DOI:
10.1039/C6DT01756H
(Paper)
Dalton Trans., 2016,
45, 16575-16584
Chiral heterobimetallic chains from a dicyanideferrite building block including a π-conjugated TTF annulated ligand†
Received
5th May 2016
, Accepted 9th June 2016
First published on 9th June 2016
Abstract
The π-conjugated tetrathiafulvalene (TTF) annulated ligand was introduced into a dicyanometallate for the first time, leading to the synthesis of the versatile redox-active dicyanideferrite building block [(n-Bu)4N][Fe(TTFbp)(CN)2] (H2TTFbp = N-(2-(4,5-bis(methylthio)-1,3-dithiol-2-ylidene)-5-(picolinamido) benzo[d][1,3]dithiol-6-yl) picol inamide). The incorporation of the new precursor with chiral MnIII Schiff-base complexes resulted in two enantiopure one-dimensional complexes, [Mn((R,R)-salphen)Fe(TTFbp)(CN)2]n (2-(RR)) and [Mn((S,S)-salphen)Fe(TTFbp)(CN)2]n (2-(SS)) (Salphen = N,N′-1,2-diphenylethylene-bis(salicylideneiminato) dianion), which were synthesized and structurally characterized. Circular dichroism (CD) and vibrational circular dichroism (VCD) spectra confirmed the enantiomeric nature of the optically active complexes, and structural analyses revealed the formation of neutral cyanide-bridged double chains in 2-(RR) and 2-(SS). Solution and solid state CV studies revealed the redox-active characteristics of the complexes. Antiferromagnetic couplings were detected between FeIII and MnIII centers within a chain, and a field-induced magnetic phase transition was observed (TN = 4.8 K). The introduction of electroactivity and chirality into cyanide-bridged complexes with interesting magnetic properties leads the way towards new multifunctional materials.
Introduction
Over the last three decades, molecule-based magnetic materials exhibiting intriguing magnetic behaviors, such as magnetic ordering with high ordering/critical temperatures (Tc), single-molecule magnets and single-chain magnets, have attracted significant attention due to their potential applications in memory storage, quantum computers and spin-based molecular electronics.1 With the ever-increasing demands for the development of new technologies, the exploration of new materials with diverse properties is increasing.2 One of the major goals is the generation of magnetic materials that not only possess expected properties, but also combine two or more characteristics into a multifunctional system.3
Since the first observation of magneto-chiral dichroism in 1997,4 intense investigation has been devoted to enantiopure chiral magnets5–7 which are hybrid materials consisting of natural optical activity and magnetic ordering. In this field, the class of building blocks known as cyanometallates has been used to prepare chiral magnetic compounds.8 Several such complexes with controlled structures have been successfully synthesized by selection of appropriate cyanide building blocks and unsaturated transition-metal complexes containing chiral ligands.8b,e
As an organic donor, tetrathiafulvalene (TTF) and its derivatives are well known as essential building blocks for their strongly electron-donating and attractive reversible redox properties.9 They have been associated with d or f metal ions to produce multifunctional materials which possess magnetic and electrical properties.10–12 One effective strategy to achieve π-d systems is to elaborate a coupling between mobile and localized electrons through π-conjugated linkages.13 Using this approach, our group have concentrated significant effort on building molecular systems incorporating TTF-based ligands to generate subsequent properties such as redox activity and electronic conductivity.14
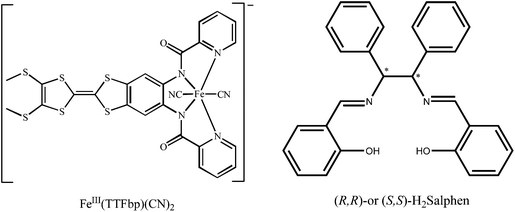
With the goal of increasing the functionality of the system, the present work investigates the introduction of a planar π-conjugated TTF annulated ligand into the dicyanideferrite building block, [(n-Bu)4N][Fe(TTFbp)(CN)2] (H2TTFbp = N-(2-(4,5-bis(methylthio)-1,3-dithiol-2-ylidene)-5-(picolinamido)benzo[d][1,3]dithiol-6-yl) picol inamide) (Scheme 1), for the first time. Chirality can be introduced into such systems by using the appropriate chiral ancillary ligand for coordination with paramagnetic ions. Herein, with the use of the new redox-active dicyanideferrite building block and enantiopure MnIII precursors including a Schiff-base ligand, two chiral one-dimensional complexes [Mn((R,R)-salphen)Fe(TTFbp)(CN)2]n (2-(RR)) and [Mn((S,S)-salphen)Fe(TTFbp)(CN)2]n (2-(SS)) (Salphen = N,N′-1,2-diphenylethylene-bis(salicylideneiminato) dianion), were synthesized. Their crystal structures, CD spectra, solid state electrochemistry and magnetic properties are described. Compound 2-(SS) is an antiferromagnet (TN = 4.8 K) with the coexistence of both redox activity and chirality, and also shows a field-induced magnetic phase transition.
 |
| | Scheme 1 | |
Experimental section
Starting materials
All reagents and solvents were commercially available and were used as received without further purification. The ligand H2TTFbp and [(n-Bu)4N][Fe(TTFbp)(CN)2] were prepared according to literature procedures.15 (R,R)- and (S,S)-[Mn(salphen)(H2O)2]ClO4 were prepared according to the published methods.16 (R,R)- and (S,S)-H2salphen (Scheme 1), were synthesized from the condensation of salicylaldehyde with 1,2-diphenylethylenediamine in a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in ethanol, respectively.
:1 in ethanol, respectively.
Caution! Although no problems were encountered in this work, perchlorate salts are potentially explosive and cyanides are very toxic. Thus, these starting materials should be handled in small quantities and with great care.
Preparation of H2TTFbp (L).
A solution of picolinic acid (246 mg, 2.00 mmol) in pyridine (3 mL) was added to a solution of 5,6-diamino-2-(4,5-bis(methylthio)-1,3-dithio-2-ylidene)-benzo[d]-1,3-dithiole (432 mg, 1.00 mmol) in pyridine (3 mL). To this reaction mixture, triphenyl phosphite (621 mg, 2.00 mmol) was added dropwise. After heating of the mixture to 100 °C for 8 h and cooling to room temperature, a yellow precipitate was isolated by filtration and washed with cold ethanol. Yield: 427 mg (73%). Anal. Calcd for C24H18N4O2S6: C, 49.12; H, 3.09; N, 9.55. Found: C, 49.24; H, 3.01; N, 9.33. 1H NMR (500 MHz, CD2Cl2, 298 K) δ (ppm) = 2.44 (s, 6H, CH3), 7.53–7.47 (m, 2H, ArH), 7.89 (s, 2H, ArH), 7.93 (td, 2H, J = 7.7, 1.5 Hz, ArH), 8.26 (d, 2H, J = 7.8 Hz, ArH), 8.55 (d, 2H, J = 4.6 Hz, ArH), 10.17 (s, 2H, NH). m/z (MALDI-TOF): 586.236.
Preparation of [(n-Bu)4N][Fe(TTFbp)(CN)2] (1).
