
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A cobalt(II) iminoiodane complex and its scandium adduct: mechanistic promiscuity in hydrogen atom abstraction reactions†
Subrata
Kundu
a,
Petko
Chernev
b,
Xenia
Engelmann
a,
Chan Siu
Chung
c,
Holger
Dau
b,
Eckhard
Bill
d,
Jason
England
*c,
Wonwoo
Nam
*e and
Kallol
Ray
*a
aHumboldt-Universität zu Berlin, Institut für Chemie, Brook-Taylor-Straße 2, D-12489 Berlin, Germany. E-mail: kallol.ray@chemie.hu-berlin.de; Fax: +49 30 2093 7387; Tel: +49 30 2093 7385
bFreie Universität Berlin, FB Physik, Arnimallee 14, D-14195-Berlin, Germany
cDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link, Singapore 637371. E-mail: jengland@ntu.edu.sg
dMax-Plank-Institut für Chemische Energiekonversion, Stiftstraße 34-36, D-45470 Mülheim an der Ruhr, Germany
eDepartment of Chemistry and Nano Science, Center for Biomimetic System, Ewha Womans University, Seoul 120-750, Korea. E-mail: wwnam@ewha.ac.kr
First published on 14th July 2016
Abstract
In addition to oxometal [Mn+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] and imidometal [Mn+NR] units, transient metal–iodosylarene [M(n−2)+–OIPh] and metal–iminoiodane [M(n−2)+–N(R)IPh] adducts are often invoked as a possible “second oxidant” responsible for the oxo and imido group transfer reactivity. Although a few metal–iodosylarene adducts have been recently isolated and/or spectroscopically characterized, metal–iminoiodane adducts have remained elusive. Herein, we provide UV-Vis, EPR, NMR, XAS and DFT evidence supporting the formation of a metal–iminoiodane complex 2 and its scandium adduct 2-Sc. 2 and 2-Sc are reactive toward substrates in the hydrogen-atom and nitrene transfer reactions, which confirm their potential as active oxidants in metal-catalyzed oxidative transformations. Oxidation of para-substituted 2,6-di-tert-butylphenols by 2 and 2-Sc can occur by both coupled and uncoupled proton and electron transfer mechanisms; the exact mechanism depends on the nature of the para substituent.
O] and imidometal [Mn+NR] units, transient metal–iodosylarene [M(n−2)+–OIPh] and metal–iminoiodane [M(n−2)+–N(R)IPh] adducts are often invoked as a possible “second oxidant” responsible for the oxo and imido group transfer reactivity. Although a few metal–iodosylarene adducts have been recently isolated and/or spectroscopically characterized, metal–iminoiodane adducts have remained elusive. Herein, we provide UV-Vis, EPR, NMR, XAS and DFT evidence supporting the formation of a metal–iminoiodane complex 2 and its scandium adduct 2-Sc. 2 and 2-Sc are reactive toward substrates in the hydrogen-atom and nitrene transfer reactions, which confirm their potential as active oxidants in metal-catalyzed oxidative transformations. Oxidation of para-substituted 2,6-di-tert-butylphenols by 2 and 2-Sc can occur by both coupled and uncoupled proton and electron transfer mechanisms; the exact mechanism depends on the nature of the para substituent.
Iodosobenzene (PhI
O) and iminoiodanes (PhINR) are an important class of group transfer reagents in organic synthesis, and they are often used in conjunction with transition metal-based catalysts.1 High-valent oxometal [Mn+O] and imidometal [Mn+NR] units are generally accepted to be key reactive intermediates in these reactions.2 However, transient metal–iodosylarene [M(n−2)+–OIPh]3 and metal–iminoiodane [M(n−2)+–N(R)IPh]4 adducts have also been suggested as a possible “second oxidant” responsible for this oxo and imido group transfer reactivity. Nam et al. reported the first isolation and spectroscopic characterization of an iodosobenzene adduct, namely [(porph)FeIII(OIPh)]+, from the reaction of [(porph+˙)FeIVO]+ with iodobenzene.3c Very recently, McKenzie et al. provided the molecular structure of a non-heme iron(III) iodosylarene adduct by X-ray crystallography.3d Fujii et al.3e,f also reported structural and spectroscopic evidence in support of bis(iodosylarene) coordination to a manganese(IV)–salen center. Notably, the aforementioned isolated [M(n−2)+–OIPh] adducts were all found to be reactive toward substrates in both hydrogen- and oxygen-atom transfer reactions, which confirms their potential as active oxidants in metal-catalyzed oxidative transformations.3 In contrast, isoelectronic [M(n−2)+–N(R)IPh] complexes have remained elusive, and their presence as transient reactive intermediates has been forwarded on the basis of labelling experiments alone.4a
Herein, we describe the spectroscopic characterization of a novel [(TMG3tren)CoII–(sPhINTs)]2+ (2) complex [sPhINTs = {N-(p-toluenesulfonyl)imino}(2-tert-butylsulfonyl)phenyliodinane;5 TMG3tren = tris[2-{N-tetramethylguanidyl}ethyl]amine] and its scandium adduct [(TMG3tren)CoII–sPhINTs(Sc(OTf)3)]2+ (2-Sc, OTf = CF3SO2−), which are able to oxidize the C–H and O–H bonds of a variety of substrates. Additionally, we demonstrate that the presence of Sc3+ in 2-Sc initiates a two-step reaction mechanism for the oxidation of benzyl alcohol (PhCH2OH), involving a [CoII–(sPhINTs)⋯Sc3+⋯O(H)–CH2Ph] association event prior to C–H bond cleavage. In the case of para-substituted phenols two different mechanistic pathways have been established for reactions with 2 and 2-Sc: a concerted proton-coupled electron transfer (PCET), and a proton transfer followed by electron transfer (PT-ET). The nature of the phenol para-substituent is shown to control the mechanism of oxidation.
