
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Versatile bonding and coordination modes of ditriazolylidene ligands in rhodium(III) and iridium(III) complexes†
Kevin
Farrell
a,
Helge
Müller-Bunz
a and
Martin
Albrecht
*ab
aSchool of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: martin.albrecht@dcb.unibe.ch
bDepartment of Chemistry & Biochemistry, University of Bern, Freiestrasse 3, 3012 Bern, Switzerland
First published on 3rd June 2016
Abstract
Metalation of novel ditriazolium salts containing a trimethylene (–CH2CH2CH2–) or dimethylether linker (–CH2OCH2–) was probed with different rhodium(III) and iridium(III) precursors. When using [MCp*Cl2]2, a transmetalation protocol via a triazolylidene silver intermediate was effective, while base-assisted metalation with MCl3via sequential deprotonation of the triazolium salt with KOtBu and addition of the metal precursor afforded homoleptic complexes. The N-substituent on the triazole heterocycle directed the metalation process and led to Ctrz,Ctrz,CPh-tridentate chelating ditriazolylidene complexes for N-phenyl substituents. With ethyl substituents, only Ctrz,Ctrz-bidentate complexes were formed, while metalation with mesityl substituents was unsuccessful, presumably due to steric constraints. Through modification of the reaction conditions for the metalation step, an intermediate species was isolated that contains a Ctrz,CPh-bidentate chelate en route to the formation of the tridentate ligand system. Accordingly, Cphenyl–H bond activation occurs prior to formation of the second metal–triazolylidene bond. Stability studies with a Ctrz,Ctrz,CPh-tridentate chelating ditriazolylidene iridium complex towards DCl showed deuterium incorporation at both N-phenyl groups and indicate that Cphenyl–H bond activation is reversible while the Ctrz–Ir bond is robust. The flexible linker between the two triazolylidene donor sites provides access to both facial and meridional coordination modes.
Introduction
The discovery of N-heterocyclic carbenes (NHCs) as ligands for transition metals has fundamentally transformed organometallic chemistry1 and has spurred in particular2 the development of new generations of homogeneous catalysts.3 As a consequence of this success, the NHC theme has been varied in all dimensions, including the development of chiral systems,4 of non-cyclic analogues,5 of carbenes with reduced heteroatom stabilization6 and of course the combination of different privileged ligands together with carbenes.7 1,2,3-Triazolylidenes are a subclass of NHCs that have received considerable attention over the last few years,8 and which offer vast opportunities for catalysis9 and beyond.10 Their mesoionic character11 and strong σ-donor ability are attractive features for tailoring the properties of the coordinated metal center.12 Moreover, these heterocycles are accessible via copper(I) catalyzed ‘click’ [2 + 3] cycloaddition of an alkyne and azide (CuAAC),13 a reaction that stands out for its versatility and its exceptionally broad functional group tolerance.14 As a consequence, various donor-functionalized triazolylidene complexes with a wide range of chelating groups have been developed.8 Surprisingly however, triazolylidene-based chelating dicarbenes have not been studied extensively, despite the kinetic and thermodynamic stability imparted by chelation. To date, two approaches for linking 1,2,3-triazolium salts via either the triazole C4 or N3 position were reported, involving a di-alkyne and a di-azide precursor, respectively. For example, C4-linked ditriazolium salts with rigid aryl-bridges have been metalated with rhodium,15 ruthenium,16 nickel,17 and palladium,18 leading to bimetallic systems (Fig. 1, A) or CNC-type pincer complexes (B). Directly linked ditriazolylidenes were coordinated to rhodium,19 iridium, and ruthenium (C).20 Similarly, alkyl- or aryl-linkages via N3 have led to bimetallic complexes of ruthenium21 and iridium22 in which the ligand adopts a bridging rather than chelating binding mode (D, E). | ||
| Fig. 1 Known ditriazolylidene complexes including C,C′-linked ditriazolylidene ligands (A, B, C) and N,N′-linked ditriazolylidene scaffolds (D, E). | ||
Trimethylene-linked dicarbene ligands similar to those present in complex E, yet comprised of imidazolylidenes rather than triazolylidene heterocycles, induced alkyl C–H bond activation of the central methylene unit by rhodium or ruthenium, thus forming a tridentate dicarbene species.23 The Calkyl–H bond activation was significantly faster when the imidazolylidene is bound to rhodium via C4 as a mesoionic carbene rather than in the normal C2-bonding mode.23a Based on these considerations, we were interested to investigate the reactivity of analogous di-1,2,3-triazolylidene systems that are mesoionic as well. In addition, heteroatoms can easily be introduced into the triazolylidene linker, which may provide further reactivity patterns. Here, we report the synthesis of chelating ditriazolylidene rhodium(III) and iridium(III) complexes which contain flexible C,C′-linked ditriazolylidenes. The flexibility accommodates both facial and meridional coordination modes and the peripheral substituents at the heterocycle dictate the coordination mode of the triazolylidenes, thus demonstrating strong reactivity control by ligand design.
Results and discussion
Synthesis of the ligand precursors
The C,C′-linked ditriazoles 3 and 4 were synthesised in good to excellent yields (71–95%) via the ‘click’ cycloaddition of the commercially available diynes 1 and 2, and the corresponding azide, (Scheme 1). The alkyl linker was identified by a characteristic triplet for the CtrzCH2 group and a quintet for the central methylene unit. For example for compound 3a, these multiplets appeared at δH 2.67 and 1.94 in the 1H NMR spectrum respectively, while the ether linker showed as a singlet, e.g. at δH 4.71 for 4a. The versatility of the ‘click’ reaction allows for the facile variation of the N-bound substituent in the ligand precursor. This versatility is beneficial for the steric and electronic tailoring of the ligand environment when bound to the metal which is particularly useful for bond activation reactions. Alkylation of the ditriazoles with methyl iodide gave the ditriazolium salts 5(I) and 6(I) in good yields (Scheme 1). After quaternization, the 1H NMR signal of the heterocyclic proton shifted downfield by almost 1 ppm. For example the Ctrz–H in 5a(I) appeared as a singlet at δH 8.87 compared to δH 7.54 ppm in the precursor 3a. In addition, a new signal emerged in the 1H and 13C NMR spectrum for the N-bound methyl group (δH 4.20, δC 38.2 for 5a(I)). Anion substitution with AgBF4 afforded the corresponding ditriazolium salts 5(BF4) and 6(BF4). Successful anion exchange was indicated by a diagnostic ∼0.5 ppm upfield shift of the Ctrz–H resonance in the 1H NMR spectrum, presumably due to less pronounced X⋯H hydrogen bonding.24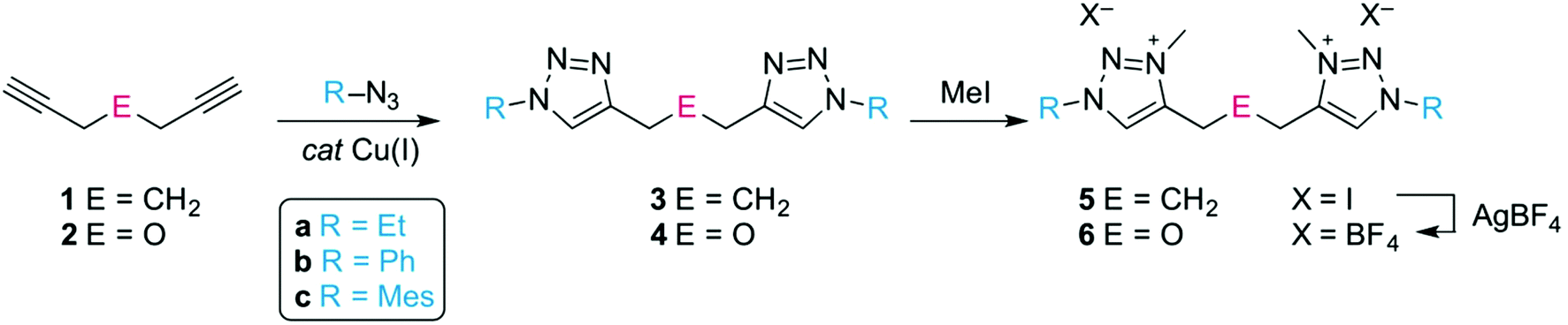 | ||
| Scheme 1 Ligand precursor synthesis via ‘click’ cycloaddition and alkylation. | ||
Metalation of ditriazolium salts with [MCp*Cl2]2
In an initial set of experiments, the well-established transmetalation procedure involving the formation of a triazolylidene silver(I) intermediate was utilised for the formation of triazolylidene rhodium(III) and iridium(III) complexes.8d,25 Successful metalation of the triazolium salts using Ag2O in the presence of KCl was surmised by the disappearance of the 1H NMR signal of the Ctrz–H unit, and by the appearance of a mass signal which is in agreement with the [Ag(ditriazolylidene)]+ fragment. The 1H NMR spectrum of the triazolylidene silver complexes showed a symmetric linker which suggests a dimeric [Ag2(trz^E^trz)2]2+ species rather than a chelating [Ag(trz^E^trz)]+ monomer, in agreement with results by Crudden and co-workers.15 The formation of the silver triazolylidene proceeded at elevated temperatures when using the N-ethyl triazolium salts (refluxing MeCN), while N-aryl triazolium salts reacted under milder conditions and provided the desired triazolylidene silver intermediate at room temperature. Triazolylidene silver complexes were also formed in the absence of a chloride additive, though significant decomposition was observed and yields were typically much lower, presumably because the triazolylidene silver unit is less stabilized by tetrafluoroborate as compared to chlorides.9e,21,26 Likewise, the triazolylidene silver intermediates were accessible also from the triazolium iodide salts 5(I), but in agreement with previous reports,27 the reaction was generally less clean than when starting from 5(BF4). Carbene silver complexes are known to exist in an equilibrium between ionic species of type [Ag(carbene)2]AgX2 and the neutral isomer [AgX(carbene)],28 which is delicately dependent on conditions and will lead to oligo- or polymeric systems with ditopic carbenes as used here. Due to this fluxionality, no attempts have been made to structurally characterize these intermediates.The triazolylidene silver complexes were used without further purification for transmetalation with [MCp*Cl2]2 (M = Rh, Ir), and afforded the chelating ditriazolylidene complexes 7–10 (Scheme 2). All complexes were air- and moisture stable and were isolated by standard column chromatography as red (7, 8) or orange solids (9, 10) that are moderately soluble in CH2Cl2 and readily soluble in MeCN.