To a solution of H2TTFbp (586 mg, 1.00 mmol) and NaOH (80 mg, 2.00 mmol) in 100 mL of methanol, a solution of FeCl3·6H2O (270 mg, 1.00 mmol) in 10 mL of methanol was added. After 3 h of refluxing, a solution of [(n-Bu)4N][CN] (540 mg, 2.00 mmol) in methanol was added. The resulting mixture was heated to reflux for 24 h and then filtered to remove the insoluble solid. The filtrate was concentrated under vacuum to a small volume. After cooling down, black microcrystals of [(n-Bu)4N][Fe(TTFbp)(CN)2] precipitated. Single crystals suitable for X-ray crystallography were obtained by slow recrystallization of this material from a CH3OH solution. Yield: 452 mg (48%). Anal. Calcd for C42H52FeN7O2S6: C, 53.95; H, 5.61; N, 10.49. Found: C, 53.88; H, 5.34; N, 10.65. Selected IR data (KBr, cm−1): 2123 (v(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N)). m/z (ESI): 692.00 (M−).
N)). m/z (ESI): 692.00 (M−).
Preparation of [Mn((R,R)-salphen)Fe(TTFbp)(CN)2]n (2-(RR)).
A solution of [Mn((R,R)-salphen)(H2O)2]ClO4 (24.8 mg, 0.04 mmol) in 4 mL of acetonitrile was added to a solution of [(n-Bu)4N][Fe(TTFbp)(CN)2] (37.40 mg, 0.04 mmol) in 20 mL of methanol. After 30 min of stirring, the resulting solution was filtered and then left undisturbed. The slow evaporation of the filtrate at room temperature yielded black needle crystals of 2-(RR) after two weeks. Yield: 43%. Anal. Calcd for C54H38FeMn N8O4S6: C, 55.62; H, 3.28; N, 9.61. Found: C, 55.52; H, 3.43; N, 9.46. Selected IR data (KBr, cm−1): 2126 (v(CN)).
Preparation of [Mn((S,S)-salphen)Fe(TTFbp)(CN)2]n (2-(SS)).
The preparation was similar to that of 2-(RR) with the exception that [Mn((S,S)-salphen)(H2O)2]ClO4 was used. After 30 min of stirring, the resulting solution was filtered and then left undisturbed. The slow evaporation of the filtrate at room temperature gave black needle crystals of 2-(SS) after two weeks. Yield: 43%. Anal. Calcd for C54H38FeMn N8O4S6: C, 55.62; H, 3.28; N, 9.61. Found: C, 55.54; H, 3.41; N, 9.47. Selected IR data (KBr, cm−1): 2126 (v(CN)).
X-ray structure determination
The crystal structures were determined with a Siemens (Bruker) SMART CCD diffractometer using monochromated Mo Kα radiation (λ = 0.71073 Å). The cell parameters were retrieved using SMART software and refined using SAINT17 for all observed reflections. Data was collected using a narrow-frame method with scan widths of 0.30° in ω and an exposure time of 10 s per frame. The absorption corrections were applied using SADABS18 supplied by Bruker. Structures were solved by direct methods using the program SHELXL-97.19 The positions of metal atoms and their first coordination spheres were located from direct methods E-maps. The other non-hydrogen atoms were found in alternating difference Fourier syntheses and least-squares refinement cycles and, during the final cycles, refined anisotropically. Hydrogen atoms were placed in calculated positions and refined as riding atoms with a uniform value of Uiso. Final crystallographic data and values of R1 and wR2 are listed in Table 1. Selected bond distances and angles for complexes 1, 2-(RR) and 2-(SS) are listed in Tables 2 and 3 and S1,† respectively. CCDC reference numbers are 1454969 (1), 1454970 (2-(RR)), 1454971 (2-(SS)).
Table 1 Summary of crystallographic data for all complexes
| |
1
|
2-(RR) |
2-(SS) |
|
R
1 = ∑||Fo| − |Fc||/∑Fo|.
wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2.
|
| Formula |
C42H52FeN7O2S6 |
C54H38FeMnN8O4S6 |
C54H38FeMnN8O4S6 |
|
F
w
|
935.12 |
1166.07 |
1166.07 |
| Crystal system |
Triclinic |
Triclinic |
Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P1 |
P1 |
|
a, Å |
10.823(2) |
10.5144(13) |
10.5901(15) |
|
b, Å |
20.388(4) |
12.1188(15) |
13.2993(19) |
|
c, Å |
24.414(5) |
22.247(3) |
21.701(3) |
|
α, ° |
83.518(4) |
96.993(2) |
97.500(3) |
|
β, ° |
82.033(4) |
94.9850(10) |
93.232(2) |
|
γ, ° |
78.424(4) |
96.292(2) |
94.499(3) |
|
V, Å3 |
5206(2) |
2782.3(6) |
3014.0(7) |
|
Z
|
4 |
2 |
2 |
|
ρ
calcd, g cm−3 |
1.193 |
1.392 |
1.285 |
|
T/K |
296(2) |
293(2) |
296(2) |
|
μ, mm−1 |
0.569 |
0.764 |
0.706 |
|
θ, ° |
0.85 to 27.39 |
0.93 to 25.50 |
1.32 to 25.00 |
|
F(000) |
1964 |
1194 |
1194 |
| Index ranges |
−13 ≤ h ≤ 13 |
−12 ≤ h ≤ 8 |
−12 ≤ h ≤ 12 |
| −24 ≤ k ≤ 26 |
−14 ≤ k ≤ 14 |
−15 ≤ k ≤ 14 |
| −31 ≤ l ≤ 30 |
−26 ≤ l ≤ 26 |
−25 ≤ l ≤ 25 |
| Data/restraints/parameters |
23223/29/1009 |
14965/3/1338 |
13352/3/1333 |
| GOF (F2) |
0.953 |
1.044 |
1.499 |
|
R
1a, wR2b (I > 2σ(I)) |
0.0674, 0.1755 |
0.0901, 0.2306 |
0.1338, 0.3477 |
|
R
1a, wR2b (all data) |
0.1464, 0.1968 |
0.1174, 0.2539 |
0.1579, 0.3758 |
Table 2 Selected bond lengths (Å) and angles (°) for complex 1
| Bond distances (Å) |
| Fe(1)–N(5) |
1.894(3) |
Fe(1)–N(6) |
1.901(3) |
| Fe(1)–N(7) |
2.003(3) |
Fe(1)–N(8) |
2.000(3) |
| Fe(1)–C(51) |
1.982(5) |
Fe(1)–C(52) |
1.974(5) |
| C(31)–C(32) |
1.335(6) |
|
|
| Bond angles (°) |
| N(11)–C(51)–Fe(1) |
178.9(4) |
N(12)–C(52)–Fe(1) |
178.8(4) |
Table 3 Selected bond lengths (Å) and angles (°) for complex 2-(SS)
| Symmetry transformations used to generate equivalent atoms are given in footnotes #1 to #2. #1x − 1, y, z. #2x + 1, y, z. |
| Bond distances (Å) |
| Fe(1)–N(1) |
1.895(15) |
Fe(1)–N(2) |
1.862(18) |
| Fe(1)–N(3) |
1.980(18) |
Fe(1)–N(4) |
2.021(16) |
| Fe(1)–C(53) |
1.964(18) |
Fe(1)–C(54) |
1.928(19) |
| Fe(2)–N(9) |
1.914(17) |
Fe(2)–N(10) |
1.835(16) |
| Fe(2)–N(11) |
1.966(17) |
Fe(2)–N(12) |
2.016(19) |
| Fe(2)–C(107) |