A combination of [(TMG3tren)CoII–OTf]+ (1) with sPhINTs5 in acetone at −40 °C led after 6000 seconds to the formation of an orange complex 2 with absorption maxima λmax [εmax] centered at 420 nm [1150 M−1 cm−1] and 820 nm [130 M−1 cm−1] (Fig. S1†). The absorption spectrum of 2 is clearly distinct from 1 + sPhINTs (Fig. 1), and titration experiments (Fig. 1 inset) show that addition of only 1 equiv. sPhINTs to 1 is necessary to maximize the yield of 2. Similarly, the 1H-NMR spectrum of 2 is distinct from that of 1 + free sPhINTs (Fig. S2 and S3†), and does not show the presence of any unreacted starting materials in significant amounts. Moreover, in the 1H-NMR spectrum of 2 the complete set of signals expected for sPhINTs are observed (Fig. S3†), which are broadened and shifted to slightly higher fields relative to free sPhINTs; this can be attributed to binding to the paramagnetic cobalt centre.
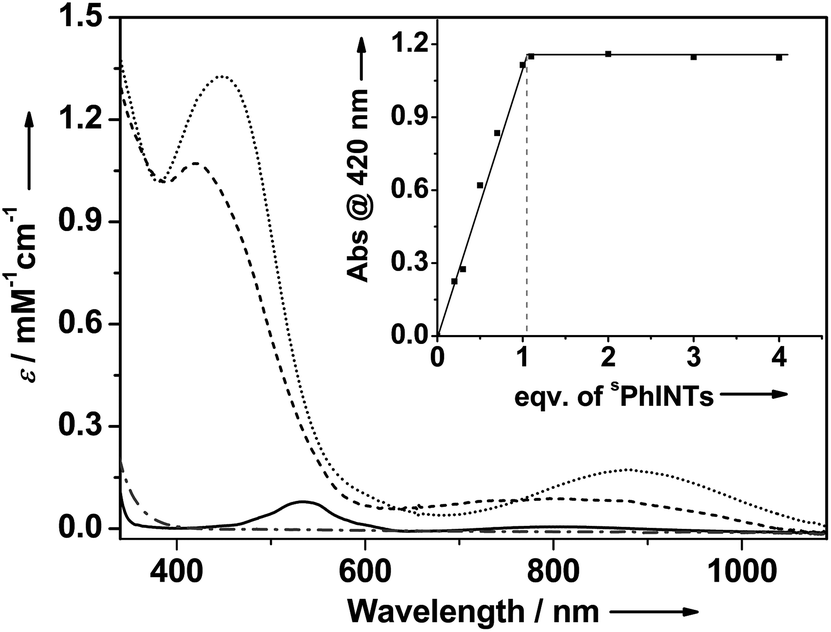 | ||
| Fig. 1 Main: Electronic spectra of 1 (solid trace), sPhINTs (dash-dotted trace), 2 (dashed trace), and 2-Sc (dotted trace) in acetone at −40 °C. Inset: Plot of the absorbance of the 420 nm band of 2 against the equivalents of sPhINTs added to 1. Notably, 2 and 2-Sc can also be generated in CH2Cl2 with λmax [εmax] values identical to that observed in acetone. | ||
Upon addition of scandium triflate (Sc(OTf)3) at −40 °C, 2 is instantaneously converted to a new species 2-Sc (λmax [εmax] = 450 nm [1350 M−1 cm−1] and 885 nm [180 M−1 cm−1]) (Fig. 1). As was the case for sPhINTs, only 1 equiv. Sc3+ is required for complete conversion of 2 to 2-Sc (Fig. S4†). Notably, complex 1 can also be converted directly to 2-Sc by reacting with sPhINTs in the presence of 1 equiv. Sc(OTf)3. Both 2 and 2-Sc are meta-stable intermediates with half-lives (t1/2) of 3600 and 720 seconds, respectively, at 25 °C.
The electronic structures of 2 and 2-Sc were probed by EPR spectroscopy. Surprisingly, in spite of the significant differences in their UV-Vis spectra, the X-band EPR spectra of 2 and 2-Sc (Fig. S5†) exhibit identical axial S = 3/2 signals, with effective g values of g⊥ = 4.40 and g‖ = 2.09 that are strikingly similar to the previously reported6 axial EPR signals of 1. Furthermore, the EPR spectra of the series of [(TMG3tren)CoIIX]+ (X = OTf, Cl, SCN, N3, CH3COO, CN) complexes all exhibit axial S = 3/2 signals identical to 2, with effective g values of g⊥ = 4.40 and g‖ = 2.09, even though their UV-Vis spectra are distinct from one another (Fig. S6†). This shows that the EPR parameters of the {(TMG3tren)CoII} moiety are effectively independent of the nature of the fifth ligand, and appear to be dictated solely by the strong binding of the TMG3tren ligand to the CoII ion. The EPR parameters change only when the formal oxidation state of cobalt is altered. For example, a rhombic EPR spectrum (E/D = 0.15 ± 0.01) with split effective g⊥ = 5.60, 3.60, and g‖ = 1.96 was observed for the unusual {(TMG3tren)CoIV–O–Sc3+} (3-Sc) core containing a cobalt center with a formal oxidation state of +4.6 Taken together, the near-identical EPR spectra of 2, 2-Sc, and 1 indicate that the cobalt centers in 2 and 2-Sc retain the +2 oxidation state of 1.