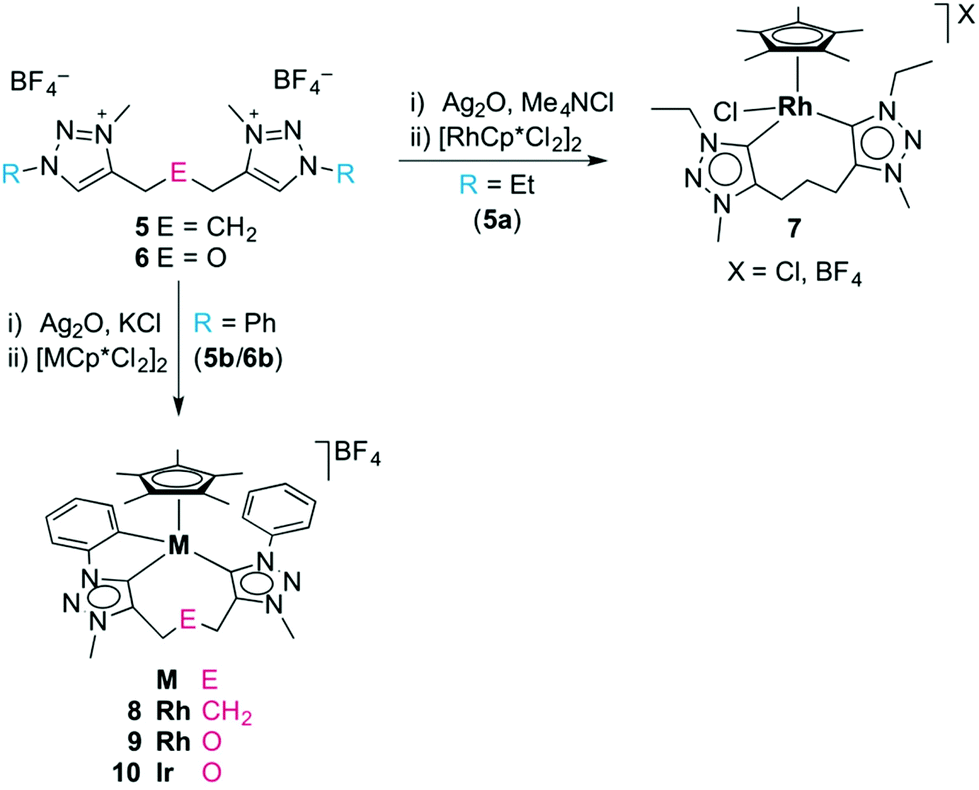 | ||
| Scheme 2 Formation of complexes 7–10 by transmetalation via a silver carbene intermediate. | ||
Chelation in complex 7 was indicated by the presence of symmetry-related triazolylidene units and the pertinent 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 integral ratio of the resonances due to the Cp* protons and the N–CH3 unit. The carbenic 13C NMR resonance is deshielded and appears at δC 154.4 as a doublet due to characteristic 103Rh coupling (1JCRh = 50.3 Hz). Furthermore, chelation led to diastereotopic methylene protons in the ethyl wingtip group as indicated by the two doublet of quartets at δH 4.81 and 4.42 (2JHH = 12.9 Hz, 3JHH = 7.2 Hz). Similarly, the triazolylidene bound CH2 group of the trimethylene linker appeared as two doublets of doublets of doublets (2JHH = 14.9 Hz, 3JHH = 8.6 Hz, 3JHH = 4.6 Hz). Of note, a multiplet in the 1.88–1.69 ppm range integrated for two protons and was attributed to the central CH2 group of the linker, thus suggesting that the linker has not been affected. This reactivity differs from that of related rhodium(III) complexes containing mesoionic diimidazolylidene ligands, in which C–H bond activation of the linker methylene group is spontaneous.23 X-ray crystallographic analysis of complex 7 unambiguously confirmed the ligand bonding mode surmised from spectroscopic studies (Fig. 2). The complex cation shows the typical three-legged piano-stool geometry with two triazolylidenes and one chloride ligand site forming the ‘legs’. The carbene bite angle is relatively wide, Ctrz–Rh–Ctrz = 91.21(9)°.29 While the triazolylidene units are symmetrical in solution, the Ctrz–Rh bond lengths of complex 7 differ considerably in the solid state (C1–Rh 2.026(2) Å, C11–Rh 2.082(2) Å). This difference may be a direct consequence of the different orientation of the triazolylidene heterocycles in the solid state with respect to the rhodium coordination geometry, e.g. the Cl–Rh–C–N dihedral angle is 48.4(2)° for the heterocycle containing C1 and 71.1(2)° for the heterocycle containing C7. In solution, these distinct arrangements average out as indicated by the symmetric NMR pattern observed for this complex.
:2 integral ratio of the resonances due to the Cp* protons and the N–CH3 unit. The carbenic 13C NMR resonance is deshielded and appears at δC 154.4 as a doublet due to characteristic 103Rh coupling (1JCRh = 50.3 Hz). Furthermore, chelation led to diastereotopic methylene protons in the ethyl wingtip group as indicated by the two doublet of quartets at δH 4.81 and 4.42 (2JHH = 12.9 Hz, 3JHH = 7.2 Hz). Similarly, the triazolylidene bound CH2 group of the trimethylene linker appeared as two doublets of doublets of doublets (2JHH = 14.9 Hz, 3JHH = 8.6 Hz, 3JHH = 4.6 Hz). Of note, a multiplet in the 1.88–1.69 ppm range integrated for two protons and was attributed to the central CH2 group of the linker, thus suggesting that the linker has not been affected. This reactivity differs from that of related rhodium(III) complexes containing mesoionic diimidazolylidene ligands, in which C–H bond activation of the linker methylene group is spontaneous.23 X-ray crystallographic analysis of complex 7 unambiguously confirmed the ligand bonding mode surmised from spectroscopic studies (Fig. 2). The complex cation shows the typical three-legged piano-stool geometry with two triazolylidenes and one chloride ligand site forming the ‘legs’. The carbene bite angle is relatively wide, Ctrz–Rh–Ctrz = 91.21(9)°.29 While the triazolylidene units are symmetrical in solution, the Ctrz–Rh bond lengths of complex 7 differ considerably in the solid state (C1–Rh 2.026(2) Å, C11–Rh 2.082(2) Å). This difference may be a direct consequence of the different orientation of the triazolylidene heterocycles in the solid state with respect to the rhodium coordination geometry, e.g. the Cl–Rh–C–N dihedral angle is 48.4(2)° for the heterocycle containing C1 and 71.1(2)° for the heterocycle containing C7. In solution, these distinct arrangements average out as indicated by the symmetric NMR pattern observed for this complex.
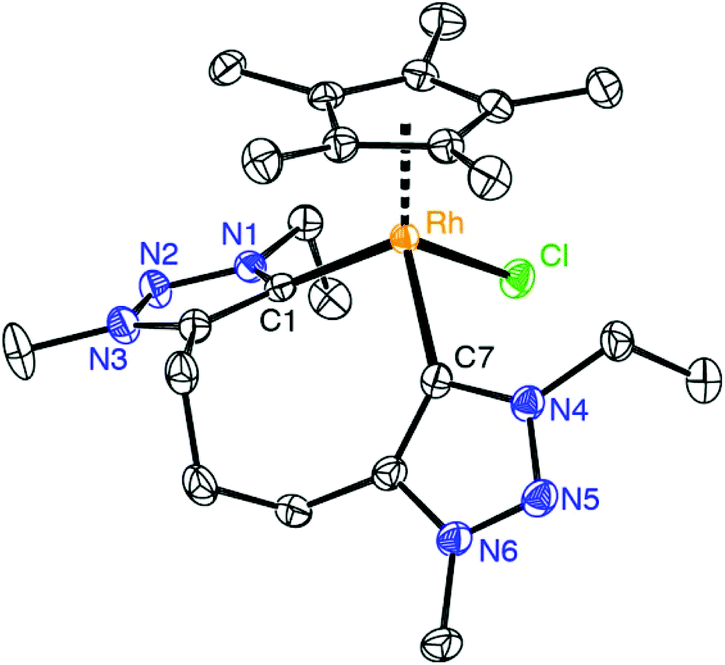 | ||
| Fig. 2 ORTEP representation of the complex cation complex 7 (50% probability ellipsoids, hydrogens omitted for clarity). Selected bond lengths: Rh–C1 2.026(2) Å, Rh–C7 2.082(2) Å, Rh–Cl 2.410(4) Å, Rh–Cpcentroid 1.862(4) Å; selected bond angles: C1–Rh–C7 91.21(9)°; C1–Rh–Cl 96.16(6)°, C7–Rh–Cl 84.74(7)°. | ||
It is worth noting that the analogous rhodation of the ether-linked ditriazolium salt 6a(BF4) has failed in our hands so far. Formation of the carbene silver intermediate was confirmed by the pertinent NMR and MS data, though substantial amounts of impurities were observed. Possibly, the ether linkage interacts with the silver ion and destabilizes this intermediate under the applied reaction conditions.
