
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pentiptycene-based concave NHC–metal complexes†
Roman
Savka
,
Sabine
Foro
and
Herbert
Plenio
*
Organometallic Chemistry, Alarich-Weiss-Str. 12, TU Darmstadt, 64287 Darmstadt, Germany. E-mail: plenio@tu-darmstadt.de
First published on 17th June 2016
Abstract
The reaction of 1-amino,4-hydroxy-pentiptycene with diacetyl or acenaphthene-1,2-dione gave the respective diimines, followed by alkylation of the hydroxyl groups, and cyclization of the alkylated diimines to the respective bispentiptycene-imidazolium salts NHC·HCl. The azolium salts, being precursors to N-heterocyclic carbenes, were converted into metal complexes [(NHC)MX] (MX = CuI, AgCl, AuCl) and [(NHC)IrCl(cod)] and [(NHC)IrCl(CO)2] in good yields. In the solid state [(NHC)AgCl] displays a bowl-shaped structure of the ligand with the metal center buried within the concave unit.
Introduction
Iptycenes are a class of aromatic molecules with arenes fused to a bicyclo[2.2.2]octane framework,1 which have found numerous applications in polymer chemistry,2 molecular machines,3,4 materials science and in host–guest chemistry.5–7 Triptycene, the first representative of this family of compounds was published by Bartlett et al. in 1942 (Scheme 1).8 The synthesis of pentiptycene was reported in 1974 by Skvarche and Shalaev,9 but despite significant improvements,10 its synthesis remains cumbersome.1 Pentiptycene quinone, on the other hand, is easily available from the reaction of anthracene and benzoquinone followed by the oxidation of the initial Diels–Alder product.11,12 This quinone can be converted into the respective amino-pentiptycene (Scheme 2), which (like most anilines)13 should serve as an excellent precursor for N-heterocyclic carbenes and the respective transition metal complexes.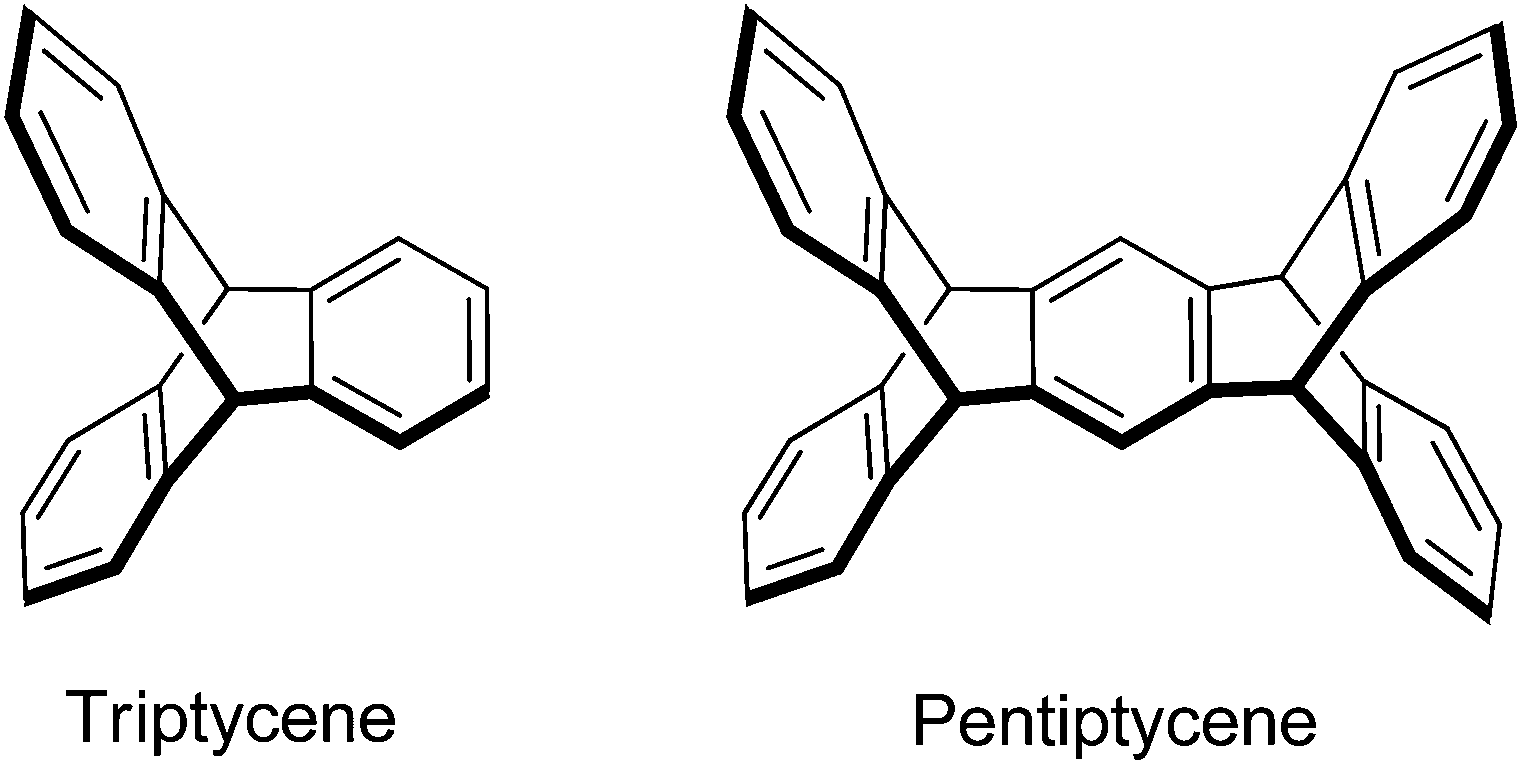 | ||
| Scheme 1 Structures of triptycene and pentiptycene. | ||
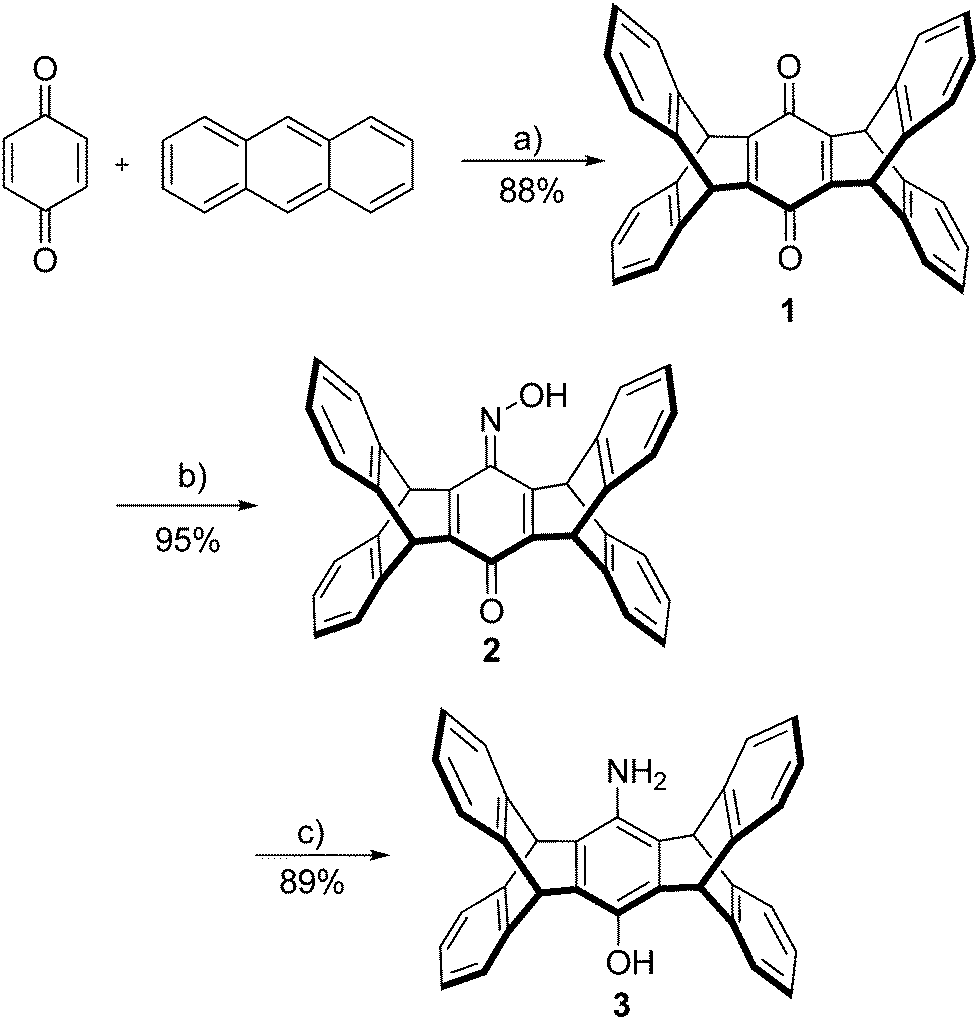 | ||
| Scheme 2 Three-step synthesis of pentiptycene aminophenol. Reagents and conditions: (a) p-chloranil, AcOH, reflux 16 h; (b) NH2OH·HCl, 4% aq. HCl, THF, reflux 3 d; (c) N2H4·H2O, Pd/C (5%), THF reflux 3 h. | ||
Triptycene phosphines have been used in organometallic chemistry,14–19 as well as the recently reported bistriptycene-based NHC ligands,20 but to the best of our knowledge, pentiptycene-based NHC ligands have not been reported. The use of pentiptycenes for NHC ligands offers interesting prospects, since the pentiptycene unit confers a bowl type shape to the respective NHC. In this contribution, we want to report on the synthesis of bispentiptycene-based NHC ligands and metal complexes with this new ligand as well as preliminary catalytic studies.