1.96(2) |
Fe(2)–C(108) |
1.92(2) |
| C(19)–C(20) |
1.36(3) |
C(73)–C(74) |
1.37(3) |
| Mn(1)–O(3) |
1.928(13) |
Mn(1)–O(4) |
1.860(13) |
| Mn(1)–N(5) |
1.964(15) |
Mn(1)–N(6) |
1.970(17) |
| Mn(1)–N(7) |
2.245(17) |
Mn(1)–N(8) |
2.310(15) |
| Mn(2)–O(7) |
1.880(12) |
Mn(2)–O(8) |
1.855(13) |
| Mn(2)–N(13) |
1.964(16) |
Mn(2)–N(14) |
2.029(16) |
| Mn(2)–N(15) |
2.336(17) |
Mn(2)–N(16) |
2.267(16) |
| Bond angles (°) |
| N(7)–C(53)–Fe(1) |
176(2) |
N(8)#2–C(54)–Fe(1) |
178.5(19) |
| N(15)–C(107)–Fe(2) |
169.9(18) |
N(16)#1–C(108)–Fe(2) |
177(2) |
| N(1)–Fe(1)–C(53) |
95.4(7) |
N(2)–Fe(1)–C(53) |
91.6(8) |
| N(1)–Fe(1)–C(54) |
93.0(7) |
N(2)–Fe(1)–C(54) |
94.6(8) |
| C(54)–Fe(1)–C(53) |
170.0(8) |
C(53)–Fe(1)–N(4) |
84.2(7) |
| C(54)–Fe(1)–N(4) |
89.0(7) |
C(53)–Fe(1)–N(3) |
88.1(8) |
| C(54)–Fe(1)–N(3) |
88.0(8) |
N(10)–Fe(2)–C(108) |
89.7(7) |
| N(9)–Fe(2)–C(108) |
91.1(8) |
N(10)–Fe(2)–C(107) |
96.0(7) |
| N(9)–Fe(2)–C(107) |
95.5(8) |
C(108)–Fe(2)–C(107) |
171.7(8) |
| C(53)–N(7)–Mn(1) |
165.1(18) |
C(54)#1–N(8)–Mn(1) |
163.0(17) |
| C(107)–N(15)–Mn(2) |
157.1(18) |
C(108)#2–N(16)–Mn(2) |
153.1(17) |
| O(4)–Mn(1)–N(8) |
93.2(6) |
O(3)–Mn(1)–N(8) |
90.0(6) |
| N(5)–Mn(1)–N(8) |
86.8(6) |
N(6)–Mn(1)–N(8) |
86.8(6) |
| O(4)–Mn(1)–N(7) |
93.5(6) |
O(3)–Mn(1)–N(7) |
94.4(6) |
| N(5)–Mn(1)–N(7) |
85.9(6) |
N(6)–Mn(1)–N(7) |
88.3(7) |
| N(8)–Mn(1)–N(7) |
171.7(5) |
O(8)–Mn(2)–N(16) |
90.0(6) |
| O(7)–Mn(2)–N(16) |
91.2(6) |
N(14)–Mn(2)–N(16) |
87.6(6) |
| N(13)–Mn(2)–N(16) |
89.1(6) |
O(8)–Mn(2)–N(15) |
92.9(6) |
| O(7)–Mn(2)–N(15) |
93.8(6) |
N(14)–Mn(2)–N(15) |
87.1(6) |
| N(13)–Mn(2)–N(15) |
87.6(7) |
N(16)–Mn(2)–N(15) |
174.0(5) |
Physical measurements
Elemental analyses for C, H and N were performed on a Perkin-Elmer 240C analyzer. 1H NMR spectra were measured on a Bruker AM 500 spectrometer. Mass spectra were recorded on a Bruker Autoflex IITM instrument for MALDI-TOF-MS or on a Varian MAT 311A instrument for ESI-MS. CD spectra were recorded on a Jasco J-810 spectropolarimeter. IR and VCD spectra in the region of 1800–800 cm−1 were recorded on a VERTEX 80v Fourier transform infrared spectrometer equipped with a PMA 50 VCD/IRRAS module (Bruker, Germany) using previous procedures.20 The solid samples were prepared by mixing the compound and KBr in a 1:200 ratio, and pressing the pellet in a Perkin Elmer hydraulic pellet press for 5 min under 10 tons of pressure. The photo elastic modulator (PEM) was set to 1500 cm−1, and the spectral resolution was 4 cm−1. All VCD measurements were collected for 1 h composed of 4 blocks, each of 15 min duration. Baseline correction was performed with the spectrum of a pure KBr pellet using the same measurement setup. Magnetic susceptibilities for polycrystalline samples were measured with the use of a Quantum Design MPMS-SQUID-VSM magnetometer in the temperature range 1.9–300 K. Field dependences of magnetization were measured in an applied field up to 70 kOe. Solid state cyclic voltammetry (CV) was performed using a BASi Epsilon Electrochemical Analyzer with ferrocene (Fc) as an internal reference. Measurements were conducted under an inert Ar atmosphere using a conventional 3-electrode cell with a glassy carbon working electrode containing the immobilized solid, a Pt wire auxiliary electrode and an Ag/Ag+ quasi reference electrode. A 0.1 M tetrabutylammonium hexafluorophosphate ([(n-Bu)4N]PF6/CH3CN or [(n-Bu)4N]PF6/DMF) supporting electrolyte was employed.
Results and discussion
Synthesis and characterization
The versatile dicyanideferrite building block [(n-Bu)4N][Fe(TTFbp)(CN)2] with a planar π-conjugated TTF annulated ligand was successfully synthesized from the H2TTFbp ligand, FeCl3·6H2O and [(n-Bu)4N][CN] in methanol. The use of equimolar amounts of [(n-Bu)4N][Fe(TTFbp)(CN)2] and the enantiopure precursors [Mn((R,R)-salphen)(H2O)2][ClO4] or [Mn((S,S)-salphen)(H2O)2][ClO4], afforded two chiral one-dimensional complexes. The characterization of these complexes has been accomplished by CD and VCD spectra, cyclic voltammetry, X-ray diffraction analysis and magnetic susceptibility measurements.
Spectroscopic studies
The optical activity and enantiomeric nature of 2-(RR) and 2-(SS) were confirmed by circular dichroism (CD) spectroscopy measurements in KBr pellets (Fig. 1). The solid-state CD spectrum of 2-(RR) (R isomer) exhibits a negative Cotton effect at λmax = 247 and 318 nm, while 2-(SS) (S isomer) shows Cotton effects of the opposite sign at the same wavelengths. The CD signals for the two compounds are a clear mirror image.
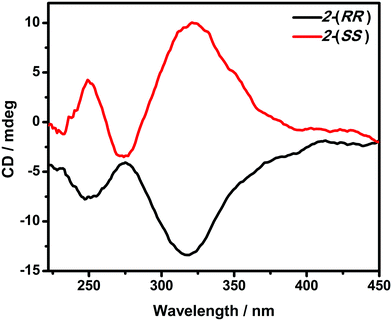 |
| | Fig. 1 CD spectra of 2-(RR) and 2-(SS) in KBr pellets. | |
Vibrational circular dichroism (VCD) is a powerful technique in the study of chiral molecules. It is the extension of CD into the infrared region of a spectrum which reflects vibrational transitions, and has aided in reaching a deeper understanding of the configuration of chiral molecules.8b,20 As expected, no noticeable difference in the signals of the enantiomers were detected from the IR, while the VCD spectra appear as approximately mirror image forms (Fig. 2). In the IR spectra, the absorption features arise from the imine double bond stretch vibrations (1625, 1601 cm−1 for 2-(RR) and 2-(SS) and C–H deformations (1444, 1306 and 1294 cm−1 for 2-(RR) and 2-(SS)). In the corresponding VCD spectra, the two imine chromophores manifest as a couplet curve, while the latter appear as incisive peaks.