The presence of phenyl substituents at the triazole heterocycle altered the outcome of the reaction and induced cyclometalation through activation of one of the ortho Cphenyl–H bonds in 5b and 6b to yield complexes 8–10 containing a C,C,C-tridentate coordinated ditriazolylidene ligand (Scheme 2). Similar C–H bond activation was observed previously in mono-triazolylidene complexes containing N-bound phenyl-substituents.30 Apparently the Cphenyl–H bond activation process is preferred over heteroatom coordination and is spontaneous even at −30 °C both with rhodium and iridium precursors. No trace of a coordinated triazolylidene with a non-cyclometalated phenyl group was observed by NMR spectroscopy. Cyclometalation was indicated by the ligand desymmetrization as revealed by the presence of seven distinct phenyl proton resonances in the low-field section of the 1H NMR spectrum integrating for 9 protons. While one set features the typical 2:2:1 pattern of a pristine phenyl group, the second set is constituted of four signals that are characteristic for a ortho disubstituted phenylene system, with two doublets and two multiplets that are correlated and integrating for one proton each. Ligand desymmetrization was also apparent from the two distinct singlets due to inequivalent NCH3 groups. Similarly, the OCH2 groups of the linker of complexes 9 and 10 were inequivalent and appeared as two sets of AB doublets with characteristic 2JHH coupling constants around 15 Hz. Three low-field 13C signals were observed in the 13C{1H} NMR spectrum for all three complexes 8–10 and were attributed to the two metal-bound carbenic carbons and the phenyl carbon. For example, the rhodium complex 8 showed three doublets at δC 164.2 (Ctrz, 1JCRh = 47.0 Hz), 159.5 (Cphenyl, 1JCRh = 36.2 Hz) and 158.2 (Ctrz, 1JCRh = 50.3 Hz). The smaller 1JCRh coupling constant was attributed to the Rh–Cphenyl bond,30b,f and the larger coupling constants to Rh–Ctrz interactions. The larger coupling constant at δC 158.2 is identical to the coupling observed in complex 7 (1JCRh = 50.3 Hz, vide supra), therefore suggesting this resonance to be due to the triazolylidene unit that contains the non-cyclometalated phenyl substituent. Accordingly, the resonance at δC 164.2 with a slightly smaller coupling constant was tentatively attributed to the carbenic carbon of the triazolylidene containing the metal-bound phenylene fragment.
Further evidence for the tridentate bonding of the ligand was obtained by single crystal X-ray diffraction of complexes 8 and 9 (Fig. 3, Table 1). Both structures are identical within errors and unambiguously reveal a Rh–Cphenyl bond in addition to the dicarbene bonding. All three Rh–C bond lengths are within expectation and in the 2.00–2.06 Å range. The average carbene bite angle is 81.5(2)° and thus considerably more acute than in the bidentate carbene complex 7 (Ctrz–Rh–Ctrz = 91.21(9)°).
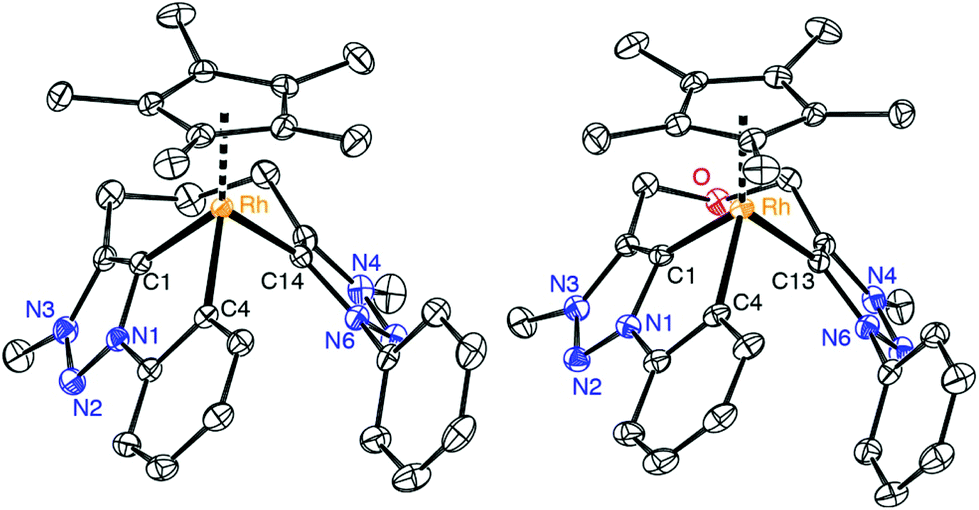 | ||
| Fig. 3 ORTEP representation of the complex cations of 8 (a), 9 (50% probability ellipsoids, hydrogen atoms omitted for clarity). | ||
| 8 | 9 | |
|---|---|---|
| Rh–C1 | 2.026(12) | 2.004(2) |
| Rh–C13/14 | 2.033(12) | 2.054(2) |
| Rh–C4 | 2.050(12) | 2.057(2) |
| Rh–Cpcentroid | 1.889(1) | 1.899(2) |
| C1–Rh–C13/14 | 81.62(5) | 81.43(8) |
| C1–Rh–C4 | 78.97(5) | 78.86(9) |
| C4–Rh–C13/14 | 92.85(5) | 92.92(8) |
Minor by-products were observed in the crude reaction mixture of the iridium complex 10 by 1H NMR spectroscopy, which revealed the characteristic pattern of a cyclometalated phenyl group. Separation by column chromatography yielded traces of the bimetallic complex 11 (Fig. 4), in which both N-phenyl groups are ortho-metalated to different iridium centers. The 1H NMR spectrum indicated symmetry-related triazolylidene and phenyl groups, and a diagnostic 2:1 Cp*/ligand ratio. The bridging coordination mode was further supported by the single AB doublet (2JHH = 12.6 Hz) for the OCH2 group, while the chelating complex 10 featured two AB doublets because of the lack of symmetry in the ligand. The carbonic resonance appeared at δC 153.2 in the 13C{1H} NMR spectrum. A high field resonance at δC 114.0, attributed to the Cphenyl–H nucleus ortho to the Ntrz (i.e. formally C3 of the phenylene unit), is diagnostic for cyclometalation. In the chelate complexes 8–10, this nucleus appears at essentially the same frequency (approximately 113.5 ppm). While NMR studies strongly support the bridging coordination mode of the bis(triazolylidene) ligand, we were unable to purify this complex sufficiently for elemental analysis or X-ray diffraction analysis, which has prevented the unambiguous identification of the spectator ligand so far. Attempts to selectively form the bimetallic complex by adding the preformed triazolylidene silver complex dropwise to a concentrated solution of [IrCp*Cl2]2 cleanly produced complex 10 together with unreacted [IrCp*Cl2]2, yet no bimetallic species was observed.
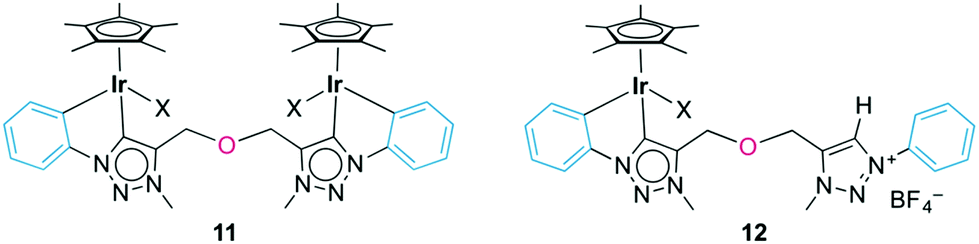 | ||
| Fig. 4 Complexes 11 and 12 as minor products from transmetalation reaction. | ||
When reacting 6b(BF4), Ag2O, and [IrCp*Cl2]2 simultaneously rather than sequentially (vide supra), a yellow mixture was obtained. NMR analysis of this crude mixture identified four distinct species including complexes 10 and 11 and two species, which were isolated by successive precipitation. Spectroscopic analysis indicated a mixture of two strongly related species in approximate 2:1 molar ratio and with a general bonding pattern that is in agreement with the one depicted for complex 12 (Fig. 4). The two species were tentatively assigned as the chloro and the solvento analogues (X = Cl, NCMe). Both species feature a low-field resonance (δH 9.10 and 8.87) for the triazolium proton and two AB systems for the OCH2 groups, indicating desymmetrization of the linker.31 Likewise, the NCH3 groups appeared as distinct singlets. The 13C{1H} NMR spectrum of this mixture showed two Ctrz–Ir signals at δC 153.4 or 146.6 and two Ctrz–H carbon resonances (δC 128.9 or 128.8). Heteronuclear multiple bond correlation (HMBC) spectroscopy identified for each of the two species an interaction of one OCH2 AB set with the iridium-bound triazolylidene unit, and a correlation of the second OCH2 resonances with the triazolium Ctrz–H unit. While the aromatic region in the 1H NMR spectrum was inconclusive due to significant signal overlap, two high field aryl 13C NMR resonances at δC 114.7 and 114.0 are in agreement with cyclometalation of the phenyl ring and Ctrz,Cphenyl-bidentate coordination of the ligand. In addition, two resonances at δC 138.6 and 138.5 have been attributed to the iridium-bound Cphenyl nucleus (cf. δC 139.5 in 10). Mass spectrometry was consistent with the NMR data and showed a m/z signal at 344 amu, in line with formation of complex 12 (expected m/z for [C30H35IrN6O]2+ is 344.12 amu). The isolation of Ctrz,Cphenyl-bidentate ligated complex 12 in combination with the absence of any Ctrz,Ctrz-bidentately bound iridium complex akin to complex 7 strongly suggests that Cphenyl–H bond activation is spontaneous. Formation of complex 12 may be a consequence of incomplete triazolylidene silver formation or due to partial hydrolysis of the semi-transmetalated intermediate prior to chelation of the second carbene unit.
