
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Azoimidazole functionalized Ni-porphyrins for molecular spin switching and light responsive MRI contrast agents†
Gernot
Heitmann
a,
Christian
Schütt
a,
Jens
Gröbner
a,
Lukas
Huber
b and
Rainer
Herges
*a
aOtto Diels-Institut für Organische Chemie, Christian-Albrechts-Universität zu Kiel, Otto-Hahn-Platz 4, D-24098 Kiel, Germany. E-mail: rherges@oc.uni-kiel.de
bMolecular Imaging North Competence Center, Christian-Albrechts-Universität zu Kiel, Am Botanischen Garten 14, 24118 Kiel, Germany
First published on 16th June 2016
Abstract
Azo-N-methylimidazole functionalized Ni(II)porphyrins were rationally designed and synthesized and their performance as molecular spin switches was investigated. They perform intramolecular light-driven coordination-induced spin state switching (LD-CISSS) in the presence of water and therefore are an important step towards spin switches for medicinal applications, particularly functional MRI contrast agents.
Introduction
The manipulation of magnetic properties in bulk materials as well as in molecular systems has gained increasing attention due to numerous promising applications,1,2e.g. spintronics, data storage,3,4 switchable diamagnetic levitation5 or responsive (“smart”) contrast agents for magnetic resonance imaging (MRI).6–10 However, spin switching of single molecules in solution or on surfaces at ambient temperatures is still challenging and only a few examples are known in the literature.11–14 Ni2+ complexes change their spin state from diamagnetic (S = 0) to paramagnetic (S = 1) when the coordination sphere is changed from square planar (CN = 4) to square pyramidal (CN = 5) or square bipyramidal (CN = 6).15–21 We have demonstrated highly efficient and fatigue resistant light-driven coordination-induced spin state switching (LD-CISSS) by adding photodissociable ligands22–24 to a solution of a Ni2+-porphyrin (PDL approach) as well as by tethering azopyridines directly to the porphyrin (“record player” approach).10,25–27 In both concepts the photoresponsive axial ligands are designed in such a way that only one photo isomer is capable of coordinating to the Ni2+. Recently, we were able to show the photo switching of MRI contrasts with record player molecules.10 Thus, the “record player” concept is a promising approach for the development of “smart” MRI contrast agents that can be switched on and off with light for interventional radiology. A number of diseases such as stroke and heart attack are increasingly treated with minimal invasive, catheter-based interventions under X-ray imaging control (e.g. placing stents). Replacing X-ray imaging by MRI would have several advantages: avoidance of ionizing radiation, better soft tissue contrast, and 3D control.28 However, for MRI guided interventions switchable contrast agents are needed to continuously check the dynamic blood flow. Light is the ideal stimulus for switching because it can be applied with temporal and spatial control, it is traceless and, regarding practical applications, even a micro-catheter can easily accommodate optical fibers that carry sufficient light energy to the site of intervention.29 Our “record player” molecules are the most promising candidates for light-controlled contrast switching, however, their function is restricted to organic solvents, thus precluding medical applications. Aiming at the extension of the “record player” concept towards operation in aqueous solution one has to consider that water severely reduces the ligand's donor strength by hydrogen bonding or, in the worst case, by protonation. An appropriate ligand for coordination-induced spin state switching (CISSS) in water30 therefore has to provide a high binding affinity (KL) to the metal centre while exhibiting a rather low basicity (corresponding to a low pKa). In toluene, the coordination strength KL of pyridine based ligands to the highly electron deficient Ni2+-porphyrin NiTPPF20 can be increased effectively by introducing electron releasing groups in para position to the coordinating nitrogen.21 However, electron releasing substituents also increase the basicity of the pyridine. The pKa and KL follow a linear free energy relationship (see Fig. 1). To avoid protonation (e.g. in blood), the pKa of the ligand should be <7. Methoxypyridine (1) (pKa 6.55) is just within this range, however, dimethylamino pyridine (2) which is an extremely strong ligand in toluene31 would be completely protonated