2 and 2-Sc were further characterized by X-ray absorption spectroscopy (XAS). The positions of the Co K-edges of these complexes do not differ from that of 1 (Fig. S7†), which indicates that the reaction of 1 with sPhINTs does not cause a change in the CoII oxidation state. This result is in agreement with our EPR study and is in stark contrast to 3-Sc,6 whose Co K-edge energy was found to be blue-shifted by 1.3 eV relative to 1.
The principal feature of the inner-sphere scattering peaks in the extended X-ray absorption fine structure (EXAFS) spectra of 2 and 2-Sc (Fig. 2, S8 and Table S1†) at R′ ∼ 1.67 Å (similar to that previously reported for 1)6 can be fitted in both cases with a single shell of 5 O/N scatterers at a distance of ∼2.05 Å, and corresponds to the donor atoms of TMG3tren and sPhINTs. Notably, in contrast to 3-Sc,6 no evidence of a short Co–N/O distance could be obtained from the fit of the EXAFS data of either 2 or 2-Sc. The outer-shell features in the EXAFS spectra of 2 and 2-Sc can be satisfactorily accounted for by considering single scattering paths involving 8 C atoms at 2.9–3.0 Å and 6 C/N atoms at 3.4–3.5 Å. Satisfactory fitting of the EXAFS data of 2 requires inclusion of an additional outer-shell scatterer at 3.78 Å corresponding to a single sulfur or iodine atom (fits 4 and 5 in Table S2†). The removal of this shell significantly lowers the quality of the fit (fit 3 in Table S2†). The best fit of the EXAFS data of 2-Sc (fit 8 in Table S3†) requires an outer-shell scatterer at 3.62 Å, which can be assigned to a Sc atom, in addition to the Co–S scatterer at 3.66 Å.
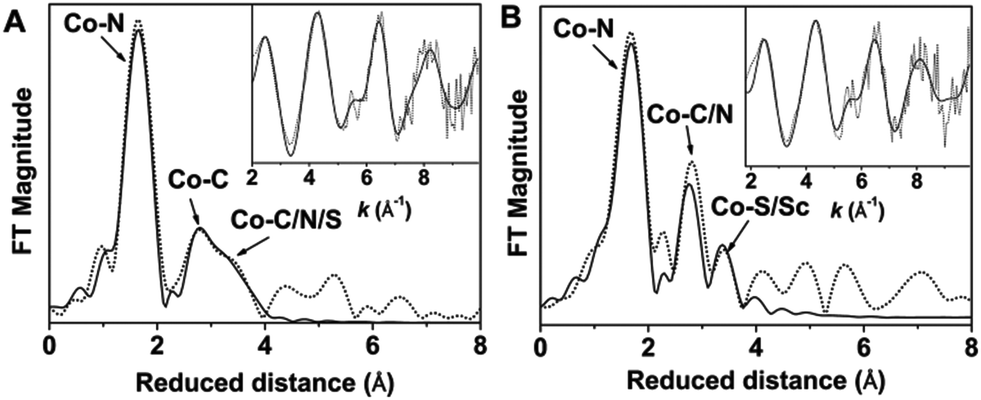 | ||
| Fig. 2 Fourier-transformed Co K-edge EXAFS spectra of 2 (A) and 2-Sc (B) [experimental data: dotted line, simulation: solid line]. Insets show the corresponding EXAFS data in a wave-vector scale before the Fourier transformations. | ||
Taken together, the XAS, EPR, NMR, and UV-Vis data of 1, 2 and 2-Sc conclusively indicate that a CoII oxidation state is retained throughout this series. Complex 2 is, therefore, formulated as the five-coordinate cobalt(II)–iminoiodane adduct [(TMG3tren)CoII–(sPhINTs)]2+ (Scheme 1), and 2-Sc is proposed to be [(TMG3tren)CoII–(sPhINTs)(Sc(OTf)3)]2+, wherein a single Sc3+ ion binds directly to the [(TMG3tren)CoII–(sPhINTs)]2+ complex.8 Consistent with this assignment the electrospray mass spectrum (ESI-MS; Fig. S9†) of both 2 and 2-Sc exhibits a prominent peak at m/z = 496.15, with a mass and isotope distribution pattern corresponding to [(TMG3tren)CoII–(sPhINTs)]2+.