The Cphenyl–H bond activation is reversible. Under acidic conditions (20 equiv. DCl in CD3CN) and at room temperature, the iridium complex 10 incorporated approximately 7% deuterium at the ortho positions of both the cyclometalated and the non-cyclometalated phenyl group (24 h). Heating accelerates this isotope exchange, reaching 27% deuterium incorporation at 50 °C (16 h) and up to 60% when heated to 80 °C for 10 h. Gradual decomposition to the triazolium salt was observed under these relatively harsh conditions, and the reaction was therefore aborted after 10 h, presumambly due to hydrolysis of the Rh–Ctrz bond. Deuteration of the ortho positions of both phenyl rings occur initially to the same extent (e.g. 27% D-incorporation in the cyclometalated phenyl group vs. 26% H/D exchange in the non-cyclometalated residue after 16 h at 50 °C). At higher conversion, the incorporation in the non-cyclometallated ring is slightly higher (68% vs. 54% in the cyclometalated phenyl group). The isotope exchange on both phenyl groups indicates that cyclometalation is reversible, involving a bidentate ditriazolylidene species with two free phenyl groups as a potential transient intermediate which undergoes cyclometalation of either of the two phenyl groups. Attempts to isolate this species have not been successful so far, suggesting that H(D)Cl elimination and cyclometalation are thermodynamically favoured. It is worth noting that the rhodium analogue 9 is much less stable under acidic conditions and rapidly decomposes to the triazolium salt and an inorganic rhodium species.
Transmetalation of the silver triazolylidene with N-mesityl substituents derived from 5c(BF4) and 6c(BF4) with [RhCp*Cl2]2 did not proceed. While the formation of a white precipitate and a color change from dark red to orange suggested transfer of the carbene to rhodium, 1H NMR analysis of the crude product showed broad resonances which were not conclusive. Unambiguous identification of the species was not achieved due to rapid decomposition of the product, both in solution and in the solid state, yielding triazolium starting material. Presumably the mesityl group inhibits chelation of the ditriazolylidene ligand because of steric interactions with the Cp* ligand. With N-phenyl substituents, such instability issues are circumvented through C–H bond activation and cyclometalation.
Base-mediated metalation
When ligand precursor 5b(I) was reacted directly with MCl3 (M = Rh or Ir) in the presence of KOtBu as base to deprotonate the triazolium salt, the Cp*-free bis(homoleptic) complexes 13 and 14 were obtained (Scheme 3). Chelation of the triazolylidene was supported by the desymmetrization of the linker CH2 groups, which appeared as multiplets between 3.1 and 2.0 ppm in the 1H NMR spectrum. Similarly to complexes 8–10, two sets of signals for the phenyl groups indicated that one phenyl substituent is cyclometalated while the other is not. The 13C{1H} NMR spectrum of the rhodium complex 13 showed three doublet resonances in the low field region. Based on the larger coupling constant, the most deshielded signals at δC 176.2 and 171.3 (1JCRh = 34.3 and 30.2 Hz, respectively) were attributed to the triazolylidene carbon, while the resonance at δC 164.9 (1JCRh = 25.7 Hz) was assigned to the cyclometalated phenyl carbon. The Rh–C coupling constants for 13 are smaller than those of 8, which is probably a direct consequence of the stronger trans influence of the C,C,C-tridentate ligand in octahedral 13 as compared to Cp* in complex 8. A meridional coordination mode of the ligand is tentatively surmised from the small chemical shift difference of the central methylene group in the linker (multiplet at δH 2.08). When coordinating facially as in complex 8, these protons are magnetically more distinct and appear at 2.33 and 1.62 ppm. The 1H NMR spectrum of 14 is very similar to that of 13 although the Hortho and Hmeta signals are broad for 14. The metal-bound carbons are more shielded than those of 13 and resonate at δC 158.9 and 152.4 (Ctrz) and at δC 147.9 (Cphenyl).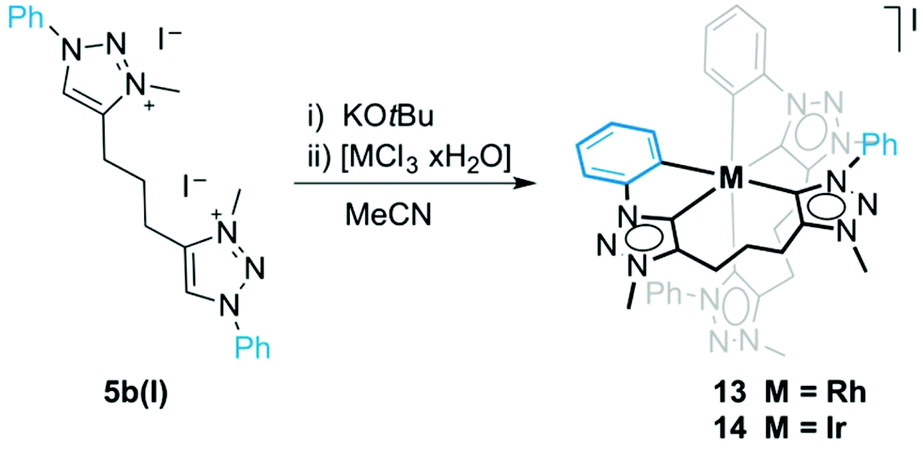 | ||
| Scheme 3 Synthesis of complex 13 and 14 by base-mediated metalation. | ||
A single crystal structure determination of complex 13 confirmed the bis-homoleptic structure with a cyclometalated phenyl group for each ditriazolylidene ligand, thus producing a distorted octahedral geometry around the rhodium center (Fig. 5). In contrast to complexes 8–10 with a facially coordinating dicarbene ligand, the same ligand now adopts a meridional coordination mode in complex 13 with the two cyclometalated phenyl units in mutual cis position. The cyclometalated phenyl ring is coplanar with the adjacent triazolylidene heterocycle. The Ctrz–Rh bond trans to Cphenyl is 0.05 Å longer than the Ctrz–Rh bond trans to a carbene, which is presumably imposed by steric constraints imparted by the chelating bonding mode rather than distinct trans influences. One triazolylidene ring is substantially more constrained by the rigid five-membered metalacycle, while the other carbene is part of a more flexible eight-membered metalacyclic motif.
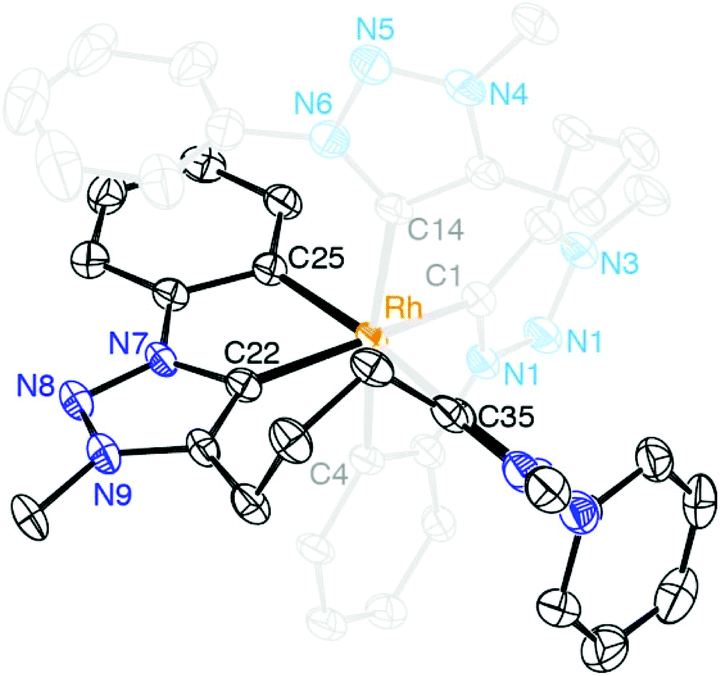 | ||
| Fig. 5 ORTEP representation of complex 13 (50% probability ellipsoids, I− counterion, hydrogen atoms, and co-crystallized CH2Cl2 molecule omitted for clarity); selected bond lengths: Rh–C1 2.041(3) Å, Rh–C4 2.071(3) Å, Rh–C14 2.098(3) Å; selected bond angles: C1–Rh–C4 80.22(13)°, C1–Rh–C14 91.79(14)°, C4–Rh–C14 171.87(13)°, C1–Rh–Ctrans 164.32(13)°. | ||
Formation of complex 13 was investigated in more detail by performing the synthesis at different temperatures. When the reaction was carried out at room temperature instead of MeCN reflux temperature, the triazolium salt 5b(I) was the only species observed by 1H NMR spectroscopy after 18 h. Upon heating to 60 °C, complex 13 started to form, and in addition, an intermediate was detected by NMR spectroscopy that was identified as complex 15 (Fig. 6). The triazolium proton of 15 appeared in the 1H NMR spectrum at δH 8.79 while the phenyl proton signals showed the diagnostic signature of both a pending phenyl group and an ortho-metalated phenylene unit, which were in equal intensity yet at distinct chemical shift from the Cphenyl resonances of the tridentate ligand. Moreover, mass spectrometry showed a signal at 409 amu, which corroborates the calculated mass for the dicationic fragment of [15–X]2+ (m/z = 409.4 calculated for [C42H43RhN12]2+).32 Upon further heating, this intermediate 15 evolved to the bis(homoleptic) complex 13 exclusively. Complex 15 is related to intermediate 12 detected en route to the tridentate coordinating dicarbene iridium complex 10 (cf.Fig. 4). The formation of a Ctrz,Cphenyl-bidentate coordinated complex with a pendant triazolium species supports our previous observation that cyclometalation and Cphenyl–H bond activation take place prior to coordination of the second triazolylidene ligand. Cyclometalation may be a key driver in the formation of complex 13, since the base-mediated metalation reaction did not proceed with the N-ethyl or N-mesityl analogues 5a(I) and 5c(I), respectively under otherwise identical conditions. Instead, only triazolium starting materials were observed by NMR spectroscopy.
