, 11017–11018 CrossRef CAS; (b) A. V. Polukeev, R. Marcos, M. S. G. Ahlquist and O. F. Wendt, Chem. Sci., 2015, 6, 2060–2067 RSC.
- I. Göttker-Schnetmann and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 9330–9338 CrossRef PubMed.
- J. Choi, A. H. R. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed.
- (a) A. Kumar, T. Zhou, T. J. Emge, O. Mironov, R. J. Saxton, K. Krogh-Jespersen and A. S. Goldman, J. Am. Chem. Soc., 2015, 137, 9894–9911 CrossRef CAS PubMed; (b) A. Brück, D. Gallego, W. Wang, E. Irran, M. Driess and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 11478–11482 CrossRef PubMed; (c) Z. Huang, P. S. White and M. Brookhart, Nature, 2010, 465, 598–601 CrossRef CAS PubMed.
- A. V. Polukeev and O. F. Wendt, Organometallics, 2015, 34, 4262–4271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra. CCDC 1478500–1478503. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01816e |
| This journal is © The Royal Society of Chemistry 2016 |