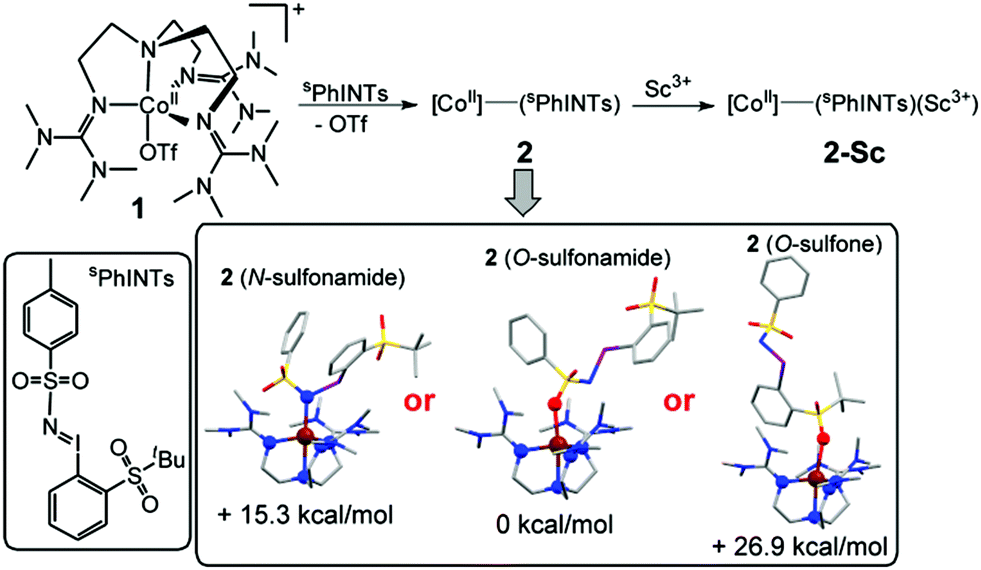 | ||
| Scheme 1 Products from reactions of 1 with sPhINTs, in the presence and absence of Sc3+ ions. The DFT calculated possible structures of 2 and their relative energies in the S = 3/2 ground state are shown in the inset. Color code: C, grey; O, red; N, blue; Co, brown; sulfur, yellow; iodine, violet. | ||
DFT calculations were also performed in order to obtain further insights into the electronic and geometric structures of 2.7 The binding of sPhINTs to the cobalt center in 2 can in principle occur either via the sulfonamide group (O-bound or N-bound) or alternatively via one of the O-atoms of the sulfone group (Scheme 1 and Table S4†). The most stable structure of the experimentally observed spin-quartet ground (S = 3/2) state was calculated to contain the O-bound sulfonamide binding mode of sPhINTs (Scheme 1, inset), with the respective energies (Table S5†) of the geometry optimized structures of the more sterically disfavoured N-bound sulfonamide and O-bound sulfone isomers being higher by 15.3 and 26.9 kcal mol−1 respectively. Consistent with this conclusion, the DFT calculated average Co–N/O distance of 2.10 Å (corresponding to four N-donors of TMG3tren and one O-donor of sPhINTs) in an S = 3/2 O-bound sulfonamide [(TMG3tren)CoII–(sPhINTs)]2+ complex (Tables S1 and S4†) best matches the EXAFS determined Co–N/O distance of 2.05 Å for 2. The calculated average Co–N distances for the corresponding N-bound sulfonamide and O-bound sulfone structures are significantly longer at 2.17 Å and 2.14 Å, respectively.
We then compared the reactivities of 2 and 2-Sc in hydrogen atom transfer (HAT) reactions from the hydrocarbon substrates xanthene, 9,10-dihydroanthracene (DHA), 1,4-cyclohexadiene (CHD), benzyl alcohol (PhCH2OH) and 1-benzyl-1,4-dihydronicotinamide (BNAH). In all cases, addition of a substrate led to significantly accelerated decay of the chromophore associated with 2 and 2-Sc. In the presence of an excess substrate this proceeded with first-order kinetics and the resulting effective rate constants (keff) were found to be linearly dependent upon substrate concentration, thereby yielding second-order rate constants. Such reaction kinetics are consistent with the direct reaction of 2 and 2-Sc with a substrate or alternatively a rapid pre-equilibrium yielding an alternative active oxidant. Intermediacy of a more traditional imidometal [(TMG3tren)CoIV–(NTs)]2+ oxidant would, therefore, require rapid reversible N–I bond scission. Given that there is no precedence for this and the high energy calculated for the N-bound isomer, which would be a necessary intermediate in its formation, this seems highly improbable. As a consequence, we postulate that the O-bound sulphonamide isomer is the active oxidant.
Product analysis of the reaction mixtures of DHA, CHD and PhCH2OH reactions revealed the formation of the corresponding dehydrogenated products (Table 1) in 52–65% yields; p-toluenesulfonamide (ArSO2NH2; see Fig. 3 and 4) is also detected as a reaction product (80–85% yield) in each case. Control reactions of the substrate with sPhINTs alone or with sPhINTs + Sc3+ did not lead to any oxidation products. Thus, sPhINTs is activated upon binding to the {(TMG3tren)CoII} core. Interestingly, the second order rate constants (k2) determined at −40 °C are comparable both in the absence and presence of Sc3+ ions (Table 1). This is in contrast to the previous report of the enhancement of the oxidizing capabilities of the corresponding metal-oxo intermediates6,8 in the presence of various Lewis-acids. Furthermore, the k2 values form a linear correlation (Fig. S10†) with the C–H bond dissociation energies (BDEC–H)9 of the substrates, which indicates that C–H bond homolysis is the rate determining step (r.d.s) in these reactions.