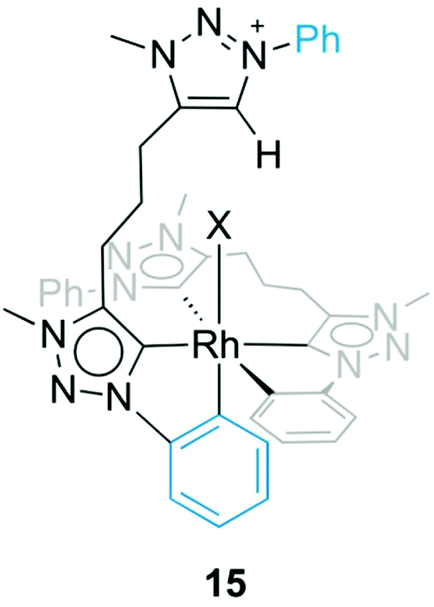 | ||
| Fig. 6 Intermediate species 15 with a Ctrz,CPh-bidentate coordinating ligand containing a pendant triazolium unit. | ||
Conclusions
A series of ditriazolylidene rhodium and iridium complexes containing alkyl- or ether-linkers between the triazolylidene bonding sites were derived from commercially available diynes. Ligand parameters and specifically the N-substituents on the triazole heterocycle critically dictate the bonding mode of the dicarbene ligand. In particular, N-phenyl substituted triazolylidenes lead in all cases to ortho-metalation of one of the phenyl rings and to a Ctrz,Ctrz,CPh-tridentate ligand coordination mode. The CPh–H bond activation process is spontaneous and the isolation of a Ctrz,CPh-bidentated intermediate suggests that this bond activation occurs prior to chelation of the second carbene. Stability studies under acidic conditions involving the Ctrz,Ctrz,CPh-tridentate coordinated iridium complex indicate that the Ir–Ctrz bonds are robust while the Ir–CPh bond is kinetically labile and formed reversibly. Moreover, depending on the applied metalation procedure, the ligand coordinates either meridionally or facially. This coordinative flexibility paired with the hemilability of the CPh–Ir bond are attractive features for catalysis. The potential of these complexes as catalysts in, for example, hydrogen transfer reactions is currently under investigation.Experimental section
General comments
All reagents were used as received from commercial suppliers. Unless specified, NMR spectra were recorded at 25 °C on Varian spectrometers operating at 300, 400 or 500 MHz (1H NMR) and 75, 100 or 125 MHz (13C{1H} NMR) respectively. Chemical shifts (δ in ppm, coupling constants J in Hz) were referenced to external SiMe4 (1H, 13C{1H}). Assignments are based on homo- and hetero-nuclear shift correlation spectroscopy. High-resolution mass spectrometry was carried out with a Micromass/Waters Corp. USA liquid chromatography with an electrospray source. Elemental analyses were performed at UCD Microanalytic Laboratory using an Exeter Analytical CE-440 elemental analyser. Residual solvents were identified by NMR spectroscopy.General procedure for the synthesis of ditriazoles 3 and 4
Method A: A mixture of the corresponding diyne (1 mol equiv.), azide (2.2 mol equiv.), CuSO4 (2 mol%) and sodium ascorbate (20 mol%) in a 1:1 mixture of THF:H2O was irradiated in a microwave reactor at 100 °C for 2 h using high absorption. The THF was removed under reduced pressure and the organics were extracted with CH2Cl2 (2 × 50 mL). The combined organics were washed with dilute (NH4)(OH) (aq., 50 mL), water (2 × 50 mL) and saturated NaCl solution (aq. 1 × 40 mL), dried over anhydrous Na2SO4, filtered and all volatiles removed under reduced pressure yielding the ditriazole product.
Method B: EtI (1 mol equiv.) and NaN3 (4 mol equiv.) were added to a microwave vessel with 1:1 of THF:H2O (20 mL) and stirred at room temperature for 48 h. To this mixture the corresponding diyne (0.33 mol equiv.), CuSO4 (14 mol%) and copper powder (5 mol%) were added and the mixture was irradiated in a microwave reactor at 100 °C for 2 h using high absorption. The THF was removed under reduced pressure and the organic residue was extracted with CH2Cl2 (2 × 50 mL). The crude reaction mixture was filtered through a short column of silica with a CH2Cl2/acetone solution (1:1, 300 mL) until all product was collected as indicated by TLC. The solvent was removed under reduced pressure giving the ditriazole product as an oil.
:1). 1H NMR (CDCl3, 400 MHz): δ 7.80 (s, 2H, Htrz), 7.70–7.66 (m, 4H, Hortho), 7.49–7.43 (m, 4H, Hmeta), 7.37 (tt, 2H, 3JHH = 7.4 Hz, 4JHH = 1.2 Hz, Hpara), 2.89 (t, 4H, 3JHH = 7.4 Hz, CtrzCH2), 2.18 (quintet, 2H, 3JHH = 7.4 Hz, CCH2CH2). 13C{1H} NMR (CDCl3, 100 MHz): δ 148.3 (Ctrz), 137.2 (Cipso) 129.7 (Cmeta), 128.5 (Cpara), 120.4 (Cortho), 119.4 (Ctrz–H), 29.0 (CtrzCH2CH2), 24.9 (CtrzCH2). ESI-MS (m/z): 331.1677, calcd for [C19H19N6]+ 331.1671. Anal calcd for C19H18N6 (330.39): C, 69.07; H, 5.49; N, 25.44%. Found: C, 68.58; H, 5.49; N, 25.19%.
1H NMR (CDCl3, 400 MHz): δ 7.41 (s, 2H, Htrz), 6.96 (s, 4H, HMes), 2.92 (t, 4H, 3JHH = 7.6 Hz, CtrzCH2), 2.33 (s, 6H, CH3-para), 2.23 (quintet, 2H, 3JHH = 7.6 Hz, CCH2CH2), 1.95 (s, 12H, CH3-ortho). 13C{1H} NMR (CDCl3, 100 MHz): δ 147.3 (Ctrz), 139.9 (Cpara), 135.2 (Cortho), 133.8 (Cipso), 129.1 (Cmeta), 123.0 (Ctrz–H), 29.3 (CCH2CH2), 25.1 (CtrzCH2), 21.2 (CH3-para), 17.4 (CH3-ortho). ESI-MS (m/z): 415.2600, calcd for [C25H31N6]+ 415.2610. Anal calcd for C25H30N6 (414.55): C, 72.43; H, 7.29; N, 20.27%. Found: C, 72.12; H, 7.38; N, 19.99%.
:1). 1H NMR (CDCl3, 400 MHz): δ 8.05 (s, 2H, Htrz), 7.73–7.69 (m, 4H, Hortho), 7.52–7.47 (m, 4H, Hmeta), 7.44–7.39 (m, 2H, 3JHH = 7.4 Hz, 4JHH = 1.2 Hz, Hpara), 4.84 (s, 4H, OCH2). 13C{1H} NMR (CDCl3, 100 MHz): δ 145.4 (Ctrz), 137.0 (Cipso) 129.8 (Cmeta), 128.9 (Cpara), 121.3 (Ctrz–H), 120.6 (Cortho), 63.7 (OCH2). ESI-MS (m/z): 333.1473, calcd for [C18H17N6O]+ 333.1464. Anal calcd for C18H16N6O (332.36): C, 65.05; H, 4.85; N, 25.29%. Found: C, 64.64; H, 4.58; N, 24.97%.