in water (pKa 9.7), and not bind at all. We therefore decided to replace pyridine by 1-methylimidazole (3) in our record player design. While having a similar basicity as methoxy pyridine (1) (pKa 6.55), 1-methylimidazole (3) (pKa 7.12) has an almost 12-fold higher binding affinity to NiTPPF20.22 We attribute this fact to a reduced steric hindrance of the hydrogen atoms in ortho position with respect to the coordinating nitrogen in the 5-membered ring. The same trends were also observed with the photoswitchable 3-phenylazo-substituted pyridines24 and 5-phenylazo-substituted imidazoles22 (PDLs, see the ESI†). The (electron withdrawing) azo group in 5-position of the 1-methylimidazol (3) should further reduce the pKa to <6, and protonation under physiological conditions should not occur. This provides further arguments to replace the phenylazopyridine by a phenylazoimidazole unit in our “record player” approach. Consequently, we set out to design new “record player” structures that allow optimal intramolecular coordination of the imidazole of the cis isomer, and a weak or no coordination in the trans form.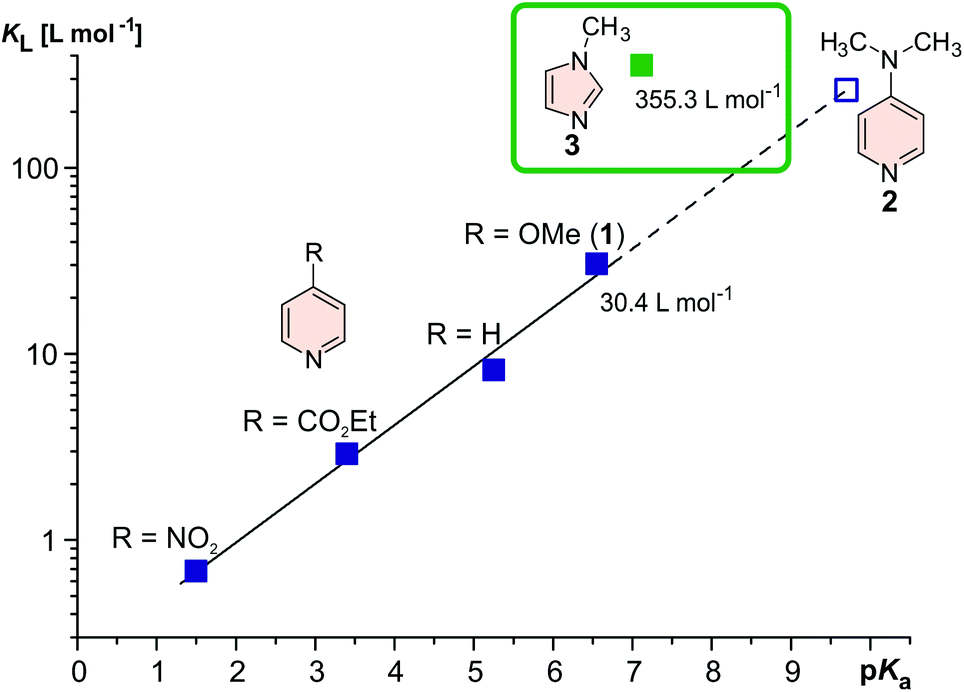 | ||
| Fig. 1 Association constants of pyridine derivatives21 and 1-methylimidazole (3)22 with Ni(II)TPPF20 (in toluene) as a function of their pKa value. Electron releasing para substituents at the pyridine ring improve the association to the nickel but also give rise to substantially higher basicity. DMAP31 strongly coordinates to Ni2+ in organic solvents, however, in water at pH 7 it would be almost completely protonated and not bind at all. 1-Methylimidazole (3) on the other hand exhibits an extremely high association constant and nevertheless a low basicity (pKa 7.12). Considering the fact that the imidazole ring would bear an additional electron withdrawing azo group, the record player molecule would not be protonated at pH 7. | ||
Results and discussion
Previous ligand design studies have shown that even small changes in geometry (bond lengths ∼0.01 Å, bond angles ∼5°) can have a large effect on functionality and switching efficiency. So, it was to be expected that a simple replacement of the pyridine ring by an imidazole ring would not necessarily lead to a successful spin switch, because the change in ring size would change the length and the angle of the coordinative bond to the Ni2+. Moreover, cis-azopyridines prefer a conformation with the phenyl and pyridine ring only slightly twisted with respect to each other, whereas in the cis-azoimidazoles the phenyl and the heterocyclic ring are exactly orthogonal.32 Our record player design is based on three structural elements (Fig. 2): the porphyrin platform (beige), the switchable ligand (red) and the tether (cyan), connecting the former two units.26 Porphyrin (Ni-tris(pentafluorophenyl)porphyrin) and the photoswitchable ligand (phenylazoimidazole) are already set. So, the tether remains to be optimized. Quantum chemical calculations were performed using the program TURBOMOLE 6.6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 at the TPSSh/def2TZVP//TPSSh/SVP level of density functional theory (DFT). This level of theory was chosen because, according to an empirical study, it performed best in predicting the coordination energy of axial ligands to nickel porphyrins.34 Among a number of promising candidates that were initially considered, two structures were selected (Fig. 2, middle and right). A reference structure was calculated as well (Fig. 2, left). In the reference structure ligand and porphyrin are not covalently connected. The reference structure, therefore, exhibits an unstrained geometry with the maximum binding energy. The tether was designed in such a way that this optimum geometry for binding is retained. Structure 4 (Fig. 2, middle) corresponds to the original design.25,26 The pyridine ring is just replaced by an imidazole ring. The geometry exhibits a strong deviation from ideal binding (cf. reference compound) which also leads to a weaker coordination (ΔHcalc = −2.15 kcal mol−1) as compared to the reference compound (−6.28 kcal mol−1). Inspection of structure 4 reveals that the tether is obviously too short. Therefore we inserted several different atoms and groups into the tether of which sulphur between the two phenyl rings proved to be optimal (Fig. 2, structure 5). This structure matches the ideal binding conformation almost perfectly and the phenylazoimidazole moiety can obtain a relaxed binding conformation, giving rise to a considerably higher binding energy (−4.04 kcal mol−1). However, an additional rotational degree of freedom is introduced which might disfavour the coordinating cis species entropically. Since both structures have pros and cons we were not able to decide at this point which design would be more advantageous. Therefore, we decided to synthesize both “record players” 4 and 5 following the synthetic protocols for the pyridine “record player”10,25 and for the imidazole based PDLs.22,32