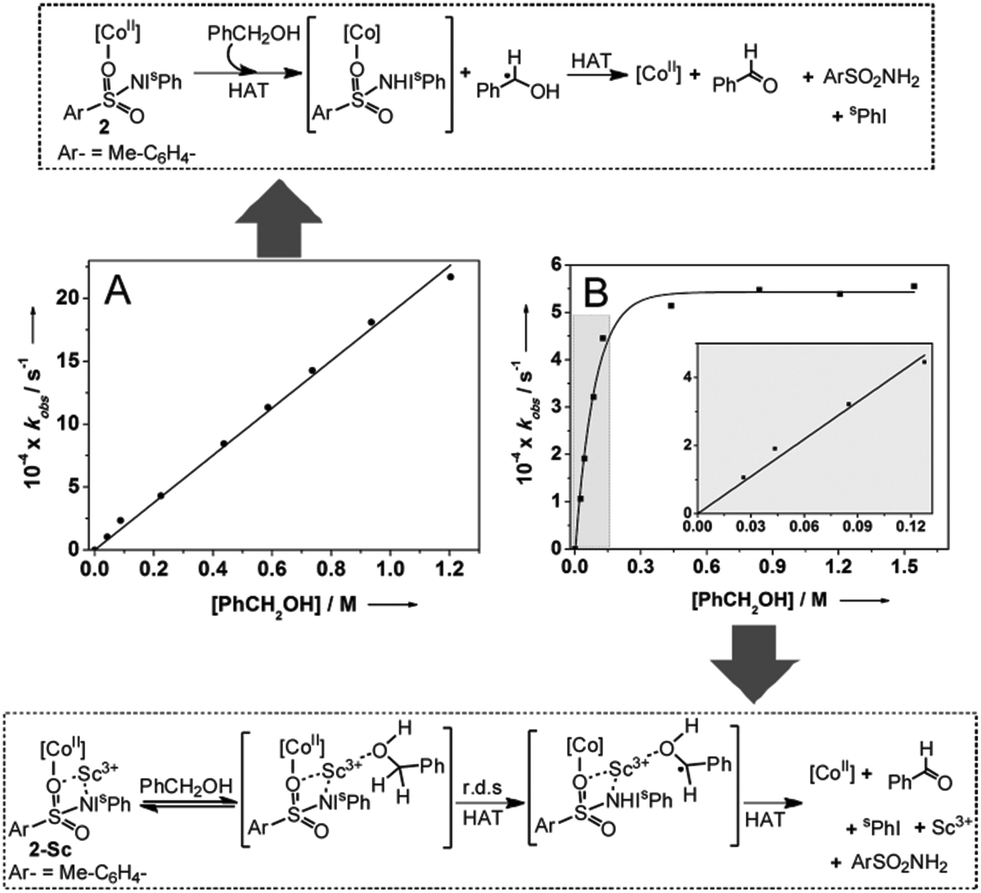 | ||
| Fig. 3 Proposed mechanisms for hydrogen atom abstraction from benzyl alcohol based on the dependence of the kobs values of complexes 2 (A) and 2-Sc (B) at −40 °C on the concentrations of benzyl alcohol. B inset: expansion of the highlighted region for low concentrations of PhCH2OH. r.d.s = rate determining step; HAT = hydrogen atom transfer. | ||
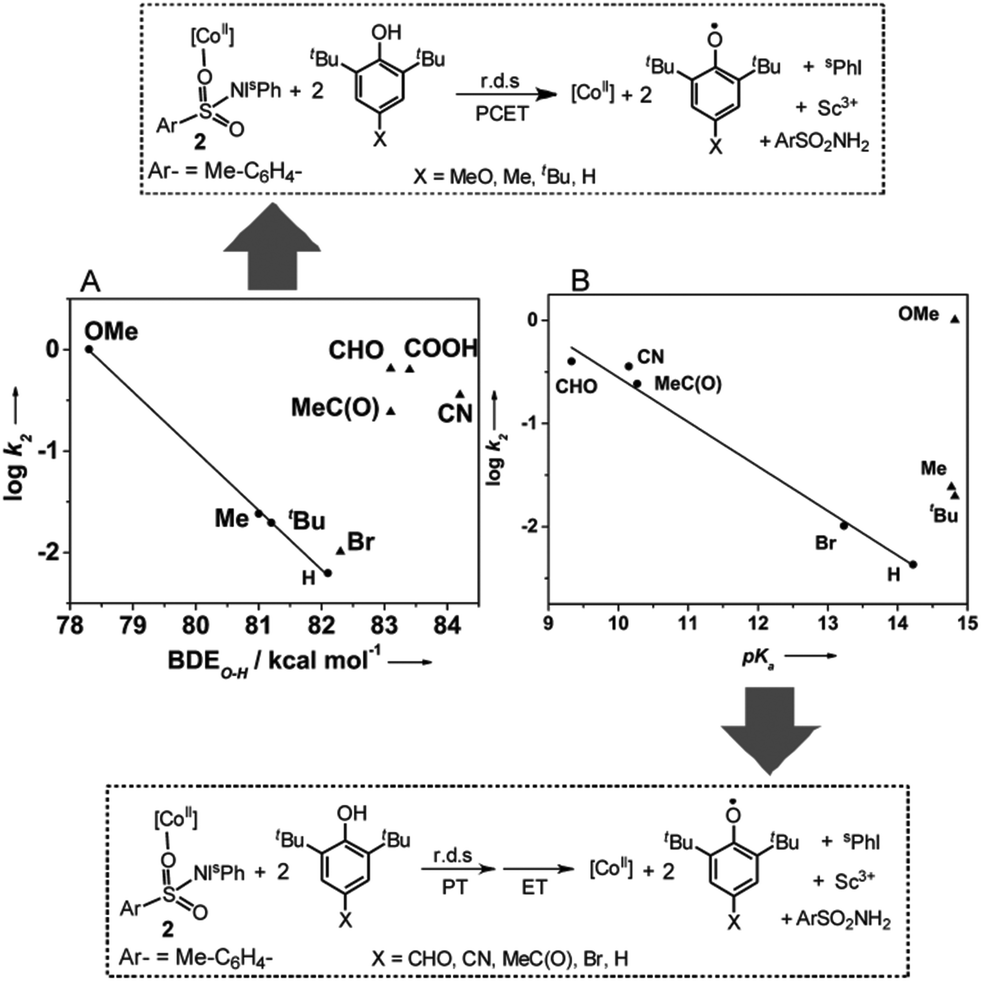 | ||
| Fig. 4 Proposed mechanisms for the hydrogen atom transfer reactions from different substituted phenols based on the dependence of the kobs values of complex 2 on the O—H bond dissociation energies (A), as well as, the pKa (B) of the phenols. PCET = proton coupled electron transfer; PT = proton transfer; ET = electron transfer; r.d.s = rate determining step. | ||
| X-DTBP X = | BDEO–H (kcal mol−1) | σ | pKa | k 2 (10−2 M−1 s−1) at −20 °C | |
|---|---|---|---|---|---|
| 2 | 2-Sc | ||||
| a Yields are for complex 2 and calculated based on the amount of the starting complex 1; yields for reactions with 2-Sc are comparable to 2. b The rate was determined using less than 150 equiv. of benzyl alcohol. | |||||
| MeO | 78.3 | −0.78 | 14.82 | 100.8 | 208.7 |
| Me | 81 | −0.31 | 14.77 | 2.42 | 0.70 |
| t Bu | 81.2 | −0.26 | 14.82 | 1.97 | 1.06 |
| H | 82.1 | 0 | 14.22 | 0.43 | 0.05 |
| CHO | 83.1 | 0.40 | 9.33 | 40.12 | 6.61 |
| MeC(O) | 83.1 | 0.48 | 10.27 | 31.29 | 4.32 |
| COOH | 83.4 | 0.55 | — | 63.47 | 2.84 |
| CN | 84.2 | 0.66 | 10.15 | 35.82 | 2.67 |
In order to obtain additional mechanistic insights, deuterium kinetic isotope effects (KIE) on the second-order rate constant k2(C–H)/k2(C–D) were measured for reactions of xanthene and DHA with 2. Values of 1.87 and 1.66 were determined at −40 °C for xanthene and DHA (Fig. S11A and B†), respectively, indicating that proton transfer is involved in the rate determining step. These KIE values are significantly smaller than those typically seen for high-valent metal-oxo and metal-imido mediated HAT reactions (3–25).1,2 This is again consistent with the [(TMG3tren)CoII–(sPhINTs)]2+ core in 2 not evolving to a reactive high-valent [(TMG3tren)CoIV–(NTs)]2+ species prior to the reaction, and supports the suggestion that it is able to act directly as the reactive species in these oxygenation reactions.