Results and discussion
Synthesis of pentiptycene NHC ligands
The reaction of two equivalents of anthracene with 1,4-benzoquinone in the presence of chloranil leads to pentiptycene quinone 1 according to a procedure from Chen et al. (Scheme 2).12In order to obtain a sufficient amount of pentiptycene for the synthesis of the NHC ligands, the scale of the reaction was considerably increased (almost 20-fold) with respect to the literature procedure. This scaling up turned out to be unproblematic and it is now possible to synthesize 75 g of pentiptycene quinone 1 in a single run, using cheap commercially available starting materials. Quinone 1 was converted into monooxime 2 employing a literature procedure. In contrast to literature reports claiming low stability of this compound,21 in our hands 2 turned out to be stable. Monooxime 2 was reduced to the amino-hydroxy-pentiptycene 3 using hydrazine hydrate and Pd/C. This procedure appears to be more convenient than the literature procedure using SnCl2/HCl, which in our hands provided impure materials, whose purification required significant effort. In contrast to literature reports,21 the pentiptycene aminophenol 3 displays reasonable solubility in several organic solvents and recording 1H and 13C NMR spectra poses no problem. In general, we notice that the pentiptycene derivatives display significantly better solubility than the related triptycene compounds also reported by us.20
Our attempts to obtain the respective diimines with glyoxal and aminophenol 3 appeared promising. However, an impure product was always obtained, which could neither be purified to a satisfactory level nor cleanly converted into the desired azolium salts. Based on the idea, that this might be due to the incomplete conversion of reactants and the formation of diimine isomers with extremely bulky pentiptycene units, we tried the analogous reactions of aminophenol 3 with acenaphthene-1,2-dione. The clean formation of such diimines with conventional anilines22 as well as their cyclization to azolium salts has been shown before.23,24 Furthermore, the acenaphthene backbone enforced Z-geometry of the diimines facilitates the formation of the respective azolium salts with sterically highly demanding N-aryl substituents,25 whose formation can be very difficult otherwise.26 The respective azolium salts were obtained in good yields as shown in Scheme 3.
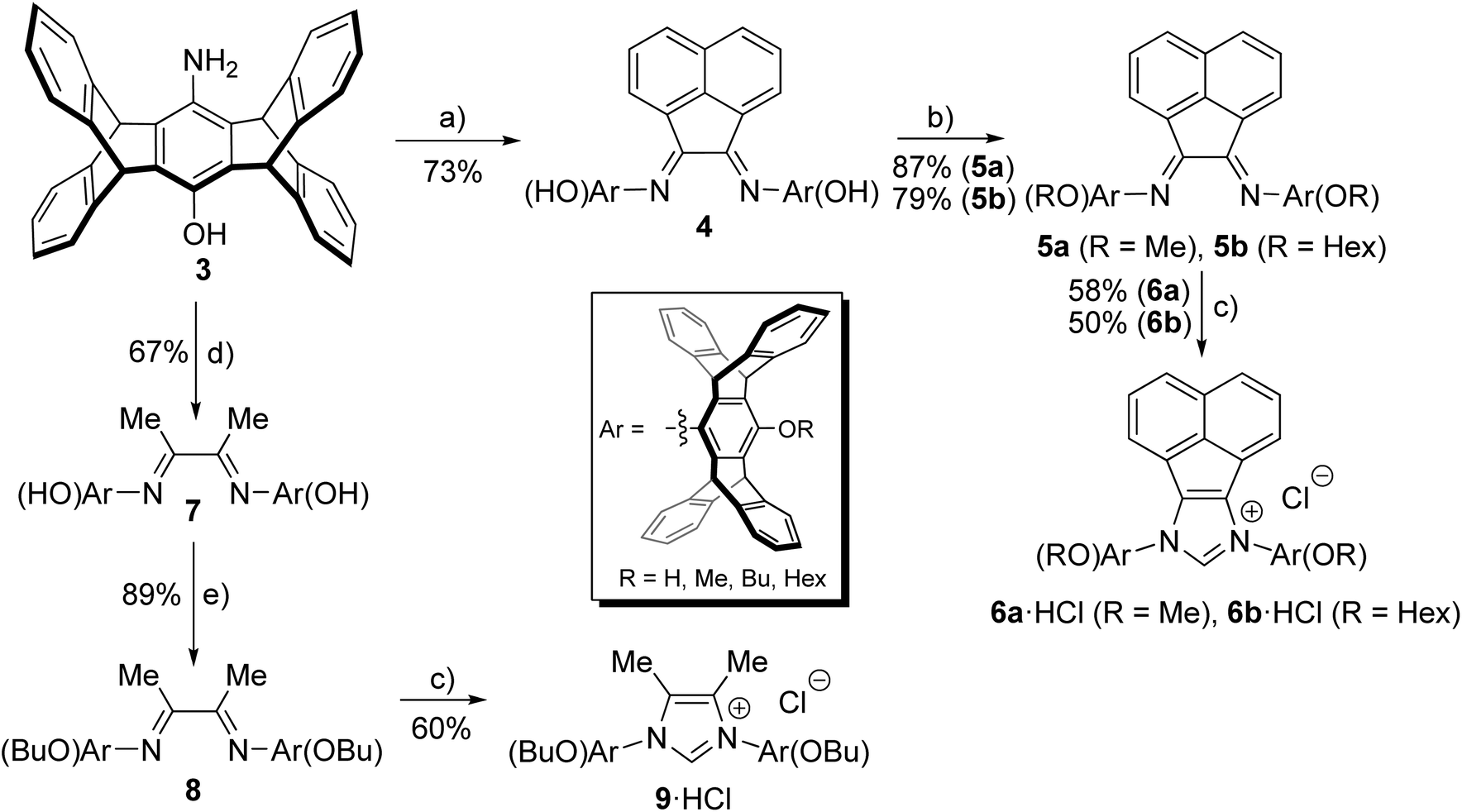 | ||
| Scheme 3 Synthesis of bispentiptycene-NHC·HCl ligands. Reagents and conditions: (a) acenaphthene-1,2-dione, MeCN, AcOH, 90 °C; (b) MeI, K2CO3, DMF, 50 °C or hexyliodide, K2CO3, DMF, 70 °C; (c) EtOCH2Cl, 80 °C; (d) 2,3-butanedione, iPrOH, HCOOH, 80 °C; (e) butyliodide, K2CO3, DMF, 70 °C. | ||
We next tested the reaction of aminophenol 3 with diacetyl according to standard procedures,27,28 which also leads to the respective diimine in good yields (Scheme 3). Following alkylation of the phenolic –OH group with MeI or BuI, the diimines were cyclized to the respective azolium salts 6a/6b·HCl or 9·HCl using ClCH2OEt.25,29 The use of this alkylating agent provides much better yields than with paraformaldehyde employed previously for the cyclization of related diimines.30
Synthesis of metal complexes
In order to evaluate the coordinating properties of the new NHC ligands, several new metal complexes were synthesized. The synthesis of metal complexes with the new carbenes 6a, 6b and 9 turned out to be unproblematic (Schemes 4 and 5). Several metal complexes with Cu, Ag and Au with coordination number 2 were prepared in good yields according to standard procedures for Cu,31 Ag32 and Au.33,34 The crystal structure of [(9)AgCl] indicated that due to the pronounced steric bulk of NHC 9 (see below), metal complexes with higher coordination numbers might be difficult to obtain. Despite testing several different approaches the respective Pd(II) complex with carbene 9 could not be synthesized. On the other