33 at the TPSSh/def2TZVP//TPSSh/SVP level of density functional theory (DFT). This level of theory was chosen because, according to an empirical study, it performed best in predicting the coordination energy of axial ligands to nickel porphyrins.34 Among a number of promising candidates that were initially considered, two structures were selected (Fig. 2, middle and right). A reference structure was calculated as well (Fig. 2, left). In the reference structure ligand and porphyrin are not covalently connected. The reference structure, therefore, exhibits an unstrained geometry with the maximum binding energy. The tether was designed in such a way that this optimum geometry for binding is retained. Structure 4 (Fig. 2, middle) corresponds to the original design.25,26 The pyridine ring is just replaced by an imidazole ring. The geometry exhibits a strong deviation from ideal binding (cf. reference compound) which also leads to a weaker coordination (ΔHcalc = −2.15 kcal mol−1) as compared to the reference compound (−6.28 kcal mol−1). Inspection of structure 4 reveals that the tether is obviously too short. Therefore we inserted several different atoms and groups into the tether of which sulphur between the two phenyl rings proved to be optimal (Fig. 2, structure 5). This structure matches the ideal binding conformation almost perfectly and the phenylazoimidazole moiety can obtain a relaxed binding conformation, giving rise to a considerably higher binding energy (−4.04 kcal mol−1). However, an additional rotational degree of freedom is introduced which might disfavour the coordinating cis species entropically. Since both structures have pros and cons we were not able to decide at this point which design would be more advantageous. Therefore, we decided to synthesize both “record players” 4 and 5 following the synthetic protocols for the pyridine “record player”10,25 and for the imidazole based PDLs.22,32
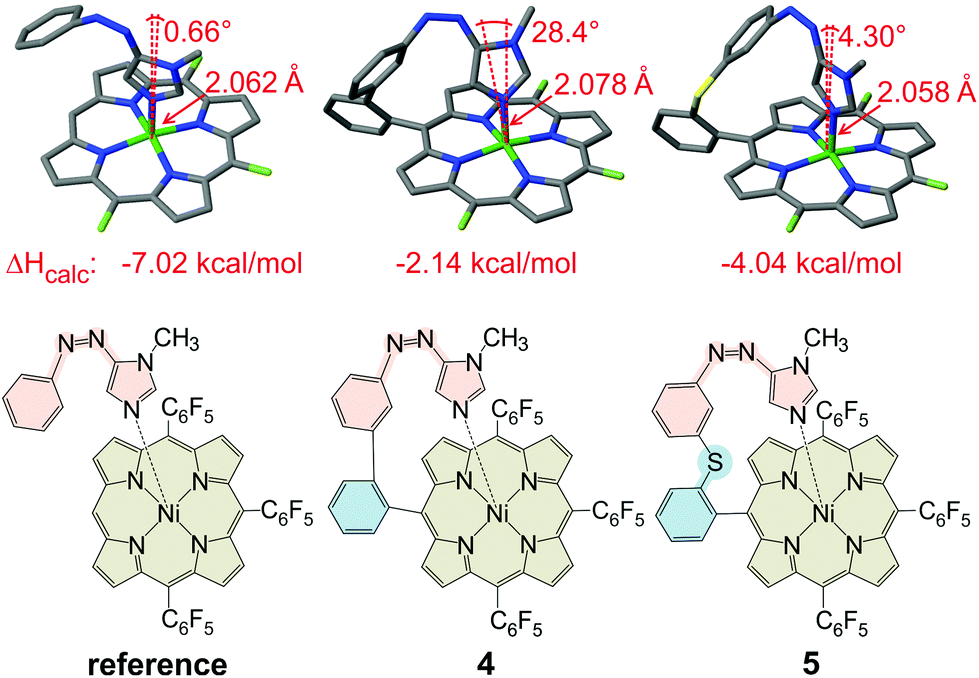 | ||
| Fig. 2 Calculated (TPSSh/SVP) geometries, ligand binding energies (ΔHcalc, TPSSh/def2TZVP//TPSSh/SVP) and schematic 2D structures for the reference structure (left, without a tether) imidazole record player structure 4 (middle, phenyl tether) and 5 (left, thiophenol tether). The reference structure with no constraints represents the ideal binding situation, and therefore exhibits the highest binding energy (ΔHcalc). The phenyl tether in 4 imposes a large deviation from the ideal binding geometry and therefore has the lowest binding energy. Obviously the tether is too short. Insertion of a sulphur atom into the tether almost restores the ideal binding geometry. Consequently, the binding energy increases substantially. For the sake of clarity, the pentafluorophenyl substituents are depicted as fluorine substituents in the geometry plots. | ||
The porphyrins were prepared via mixed aldehyde syntheses (“tone arm” approach; see Scheme 1; for experimental details see the ESI†). Intermediate for both asymmetric aldehydes (“record player tone arms”) 13 and 16 is the tritylated azoimidazole 11 which is prepared from 3-bromoaniline (6) and N,N-dimethylsulfamoylimidazole (7) in 8 steps (4 pots) according to a general procedure that we published recently.22,32 The azo moiety is then connected to the respective benzaldehyde, either by Suzuki cross coupling with 2-formylphenylboronic acid (for biphenyl tone arm 13), or by Pd-catalyzed thiol functionalization followed by nucleophilic aromatic substitution with 2-fluorobenzaldehyde (for thioether tone arm 16). Afterwards, the tritylated “tone arms” 12 and 15 are methylated and trityl-deprotected in a one-pot procedure to obtain 13 and 16. Mixed aldehyde porphyrin syntheses under Lindsey conditions with 13 or 16, respectively, pentafluoro-benzaldehyde and pentafluorophenyl dipyrromethane gave the porphyrins as free bases which were then treated with nickel(II) acetylacetonate to obtain N-methylimidazole “record players” 4 and 5.
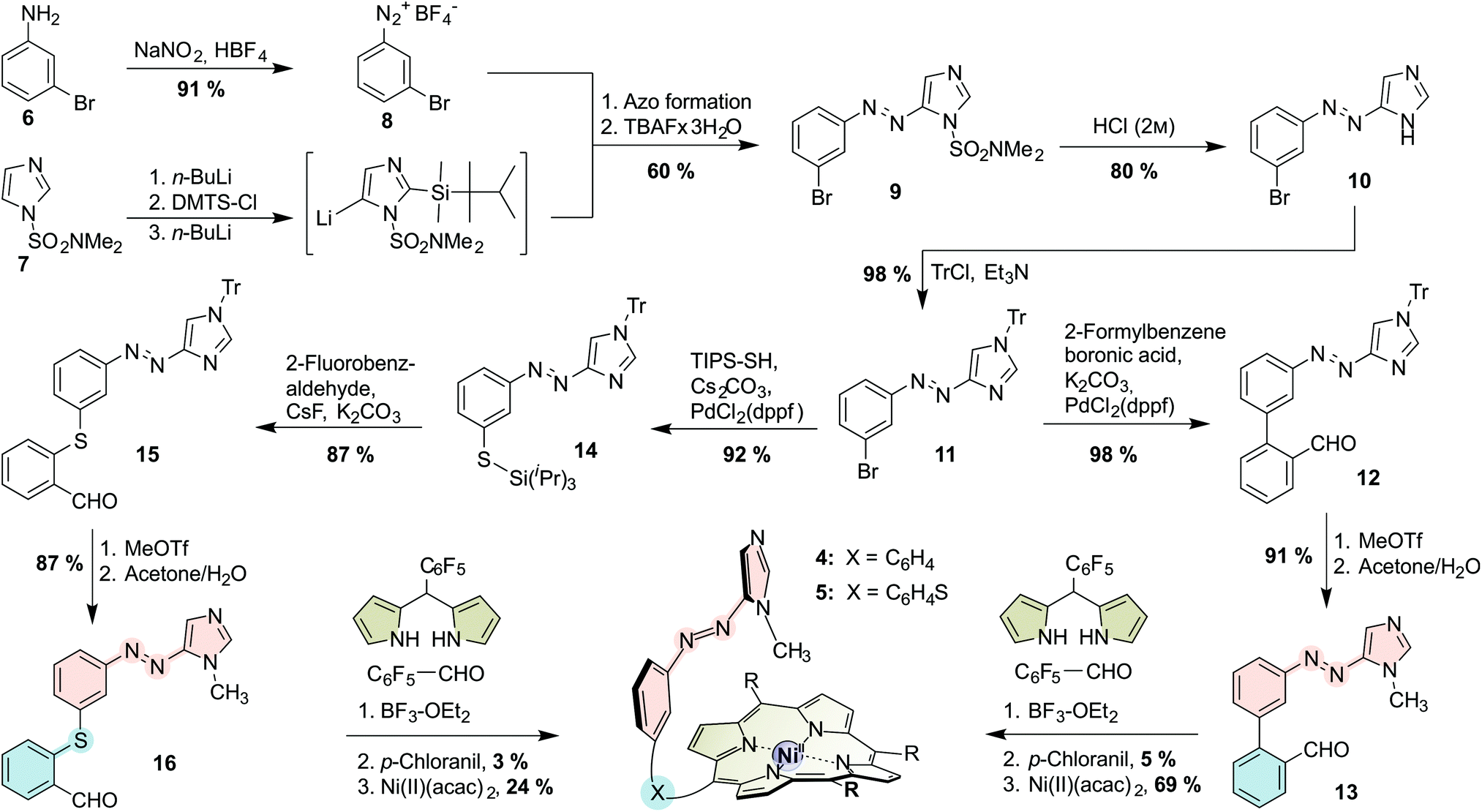 | ||
| Scheme 1 Synthesis of 1-methylimidazole “record player” molecules 4 and 5. | ||
Imidazole “record players” 4 and 5 exhibit considerable differences regarding the photochromism compared to the already known pyridine derivatives (for UV-vis spectra see the ESI†). Pyridine “record players” are switching to the coordinating cis isomer upon irradiation into the porphyrin Q bands (∼500 nm).10,25–27 At 365 nm no trans → cis switching is observed which is peculiar because 365 nm is the usual wavelength for trans → cis isomerization of azobenzenes and azopyridines. Imidazole “record players” 4 and 5, however, switch both at 505 nm and 365 nm. Direct excitation of the azo unit with 365 nm or 385 nm is much more efficient (up to 85% cis). We assign this improved trans → cis photoisomerization of 4 and 5 to the bathochromic shift of the azoimidazole π–π* bands in comparison with azopyridines. Unfortunately, this shift also gives rise to a somewhat impaired back-switching to the trans-species by irradiation into the cis Soret band with 435 nm (14% residual cis for 4; 13% residual cis for 5) which surprisingly is even more pronounced at higher concentrations (for details see the ESI†). The thermal half-life times of cis-4 (t1/2 = 69 d) and cis-5 (t1/2 = 23 d) exceed those of the free ligands 13 and 16 (18.9 and 19.4 d, measured with 1H NMR, for details see the ESI†). Obviously, coordination increases the stability of the cis isomer.
The switching performance of our spin switches does not only depend on the conversion rate of trans to cis and back to the trans isomer (complete conversion in an ideal system) but also to what extend the cis isomer would be paramagnetic (100% in an ideal system with a strongly coordinating ligand). As we described earlier, the ratio of paramagnetic to diamagnetic cis-species can be easily derived from the NMR shift (δ) of the pyrrole protons of the porphyrin system.23 As expected for the strongly coordinating imidazole ligand, we determined a high amount of paramagnetic cis species for 5 (cis-5para: 77%) whereas the cis isomer of 4 with its more strained binding geometry (Fig. 2 middle) exhibits a lower percentage of paramagnetic species (cis-4para: 72%; for details see the ESI†). The measured ratios of paramagnetic cis species are in good agreement to the results of our computational studies; however, the performance of thioether “record player” 5 fell somewhat short of our expectations. Temperature dependent NMR experiments provided the thermodynamic parameters ΔH and ΔS of the coordination event, and revealed the reason for this less than optimum performance of 5 (see Table 1, for details see the ESI†). The binding enthalpy ΔHexp for cis-5, indeed, is much higher than for cis-4. However, part of the enthalpy gain is cancelled by a more negative coordination entropy ΔS. Insertion of the sulphur atom into the tether introduces an additional degree of conformational freedom in the unbound state (rotation around the C–S bond) which is frozen upon coordination. At 300 K, the free Gibbs enthalpy of coordination (ΔG300) of 5 is only 0.21 kcal mol−1 more negative than in 4. Note that the experimentally determined binding enthalpies of 4 and 5 are in good agreement with the theoretically obtained values (ΔHcalc, for details see the ESI†).
|
|
||
|---|---|---|
| 4 (X = C6H4) | 5 (X = C6H4S) | |
| ΔHexp/kcal mol−1 | −1.96 | −2.76 |
| ΔS/cal mol−1 | −4.64 | −6.84 |
| ΔG300/kcal mol−1 | −0.57 | −0.71 |
| ΔHcalc/kcal mol−1 | −2.14 | −4.04 |
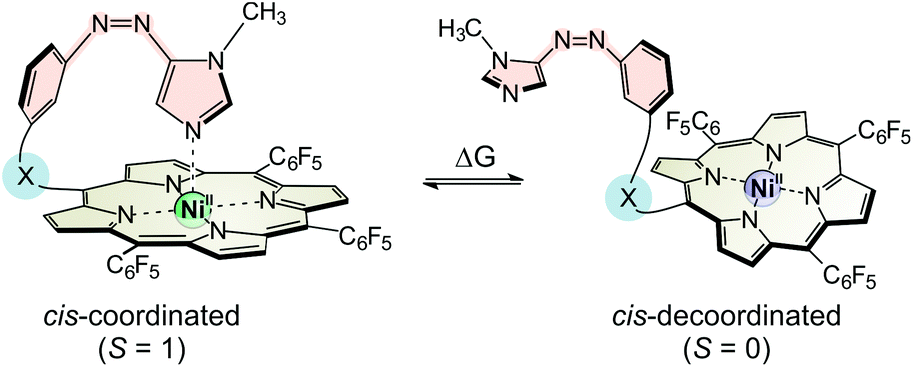
The overall performance of a molecular switch is defined as switching efficiency (SE), which in the case of “record player” molecules is the difference of the percentage of the paramagnetic nickel ions in both switching states. The percentage of paramagnetic Ni2+ in 4 can be switched between 10% and 61% (SE = 51%) whereas the sulphur derivative is superior with a SE of 55% (10% to 65% paramagnetic Ni2+) (see Fig. 3). Thus, the SE of the imidazole record player 5 in acetonitrile is higher than the SE of the parent pyridine record player in the same solvent (52%).26
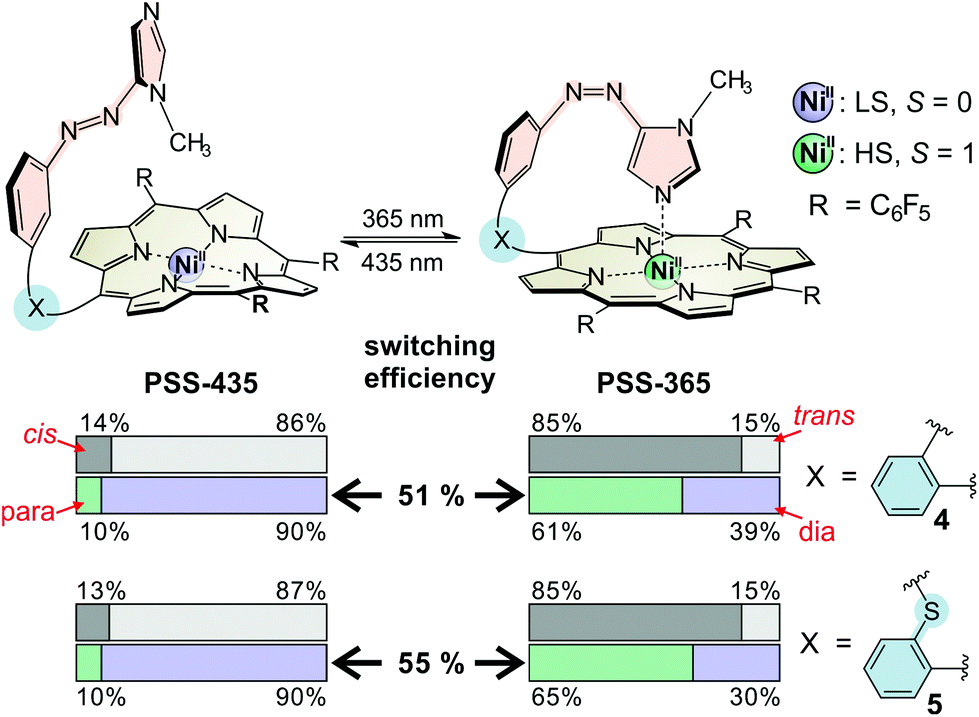 | ||
| Fig. 3 Switching parameters and switching efficiencies for imidazole record players 4 and 5 in acetonitrile. For the sake of simplicity only the predominant species of each PSS is shown as molecular structure. | ||
Encouraged by these results and in view of the development of water soluble spin switches and particularly of functional MRI contrast agents, we performed UV-vis switching experiments with 4 and 5 in a 1:1 mixture of phosphate-buffered saline (PBS, pH 7.4) and acetonitrile. The UV-vis spectra of thioether “record player” 5 are depicted in Fig. 4 (for spectra of 4, see the ESI†). With respect to measurements in pure acetonitrile the MRI “off-state” (PSS-435) is nearly unchanged (∼10% para) whereas the amount of paramagnetic species in the MRI “on-state” (PSS-365) is somewhat reduced. However, since the paramagnetic species still is formed to 47–48% in 5 (estimated from UV-vis extinction, see the ESI†) we are confident that switching under physiological conditions will be feasible if the solubility in water would be improved.
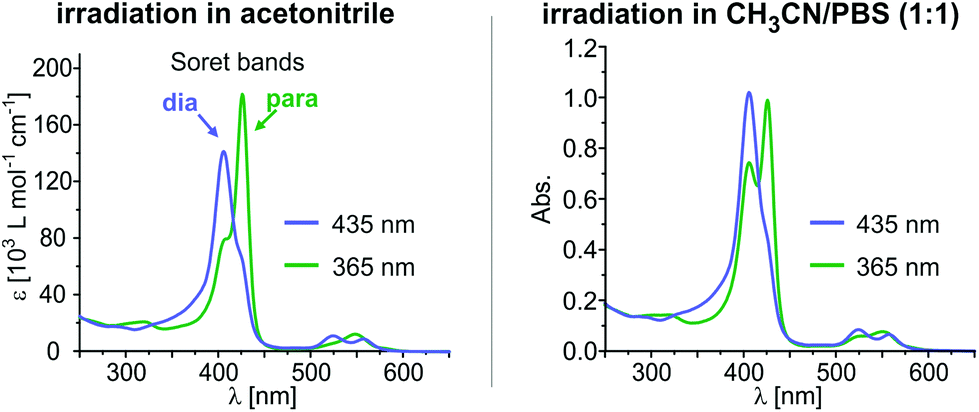 | ||
| Fig. 4 UV-vis switching experiments of 5 in acetonitrile (left) and in acetonitrile/PBS (1:1) (right). Intramolecular coordination of the cis species is largely maintained in the presence of water (pH 7.4). | ||
To take a further step towards application as photoswitchable MRI contrast agents we performed magnetic resonance measurements with 4 and 5 in acetonitrile (∼2 mM solutions) in a “small animal” 7T MRI system. The contrast enhancement (corresponding to an accelerated relaxation time of the solvent protons) in the “on-state” (PSS at 365 nm) is clearly visible, proving the applicability of our concept for functional MRI (see Fig. 5, for details see the ESI†). It is noteworthy that acetonitrile is in fact a rather adverse solvent for MRI because of its low proton density, its low coordination affinity to the paramagnetic Ni2+, and the large distance of the methyl protons from the paramagnetic metal ion during coordination. Nevertheless, the MRI contrast of a 2 mM solution of 5 can be switched with UV and visible light by 24%. Previously reported light-responsive MRI contrast agents exhibit switching efficiencies of ∼20% in water.35,36 Corresponding water soluble record player molecules can be expected to be considerably more effective.
 | ||
| Fig. 5 Left: 7T magnetic resonance image (gradient echo sequence, for details see the ESI†) of record players 4 and 5 in both switching states. The contrast enhancement (corresponding to an accelerated relaxation time of the solvent protons) in the “on-state” (PSS-365 nm) of 4 and 5 is clearly visible. Right: Experimental setup for the MRI measurements. NMR tubes were used as sample carriers and immobilized in an agarose phantom. MRI measurements were carried out on a 7T MRI system (ClinScan by co. Bruker) for “small animals” with a mouse body coil. | ||
Conclusions
In summary, we present two new and very efficient molecular spin switches (4 and 5) based on the light-driven coordination-induced spin state switching (LD-CISSS) principle. The molecules are constructed from three building blocks: (a) Ni-porphyrin (disk), (b) azoimidazole (tone arm), (c) tether that connects both units. Upon irradiation with 365 nm the azoimidazole undergoes trans–cis isomerization. The associated geometry movement puts the imidazole nitrogen on top of the Ni ion (“record player” design) which changes its coordination number (from 4 to 5), and in turn changes the spin state from diamagnetic to paramagnetic. Irradiation with 435 nm reverses the process. The light-driven spin state switching is completely reversible at room temperature, in solution, and under air. Azo-methylimidazole is a hitherto scarcely used photochromic compound but it combines two properties that are important for the design of spin switches: it's a very efficient photoswitch and the imidazole unit is a very strong ligand that binds to Ni2+ even in water. Equally important for the construction of efficient molecular spin switches is a careful design of the tether connecting the azoimidazole with the porphyrin. Quantum chemical calculations and experimental properties of 4 and 5 prove that even very small changes in geometry have a large impact on the switching efficiency. Particularly, the carefully designed “record player” molecule 5 exhibits most prerequisites that are necessary to perform spin switching in water. The spin switching efficiency in a 1:1 mixture of water/acetonitrile was determined as 40%. There are a number of potential applications for spin switching in solution. Most obvious is the control of relaxation times of protons. In NMR this could be used to reduce acquisition times, particularly in 2D experiments, whereas in magnetic resonance imaging (MRI) our compounds could be applied as switchable contrast agents in catheter based interventions.37,38 To demonstrate the efficiency of our “record player” molecules as light controlled contrast agents we performed MRI experiments. Upon irradiation of a 2 mM solution of 5 in acetonitrile the proton relaxation time of the bulk solvent could be reversibly changed, giving rise to a contrast change of 24%.
Acknowledgements
The authors thank Claus Gernert and Jürgen Grotemeyer (Institute for Physical Chemistry, Christian-Albrechts-Universität zu Kiel) for the measurement of high-resolution mass spectra of the compounds 4 and 5.This work was funded by the “Deutsche Forschungs-gemeinschaft” (DFG) via the collaborative research center 677 (SFB 677) “Function by Switching”.
Notes and references
- S. Hayami, S. M. Holmes and M. A. Halcrow, J. Mater. Chem. C, 2015, 3, 7775–7778 RSC.
- M. A. Halcrow, Spin-Crossover Materials: Properties and Applications, John Wiley & Sons Ltd, 2013 Search PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- J. Mater. Chem., 2009, 19, 1670–1695, themed issue on “Molecular Spintronics and Quantum Computing”.
- K. A. Mirica, S. S. Shevkoplyas, S. T. Phillips, M. Gupta and G. M. Whitesides, J. Am. Chem. Soc., 2009, 131, 10049–10058 CrossRef CAS PubMed.
- J. Hasserodt, New J. Chem., 2012, 36, 1707–1712 RSC.
- R. Herges, Nachr. Chem., 2011, 59, 817–821 CrossRef CAS.
- I.-R. Jeon, J. G. Park, C. R. Haney and T. D. Harris, Chem. Sci., 2014, 5, 2461–2465 RSC.
- R. N. Muller, L. Vander Elst and S. Laurent, J. Am. Chem. Soc., 2003, 125, 8405–8407 CrossRef CAS PubMed.
- M. Dommaschk, M. Peters, F. Gutzeit, C. Schütt, C. Näther, F. D. Sönnichsen, S. Tiwari, C. Riedel, S. Boretius and R. Herges, J. Am. Chem. Soc., 2015, 137, 7552–7555 CrossRef CAS PubMed.
- A. Witt, F. W. Heinemann and M. M. Khusniyarov, Chem. Sci., 2015, 6, 4599–4609 RSC.
- M.-L. Boillot, S. Chantraine, J. Zarembowitch, J.-Y. Lallemand and J. Prunet, New J. Chem., 1999, 23, 179–184 RSC.
- A. Sour, M.-L. Boillot, E. Rivière and P. Lesot, Eur. J. Inorg. Chem., 1999, 1999, 2117–2119 CrossRef.
- Y. Hasegawa, S. Kume and H. Nishihara, Dalton Trans., 2009, 280–284 RSC.
- W. S. Caughey, R. M. Deal, B. D. McLees and J. O. Alben, J. Am. Chem. Soc., 1962, 84, 1735–1736 CrossRef CAS.
- W. S. Caughey, W. Y. Fujimoto and B. P. Johnson, Biochemistry, 1966, 5, 3830–3843 CrossRef CAS.
- S. J. Cole, G. C. Curthoys, E. A. Magnusson and J. N. Phillips, Inorg. Chem., 1972, 11, 1024–1028 CrossRef CAS.
- D. Kim, Y. O. Su and T. G. Spiro, Inorg. Chem., 1986, 25, 3988–3993 CrossRef CAS.
- B. D. McLees and W. S. Caughey, Biochemistry, 1968, 7, 642–652 CrossRef CAS PubMed.
- Y. Song, R. E. Haddad, S.-L. Jia, S. Hok, M. M. Olmstead, D. J. Nurco, N. E. Schore, J. Zhang, J.-G. Ma, K. M. Smith, S. Gazeau, J. Pécaut, J.-C. Marchon, C. J. Medforth and J. A. Shelnutt, J. Am. Chem. Soc., 2005, 127, 1179–1192 CrossRef CAS PubMed.
- S. Thies, C. Bornholdt, F. Köhler, F. D. Sönnichsen, C. Näther, F. Tuczek and R. Herges, Chem. – Eur. J., 2010, 16, 10074–10083 CrossRef CAS PubMed.
- C. Schütt, G. Heitmann, T. Wendler, B. Krahwinkel and R. Herges, J. Org. Chem., 2016, 81, 1206–1215 CrossRef PubMed.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczek and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- S. Thies, H. Sell, C. Bornholdt, C. Schütt, F. Köhler, F. Tuczek and R. Herges, Chem. – Eur. J., 2012, 18, 16358–16368 CrossRef CAS PubMed.
- S. Venkataramani, U. Jana, M. Dommaschk, F. D. Sönnichsen, F. Tuczek and R. Herges, Science, 2011, 331, 445–448 CrossRef CAS PubMed.
- M. Dommaschk, C. Schütt, S. Venkataramani, U. Jana, C. Näther, F. D. Sönnichsen and R. Herges, Dalton Trans., 2014, 43, 17395–17405 RSC.
- M. Dommaschk, C. Näther and R. Herges, J. Org. Chem., 2015, 80, 8496–8500 CrossRef CAS PubMed.
- J.-M. Serfaty, X. Yang, P. Aksit, H. H. Quick, M. Solaiyappan and E. Atalar, J. Magn. Reson. Imaging, 2000, 12, 590–594 CrossRef CAS PubMed.
- K. D. Taylor and C. Reiser, in Lasers in Cardiovascular Interventions, ed. O. Topaz, Springer-Verlag, London, 2015, ch. 1, pp. 1–14 Search PubMed.
- M. Dommaschk, F. Gutzeit, S. Boretius, R. Haag and R. Herges, Chem. Commun., 2014, 50, 12476–12478 RSC.
- The association constant (KL) of DMAP (2) to NiTPPF20 in toluene could not be measured due to solubility problems and was extrapolated from the values of the other pyridine derivatives.
- T. Wendler, C. Schütt, C. Näther and R. Herges, J. Org. Chem., 2012, 77, 3284–3287 CrossRef CAS PubMed.
- TURBOMOLE V6.6 2014, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available from http://www.turbomole.com.
- Unpublished results.
- C. Tu and A. Y. Louie, Chem. Commun., 2007, 1331–1333 RSC.
- C. Tu, E. A. Osborne and A. Y. Louie, Tetrahedron, 2009, 65, 1241–1246 CrossRef CAS PubMed.
- R. Herges, O. Jansen, F. Tuczek and S. Venkatamarani, Molecular Switch., WO Pat., 2012022299 A1, 2012 Search PubMed.
- R. Herges, O. Jansen, F. Tuczek and S. Venkatamarani, Photosensitive metal porphyrin complexes with pendant photoisomerizable chelate arm as photochromic molecular switches undergoing photoinduced spin transition, DE Pat., 102010034 A1, 2012 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of computational studies, experimental procedures, analytical data. See DOI: 10.1039/c6dt01727d |
| This journal is © The Royal Society of Chemistry 2016 |