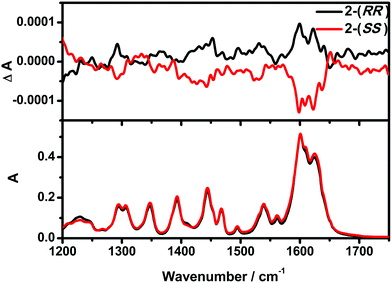 |
| | Fig. 2 VCD (top) and IR absorption (bottom) spectra of 2-(RR) and 2-(SS). | |
Structural description
The crystal structure of the precursor (complex 1) is shown in Fig. 3. It consists of one mononuclear [Fe(TTFbp)(CN)2]− unit and one [(n-Bu)4N]+ cation. In each [Fe(TTFbp)(CN)2]− anion, the central iron(III) ion is coordinated by two carbon atoms from cyanide units, two amide-nitrogen atoms and two pyridine nitrogen atoms from the TTFbp moieties, giving a distorted octahedral coordination structure. The Fe–N (1.894(3)–2.002(3) Å) and Fe–C (1.974(5)–1.982(5) Å) bond lengths are in good agreement with those observed previously in related compounds.15 The Fe–CN angles for the cyanide groups (178.9(4) and 178.8(4)°) depart slightly from linearity. The central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the TTF core is 1.335(6) Å in length, confirming the neutral form of the TTF moiety.11a
C bond of the TTF core is 1.335(6) Å in length, confirming the neutral form of the TTF moiety.11a
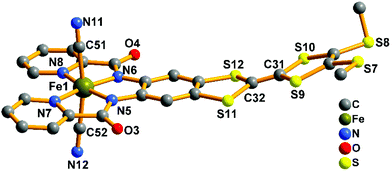 |
| | Fig. 3 View of the [Fe(TTFbp)(CN)2]− unit. The [(n-Bu)4N]+ cation and hydrogen atoms are omitted for clarity. | |
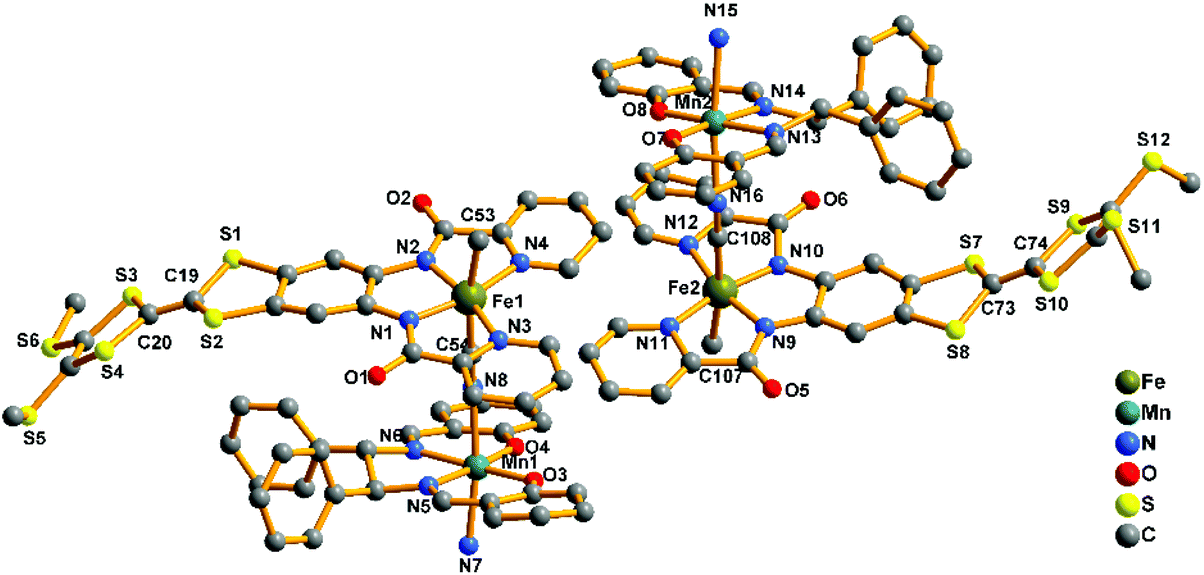
2-(RR) and 2-(SS) are a pair of enantiomers and crystallize in the chiral space group P1 (Fig. 4 and S1†). They are composed of a [Fe(TTFbp)(CN)2]− anion and a [Mn(salphen)]+ cation, forming neutral cyano-bridged linear-like double chains with the repeating units (–MnIII–NC–FeIII–CN–)n. As shown in Fig. 5, complex 2-(SS) has two types of [Fe(TTFbp)(CN)2]− (including Fe1 and Fe2 atoms, respectively). Both FeIII centers of the compounds are coordinated by four nitrogen atoms from the TTFbp2− ligand located at its equatorial plane and two trans cyanide carbon atoms, yielding a distorted octahedral coordination geometry. The Fe–C (1.92(2)–1.964(18) Å) and Fe–N (1.835(16)–2.021(16) Å) bond lengths are in good agreement with those observed previously in related compounds.21 The Fe–CN angles for the bridging cyanide groups (169.9(18)–178.5(19)°) depart slightly from linearity. There are also two crystallographically independent MnIII ions in 2-(SS), Mn1 and Mn2. The MnIII ions adopt the same coordination mode, which consists of two axially-ligated cyanide groups (2.245(17) and 2.310(15) Å for Mn1, 2.336(17) and 2.267(16) Å for Mn2), two imino-nitrogen atoms and two phenolic oxygen atoms from the salphen ligand (1.964(15) and 1.970(17) Å for Mn1–N, 1.928(13) and 1.860(13) Å for Mn1–O, 1.964(16) and 2.029(16) Å for Mn2–N, and 1.855(13) and 1.880(12) Å for Mn2–O). The axial elongation results from the well-known Jahn–Teller effect on an octahedral high-spin MnIII ion.22 The Mn–NC angles range from 153.1(17) to 165.1(18)°, deviating significantly from linearity. The chains extend along the a axis with an intrachain Fe–Mn separation of 5.284–5.286 Å through bridging cyanide. The shortest interchain Fe⋯Fe, Mn⋯Mn, and Fe⋯Mn distances are 8.615, 7.846 and 8.464 Å, respectively – longer than the intrachain metal–metal distance. Each chain interacts with two other adjacent chains by nonclassical hydrogen bonds C(58)–H(58)⋯O(3) (H⋯O distance, 2.44 Å) and C(31)–H(31)⋯S(12) (H⋯S distance, 2.85 Å), thus forming a two-dimensional (2D) layer (ESI, Fig. S2†). The layers stack to form a three-dimensional (3D) structure along the b axis via weak H-bonding interactions (ESI, Fig. S3†).