Although the presence of Sc3+ did not significantly perturb the rate of C–H bond homolysis mediated by the [(TMG3tren)CoII–(sPhINTs)]2+ core, it does affect a change in the mechanism in the reaction with PhCH2OH. As expected, the addition of large excess of PhCH2OH (30–1000 equivalents) to preformed solutions of 2 at −40 °C caused pseudo-first order decay (Fig. S12†) of the absorption bands associated with this complex at 420 and 820 nm. Furthermore, the rate of decay was found to increase linearly with increasing PhCH2OH concentration (Fig. 3A), thereby affording a second order rate constant (k2) of 0.0020 M−1 s−1. The pseudo-first order rate constants (kobs) for the reaction of 2-Sc with 30–150 equivalents of PhCH2OH were also observed to follow a linear correlation, and a second order rate constant (k2) of 0.0037 M−1 s−1 at −40 °C was measured (Table 1; Fig. 3B inset). However, at higher concentrations (>150 equivalents) the correlation became non-linear with saturation behavior being obtained (Fig. 3B). The simplest model to account for these findings is to invoke the presence of a relatively fast equilibrium preceding the rate-determining step.10 More specifically, PhCH2OH reversibly binds to 2-Sc (Fig. 3) to yield a substrate bound intermediate [(TMG3tren)CoII–(sPhINTs)⋯Sc3+⋯O(H)–CH2Ph], which then undergoes C–H bond cleavage by a rate determining H-atom abstraction process. At high PhCH2OH concentrations (>150 equivalents), the equilibrium shifts completely towards the substrate-bound species and the rate becomes independent of PhCH2OH concentration, thereby explaining the saturation kinetics.
This notion is further reinforced by the absence of saturation kinetics for the other substrates, which do not possess a ligating atom, and 2, which lacks a Lewis acidic binding site.
Mechanistic studies for the reaction of 2 and 2-Sc with para-substituted 2,6-di-tert-butylphenols [X-DTBP; X = –OMe, –Me, –tBu, –H, –CN, –CHO, –COOH, –MeC(O)] reveal that the second-order rate constants depend markedly on the electron-donating/-withdrawing properties of the para-substituents (Table 1). For all the substituted phenols studied, second order kinetic behavior (Fig. S13†) and identical reaction products, namely ArSO2NH2 and two equivalents of the corresponding phenoxyl radical, were observed. However, when the rates obtained for both 2 and 2-Sc are plotted as a function of σ of the para-substituents a linear correlation with a negative Hammett slope (Fig. S14†), as previously reported for the reaction of metal-oxo11a and -superoxo11b intermediates with phenol substrates, is obtained only for the electron-donating substituents (X = –OMe, –tBu, –Me, and –H). The k2 values for the electron withdrawing substituents [X = –CN, –CHO, –COOH, –MeC(O), –Br] appear significantly above the trend line, thereby hinting at a change in the mechanism for these substrates.