:1). 1H NMR (CDCl3, 400 MHz): δ 7.67 (s, 2H, Htrz), 6.98 (s, 4H, HMes), 4.87 (s, 4H, OCH2), 2.34 (s, 6H, CH3-para), 1.96 (s, 12H, CH3-ortho). 13C{1H} NMR (CDCl3, 100 MHz): δ 144.6 (Ctrz), 140.1 (Cpara) 135.2 (Cortho), 133.5 (Cipso), 129.2 (Cmeta), 124.9 (Ctrz–H), 63.8 (OCH2), 21.2 (CH3-para), 17.4 (CH3-ortho). ESI-MS (m/z): 417.2398, calcd for [C24H29N6O]+ 417.2403. Anal calcd for C24H28N6O (416.52): C, 69.21; H, 6.78; N, 20.18%. Found: C, 69.44; H, 6.90; N, 19.93%.
General procedure for alkylation of ditriazoles (5a–c(I), 6a–c(I))
A mixture of ditriazole (1 mol equiv.) and MeI (excess) were added to a pressure tube with MeCN (10 mL). The mixture was heated for a set time at reflux. After cooling to room temperature the product was precipitated by addition of Et2O (80 ml) and the ditriazolium salt was isolated by centrifugation. The crude product was suspended in MeCN (10 mL) and precipitated again with Et2O (80 mL) and isolated by centrifugation. This purification procedure was repeated once more.General procedure for halide exchange and formation of ditriazolium salts 5(BF4) and 6(BF4)
The ditriazolium iodide was suspended in MeCN (10 mL) and AgBF4 (2 mol equiv.) was added. The reaction mixture was stirred at room temperature for a set time. The crude reaction mixture was filtered through a short pad of Celite and evaporated to dryness.:1) yielding the title complex as an orange solid (75 mg, 85%). Crystals of 7 suitable for X-ray diffraction studies were obtained by diffusion of pentane into a CH2Cl2/MeCN (20:1) solution of the complex.
1H NMR (CD3CN, 400 MHz): δ 4.81, 4.42 (2 × dq, 2H, 2JHH = 12.9 Hz, 3JHH = 7.2 Hz, NCH2), 4.06 (s, 6H, NCH3), 2.70, 2.40 (2 × ddd, 2H, 2JHH = 14.9 Hz, 3JHH = 8.6 Hz, 3JHH = 4.6 Hz, CtrzCH2), 1.88–1.69 (m, 2H, CCH2CH2) 1.45 (t, 6H, 3JHH = 7.1 Hz, NCH2CH3), 1.43 (s, 15H, Cp–CH3). 13C{1H} NMR (CD3CN, 100 MHz): δ 154.4 (d, 1JCRh = 50.3 Hz, Ctrz–Rh), 145.5 (d, 2JCRh = 3.3 Hz, Ctrz), 100.2 (d, 1JCRh = 5.2 Hz, CCp), 50.2 (NCH2), 37.5 (NCH3), 24.4 (CCH2CH2), 22.5 (CtrzCH2), 17.0 (NCH2CH3), 9.5 (Cp–CH3). ESI-MS (m/z): 535.1844, calcd for [C23H37N6RhCl]+ 535.1823. Anal calcd for C23H37B0.21F0.84Cl1.79N6Rh (582.17) × 0.3 CH2Cl2: C, 46.05; H, 6.24; N, 13.83%. Found: C, 46.44; H, 5.85; N, 13.44%.
:1) yielding the title complex as a yellow solid (38 mg, 35%). Crystals of 8 suitable for X-ray diffraction studies were obtained by diffusion of Et2O into a CH2Cl2 solution of the complex.
1H NMR (CD3CN, 400 MHz): δ 7.47 (tt, 1H, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, HPh), 7.37 (dd, 1H, 3JHH = 7.8 Hz, 4JHH = 1.3 Hz, HPh–Rh), 7.28 (t, 3JHH = 7.9 Hz, 2H, HPh), 6.82 (ddd, 1H, 3JHH = 7.8 Hz, 3JHH = 7.2 Hz, 4JHH = 1.3 Hz, HPh–Rh), 6.64 (d, 2H, 3JHH = 7.3 Hz, HPh), 6.59 (d, 1H, 3JHH = 7.7 Hz, HPh–Rh), 6.46 (dt, 1H, 3JHH = 7.5 Hz, 4JHH = 1.4 Hz, HPh–Rh), 4.01, 3.86 (2 × s, 3H, NCH3), 3.32–3.11 (m, 4H, CtrzCH2), 2.38–2.29 (m, 1H, CtrzCH2CH2), 1.76 (s, 15H, Cp–CH3), 1.68–1.56 (m, 1H, CtrzCH2CH2). 13C{1H} NMR (CD3CN, 100 MHz): δ 164.2 (d, 1JCRh = 47.0 Hz, Ctrz–Rh), 159.5 (d, 1JCRh = 36.2 Hz, CPh–Rh), 158.2 (d, 1JCRh = 50.3 Hz, Ctrz–Rh), 149.6 (d, 2JCRh = 1.4 Hz, Ctrz), 146.2 (d, 2JCRh = 3.7 Hz, Ctrz), 144.0 (CPh–Rh), 141.3 (CPh), 139.6 (CPh–Rh), 130.4 (CPh), 129.9 (CPh), 127.9 (d, 3JCRh = 1.7 Hz, CPh–Rh), 126.5 (CPh), 122.5 (CPh–Rh), 113.4 (d, 3JCRh = 1.2 Hz, CPh–Rh), 99.2 (d, J = 4.1 Hz, CCp), 37.1, 37.0 (2 × NCH3), 26.4 (CtrzCH2CH2), 26.0, 25.7 (2 × CtrzCH2), 10.7 (Cp–CH3). ESI-MS (m/z): 595.2040, calcd for [C31H36N6Rh]+ 595.2056. Anal calcd for C31H36BF4N6Rh (595.56) × 0.2 CH2Cl2: C, 53.58; H, 5.25; N, 12.02%. Found: C, 53.69; H, 5.13; N, 11.95%.
:1) yielding complex 9 as a pale yellow solid (93 mg, 36%). Crystals suitable for X-ray diffraction studies were obtained by diffusion of Et2O into a CH2Cl2 solution of the complex.
1H NMR (CD3CN, 400 MHz): δ 7.50 (tt, 1H, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, HPh), 7.37 (dd, 1H, 3JHH = 7.8 Hz, 4JHH = 1.3 Hz, HPh), 7.34–7.29 (t, 3JHH = 8.0 Hz, 2H, HPh), 6.86 (ddd, 1H, 3JHH = 7.9 Hz, 3JHH = 7.2 Hz, 4JHH = 1.3 Hz, HPh–Rh), 6.70 (dd, 2H, 3JHH = 8.3 Hz, 4JHH = 1.1 Hz, HPh), 6.57 (d, 1H, 3JHH = 7.7 Hz, HPh–Rh), 6.50 (dt, 1H, 3JHH = 7.2 Hz, 4JHH = 1.4 Hz, HPh–Rh), 5.12 (d, 1H, 2JHH = 14.7 Hz, OCH2), 5.10, 4.99 (2 × d, 1H, 2JHH = 14.9 Hz, OCH2), 4.96 (d, 1H, 2JHH = 14.7 Hz, OCH2), 4.08, 3.95 (2 × s, 3H, NCH3), 1.75 (s, 15H, Cp-CH3). 13C{1H} NMR (CD3CN, 100 MHz): δ 167.9 (d, 1JCRh = 46.9 Hz, Ctrz–Rh), 160 (Ctrz–Rh), 158.7 (d, 1JCRh = 46.9 Hz, CPh–Rh), 147.1 (d, 2JCRh = 1.3 Hz, Ctrz), 144.1 (d, 2JCRh = 3.8 Hz, Ctrz), 144.0 (CPh–Rh), 140.9 (CPh), 139.5 (CPh–Rh), 130.7 (CPh), 129.4 (CPh), 128.0 (d, 3JCRh = 1.6 Hz, CPh–Rh), 126.6 (CPh), 122.8 (CPh–Rh), 113.6 (d, 3JCRh = 1.3 Hz, CPh–Rh), 99.4 (d, J = 4.0 Hz, CCp), 65.3, 64.8 (2 × OCH2), 37.4, 37.3 (2 × NCH3), 10.6 (Cp–CH3). ESI-MS (m/z): 597.1878, calcd for [C30H34N6ORh]+ 597.1849. Anal calcd for C30H34BF4N6ORh (684.34) × 0.25 CH2Cl2: C, 51.49; H, 4.93; N, 11.91%. Found: C, 51.53; H, 4.88; N, 11.84%.
:1) yielded complex 10 as a pale yellow solid (113 mg, 39%).
1H NMR (CD3CN, 400 MHz): δ 7.50 (tt, 1H, 3JHH = 7.5 Hz, 4JHH = 1.2 Hz, HPh), 7.37 (dd, 1H, 3JHH = 7.8 Hz, 4JHH = 1.2 Hz, HPh–Ir), 7.33–7.28 (m, 2H, HPh), 6.85–6.80 (m, 1H, HPh–Ir), 6.70–6.67 (m, 2H, HPh), 6.51 (dd, 1H, 3JHH = 7.7 Hz, 4JHH = 1.1 Hz, HPh–Ir), 6.46–6.41 (m, 1H, HPh–Ir), 5.12 (d, 1H, 2JHH = 14.7 Hz, OCH2), 5.11, 4.90 (2 × d, 1H, 2JHH = 14.9 Hz, OCH2), 4.82 (d, 1H, 2JHH = 14.7 Hz, OCH2), 4.09, 3.95 (2 × s, 3H, NCH3), 1.81 (s, 15H, Cp–CH3). 13C{1H} NMR (CD3CN, 100 MHz): δ 149.0 (Ctrz), 148.8 (Ctrz–Ir), 143.9 (CPh–Ir), 143.1 (Ctrz), 141 (Ctrz–Ir), 140.7 (CPh), 139.5, 138.6 (2 × CPh–Ir), 130.7 (CPh), 129.9 (CPh), 128.4 (CPh–Ir), 126.7 (CPh), 121.9, 113.5 (2 × CPh–Ir), 93.9 (CCp), 65.2, 64.3 (2 × OCH2), 37.5, 37.3 (2 × NCH3), 10.3 (Cp–CH3). ESI-MS (m/z): 687.2411, calcd for [C30H34N6OIr]+ 687.2424. Anal calcd for C30H34BF4N6OIr (773.65): C, 46.57; H, 4.43; N, 10.86%. Found: C, 46.15; H, 4.31; N, 10.60%.