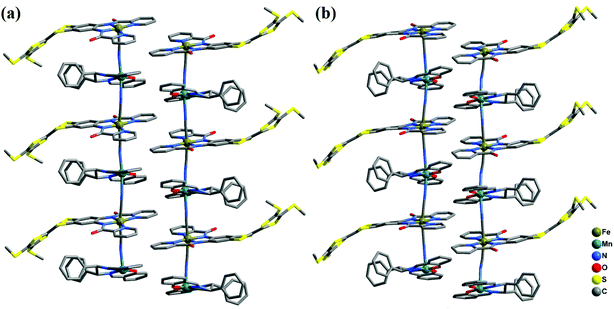 |
| | Fig. 4 Perspective view of the infinite one-dimensional zigzag chains of 2-(RR) (a) and 2-(SS) (b). Hydrogen atoms are omitted for clarity. | |
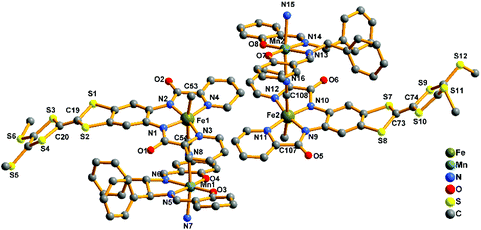 |
| | Fig. 5 Perspective drawing of the crystallographic structural unit of 2-(SS) showing the atom numbering. Hydrogen atoms are omitted for clarity. | |
Electrochemical properties
The cyclic voltammogram (CV) of H2TTFbp (Fig. 6a) displays two distinct, reversible oxidation waves at E1/2 = 0.15 and 0.31 V which are assigned to the TTF/TTF˙+ and TTF˙+/TTF2+ redox couples, respectively. The cathodic region reveals one irreversible reduction process at −2.26 V which can assigned to the reduction of the amide functionality. Furthermore, two very close, quasi-reversible reduction processes at E1/2 = −2.43 and −2.54 V are tentatively assigned to the reductions of the two pyridyl rings. CVs collected over a range of scan rates (Fig. 6b) indicate that the electrochemical processes occur independent of the scan rate.
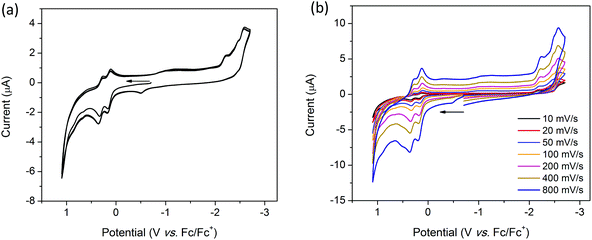 |
| | Fig. 6 Solution state CV of H2TTFbp (a) over 3 full cycles at 100 mV s−1 and, (b) single scans over multiple scan rates. Measurements were performed in 0.1 M [(n-Bu)4N]PF6/DMF electrolyte and were referenced to Fc/Fc+. Arrows indicate the direction of the forward scan. | |
Solution state cyclic voltammetry of [(n-Bu)4N][Fe(TTFbp)(CN)2] reveals two reversible oxidation waves at E1/2 = −0.06 and 0.41 V which were assigned to the TTF/TTF˙+ and TTF˙+/TTF2+ couples, respectively (Fig. 7a). The spike at 0.72 V observed at scan rates over the range 10–400 mV s−1 is tentatively assigned to chemical interaction between the compound and the glassy carbon working electrode, as this feature was not observed at a faster scan rate of 800 mV s−1. A reversible oxidation wave was observed at E1/2 = 0.78 V which may be due to oxidation of FeIII to FeIV. At E1/2 = −1.16 V, a reversible reduction wave due to the FeIII/II redox couple was observed. Cycling of the redox potential between 1.5 and −2.5 V revealed reproducible CVs, demonstrating the reversibility and stability of the compound in its various redox states. Over multiple scan rates, the positions of the aforementioned redox processes did not shift in potential, indicating the scan rate independence of [(n-Bu)4N][Fe(TTFbp)(CN)2] (Fig. 7b). The absence of any distinct reduction processes reinforces the assignment of the reductions in H2TTFbp to a chemical change to the amide and pyridyl moieties which become less accessible once coordinated to the FeIII center.
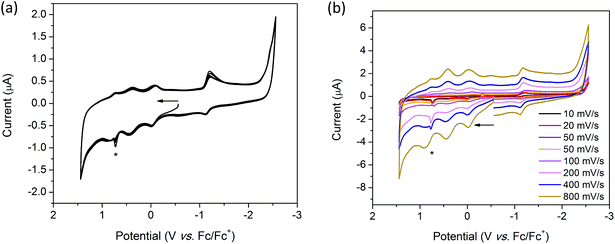 |
| | Fig. 7 Solution state CV of [(n-Bu)4N][Fe(TTFbp)(CN)2] showing (a) 3 successive cycles at 100 mV s−1 and (b) single scans over multiple scan rates. The asterisk marks a current spike due to solute-electrode interactions. Measurements were performed in 0.1 M [(n-Bu)4N]PF6/CH3CN electrolyte and were referenced to Fc/Fc+. Arrows indicate the direction of the forward scan. | |
Coordination of [(n-Bu)4N][Fe(TTFbp)(CN)2] to a [Mn(salphen)]+ cation results in the appearance of new features in the oxidative region of the CV for 2-(SS). The solid state CV of 2-(SS) reveals quasi-reversible oxidation processes which result in the decomposition or dissolution of the complex in the [(n-Bu)4N]PF6/CH3CN electrolyte. Scan rate dependence studies reveal three irreversible reduction processes at onset potentials of 0.37, 0.89 and 1.10 V which are most prominent at scan rates of 20, 50, 200, 400 and 800 mV s−1. These processes are likely to be attributable to the TTF/TTF˙+, TTF˙+/TTF2+ and MnIII/IV redox couples, respectively (ESI, Table S2†). At 100 mV s−1 however, a fourth irreversible oxidation peak was observed at an onset potential of 0.57 V (Fig. 8a). On first inspection, the cathodic region appears to be featureless, however, upon closer examination, a quasi-reversible reduction wave is observed at E1/2 = −0.66 V, and is attributed to the FeIII/II couple. The shift in this couple to a less cathodic potential upon coordination to [Mn(salphen)]+ is consistent with electron density being redistributed across the shared cyanide ligand. Two further successive cycles were featureless owing to the irreversible nature of these redox processes.
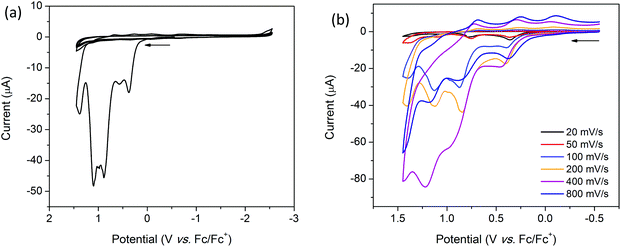 |
| | Fig. 8 Solid state CV of 2-(SS) (a) over 3 successive cycles at 100 mV s−1 and (b) single scans over multiple scan rates. Measurements were performed in 0.1 M TBAPF6/CH3CN electrolyte and were referenced to Fc/Fc+. Arrows indicate the direction of the forward scan. | |
Magnetic properties
The dc magnetic susceptibility data for complex 2-(SS) were collected at 1000 Oe in the 2–300 K range, as shown in Fig. 9. The χMT value at room temperature is 3.34 cm3 K mol−1 per FeIIIMnIII unit, which is close to the theoretical value (3.38 cm3 K mol−1) calculated for a noncoupled FeIII center (S = 1/2) and MnIII center (S = 2) assuming g = 2.00. As the temperature is lowered, the χMT value is almost constant above 50 K, and then decreases to 0.61 cm3 K mol−1 at 2 K, demonstrating the weak antiferromagnetic coupling between cyanide-bridged FeIII–MnIII. The magnetic susceptibility above 30 K obeys the Curie–Weiss law, with a Curie constant of 3.36 cm3 K mol−1 and a Weiss constant of −1.67 K (ESI, Fig. S4†). The negative θ confirms the presence of antiferromagnetic interactions among magnetic centers.