In response, we also plotted the rate constants against the O–H bond dissociation energy (BDE).9 Once again, a linear correlation with a negative slope comparable to that obtained for the metal-oxo and -dioxygen intermediates was observed for the phenols with electron-donating substituents, but the data for the phenols with electron-withdrawing groups appeared outside the correlation (Fig. 4A and S15A†). Plotting log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k2versus pKa12 (Fig. 4B and S15B†) revealed that for X-DTBP with X = –CHO, –C(O)Me, –CN, –Br, and –H, the rate (logk2) decreased linearly with decreasing acidity (increasing pKa), whereas the rates for the electron donating substituents –MeO, –Me, and –tBu scatter irregularly. From the above studies we can conclude that 2 and 2-Sc oxidize acidic phenols (X-DTBP; X = –CN, –CHO, –COOH, –C(O)Me) via a stepwise proton transfer and electron transfer mechanism, with the proton transfer (PT) being effectively rate determining. This is likely to involve an initial proton transfer to solvent (acetone) to give a phenolate anion, which may bind to the Co(II) center and is much easier to oxidize than the phenol itself. The resulting rate acceleration, therefore, parallels the extent of ionization (acidity) of the phenol. In contrast, less acidic phenols like MeO-DTBP, Me-DTBP, and tBu-DTBP, proceed via a concerted proton-coupled electron transfer (PCET) mechanism. Interestingly, data for 2,6-di-tert-butylphenol (H-DTBP) falls in all the plots of the second-order rate constant (logk2) versus the thermochemical parameters BDEO–H and pKa (Fig. 4A, B and S15A, B†), thereby locating H-DTBP on the mechanistic borderline between the concerted PCET and stepwise PT-ET reaction pathways.
k2versus pKa12 (Fig. 4B and S15B†) revealed that for X-DTBP with X = –CHO, –C(O)Me, –CN, –Br, and –H, the rate (logk2) decreased linearly with decreasing acidity (increasing pKa), whereas the rates for the electron donating substituents –MeO, –Me, and –tBu scatter irregularly. From the above studies we can conclude that 2 and 2-Sc oxidize acidic phenols (X-DTBP; X = –CN, –CHO, –COOH, –C(O)Me) via a stepwise proton transfer and electron transfer mechanism, with the proton transfer (PT) being effectively rate determining. This is likely to involve an initial proton transfer to solvent (acetone) to give a phenolate anion, which may bind to the Co(II) center and is much easier to oxidize than the phenol itself. The resulting rate acceleration, therefore, parallels the extent of ionization (acidity) of the phenol. In contrast, less acidic phenols like MeO-DTBP, Me-DTBP, and tBu-DTBP, proceed via a concerted proton-coupled electron transfer (PCET) mechanism. Interestingly, data for 2,6-di-tert-butylphenol (H-DTBP) falls in all the plots of the second-order rate constant (logk2) versus the thermochemical parameters BDEO–H and pKa (Fig. 4A, B and S15A, B†), thereby locating H-DTBP on the mechanistic borderline between the concerted PCET and stepwise PT-ET reaction pathways.
The reaction of excess triphenylphosphine (30–60 equivalents) with preformed 2 and 2-Sc at −40 °C leads to the quantitative formation of triphenylphosphonium tosyl-aza-ylide (Ph3PNTs) and regeneration of the starting CoII complex 1. Interestingly, the second order rate constants (k2) determined for the reactions of 2 and 2-Sc with PPh3 are 0.531 and 0.0321 M−1 s−1, respectively, demonstrating that the group-transfer reactivity of 2 is decelerated by factors of ∼15 upon binding of Sc3+ ions (Table 1; Fig. S16 and S17†).
Conclusions
In conclusion, we have provided spectroscopic evidence supporting formation of a metal–iminoiodane complex 2 and its scandium adduct 2-Sc. A novel binding mode of sPhINTs via the sulfonamide O-atom is predicted in 2 based on DFT calculations. Both 2 and 2-Sc possess cobalt in a +2 oxidation state. This is in sharp contrast to the analogous oxo chemistry,6 where the presence of scandium led to the stabilization of an unusual {CoIV–O–Sc3+} core. The formation of the isoelectronic {CoIV–N(Ts)–Sc3+} core in 2-Sc, which would require a short Co–N(Ts) distance, is presumably prevented by the additional steric demands of the tosyl group, which is absent in the oxo chemistry. The reaction of 2 and 2-Sc with 4-substituted 2,6-di-tert-butylphenols is shown to proceed via a stepwise PT-ET mechanism for electron withdrawing substituents. Although PT-ET mechanisms have previously been invoked for the oxidation of phenols by organic radicals,13 this is the first experimental evidence for a metal complex mediated variant. Additionally, the presence of Sc3+ in 2-Sc is shown to promote the formation of a precursor complex during the oxidation of benzyl alcohol, thereby demonstrating the cooperativity of two metal centers in promoting substrate oxidation reactions. Taken together, the present study expands our understanding of metal-mediated oxidation reactions using iminoiodanes, with metal–iminoiodane adducts being demonstrated to be a second plausible oxidant, in addition to the often invoked high-valent metal-imido reactive intermediates,2b,c,e in hydrogen atom abstraction and group transfer reactions.We gratefully acknowledge financial support of this work from the Cluster of Excellence “Unifying Concepts in Catalysis” (EXC 314/2), Berlin. K. R. also thanks the Heisenberg-Programm of the Deutsche Forschungsgemeinschaft for financial support. W. N. acknowledges financial support from the NRF of Korea through the CRI (NRF-2012R1A3A2048842) and GRL (NRF-2010-00353). JE is thankful to the NAP fellowship of the Nanyang Technological University.
Notes and references
- (a) D. Ostovic and T. C. Bruice, Acc. Chem. Res., 1992, 25, 314 CrossRef CAS; (b) M. Costas, Coord. Chem. Rev., 2011, 255, 2912 CrossRef CAS; (c) R. T. Gephart and T. H. Warren, Organometallics, 2012, 31, 7728 CrossRef CAS.
- (a) W. Nam, Acc. Chem. Res., 2007, 40, 522 CrossRef CAS PubMed; (b) J. Hohenberger, K. Ray and K. Meyer, Nat. Commun., 2012, 3, 720 CrossRef PubMed; (c) K. Ray, F. Heims and F. F. Pfaff, Eur. J. Inorg. Chem., 2013, 3784 CrossRef CAS; (d) V. Lyaskovskyy, A. I. O. Suarez, H. Lu, H. Jiang, X. P. Zhang and B. de Bruin, J. Am. Chem. Soc., 2011, 133, 12264 CrossRef CAS PubMed; (e) J. F. Berry, Dalton Trans., 2012, 41, 700 RSC.
- (a) J. A. Smegal, B. C. Schardt and C. L. Hill, J. Am. Chem. Soc., 1983, 105, 3510 CrossRef CAS; (b) S. H. Wang, B. S. Mandimutsira, R. Todd, B. Ramdhanie, J. P. Fox and D. P. Goldberg, J. Am. Chem. Soc., 2004, 126, 18 CrossRef CAS PubMed; (c) W. Nam, S. K. Choi, M. H. Lim, J. Rohde, I. Kim, J. Kim, C. Kim and L. Que, Angew. Chem., Int. Ed., 2003, 42, 109 CrossRef CAS; (d) A. Lennartson and C. J. McKenzie, Angew. Chem., Int. Ed., 2012, 124, 6871 CrossRef; (e) C. Wang, T. Kurahashi and H. Fujii, Angew. Chem., Int. Ed., 2012, 51, 7809 CrossRef CAS PubMed; (f) C. Wang, T. Kurahashi, K. Inomata, M. Hada and H. Fujii, Inorg. Chem., 2013, 52, 9557 CrossRef CAS PubMed; (g) B. Wang, Y.-M. Lee, M. S. Seo and W. Nam, Angew. Chem. Int. Ed., 2015, 54, 11740 CrossRef CAS PubMed; (h) S. Hong, B. Wang, M. S. Seo, Y.-M. Lee, M. J. Kim, H. R. Kim, T. Ogura, R. Garcia-Serres, M. Clmancey, J.-M. Latour and W. Nam, Angew. Chem. Int. Ed., 2014, 53, 6388 CrossRef CAS PubMed.
- (a) M. J. Zdilla and M. M. Abu-Omar, J. Am. Chem. Soc., 2006, 128, 16971 CrossRef CAS PubMed; (b) Z. Ke and T. R. Cundari, Organometallics, 2010, 29, 821 CrossRef CAS.
- (a) D. Macikenas, E. Skrzypczak-jankun and J. D. Protasiewicz, J. Am. Chem. Soc., 1999, 121, 7164 CrossRef CAS; (b) S. Kundu, E. Miceli, E. Farquhar, F. F. Pfaff, U. Kuhlmann, P. Hildebrandt, B. Braun, C. Greco and K. Ray, J. Am. Chem. Soc., 2012, 134, 14710 CrossRef CAS PubMed.
- F. F. Pfaff, S. Kundu, M. Risch, S. Pandian, F. Heims, I. Pryjomska-Ray, P. Haack, R. Metzinger, E. Bill, H. Dau, P. Comba and K. Ray, Angew. Chem., Int. Ed., 2011, 50, 1711 CrossRef CAS PubMed.
- Owing to the structural complexity of 2-Sc, the actual binding site of scandium is not clear at present.
- (a) J. Chen, Y.-M. Lee, K. M. Davis, X. Wu, M. S. Seo, K.-B. Cho, H. Yoon, Y. J. Park, S. Fukuzumi, Y. N. Pushkar and W. Nam, J. Am. Chem. Soc., 2013, 135, 6388 CrossRef CAS PubMed; (b) S. Fukuzumi, Coord. Chem. Rev., 2013, 257, 1564 CrossRef CAS.
- Y.-R. Luo, Comprehensive handbook of chemical bond energies, Taylor & Francis, 2007 Search PubMed.
- (a) I. Garcia-Bosch, A. Company, C. W. Cady, S. Styring, W. R. Browne, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2011, 50, 5648 CrossRef CAS PubMed; (b) J. M. Mayer, Acc. Chem. Res., 2011, 44, 36 CrossRef CAS PubMed.
- (a) D. E. Lansky and D. P. Goldberg, Inorg. Chem., 2006, 45, 5119 CrossRef CAS PubMed; (b) J. Cho, J. Woo, J. Eun Han, M. Kubo, T. Ogura and W. Nam, Chem. Sci., 2011, 2, 2057 RSC.
- L. A. Cohen and W. M. Jones, J. Am. Chem. Soc., 1963, 81, 3397 CrossRef.
- (a) G. Litwinienko and K. U. Ingold, J. Org. Chem., 2003, 68, 3433 CrossRef CAS PubMed; (b) G. Litwinienko and K. U. Ingold, J. Org. Chem., 2004, 69, 5888 CrossRef CAS PubMed; (c) M. C. Foti, C. Daquino and C. Geraci, J. Org. Chem., 2004, 69, 2309 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01815g |
| This journal is © The Royal Society of Chemistry 2016 |