:1), which afforded complex 13 as a pale yellow solid (52 mg, 56%). Crystals suitable for X-ray diffraction studies were obtained by layering a CH2Cl2/MeCN (2:1) solution with Et2O.
1H NMR (CD3CN, 400 MHz): δ 7.23 (t, 2H, 3JHH = 7.5 Hz, HPh), 6.92 (t, 4H, 3JHH = 7.5 Hz, HPh), 6.75 (d, 2H, 3JHH = 7.6 Hz, HPh–Rh), 6.57, 6.45 (2×t, 2H, 3JHH = 7.6 Hz, HPh–Rh), 6.25 (br, 4H, HPh), 6.08 (d, 2H, 3JHH = 7.6 Hz, HPh–Rh), 4.15, 4.03 (2 × s, 6H, HH), 3.06–2.97, 2.94–2.87, 2.61–2.54, 2.48–2.38 (4 × m, 2H, CtrzCH2), 2.12–2.04 (m, 4H, CtrzCH2CH2). 13C{1H} NMR (CD3CN, 100 MHz): δ 176.2 (1JCRh = 34.3 Hz, Ctrz–Rh), 171.3 (1JCRh = 30.2 Hz, Ctrz–Rh), 164.9 (1JCRh = 25.7 Hz, CPh–Rh), 146.7 (CPh–Rh), 145.5 (Ctrz), 143.9 (Ctrz), 139.9 (CPh), 137.1 (CPh–Rh), 129.2 (CPh), 128.8 (CPh), 126.7 (CPh–Rh), 126.2 (CPh), 121.3, 115.0 (2 × CPh–Rh), 36.8, 36.5 (2 × NCH3), 26.2 (CCH2CH2), 22.6, 21.3 (2 × CtrzCH2). ESI-MS (m/z): 817.2750, calcd for [C42H42N12Rh]+ 817.2710. Anal calcd for C42H42IN12Rh (944.67) × 0.75 CH2Cl2: C, 50.92; H, 4.35; N, 16.67%. Found: C, 50.95; H, 4.28; N, 16.37%.
:1) to give the title complex as a pale yellow solid (12 mg, 17%). Complex 14 decomposes upon extended time in solution. Because of this behaviour and due to the low yield and quantity, we did not succeed in purifying the compound to analytical purity (see also ESI†).
1H NMR (CD3CN, 400 MHz): δ 7.22 (tt, 2H, 3JHH = 7.5 Hz, 4JHH = 1.1 Hz, HPh), 6.90 (br, 4H, HPh), 6.72 (dd, 2H, 3JHH = 7.7 Hz, 4JHH = 1.1 Hz, HPh–Ir), 6.55 (m, 2H, HPh–Ir), 6.42 (td, 2H, 3JHH = 7.3 Hz, 4JHH = 1.3 Hz, HPh–Ir), 6.25 (br, 4H, HPh), 6.07 (dd, 2H, 3JHH = 7.3 Hz, 4JHH = 1.1 Hz, HPh–Ir), 4.15, 4.03 (2 × s, 6H, NCH3), 3.05–2.87 (m, 4H, CtrzCH2), 2.67–2.60, 2.46–2.36 (2 × m, 2H, CtrzCH2), 2.11–2.01 (m, 4H, CCH2CH2). 13C{1H} NMR (CD3CN, 100 MHz): δ 158.9 (Ctrz–Ir), 152.4 (Ctrz–Ir), 147.9 (CPh–Ir), 146.9 (Ctrz), 146.7 (CPh–Ir), 143.3 (Ctrz), 139.9 (CPh), 136.6 (CPh–Ir), 129.1 (CPh), 128.6 (CPh), 127.3, 120.7, 115.1 (3 × CPh–Ir), 36.9, 36.5 (2 × NCH3), 25.8 (CCH2CH2), 22.8, 21.1 (2 × CtrzCH2), one CPh not resolved. ESI-MS (m/z): 907.3322, calcd for [C42H42N12Ir]+ 907.3285.
Crystal structure determinations
Crystal data for 7–9, and 13 were collected using a Rigaku (former Agilent Technologies) Oxford Diffraction SuperNova A diffractometer fitted with an Atlas detector and using monochromated Mo-Kα radiation (0.71073 Å) (7–9) or Cu-Kα (1.54184 Å) (13). A complete dataset was collected, assuming that the Friedel pairs are not equivalent. The structures were solved by direct methods using SHELXS-97 and refined by full-matrix least squares fitting on F2 for all data using SHELXL-97.33 Hydrogen atoms were added at calculated positions and refined by using a riding model. Anisotropic thermal displacement parameters were used for all non-disordered nonhydrogen atoms. The solvent in 13 could not be modelled in terms of atomic sites. The SQUEEZE option as incorporated in PLATON34 was used to compensate for the spread electron density. The B–F bonds of the tetrafluoroborate anion in 7 were restrained to be equal using SADI. Further crystallographic details are compiled in Tables S1–S3.† Crystallographic data (excluding structure factors) for all three complexes have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1477830 (7), 1477829 (8), 1477828 (9) and 1477827 (13).Acknowledgements
This work has been financially supported by a NUI Traveling Scheme (K. F.), by the European Research Council (CoG 615653) and Science Foundation Ireland (RFP/2010/CHS2844).References
- (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122 CrossRef CAS PubMed; (b) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209 CrossRef CAS PubMed; (c) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef CAS PubMed.
- (a) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884 CrossRef CAS; (b) M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Dalton Trans., 2009, 6894 RSC; (c) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903 RSC; (d) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551 RSC.
- (a) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290 CrossRef CAS; (b) N-Heterocyclic Carbenes in Transition Metal Catalysis, in Topics in Organometallic Chemistry, ed. F. Glorius, Springer, Berlin, 2007 Search PubMed; (c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612 CrossRef PubMed; (d) M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677 CrossRef CAS PubMed; (e) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247 CrossRef CAS; (f) R. H. Crabtree, Coord. Chem. Rev., 2007, 251, 595 CrossRef CAS; (g) N-Heterocyclic Carbenes: from Laboratory Curiosities to Efficient Synthetic Tools, ed. S. Díez-González, RSC Publishing, Cambridge, 2011 Search PubMed; (h) N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, ed. C. S. J. Cazin, Springer, Berlin, 2011 Search PubMed.
- (a) M. C. Perry and K. Burgess, Tetrahedron: Asymmetry, 2003, 14, 951 CrossRef CAS; (b) J. J. Van Veldhuizen, J. E. Campbell, R. E. Giudici and A. H. Hoveyda, J. Am. Chem. Soc., 2005, 127, 6877 CrossRef CAS PubMed; (c) E. P. Kündig, T. M. Seidel, Y. X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484 CrossRef PubMed.
- J. Vignolle, X. Cattoën and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS PubMed.
- (a) K. Ofele, E. Tosh, C. Taubmann and W. A. Herrmann, Chem. Rev., 2009, 109, 3408 CrossRef CAS PubMed; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445 CrossRef CAS PubMed; (c) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810 CrossRef CAS PubMed.
- See for examples: (a) A. T. Normand and K. J. Cavell, Eur. J. Inorg. Chem., 2008, 2781 CrossRef CAS; (b) E. Peris and R. H. Crabtree, Coord. Chem. Rev., 2004, 248, 2239 CrossRef CAS; (c) J. F. Lefebvre, M. Lo, D. Leclercq and S. Richeter, Chem. Commun., 2011, 47, 2976 RSC; (d) K. Farrell and M. Albrecht, in The Privileged Pincer-Metal Platform: Coordination Chemistry & Applications, Topics in Organometallic Chemistry, ed. G. van Koten and R. A. Gossage, Springer, Berlin, 2016, p. 45 Search PubMed.
- (a) P. Mathew, A. Neels and M. Albrecht, J. Am. Chem. Soc., 2008, 130, 13534 CrossRef CAS PubMed; (b) G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 4759 CrossRef CAS PubMed; (c) J. D. Crowley, A. Lee and K. J. Kilpin, Aust. J. Chem., 2011, 64, 1118 CrossRef CAS; (d) K. F. Donnelly, A. Petronilho and M. Albrecht, Chem. Commun., 2013, 49, 1145 RSC; (e) B. Schulze and U. S. Schubert, Chem. Soc. Rev., 2014, 43, 2522 RSC.