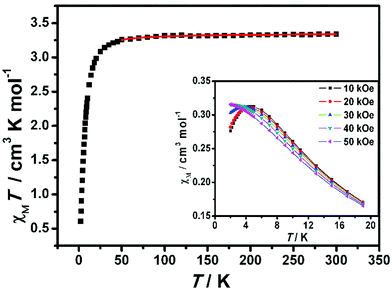 |
| | Fig. 9 Temperature dependence of the χMT product for 2-(SS) at 1 kOe. The red solid line represents the best fits of the data. (Inset) Thermal dependence of the magnetic susceptibility at T ≤ 19 K. | |
To estimate the magnetic coupling of the single chain complexes, we applied an analytical expression derived from an alternating ferrrimagnetic chain model (H = −J∑iSFei[(1 + α)SMni + (1 − α)SMni+1]).23 The best fit over the temperature range 50–300 K gives gMn = 1.99, gFe = 2.01, J = −0.96 cm−1, α = 0.34 and zJ′ = −0.34 (R = 3.21 × 10−5). The negative value of J indicates antiferromagnetic couplings between FeIII and MnIII ions within the chain.
The field dependence of magnetization was measured up to 70 kOe at 1.9 K (Fig. 10). The inconspicuous S-shaped curve increases to a maximum value of 3.40Nβ at 70 kOe which is slightly higher than the expected value of 3Nβ calculated from g = 2.00. Importantly, the magnetization will continuously increase with the field, and it is suggested that decoupling occurs at high field. This result also confirms that the antiferromagnetic coupling between spin centers is very weak. From the dM/dH vs. H plot at 1.9 K (inset of Fig. 10), the critical field is estimated to be about 35 kOe. Below 35 kOe, 2-(SS) has an antiferromagnetic spin ground state; above this field, the decoupling effect of the field leads to the paramagnetic phase in this complex. To further elucidate the magnetic behavior, we recorded χMvs. T data for 2-(SS) under several fields as shown in the inset of Fig. 9. A maximum of χM was observed at about 4.8 K under 10 kOe. The peak broadens and shifts to lower temperature as the magnetic field increases, and it finally disappears for H ≥ 40 kOe. This behavior shows the existence of a field-induced magnetic phase transition.
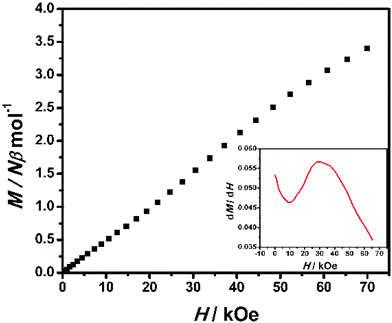 |
| | Fig. 10 Field dependence of magnetization at 1.9 K for 2-(SS). (Inset) dM/dH vs. H plot. | |
The alternating current (ac) susceptibility of complex 2-(SS) was measured over the frequency range 1–999 Hz at 2–16.0 K under zero dc field (Fig. 11). The in-phase (χ′M) susceptibility has a maximum around 4.8 K, with no peaks observed in the out-of-phase susceptibility (χ′′M) at the same temperature, suggesting an absence of single-chain magnetism below this temperature.
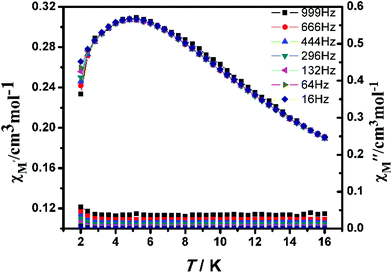 |
| | Fig. 11 Temperature dependence of the in-phase χ′M (top) and out-of-phase χ′′M (bottom) at different frequencies with a zero applied dc field for 2-(SS). | |
Magneto-structural correlation
In cyanide-bridged FeIII and MnIII systems, the magnetic coupling between low-spin FeIII (t2g5eg0) and high-spin MnIII (t2g3eg1) through cyanide is either ferro- or antiferromagnetic. Regarding the nature of the FeIII and MnIII interaction, the orthogonality of the magnetic orbitals dπ in FeIII and dz2 in MnIII will lead to ferromagnetic interactions, while the orbital overlap between dπ orbitals of FeIII and MnIII gives rise to antiferromagnetic interactions. The overall magnetic properties depend on which contribution predominates. As shown in Table 4, structural and magnetic parameters of some 1D cyanide-bridged FeIII–MnIII complexes based on [FeL(CN)2]− are summarrized for comparasion.21c,24 It is noted that ferromagnetic properties are common in cyanide-bridged FeIII–MnIII complexes, suggesting that the orbital orthogonality effect usually overwhelms the orbital overlap effect. Considering the stronger delocalization (σ-type) of the spin density from the MnIII occurring on the cyanide group, the dz2 orbital of MnIII is the major contributor to the magnetic coupling. Thus, ferromagnetic properties are more usual. By analyzing the relationship between Mn–NC bond angles and the magnetism, we concluded that the magnetic coupling of FeIII–MnIII changes from ferromagnetic to antiferromagnetic with an increasing Mn–NC angle. This feature indicates that the increase of the Mn–NC angles plays a crucial role in improving the overlap of magnetic Fe and Mn dπ orbitals, resulting in the dominance of JAF over JF, and in turn favoring antiferromagnetic couplings. Similar magnetic features were also found in other cyanide-bridged FeIII–MnIII systems.21c,25 It should be noted that the Mn–NC bond angle in this system is larger than some cyano-bridged Fe(III)–Mn(III) complexes in Table 4. We assume that the steric effect originates from TTF moieties and leads to the larger Mn–NC bond angle, since the volume of the TTFs is significantly larger than other simple ligands used in similar cyano-briged Fe(III)–Mn(III) complexes.
Table 4 Structural and magnetic parameters for some cyano-bridged FeIII–MnIII complexes
| Compound |
Structure |
Mn–Ncyano (Å) |
CN–Mn (°) |
Magnetism |
Ref. |
| [Mn(5-Cl-salen)Fe(bpClb)(CN)2]n |
1D |
2.258(4)–2.277(4) |
149.5(4), 154.3(3) |
F |
24a
|
| [Mn(5-Cl-salen)Fe(bpb)(CN)2]n |
1D |
2.270(7)–2.339(7) |
149.1(7)–160.8(7) |
F |
24a
|
| [Mn(5-Br-salen)Fe(bpb)(CN)2]n |
1D |
2.304(3)–2.372(3) |
149.0(3)–161.3(2) |
F |
24a
|
| [Mn(5-Me-salen)Fe(bpb)(CN)2]n |
1D |
2.308(3)–2.376(3) |
147.5(2)–160.9(3) |
F |
24a
|
| [Mn(saltmen)Fe(bpb)(CN)2]n |
1D |
2.242(6)–2.328(5) |
148.8(5)–166.0(6) |
F |
24b
|
| [Mn(salen)][Fe(bpb)(CN)2]n |
1D |
2.301(3)–2.439(3) |
151.8(3), 158.6(3) |
AF |
24c
|
|
2-(SS) |
1D |
2.245(17)–2.336(17) |
153.1(17)–165.1(18) |
AF |
This work |
Conclusion
In summary, we have introduced a redox-active TTF ligand into the dicyanideferrite building block [(n-Bu)4N][Fe(TTFbp)(CN)2] for the first time. Two enantiopure one-dimensional complexes, [Mn((R,R)-salphen)Fe(TTFbp)(CN)2]n (2-(RR)) and [Mn((S,S)-salphen)Fe(TTFbp)(CN)2]n (2-(SS)), were subsequently synthesized and structurally characterized. Circular dichroism and vibrational circular dichroism spectra confirm the enantiomeric nature of the optically active complexes, while solution and solid state CV studies reveal that complexes exhibit multiple redox-accessible states. Magnetic studies show that antiferromagnetic coupling is operative between FeIII and MnIII centers bridged by cyanide. The intermolecular hydrogen interactions of compound 2-(SS) lead to an antiferromagnet with TN = 4.8 K. A field-induced magnetic phase transition was also observed in 2-(SS), and was ascribed to the decoupling effect of the field. Magnetostructural correlations for some typical cyano-bridged heterobimetallic FeIII–MnIII compounds were also instructive in elucidating the origins of the magnetic behavior. The introduction of the redox-active TTF unit, coupled with chirality into cyanide-bridged complexes with interesting magnetic properties makes it possible to construct novel multifunctional materials. Further investigations into the partial oxidation of these compounds via chemical and electrochemical methods will pave the way towards applications of these new materials, and this work is currently underway in our laboratory.