hand, the synthesis of the four coordinate Ir complexes turned out to be straightforward, even though the higher coordination number of Ir in the expected complexes might have caused problems when binding to the very bulky NHC ligand. Two different established approaches (Schemes 4 and 5) were tried, both of them afforded the desired complexes. The transmetalation via in situ formed [(9)AgCl] complexes to give [(9)IrCl(cod)] requires harsh reaction conditions. The formation of [(6)IrCl(cod)] via the free carbene works under mild reaction conditions. Despite the very different reaction conditions employed, both methods produce the respective Ir complexes in good yields of 70% and 79%. Both complexes were converted into the respective [(NHC)IrCl(CO)2] complexes in excellent yields.35 In order to evaluate the donor properties of NHC ligands 6 and 9, the redox potentials of [(6a)IrCl(cod)] (E1/2 = 0.862 V) and [(9)IrCl(cod)] (E1/2 = 0.804 V) were determined and found to be comparable to those of the analogous complexes with SIMes ligands.27,36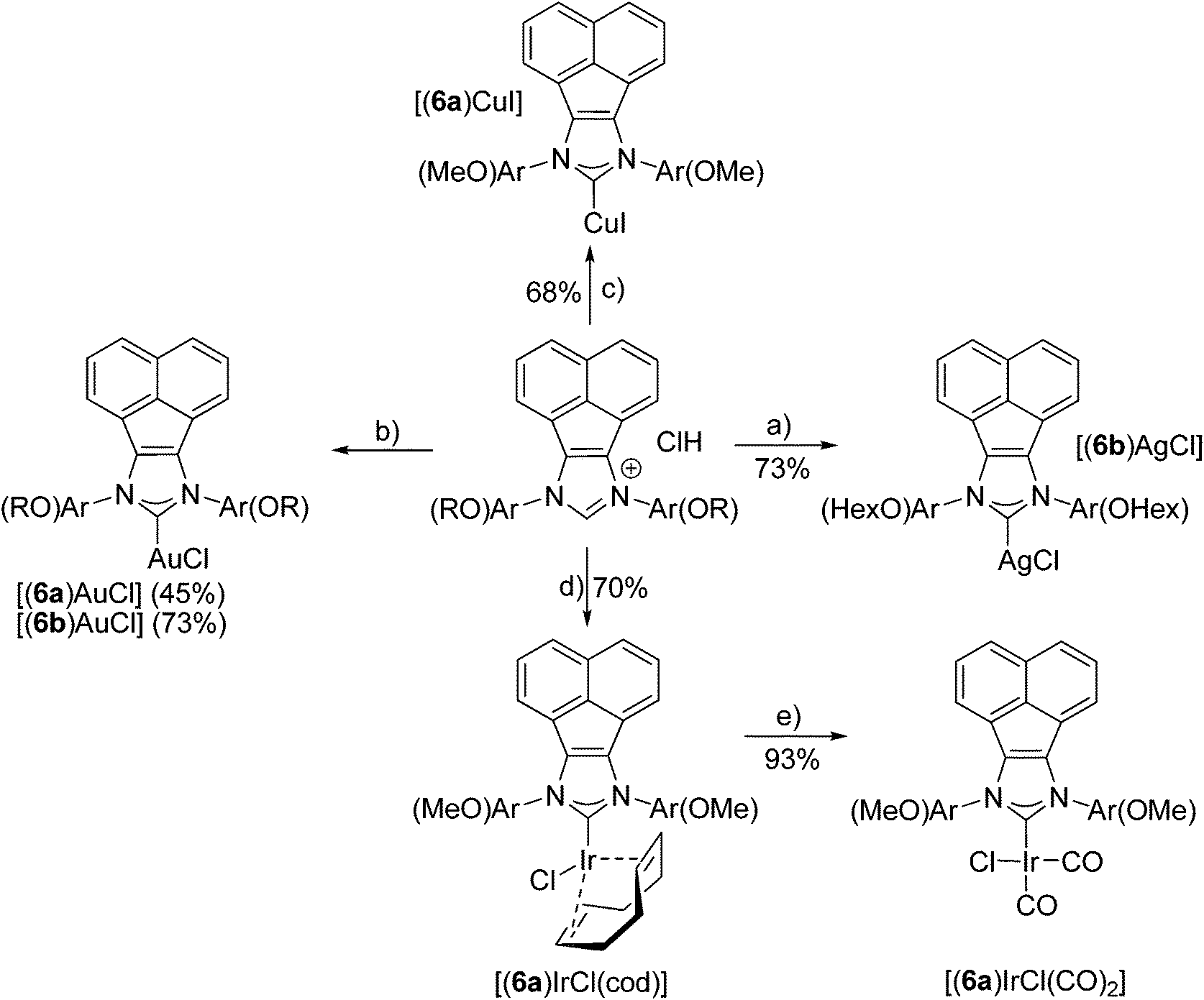 | ||
| Scheme 4 Synthesis of metal complexes with acenaphthene-based bispentiptycene NHC ligands. Reagents and conditions: (a) Ag2O, CH2Cl2, 40 °C; (b) [AuCl(Me2S)], K2CO3, acetone, 60 °C; (c) CuI, K2CO3, acetone, 60 °C; (d) [IrCl(cod)]2, sodium tert-pentoxide, THF, rt; (e) CO, CH2Cl2, rt. | ||
 | ||
| Scheme 5 Synthesis of metal complexes with diacetyl-derived bispentiptycene NHC ligands. Reagents and conditions: (a) Ag2O, CH2Cl2, 40 °C; (b) [IrCl(cod)]2, toluene, 100 °C; (c) CO, CH2Cl2, rt; (d) [AuCl(Me2S)], K2CO3, CH2Cl2, rt. | ||
Preliminary catalytic studies37 of the gold complexes [(6b)AuCl] and [(9)AuCl] show better catalytic activity in the cyclization of diethyl 2-allyl-2-(prop-2-ynyl)malonate38,39 (Table 1) than the closely related [(IMes)AuCl] (Table 1), furthermore the pentiptycene-based catalysts give a much higher selectivity for the formation of the five-membered ring than the IMes type catalysts. More detailed studies of the catalytic properties of such complexes are underway. This appears promising, since gold complexes with sterically highly demanding ligands have been shown to exhibit excellent catalytic activity.40,41
|
|
||
|---|---|---|
| Catalyst | Reaction time, h | Isolated yield (%) (I![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :II) :II) |
| [(6b)AuCl] | 16 | 91 (2.6:1) |
| [(9)AuCl] | 16 | 93 (2.4:1) |
| [(IMes)AuCl] | 18 | 35 (1:2.1) |
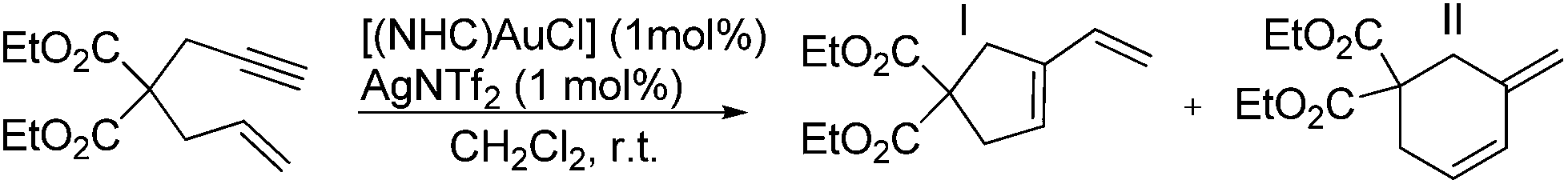
Crystal structure of [(9)AgCl]
Single crystals of [(9)AgCl] were obtained by slow evaporation of an isopropanol/CH2Cl2 solution. The NHC ligand has an approximately rectangular bowl type shape, which is defined by six phenyl rings of the two pentiptycene units and the two butoxy groups on the metal-facing side (Fig. 1 and 2). However, the angles of the planes of the six-membered N-aryl rings and of the five-membered heterocyclic ring are tilted by 17°. Since the two N-aryl units are tilted in opposite directions, the two pentiptycene units are inclined at an angle of ca. 35°. This leads to an interdigitized structure of the four phenyl rings forming the walls of the bowl. Consequently, two phenyl rings are close to the silver ion (shortest Ag–C distances ca. 350 pm), while the two closest carbon atoms of the other pair of phenyl rings are at an Ag–C distance of ca. 510 pm. Two pairs of Ag–C distances are sufficient for the description of the structure, since the silver atom and the carbene carbon of the NHC ligand both lie on a two-fold crystallographic axis. The bond distances and angles within the coordination sphere of silver are unspectacular, since the steric bulk of the NHC ligand cannot act on the two-coordinate metal center. The silver ion is deeply buried in the roughly hemispherical NHC ligand and even the chloro ligand42 hardly extends beyond the upper rim of the pentiptycene unit. The buried volume43 