- For selected and recent examples, see: (a) J. Bouffard, B. K. Keitz, R. Tonner, V. Lavallo, G. Guisado-Barrios, G. Frenking, R. H. Grubbs and G. Bertrand, Organometallics, 2011, 30, 2617 CrossRef CAS PubMed; (b) B. K. Keitz, J. Bouffard, G. Bertrand and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8498 CrossRef CAS PubMed; (c) E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 6215 CrossRef CAS; (d) D. Canseco-Gonzalez, A. Petronilho, H. Müller-Bunz, K. Ohmatsu, T. Ooi and M. Albrecht, J. Am. Chem. Soc., 2013, 135, 13193 CrossRef CAS PubMed; (e) J. R. Wright, P. C. Young, N. T. Lucas, A.-L. Lee and J. D. Crowley, Organometallics, 2013, 32, 7065 CrossRef CAS PubMed; (f) I. Corbucci, A. Petronilho, H. Müller-Bunz, L. Rocchigiani, M. Albrecht and A. Macchioni, ACS Catal., 2015, 5, 2714 CrossRef CAS; (g) R. Heath, H. Müller-Bunz and M. Albrecht, Chem. Commun., 2015, 51, 8699 RSC; (h) S. Hohloch, S. Kaiser, F. L. Duecker, A. Bolje, R. Maity, J. Košmrlj and B. Sarkar, Dalton Trans., 2015, 44, 686 RSC; (i) M. Valencia, H. Müller-Bunz, R. A. Gossage and M. Albrecht, Chem. Commun., 2016, 52, 3344 RSC.
- (a) D. G. Brown, N. Sanguantrakun, B. Schulze, U. S. Schubert and C. P. Berlinguette, J. Am. Chem. Soc., 2012, 134, 12354 CrossRef CAS PubMed; (b) V. Leigh, W. Ghattas, R. Lalrempuia, H. Müller-Bunz, M. T. Pryce and M. Albrecht, Inorg. Chem., 2013, 52, 5395 CrossRef CAS PubMed; (c) D. G. Brown, P. A. Schauer, J. Borau-Garcia, B. R. Fancy and C. P. Berlinguette, J. Am. Chem. Soc., 2013, 135, 1692 CrossRef CAS PubMed.
- (a) According to the IUPAC Golden Book, mesoionic compounds are “Dipolar five- (possibly six-) membered heterocyclic compounds in which both the negative and the positive charge are delocalized, for which a totally covalent structure cannot be written, and which cannot be represented satisfactorily by any one polar structure. The formal positive charge is associated with the ring atoms, and the formal negative charge is associated with ring atoms or an exocyclic nitrogen or chalcogen atom. Mesoionic compounds are a subclass of betains.” IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”) compiled by McNaught AD and Wilkinson A. Blackwell Oxford, UK: Scientific Publications; 1997. See also: ; (b) S. Araki, Y. Wanibe, F. Uno, A. Morikawa, K. Yamamoto, K. Chiba and Y. Butsugan, Chem. Ber., 1993, 12, 1149 CrossRef.
- (a) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755 CrossRef CAS; (b) M. Albrecht, Adv. Organomet. Chem., 2014, 62, 111 CrossRef CAS.
- (a) R. Huisgen, Angew. Chem., Int. Ed., 1963, 2, 565 CrossRef; (b) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (c) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (d) F. Himo, T. Lovell, R. Hilgraf, V. V. Rostovtsev, L. Noodleman, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 210 CrossRef CAS PubMed.
- (a) V. D. Bock, H. Hiemstra and J. H. Van Maarseveen, Eur. J. Org. Chem., 2006, 51 CrossRef CAS; (b) M. Meldal and C. W. Tornøe, Chem. Rev., 2008, 108, 2952 CrossRef CAS PubMed; (c) J. E. Hein and V. V. Fokin, Chem. Soc. Rev., 2010, 39, 1302 RSC.
- E. C. Keske, O. V. Zenkina, R. Wang and C. M. Crudden, Organometallics, 2012, 31, 456 CrossRef CAS.
- (a) B. Schulze, D. Escudero, C. Friebe, R. Siebert, H. Goerls, U. Koehn, E. Altuntas, A. Baumgaertel, M. D. Hager, A. Winter, B. Dietzek, J. Popp, L. Gonzalez and U. S. Schubert, Chem. – Eur. J., 2011, 17, 5494 CrossRef CAS PubMed; (b) S. Sinn, B. Schulze, C. Friebe, D. G. Brown, M. Jäger, E. Altuntaş, J. Kübel, O. Guntner, C. P. Berlinguette, B. Dietzek and U. S. Schubert, Inorg. Chem., 2014, 53, 2083 CrossRef CAS PubMed.
- D. I. Bezuidenhout, G. Kleinhans, G. Guisado-Barrios, D. C. Liles, G. Ung and G. Bertrand, Chem. Commun., 2014, 50, 2431 RSC.
- M. T. Zamora, M. J. Ferguson and M. Cowie, Organometallics, 2012, 31, 5384 CrossRef CAS.
- G. Guisado-Barrios, J. Bouffard, B. Donnadieu and G. Bertrand, Organometallics, 2011, 30, 6017 CrossRef CAS PubMed.
- S. Hohloch, L. Suntrup and B. Sarkar, Organometallics, 2013, 32, 7376 CrossRef CAS.
- J. Cai, X. Yang, K. Arumugam, C. W. Bielawski and J. L. Sessler, Organometallics, 2011, 30, 5033 CrossRef CAS PubMed.
- A. Petronilho, J. A. Woods, S. Bernhard and M. Albrecht, Eur. J. Inorg. Chem., 2014, 708 CrossRef CAS.
- (a) A. Krüger, L. J. L. Häller, H. Müller-Bunz, O. Serada, A. Neels, S. A. Macgregor and M. Albrecht, Dalton Trans., 2011, 40, 9911 RSC; (b) A. Krüger, A. Neels and M. Albrecht, Chem. Commun., 2010, 46, 315 RSC; (c) J. A. Cabeza, M. Damonte, P. Garcia-Alvarez and E. Perez-Carreno, Chem. Commun., 2013, 49, 2813 RSC.
- (a) K. M. Mullen, J. Mercurio, C. J. Serpell and P. D. Beer, Angew. Chem., Int. Ed., 2009, 48, 4781 CrossRef CAS PubMed; (b) J. Dupont, P. A. Z. Suarez, R. F. De Souza, R. A. Burrow and J.-P. Kintzinger, Chem. – Eur. J., 2000, 6, 2377 CrossRef CAS PubMed.
- (a) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561 CrossRef CAS PubMed; (b) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978 CrossRef CAS PubMed.
- (a) T. Nakamura, T. Terashima, K. Ogata and S.-I. Fukuzawa, Org. Lett., 2011, 13, 620 CrossRef CAS PubMed; (b) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 3164 CrossRef CAS.
- M. Iglesias, D. J. Beetstra, J. C. Knight, L.-L. Ooi, A. Stasch, S. Coles, L. Male, M. B. Hursthouse, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2008, 27, 3279 CrossRef CAS.
- (a) H.-L. Su, L. M. Pérez, S.-J. Lee, J. H. Reibenspies, H. S. Bazzi and D. E. Bergbreiter, Organometallics, 2012, 31, 4063–4071 CrossRef CAS; (b) E. Caytan and S. Roland, Organometallics, 2014, 33, 2115–2118 CrossRef CAS.
- For a related system with imidazol-2-ylidene ligands and smaller bite angles, see: J. A. Mata, A. R. Chianese, J. R. Miecznikowski, M. Poyatos, E. Peris, J. W. Faller and R. H. Crabtree, Organometallics, 2004, 23, 1253 CrossRef CAS.
- (a) R. Lalrempuia, N. D. McDaniel, H. Mueller-Bunz, S. Bernhard and M. Albrecht, Angew. Chem., Int. Ed., 2010, 49, 9765 CrossRef CAS PubMed; (b) A. Poulain, D. Canseco-Gonzalez, R. Hynes-Roche, H. Müller-Bunz, O. Schuster, H. Stoeckli-Evans, A. Neels and M. Albrecht, Organometallics, 2011, 30, 1021 CrossRef CAS; (c) R. Saravanakumar, V. Ramkumar and S. Sankararaman, Organometallics, 2011, 30, 1689 CrossRef CAS; (d) L. Bernet, R. Lalrempuia, W. Ghattas, H. Mueller-Bunz, L. Vigara, A. Llobet and M. Albrecht, Chem. Commun., 2011, 47, 8058 RSC; (e) A. Petronilho, M. Rahman, J. A. Woods, H. Al-Sayyed, H. Müller-Bunz, J. M. Don MacElroy, S. Bernhard and M. Albrecht, Dalton Trans., 2012, 41, 13074 RSC; (f) K. F. Donnelly, R. Lalrempuia, H. Müller-Bunz and M. Albrecht, Organometallics, 2012, 31, 8414 CrossRef CAS; (g) K. Ogata, S. Inomata and S. Fukuzawa, Dalton Trans., 2013, 42, 2362 RSC.
- W. A. Herrmann and M. G. Gardiner, Organometallics, 1999, 18, 4082 CrossRef CAS.
- Further attempts to characterize complex 15 included trapping of this species with PPh3. The appearance of a doublet in the 31P NMR spectrum (δP 15.2, 1JPRh = 92 Hz) as well as a MS signal at confirmed phosphine coordination to rhodium, however, crystallization efforts to obtain structural information have been unsuccessful.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of 5a and 14, and details on crystal structure determinations. CCDC 1477827–1477830. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01760f |
| This journal is © The Royal Society of Chemistry 2016 |