Acknowledgements
This work was supported by the Major State Basic Research Development Program (2013CB922101), the National Natural Science Foundation of China (91433113), and the Australian Research Council (DP110101671). We also thank Dr Tian-Wei Wang for experimental assistance on magnetic measurements.
References
-
(a) M. Mannini, F. Pineider, P. Sainctavit, C. Danieli, E. Otero, C. Sciancalepore, A. M. Talarico, M.-A. Arrio, A. Cornia and D. Gatteschi, Nat. Mater., 2009, 8, 194–197 CrossRef CAS PubMed;
(b) R. Sessoli, D. Gatteschi, A. Caneschi and M. Novak, Nature, 1993, 365, 141–143 CrossRef CAS;
(c) W. Wernsdorfer and R. Sessoli, Science, 1999, 284, 133–135 CrossRef CAS PubMed;
(d) J. S. Miller, Chem. Soc. Rev., 2011, 40, 3266–3296 RSC;
(e) D. Pinkowicz, H. I. Southerland, C. Avendaño, A. Prosvirin, C. Sanders, W. Wernsdorfer, K. S. Pedersen, J. Dreiser, R. Clérac and J. Nehrkorn, J. Am. Chem. Soc., 2015, 137, 14406–14422 CrossRef CAS PubMed;
(f) R. E. Winpenny, Angew. Chem., Int. Ed., 2008, 47, 7992–7994 CrossRef CAS PubMed.
-
(a) N. A. Spaldin and M. Fiebig, Science, 2005, 309, 391–392 CrossRef CAS PubMed;
(b) W. Eerenstein, N. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed;
(c) C.-M. Liu, R.-G. Xiong, D.-Q. Zhang and D.-B. Zhu, J. Am. Chem. Soc., 2010, 132, 4044–4045 CrossRef CAS PubMed;
(d) L. E. Darago, M. L. Aubrey, C. J. Yu, M. I. Gonzalez and J. R. Long, J. Am. Chem. Soc., 2015, 137, 15703–15711 CrossRef CAS PubMed.
-
(a) E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García and V. Laukhin, Nature, 2000, 408, 447–449 CrossRef CAS PubMed;
(b) C. C. Beedle, C. J. Stephenson, K. J. Heroux, W. Wernsdorfer and D. N. Hendrickson, Inorg. Chem., 2008, 47, 10798–10800 CrossRef CAS PubMed;
(c) C. Faulmann, K. Jacob, S. Dorbes, S. Lampert, I. Malfant, M.-L. Doublet, L. Valade and J. A. Real, Inorg. Chem., 2007, 46, 8548–8559 CrossRef CAS PubMed;
(d) S. Dorbes, L. Valade, J. A. Real and C. Faulmann, Chem. Commun., 2005, 69–71 RSC.
- G. Rikken and E. Raupach, Nature, 1997, 390, 493–494 CrossRef CAS.
-
(a) H. Imai, K. Inoue, K. Kikuchi, Y. Yoshida, M. Ito, T. Sunahara and S. Onaka, Angew. Chem., Int. Ed., 2004, 116, 5736–5739 CrossRef;
(b) J. R. Galán-Mascarós, E. Coronado, P. A. Goddard, J. Singleton, A. I. Coldea, J. D. Wallis, S. J. Coles and A. Alberola, J. Am. Chem. Soc., 2010, 132, 9271–9273 CrossRef PubMed;
(c) M. Atzori, F. Pop, P. Auban-Senzier, R. Clérac, E. Canadell, M. L. Mercuri and N. Avarvari, Inorg. Chem., 2015, 54, 3643–3653 CrossRef CAS PubMed.
-
(a) K. Inoue, K. Kikuchi, M. Ohba and H. Ōkawa, Angew. Chem., Int. Ed., 2003, 115, 4958–4961 CrossRef;
(b) R. Inglis, F. White, S. Piligkos, W. Wernsdorfer, E. K. Brechin and G. S. Papaefstathiou, Chem. Commun., 2011, 47, 3090–3092 RSC;
(c) M. Clemente-León, E. Coronado, J. C. Dias, A. Soriano-Portillo and R. D. Willett, Inorg. Chem., 2008, 47, 6458–6463 CrossRef PubMed;
(d) S. Chorazy, K. Nakabayashi, K. Imoto, J. Mlynarski, B. Sieklucka and S.-i. Ohkoshi, J. Am. Chem. Soc., 2012, 134, 16151–16154 CrossRef CAS PubMed;
(e) C. Train, R. Gheorghe, V. Krstic, L.-M. Chamoreau, N. S. Ovanesyan, G. L. Rikken, M. Gruselle and M. Verdaguer, Nat. Mater., 2008, 7, 729–734 CrossRef CAS PubMed.
-
(a) D.-H. Ren, D. Qiu, C.-Y. Pang, Z. Li and Z.-G. Gu, Chem. Commun., 2015, 51, 788–791 RSC;
(b) R. Sessoli, M.-E. Boulon, A. Caneschi, M. Mannini, L. Poggini, F. Wilhelm and A. Rogalev, Nat. Phys., 2015, 11, 69–74 Search PubMed;
(c) Y.-Y. Zhu, X. Guo, C. Cui, B.-W. Wang, Z.-M. Wang and S. Gao, Chem. Commun., 2011, 47, 8049–8051 RSC;
(d) L.-L. Fan, F.-S. Guo, L. Yun, Z.-J. Lin, R. Herchel, J.-D. Leng, Y.-C. Ou and M.-L. Tong, Dalton Trans., 2010, 39, 1771–1780 RSC.
-
(a) W. Kaneko, S. Kitagawa and M. Ohba, J. Am. Chem. Soc., 2007, 129, 248–249 CrossRef CAS PubMed;
(b) J. Ru, F. Gao, T. Wu, M.-X. Yao, Y.-Z. Li and J.-L. Zuo, Dalton Trans., 2014, 43, 933–936 RSC;
(c) J. Ru, F. Gao, M.-X. Yao, T. Wu and J.-L. Zuo, Dalton Trans., 2014, 43, 18047–18055 RSC;
(d) J. Xiang, L.-H. Jia, B.-W. Wang, S.-M. Yiu, S.-M. Peng, W.-Y. Wong, S. Gao and T. C. Lau, Dalton Trans., 2013, 42, 3876 RSC;
(e) M.-X. Yao, Q. Zheng, X.-M. Cai, Y.-Z. Li, Y. Song and J.-L. Zuo, Inorg. Chem., 2012, 51, 2140–2149 CrossRef CAS PubMed.