of the NHC ligand in complex [(9)AgCl] was calculated as Vbur = 48.8% (r = 350 pm, d = 200 pm), which is considerably larger than in the related [(iPr)AgCl] complexes (Vbur = 46.5%).44 The rigidity of the pentiptycene units in the crystal results in a porous solid structure with large empty voids into which the chloro ligands and the methyl groups of the heterocyclic unit are pointing. The formation of porous solids has been observed in other iptycene structures.45 The presence of solvent accessible pores also explains, why it is very difficult to remove solvent from such compounds – even after prolonged heating under vacuum, elemental analysis provides unsatisfactory values. | ||
| Fig. 1 X-ray crystal structure of [(9)AgCl] (space-filling plot). CCDC 1424889 contains the supplementary crystallographic data for this structure. | ||
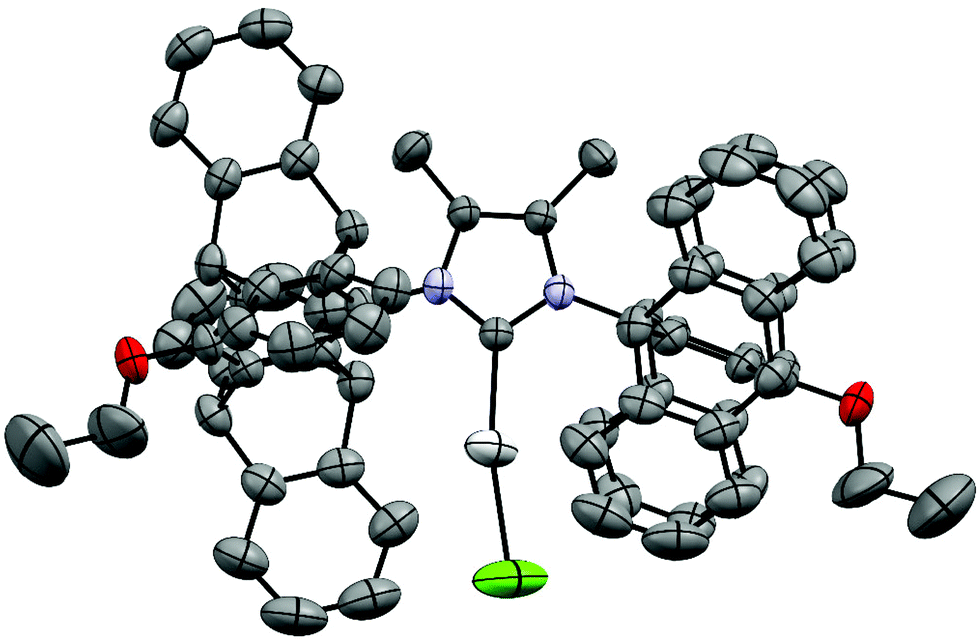 | ||
| Fig. 2 X-ray crystal structure49 of [(9)AgCl] (ORTEP-plot) (hydrogen atoms omitted, only α and β-carbon atoms of the disordered butyl chain are shown). Important bond lengths (pm): Ag–C(NHC) 209.0(5), Ag–Cl 234.8(3). | ||
Such bowl shaped NHC ligands are rare46,47 and hold potential for selective catalysis or the stabilization of otherwise unstable species.48
Summary and conclusions
Based on a large scale synthesis of 1-amino,4-hydroxy-pentiptycene this aniline was converted into various azolium salts, which are convenient precursors for the synthesis of N-heterocyclic carbenes. The steric shielding of the pentiptycene wings provides bowl-shaped NHC ligands, in which the metal center is buried in a pocket – as shown in the crystal structure of the respective (NHC)AgCl complex. Such ligands offer the possibility for the kinetic stabilization of unusual coordination geometries and could thus lead to highly active catalysts, which will be the topic of future studies. Preliminary catalytic tests employing (NHC)AuCl/AgNTf2 reveal good catalytic activities in the cyclization of diethyl 2-allyl-2-(prop-2-ynyl)malonate.Experimental
General experiments
All chemicals were purchased as reagent grade from commercial suppliers and used without further purification, unless otherwise noted. All reactions involving metal complexes were performed under an atmosphere of nitrogen (except gold and copper complexes). Tetrahydrofuran was dried over sodium, distilled under an argon atmosphere and stored over molecular sieves (4 Å). Dichloromethane and toluene were refluxed over calcium hydride, distilled under a nitrogen atmosphere and stored over molecular sieves (4 Å). 1H and 13C NMR spectra were recorded on a Bruker AC300 or a DRX500 spectrometer. The chemical shifts are given in parts per million (ppm) on the delta scale (δ) and are referenced to the residual peak of chloroform (1H NMR = 7.26, 13C NMR = 77.16 ppm), dimethyl sulfoxide (1H NMR = 2.50, 13C NMR = 39.52 ppm) or methylene chloride (1H NMR = 5.32, 13C NMR = 53.84 ppm). Abbreviations for NMR: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sep = septet, sex = sextet, m = multiplet, br = broad signal. Thin layer chromatography (TLC) was performed by using silica 60 F 254 (0.2 mm) on aluminum plates. Preparative chromatography was done on Merck silica 60 (0.063–0.2 mm). Cyclic voltammograms were recorded in dry CH2Cl2 under an argon atmosphere at ambient temperature. A three-electrode configuration was employed. The working electrode was a Pt disk (diameter 1 mm) sealed in soft glass with a Pt wire as a counter electrode. The pseudoreference electrode was an Ag wire. Potentials were calibrated internally against the formal potential of octamethylferrocene (E1/2 = −0.01 V (CH2Cl2)) and NBu4PF6 (0.1 mol L−1) was used as a supporting electrolyte. Compounds 1, 2 and 3 have been reported previously in the literature, however no 13C data were given.12,21![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 151.09 (CC), 143.83 (Ar–C), 125.61 (Ar–CH), 124.39 (Ar–CH), 47.56 (CHAr3).
O), 154.55, 148.50, 146.05, 145.14, 144.57, 144.43, 144.37, 144.25, 143.82, 125.56 (Ar–CH), 125.53 (Ar–CH), 125.28 (Ar–CH), 125.14 (Ar–CH), 124.32 (Ar–CH), 124.21 (Ar–CH), 124.01 (Ar–CH), 123.92 (Ar–CH), 52.99 (CHAr3), 49.16 (CHAr3), 47.36 (CHAr3), 47.13 (CHAr3).
N), 145.56, 145.45, 145.23, 144.85, 142.52, 141.33, 137.57, 132.58, 131.16, 130.60, 128.81, 128.48, 127.72, 125.38, 125.31, 124.75, 124.56, 124.44, 123.67, 123.44, 123.42, 49.96 (CHAr3), 47.88 (CHAr3). HRMS (EI): m/z calcd for C80H48N2O2: 1068.3710 [M]+; found: 1068.3723.
O), 151.09 (CC), 143.83 (Ar–C), 125.61 (Ar–CH), 124.39 (Ar–CH), 47.56 (CHAr3).
O), 154.55, 148.50, 146.05, 145.14, 144.57, 144.43, 144.37, 144.25, 143.82, 125.56 (Ar–CH), 125.53 (Ar–CH), 125.28 (Ar–CH), 125.14 (Ar–CH), 124.32 (Ar–CH), 124.21 (Ar–CH), 124.01 (Ar–CH), 123.92 (Ar–CH), 52.99 (CHAr3), 49.16 (CHAr3), 47.36 (CHAr3), 47.13 (CHAr3).
N), 145.56, 145.45, 145.23, 144.85, 142.52, 141.33, 137.57, 132.58, 131.16, 130.60, 128.81, 128.48, 127.72, 125.38, 125.31, 124.75, 124.56, 124.44, 123.67, 123.44, 123.42, 49.96 (CHAr3), 47.88 (CHAr3). HRMS (EI): m/z calcd for C80H48N2O2: 1068.3710 [M]+; found: 1068.3723.
5a: 1H NMR (500 MHz, CDCl3) δ 7.61 (d, J = 8.1 Hz, 2H), 7.51–7.47 (m, 4H), 7.46–7.42 (m, 4H), 7.38 (d, J = 7.4 Hz, 4H), 7.04–6.96 (m, 8H), 6.86–6.80 (m, 4H), 6.51–6.44 (m, 10H), 5.86 (s, 4H, CHAr3), 5.62 (s, 4H, CHAr3), 5.40 (d, J = 7.2 Hz, 2H), 4.11 (s, 6H, OMe). 13C NMR (126 MHz, CDCl3) δ 164.71 (CN), 147.60, 145.59, 145.39, 145.06, 143.82, 141.30, 139.81, 136.52, 132.70, 130.53, 128.81, 128.37, 127.69, 125.62, 125.44, 125.38, 124.81, 124.72, 124.45, 124.41, 123.56, 123.54, 123.33, 63.53 (OCH3), 49.96 (CHAr3), 48.55 (CHAr3). MS (ESI) calcd for C82H53N2O2 (M + H) 1097.4, found 1097.6.
5b: 1H NMR (500 MHz, CDCl3) δ 7.59 (d, J = 8.1 Hz, 2H), 7.52–7.47 (m, 4H), 7.44–7.40 (m, 4H), 7.37 (d, J = 7.2 Hz, 4H), 7.03–6.98 (m, 8H), 6.85–6.79 (m, 4H), 6.48–6.44 (m, 8H), 6.42 (t, J = 7.7 Hz, 2H), 5.83 (s, 4H, CHAr3), 5.62 (s, 4H, CHAr3), 5.33 (d, J = 7.2 Hz, 2H), 4.17 (t, J = 6.6 Hz, 4H, OHex), 2.17 (quint, J = 6.7 Hz, 4H, OHex), 1.84 (quint, J = 7.3 Hz, 4H, OHex), 1.66–1.52 (m, 8H, OHex), 1.09 (t, J = 7.1 Hz, 6H, OHex). 13C NMR (126 MHz, CDCl3) δ 164.85 (CN), 146.73, 145.56, 145.47, 145.43, 145.12, 141.26, 139.75, 136.63, 130.48, 128.74, 128.36, 127.57, 125.39, 125.34, 124.73, 124.68, 124.44, 123.60, 123.52, 123.24, 76.63 (OCH2), 49.95 (CHAr3), 48.76 (CHAr3), 32.09 (CH2), 30.83 (CH2), 26.41 (CH2), 22.98 (CH2), 14.37 (CH3). HRMS (EI): m/z calcd for C86H59N2O2: 1151.4571 [M − C6H13]+; found: 1151.4561.
N), 145.66, 145.36, 145.17, 142.14, 136.06, 131.94, 130.47, 125.41, 125.39, 123.90, 123.79, 123.68, 50.00 (CHAr3), 47.85 (CHAr3), 17.74 (CH3). HRMS (EI): m/z calcd for C72H48N2O2: 972.3710 [M]+; found: 972.3720.
1H NMR (300 MHz, CDCl3) δ 7.48–7.38 (m, 12H, HAr), 7.36 (d, J = 7.2 Hz, 4H, HAr), 7.08–6.90 (m, 16H, HAr), 5.81 (s, 4H, CHAr3), 5.34 (s, 4H, CHAr3), 4.05 (t, J = 6.7 Hz, 4H, OCH2), 2.19 (s, 6H, Me), 2.10 (dt, J = 14.5, 6.8 Hz, 4H, OCH2CH2CH2CH3), 1.82 (sep, J = 7.3 Hz, 4H, OCH2CH2CH2CH3), 1.22 (t, J = 7.3 Hz, 6H, OCH2CH2CH2CH3). 13C NMR (75 MHz, CDCl3) δ 171.96 (CN), 146.54, 145.87, 145.45, 145.29, 137.92, 135.99, 131.83, 125.43, 125.33, 123.85, 123.59, 76.31 (OCH2), 50.00 (CHAr3), 48.69 (CHAr3), 32.90 (CH2), 19.98 (CH2), 17.84 (CH3), 14.39 (CH3, hexyl). HRMS (EI): m/z calcd for C80H64N2O2: 1084.4962 [M]+; found: 1084.4965.
:1, v/v to elute pure product) provides the azolium salt 6a·HCl (363 mg, 58% yield) as a pale yellow powder. The azolium salt 6b·HCl (272 mg, 50% yield) was obtained as a pale yellow powder from diimine 5b (530 mg, 0.428 mmol) and chloromethyl ethyl ether (2.5 mL, 26.9 mmol stored overnight over K2CO3 under nitrogen) using a similar procedure. After stirring the mixture for 20 h at 80 °C, diethyl ether was added and the obtained solution was sonicated for a few minutes. The obtained precipitate was collected by filtration and washed with diethyl ether affording the pure product.
6a·HCl: 1H NMR (500 MHz, CDCl3) δ 8.11 (d, J = 8.2 Hz, 2H, acenaph.), 8.07 (s, 1H, NCHN), 7.49–7.42 (m, 12H, HAr), 7.32 (t, J = 7.3 Hz, 2H, acenaph.), 7.26 (d, J = 7.1 Hz, 4H, HAr), 7.05–6.96 (m, 8H, HAr), 6.87–6.80 (m, 8H, HAr), 6.73 (d, J = 7.0 Hz, 2H, acenaph.), 6.34 (s, 4H, CHAr3), 5.95 (s, 4H, CHAr3), 4.18 (s, 6H, OMe). 13C NMR (126 MHz, CDCl3) δ 152.70 (NCHN), 144.64, 144.32, 144.16, 143.96, 142.04, 139.81, 139.22, 135.57, 131.66, 131.33, 130.19, 127.96, 126.14, 126.04, 125.75, 125.58, 124.53, 124.16, 123.79, 122.23, 120.80, 63.23 (OCH3), 49.34 (CHAr3), 48.41 (CHAr3). MS (ESI) calcd for C83H53N2O2 (M − Cl)+ 1109.4, found 1109.8.
6b·HCl: 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 8.0 Hz, 2H), 8.08 (s, 1H, NCHN), 7.48–7.40 (m, 12H), 7.31 (t, J = 7.4 Hz, 2H), 7.27–7.20 (br, 4H), 7.02 (t, J = 7.4 Hz, 4H), 6.99 (t, J = 7.4 Hz, 4H), 6.88–6.80 (m, 8H), 6.71 (d, J = 6.9 Hz, 2H), 6.24 (s, 4H, CHAr3), 5.92 (s, 4H, CHAr3), 4.23 (t, J = 6.7 Hz, 4H, OHex), 2.19 (quint, J = 6.8 Hz, 4H, OHex), 1.81 (quint, J = 7.4 Hz, 4H, OHex), 1.65–1.51 (m, 8H, OHex), 1.07 (t, J = 7.1 Hz, 6H, OHex). 13C NMR (126 MHz, CDCl3) δ 152.17 (NCHN), 144.66, 144.34, 144.13, 143.92, 141.77, 139.74, 139.24, 135.65, 131.62, 131.45, 130.19, 127.97, 126.11, 126.02, 126.00, 125.74, 125.57, 125.49, 124.46, 124.15, 123.79, 122.12, 120.48, 76.65 (OCH2), 49.44 (CHAr3), 48.62 (CHAr3), 31.98 (CH2), 30.81 (CH2), 26.23 (CH2), 22.93 (CH2), 14.32 (CH3). HRMS (EI): m/z calcd for C87H60N2O2: 1164.4649 [M − Cl − C6H13]+; found: 1164.4620.
[(6b)AgCl]: 1H NMR (500 MHz, CDCl3) δ 7.84 (d, J = 8.2 Hz, 2H), 7.47 (d, J = 7.4 Hz, 4H), 7.43 (d, J = 7.2 Hz, 4H), 7.33 (d, J = 7.3 Hz, 4H), 7.10 (t, J = 7.6 Hz, 2H), 6.99 (q, J = 6.1 Hz, 8H), 6.93 (t, J = 7.4 Hz, 4H), 6.79–6.72 (m, 8H), 6.29 (d, J = 7.0 Hz, 2H), 5.91 (s, 4H, CHAr3), 5.41 (s, 4H, CHAr3), 4.27 (t, J = 6.7 Hz, 4H, OHex), 2.21 (quint, J = 6.8 Hz, 4H, OHex), 1.85 (quint, J = 7.4 Hz, 4H, OHex), 1.69–1.54 (m, 8H, OHex), 1.10 (t, J = 7.1 Hz, 6H, OHex). 13C NMR (126 MHz, CDCl3) δ 151.09, 145.40, 144.49, 144.24, 143.67, 141.33, 139.76, 139.71, 137.90, 130.68, 129.70, 128.81, 127.61, 126.47, 126.16, 125.68, 125.05, 124.79, 124.34, 124.24, 124.13, 123.94, 123.77, 123.69, 76.62 (OCH2), 50.59 (CHAr3), 48.74 (CHAr3), 32.07 (CH2), 30.90 (CH2), 26.28 (CH2), 23.01 (CH2), 14.38 (CH3). HRMS (EI): m/z calcd for C87H60N2O2: 1164.4649 [M − AgCl − C6H12]+; found: 1164.4630.
[(9)AgCl]: 1H NMR (500 MHz, CDCl3) δ 7.47 (d, J = 6.7 Hz, 4H, HAr), 7.41 (d, J = 7.0 Hz, 4H, HAr), 7.37 (d, J = 7.1 Hz, 4H, HAr), 7.32 (d, J = 6.6 Hz, 4H, HAr), 7.07–6.97 (m, 12H, HAr), 6.94 (t, J = 7.3 Hz, 4H, HAr), 5.85 (s, 4H, CHAr3), 5.19 (s, 4H, HAr), 4.20 (t, J = 6.7 Hz, 4H, OCH2), 2.18–2.12 (m, 4H, OCH2CH2CH2CH3), 1.88 (s, 6H, Me), 1.87–1.80 (m, 4H, OCH2CH2CH2CH3), 1.24 (t, J = 7.4 Hz, 6H, OCH2CH2CH2CH3). 13C NMR (126 MHz, CDCl3) δ 145.66, 144.74, 144.22, 143.59, 141.53, 137.46, 128.23, 128.18, 126.31, 126.19, 125.89, 125.41, 124.77, 124.34, 124.26, 124.18, 123.44, 76.35 (OCH2), 50.59 (CHAr3), 48.65 (CHAr3), 32.97 (CH2), 19.86 (CH2), 14.38 (CH3), 10.20 (CH3, butyl). HRMS (EI): m/z calcd for C81H64N2O2: 1096.4962 [M − AgCl]+; found: 1096.4950.
[(6a)AuCl]: 1H NMR (500 MHz, CD2Cl2) δ 7.84 (d, J = 8.1 Hz, 2H, acenapht.), 7.52–7.47 (m, 12H, HAr), 7.06–6.95 (m, 14H, HAr), 6.76 (dd, J = 7.3, 1.4 Hz, 4H, HAr), 6.73 (td, J = 7.2, 1.1 Hz, 4H, HAr), 6.08 (d, J = 6.9 Hz, 2H, acenapht.), 5.98 (s, 4H, CHAr3), 5.47 (s, 4H, CHAr3), 4.21 (s, 6H, OMe). 13C NMR (126 MHz, CD2Cl2) δ 180.39 (Ccarbene), 145.81, 145.02, 144.75, 144.29, 141.96, 139.08, 138.42, 130.76, 130.15, 129.45, 127.96, 126.43, 126.40, 126.20, 125.60, 125.48, 125.07, 124.59, 124.32, 123.78, 63.79 (OCH2), 51.08 (CHAr3), 48.89 (CHAr3). MS (ESI) calcd for C83H52AuClNaN2O2 (M + Na+) 1363.3, found 1363.5; calcd for C83H52AuClKN2O2 (M + K+) 1379.3, found 1379.5.
[(6b)AuCl]: 1H NMR (500 MHz, CDCl3) δ 7.80 (d, J = 8.1 Hz, 2H), 7.46 (d, J = 7.3 Hz, 8H), 7.42 (d, J = 7.1 Hz, 4H), 7.05–6.91 (m, 14H), 6.73–6.67 (m, 8H), 6.11 (d, J = 7.0 Hz, 2H), 5.90 (s, 4H, CHAr3), 5.44 (s, 4H, CHAr3), 4.27 (t, J = 6.7 Hz, 4H, OHex), 2.21 (quint, J = 6.8 Hz, 4H, OHex), 1.85 (quint, J = 7.4 Hz, 4H, OHex), 1.69–1.50 (m, 8H, OHex), 1.10 (t, J = 7.1 Hz, 6H, OHex). 13C NMR (126 MHz, CDCl3) δ 180.53 (Ccarbene), 151.03, 145.50, 144.51, 144.22, 143.68, 141.37, 138.47, 137.84, 130.20, 129.54, 128.79, 127.58, 126.11, 126.01, 125.58, 125.08, 125.04, 124.45, 124.13, 123.89, 123.57, 76.61 (OCH2), 50.71 (CHAr3), 48.75 (CHAr3), 32.07 (CH2), 30.89 (CH2), 26.28 (CH2), 23.01 (CH2), 14.38 (CH3). HRMS (EI): m/z calcd for C87H60N2O2: 1164.4649 [M − AuCl − C6H12]+; found: 1164.4640.
:1, v/v) (60 mg, 51% yield). 1H NMR (500 MHz, CDCl3) δ 7.50–7.45 (m, 8H), 7.41 (d, J = 6.7 Hz, 4H), 7.30 (d, J = 6.6 Hz, 4H), 7.07–6.93 (m, 16H), 5.85 (s, 4H, CHAr3), 5.23 (s, 4H, CHAr3), 4.20 (t, J = 6.7 Hz, 4H, OCH2), 2.19–2.12 (m, 4H, OCH2CH2CH2CH3), 1.85 (sext, J = 7.4 Hz, 4H, OCH2CH2CH2CH3), 1.77 (s, 6H, Me), 1.25 (t, J = 7.4 Hz, 6H, OCH2CH2CH2CH3). 13C NMR (126 MHz, CDCl3) δ 173.17 (Ccarbene), 150.85, 145.78, 144.78, 144.18, 143.60, 141.54, 137.38, 127.59, 126.05, 125.99, 125.84, 125.39, 124.99, 124.28, 124.23, 123.96, 123.37, 76.35 (OCH2), 50.65 (CHAr3), 48.67 (CHAr3), 32.98 (CH2), 19.87 (CH2), 14.38 (CH3), 10.20 (CH3, butyl). MS (ESI) calcd for C81H64N2O2AuClNa (M + Na+) 1351.4, found 1351.6.
:1, v/v) to afford the desired product as an orange microcrystalline powder (150 mg, 70% yield). 1H NMR (500 MHz, CDCl3) δ 8.37 (br, 2H, HAr), 7.60 (br, 2H, HAr), 7.50–7.45 (m, 4H, HAr), 7.39–7.34 (m, 6H, HAr), 7.14–7.00 (m, 8H, HAr), 6.77 (t, J = 6.8 Hz, 4H, HAr), 6.61 (br, 2H, HAr), 6.38 (dd, J = 8.2, 7.1 Hz, 2H, acenapht.), 6.31 (br, 6H), 6.15 (br, 2H, HAr), 5.93 (s, 4H, CHAr3), 5.37 (br, 2H, HAr), 4.63 (d, J = 7.0 Hz, 2H, acenapht.), 4.33–4.27 (m, 2H, cod), 4.21 (s, 6H, OMe), 3.32–3.27 (m, 2H, cod), 1.16–1.07 (m, 4H, cod), 1.06–0.96 (m, 2H, cod), 0.92–0.84 (m, 2H, cod). 13C NMR (126 MHz, CDCl3) δ 188.09 (Ccarbene), 151.06, 146.71–143.08 (br multiplet), 140.27, 128.80, 128.73, 128.61, 126.86, 125.85 (br), 125.24 (br), 124.78 (br), 123.80, 123.43 (br), 121.01, 86.12 (Hcod), 63.40 (OCH3), 52.73 (Hcod), 50.72 (br, CHAr3), 48.59 (br, CHAr3), 32.93 (Hcod), 28.31 (Hcod). HRMS (EI): m/z calcd for C83H53N2O2: 1109.4102 [M + H − IrCl(cod)]+; found: 1109.4103.
[(6a)IrCl(CO)2]: 1H NMR (500 MHz, CDCl3) δ 7.79 (d, J = 7.0 Hz, 4H, HAr), 7.65 (d, J = 8.2 Hz, 2H, acenapht.), 7.44 (d, J = 7.0 Hz, 8H, HAr), 7.01 (td, J = 7.3, 1.4 Hz, 4H, HAr), 6.97 (td, J = 7.4, 1.3 Hz, 4H, HAr), 6.90 (td, J = 7.1, 1.7 Hz, 4H, HAr), 6.76 (dd, J = 8.2, 7.0 Hz, 2H, acenapht.), 6.60–6.53 (m, 8H, HAr), 5.94 (s, 4H, CHAr3), 5.72 (s, 4H, CHAr3), 5.52 (d, J = 7.0 Hz, 2H, acenapht.), 4.19 (s, 6H, Me). 13C NMR (126 MHz, CDCl3) δ 184.63 (Ccarbene), 179.87 (CO), 167.79 (CO), 151.28, 145.99, 144.81, 144.26, 144.21, 141.95, 140.01, 137.65, 129.92, 129.26, 128.13, 127.47, 126.72, 126.58, 125.58, 125.28, 125.19, 125.03, 124.17, 123.75, 123.67, 123.33, 123.11, 63.41 (OCH3), 50.82 (CHAr3), 48.52 (CHAr3). Recording of mass spectra with complex [(6a)Ir(CO)2Cl] was unsuccessful, due to instability of this complex.
[(9)IrCl(CO)2]: 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 7.0 Hz, 4H, HAr), 7.43 (dd, J = 6.8, 1.6 Hz, 4H, HAr), 7.39 (dd, J = 6.8, 1.4 Hz, 4H, HAr), 7.33 (dd, J = 6.8, 1.7 Hz, 4H, HAr), 7.02–6.93 (m, 16H, HAr), 5.84 (s, 4H, CHAr3), 5.52 (s, 4H, CHAr3), 4.16 (t, J = 6.6 Hz, 4H, OCH2), 2.16–2.09 (m, 4H, OCH2CH2CH2CH3), 1.83 (sex, J = 7.4 Hz, 4H, OCH2CH2CH2CH3), 1.54 (s, 6H, Me), 1.22 (t, J = 7.3 Hz, 6H, OCH2CH2CH2CH3). 13C NMR (126 MHz, CDCl3) δ 179.67 (Ccarbene), 178.22 (CO), 167.89 (CO), 150.34, 146.58, 145.35, 144.26, 144.15, 141.90, 137.20, 128.95, 126.82, 126.22, 125.53, 125.51, 125.23, 125.21, 124.06, 123.57, 123.33, 76.27 (OCH2), 50.73 (CHAr3), 48.66 (CHAr3), 32.89 (CH2), 19.94 (CH2), 14.37 (CH3), 10.32 (CH3, butyl). MS (ESI) calcd for C83H64ClIrNaN2O4 (M + Na) 1403.4, found 1403.1.
General procedure for cyclization of diethyl 2-allyl-2-(prop-2-ynyl)malonate
The respective (NHC)AuCl complex (0.01 equiv.) and AgNTf2 (0.01 equiv.) were placed into a flame dried 10 mL Schlenk tube under a nitrogen atmosphere and CH2Cl2 (3 mL) was added. The mixture was stirred for 15 min at r.t. in the absence of light. Next, diethyl 2-allyl-2-(prop-2-ynyl)malonate (200 mg, 1 equiv.) was added and the mixture was stirred at r.t. in the absence of light for the indicated time. Finally, the solvent was removed on a rotary evaporator and the residue was purified by column chromatography on silica (pentane/diethyl ether, 10:1, v/v). The fraction containing the mixture of five- and six-membered products was collected and analyzed by NMR.
Acknowledgements
This work was supported by the DFG via grant Pl 178/13-2. We wish to thank Yuki Kanai and Andreas Hegelein for the synthesis of pentiptycene.References
- C. F. Chen and Y. X. Ma, Iptycenes Chemistry: From Synthesis to Applications, Springer Verlag, Berlin, 2012 Search PubMed.
- T. M. Swager, Acc. Chem. Res., 2008, 41, 1181–1190 CrossRef CAS PubMed.
- X. Jiang, B. Rodríguez-Molina, N. Nazarian and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2014, 136, 8871–8874 CrossRef CAS PubMed.
- E. R. Kay, D. A. Leigh and F. Zerbetto, Angew. Chem., Int. Ed., 2007, 46, 72–191 CrossRef CAS PubMed.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC.
- J. S. Yang and J. L. Yan, Chem. Commun., 2008, 1501–1512 RSC.
- Y. Jiang and C.-F. Chen, Eur. J. Org. Chem., 2011, 6377–6403 CrossRef CAS.
- P. D. Bartlett, M. J. Ryan and S. G. Cohen, J. Am. Chem. Soc., 1942, 64, 2649–2653 CrossRef CAS.
- R. V. Skvarche and V. K. Shalaev, Dokl. Akad. Nauk SSSR, 1974, 216, 110–112 Search PubMed.
- H. Hart, S. Shamouilian and Y. Takehira, J. Org. Chem., 1981, 46, 4427–4432 CrossRef CAS.
- J.-S. Yang and T. M. Swager, J. Am. Chem. Soc., 1998, 120, 11864–11873 CrossRef CAS.
- X.-Z. Zhu and C.-F. Chen, J. Org. Chem., 2005, 70, 917–924 CrossRef CAS PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. Cesar, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- C. Azerraf and D. Gelman, Organometallics, 2009, 28, 6578–6584 CrossRef CAS.
- O. Grossman, C. Azerraf and D. Gelman, Organometallics, 2005, 25, 375–381 CrossRef.
- C. Azerraf and D. Gelman, Chem. – Eur. J., 2008, 14, 10364–10368 CrossRef CAS PubMed.
- C. Azerraf, O. Grossman and D. Gelman, J. Organomet. Chem., 2007, 692, 761–767 CrossRef CAS.
- L. Bini, C. Müller, J. Wilting, L. von Chrzanowski, A. L. Spek and D. Vogt, J. Am. Chem. Soc., 2007, 129, 12622–12623 CrossRef CAS PubMed.
- S. Gonell, M. Poyatos and E. Peris, Angew. Chem., Int. Ed., 2013, 52, 7009–7013 CrossRef CAS PubMed.
- R. Savka, M. Bergmann, Y. Kanai, S. Foro and H. Plenio, Chem. – Eur. J., 2016, 22 DOI:10.1002/chem.201601474.
- J.-S. Yang and C.-W. Ko, J. Org. Chem., 2006, 71, 844–847 CrossRef CAS PubMed.
- C. S. Popeney, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432–2452 CrossRef CAS.
- S. Dastgir, K. S. Coleman, A. R. Cowley and M. L. H. Green, Dalton Trans., 2009, 7203–7214 RSC.
- K. V. Vasudevan, R. R. Butorac, C. D. Abernethy and A. H. Cowley, Dalton Trans., 2010, 39, 7401–7408 RSC.
- R. Savka and H. Plenio, Eur. J. Inorg. Chem., 2014, 6246–6253 CrossRef CAS.
- M. Tsimerman, D. Mallik, T. Matsuo, T. Otani, K. Tamao and M. G. Organ, Chem. Commun., 2012, 48, 10352–10354 RSC.
- S. Leuthäußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- H. Türkmen and B. Cetinkaya, J. Organomet. Chem., 2006, 691, 3749–3759 CrossRef.
- T. Tu, Z. Sun, W. Fang, M. Xu and Y. Zhou, Org. Lett., 2012, 14, 4250–4253 CrossRef CAS PubMed.
- B. R. Van Ausdall, J. L. Glass, K. M. Wiggins, A. M. Aarif and J. Louie, J. Org. Chem., 2009, 74, 7935–7942 CrossRef CAS PubMed.
- J. D. Egbert, C. S. J. Cazin and S. P. Nolan, Catal. Sci. Technol., 2013, 3, 912–926 Search PubMed.
- J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS PubMed.
- R. Visbal, A. Laguna and M. C. Gimeno, Chem. Commun., 2013, 49, 5642–5644 RSC.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
- R. Savka and H. Plenio, Dalton Trans., 2015, 44, 891–893 RSC.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef CAS.
- D. Pflasterer and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 1331–1367 RSC.
- M. Raducan, C. Rodriguez-Escrich, X. C. Cambeiro, E. C. Escudero-Adan, M. A. Pericas and A. M. Echavarren, Chem. Commun., 2011, 47, 4893–4895 RSC.
- K. Fourmy, S. Mallet-Ladeira, O. Dechy-Cabaret and M. Gouygou, Organometallics, 2013, 32, 1571–1574 CrossRef CAS.
- R. E. Ebule, D. Malhotra, G. B. Hammond and B. Xu, Adv. Synth. Catal., 2016, 358, 1478–1481 CrossRef CAS.
- M. C. Blanco Jaimes, C. R. Bohling, J. M. Serrano-Becerra and A. S. Hashmi, Angew. Chem., Int. Ed., 2013, 52, 7963–7966 CrossRef CAS PubMed.
- The chloro ligand displays a pronounced anisotropy of the displacement parameter along the axis defined by the two long Ag–C distances. It was therefore decided to refine the two chloro ligands with s.o.f = 0.5 in two separate positions, which are 105 pm apart.
- A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 1759–1766 CrossRef CAS.
- T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- O. Winkelmann, C. Näther and U. Lüning, Eur. J. Org. Chem., 2007, 981–987 CrossRef CAS.
- M. Guitet, P. Zhang, F. Marcelo, C. Tugny, J. Jiménez-Barbero, O. Buriez, C. Amatore, V. Mouriès-Mansuy, J.-P. Goddard, L. Fensterbank, Y. Zhang, S. Roland, M. Ménand and M. Sollogoub, Angew. Chem., Int. Ed., 2013, 52, 7213–7218 CrossRef CAS PubMed.
- M. W. Hussong, F. Rominger, P. Krämer and B. F. Straub, Angew. Chem., Int. Ed., 2014, 53, 9372–9375 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1424889. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01724j |
| This journal is © The Royal Society of Chemistry 2016 |