-
(a) A. Kobayashi, E. Fujiwara and H. Kobayashi, Chem. Rev., 2004, 104, 5243–5264 CrossRef CAS PubMed;
(b) T. Jørgensen, T. K. Hansen and J. Becher, Chem. Soc. Rev., 1994, 23, 41–51 RSC.
-
(a) H.-Y. Wang, Y. Wu, C. F. Leong, D. M. D'Alessandro and J.-L. Zuo, Inorg. Chem., 2015, 54, 10766–10775 CrossRef CAS PubMed;
(b) G. Cosquer, F. Pointillart, S. Golhen, O. Cador and L. Ouahab, Chem. – Eur. J., 2013, 19, 7895–7903 CrossRef CAS PubMed;
(c) M. Feng, F. Pointillart, B. Le Guennic, B. Lefeuvre, S. Golhen, O. Cador, O. Maury and L. Ouahab, Chem. – Asian J., 2014, 9, 2814–2825 CrossRef CAS PubMed.
-
(a) F. Pointillart, J. Jung, R. Berraud-Pache, B. Le Guennic, V. Dorcet, S. Golhen, O. Cador, O. Maury, Y. Guyot and S. Decurtins, Inorg. Chem., 2015, 54, 5384–5397 CrossRef CAS PubMed;
(b) H. Hiraga, H. Miyasaka, K. Nakata, T. Kajiwara, S. Takaishi, Y. Oshima, H. Nojiri and M. Yamashita, Inorg. Chem., 2007, 46, 9661–9671 CrossRef CAS PubMed;
(c) F. Pointillart, K. Bernot, S. Golhen, B. Le Guennic, T. Guizouarn, L. Ouahab and O. Cador, Angew. Chem., Int. Ed., 2015, 54, 1504–1507 CrossRef CAS PubMed;
(d) F. Pointillart, B. Le Guennic, S. Golhen, O. Cador and L. Ouahab, Chem. Commun., 2013, 49, 11632–11634 RSC;
(e) S. Ichikawa, S. Kimura, K. Takahashi, H. Mori, G. Yoshida, Y. Manabe, M. Matsuda, H. Tajima and J.-i. Yamaura, Inorg. Chem., 2008, 47, 4140–4145 CrossRef CAS PubMed.
-
(a) E. Coronado and P. Day, Chem. Rev., 2004, 104, 5419–5448 CrossRef CAS PubMed;
(b) M. Nihei, N. Takahashi, H. Nishikawa and H. Oshio, Dalton Trans., 2011, 40, 2154–2156 RSC;
(c) F. Pointillart, S. Klementieva, V. Kuropatov, Y. Le Gal, S. Golhen, O. Cador, V. Cherkasov and L. Ouahab, Chem. Commun., 2012, 48, 714–716 RSC;
(d) F. Pointillart, B. Le Guennic, S. Golhen, O. Cador, O. Maury and L. Ouahab, Inorg. Chem., 2013, 52, 1610–1620 CrossRef CAS PubMed.
- L. Ouahab and T. Enoki, Eur. J. Inorg. Chem., 2004, 2004, 933–941 CrossRef.
-
(a) L. Cui, F. Zhu, C. F. Leong, J. Ru, F. Gao, D. M. D'Alessandro and J. Zuo, Sci. China: Chem., 2015, 58, 650–657 CrossRef CAS;
(b) F. Gao, L. Cui, W. Liu, L. Hu, Y.-W. Zhong, Y.-Z. Li and J.-L. Zuo, Inorg. Chem., 2013, 52, 11164–11172 CrossRef CAS PubMed;
(c) R. Wang, L.-C. Kang, J. Xiong, X.-W. Dou, X.-Y. Chen, J.-L. Zuo and X.-Z. You, Dalton Trans., 2011, 40, 919–926 RSC;
(d) Y.-H. Peng, Y.-F. Meng, L. Hu, Q.-X. Li, Y.-Z. Li, J.-L. Zuo and X.-Z. You, Inorg. Chem., 2010, 49, 1905–1912 CrossRef CAS PubMed.
-
(a) C. Jia, S. X. Liu, C. Tanner, C. Leiggener, A. Neels, L. Sanguinet, E. Levillain, S. Leutwyler, A. Hauser and S. Decurtins, Chem. – Eur. J., 2007, 13, 3804–3812 CrossRef CAS PubMed;
(b) J.-C. Wu, S.-X. Liu, T. D. Keene, A. Neels, V. Mereacre, A. K. Powell and S. Decurtins, Inorg. Chem., 2008, 47, 3452–3459 CrossRef CAS PubMed;
(c) X. Guégano, A. L. Kanibolotsky, C. Blum, S. F. Mertens, S. X. Liu, A. Neels, H. Hagemann, P. J. Skabara, S. Leutwyler and T. Wandlowski, Chem. – Eur. J., 2009, 15, 63–66 CrossRef PubMed.
- P. Przychodzen, K. Lewinski, M. Balanda, R. Pelka, M. Rams, T. Wasiutynski, C. Guyard-Duhayon and B. Sieklucka, Inorg. Chem., 2004, 43, 2967–2974 CrossRef CAS PubMed.
-
SAINT-Plus, version 6.02, Bruker Analytical X-ray System, Madison, WI, 1999 Search PubMed.
-
G. M. Sheldrick, SADABS an empirical absorption correction program, Bruker Analytical X-ray Systems, Madison, WI, 1996 Search PubMed.
-
G. M. Sheldrick, SHELXTL-97, Universität of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- T. Wu, C.-H. Li, Y.-Z. Li, Z.-G. Zhang and X.-Z. You, Dalton Trans., 2010, 39, 3227–3232 RSC.
-
(a) D. Zhang, W. Si, P. Wang, X. Chen and J. Jiang, Inorg. Chem., 2014, 53, 3494–3502 CrossRef CAS PubMed;
(b) D. Zhang, H. Wang, Y. Chen, Z.-H. Ni, L. Tian and J. Jiang, Inorg. Chem., 2009, 48, 5488–5496 CrossRef CAS PubMed;
(c) Z.-H. Ni, J. Tao, W. Wernsdorfer, A.-L. Cui and H.-Z. Kou, Dalton Trans., 2009, 2788–2794 RSC.
- J. H. Yoon, H. S. Yoo, H. C. Kim, S. W. Yoon, B. J. Suh and C. S. Hong, Inorg. Chem., 2008, 48, 816–818 CrossRef PubMed.
-
O. Kahn, Molecular Magnetism, VCH, Weinhem, Germany, 1993 Search PubMed.
-
(a) H.-Z. Kou, Z.-H. Ni, C.-M. Liu, D.-Q. Zhang and A.-L. Cui, New J. Chem., 2009, 33, 2296–2299 RSC;
(b) T. Senapati, C. Pichon, R. Ababei, C. Mathonière and R. Clérac, Inorg. Chem., 2012, 51, 3796–3812 CrossRef CAS PubMed;
(c) Z. H. Ni, H. Z. Kou, L. F. Zhang, C. Ge, A. L. Cui, R. J. Wang, Y. Li and O. Sato, Angew. Chem., Int. Ed., 2005, 44, 7742–7745 CrossRef CAS PubMed.
- H. Y. Kwak, D. W. Ryu, J. W. Lee, J. H. Yoon, H. C. Kim, E. K. Koh, J. Krinsky and C. S. Hong, Inorg. Chem., 2010, 49, 4632–4642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional structures, electrochemical data and magnetic characterization data, and X-ray crystallographic files in CIF format for 1, 2-(RR) and 2-(SS). CCDC 1454969–1454971. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01756h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence