
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSmall-molecule activation at Au(III): metallacycle construction from ethylene, water, and acetonitrile†
Marte Sofie Martinsen
Holmsen
a,
Ainara
Nova
b,
David
Balcells
b,
Eirin
Langseth‡
a,
Sigurd
Øien-Ødegaard
a,
Eline Aasen
Tråseth
a,
Richard H.
Heyn
c and
Mats
Tilset
*ab
aDepartment of Chemistry, University of Oslo, P.O. Box 1033 Blindern, N-0315 Oslo, Norway. E-mail: mats.tilset@kjemi.uio.no
bCentre for Theoretical and Computational Chemistry (CTCC), Department of Chemistry, University of Oslo, P.O. Box 1033 Blindern, N-0315 Oslo, Norway
cSINTEF Materials and Chemistry, P.O. Box 124 Blindern, N-0314 Oslo, Norway
First published on 10th June 2016
Abstract
Incorporation of the simple, readily available, building blocks ethylene, water and acetonitrile into Au(tpy)(OCOCF3)2 (tpy = 2-(p-tolyl)pyridine) in a one-step reaction leads to high yields of a new 6-membered ring gold(III) metallacycle complex. The metallacycle has been characterized spectroscopically and crystallographically, and the mechanism of its formation has been investigated with the aid of DFT calculations.
Introduction
The functionalization of alkenes has great practical and economic value in catalysis, and as the simplest of such building blocks, ethylene is of particular interest.1 Gold is known for its ability to π-coordinate and thereby activate alkenes and alkynes towards nucleophilic attack, which is considered to be a key step in gold catalysis.2–8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) However, there are only scarce reports of Au(III)-mediated functionalization of the simplest olefin, ethylene. One important example was provided by Atwood and co-workers (Scheme 1, top),9 who demonstrated that ethylene and propylene could be functionalized stoichiometrically at the Au(III) complex Au(bipy)Cl2+ (bipy = 2,2′-bipyridine) in water to furnish Au(III) hydroxyalkyl products that were observed in solution by 1H NMR spectroscopy but not isolated. Reactions of ethylene and propylene with HAuCl3 and AuCl3(tppts) (tppts = 3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt) led to Au(III) hydroxyalkyl species, which upon heating underwent gold reduction to the metal with concomitant formation of organic oxygenated products (alcohols, aldehyde or ketone).9 More recently, Bochmann and co-workers (Scheme 1, bottom)10 showed that ethylene slowly undergoes a formal insertion into the Au–OAcF bond trans to the pyridine-N in a diarylpyridine CNC pincer complex to yield an Au(III) acetoxyalkyl complex (OAcF = OCOCF3).
However, there are only scarce reports of Au(III)-mediated functionalization of the simplest olefin, ethylene. One important example was provided by Atwood and co-workers (Scheme 1, top),9 who demonstrated that ethylene and propylene could be functionalized stoichiometrically at the Au(III) complex Au(bipy)Cl2+ (bipy = 2,2′-bipyridine) in water to furnish Au(III) hydroxyalkyl products that were observed in solution by 1H NMR spectroscopy but not isolated. Reactions of ethylene and propylene with HAuCl3 and AuCl3(tppts) (tppts = 3,3′,3′′-phosphanetriyltris(benzenesulfonic acid) trisodium salt) led to Au(III) hydroxyalkyl species, which upon heating underwent gold reduction to the metal with concomitant formation of organic oxygenated products (alcohols, aldehyde or ketone).9 More recently, Bochmann and co-workers (Scheme 1, bottom)10 showed that ethylene slowly undergoes a formal insertion into the Au–OAcF bond trans to the pyridine-N in a diarylpyridine CNC pincer complex to yield an Au(III) acetoxyalkyl complex (OAcF = OCOCF3).
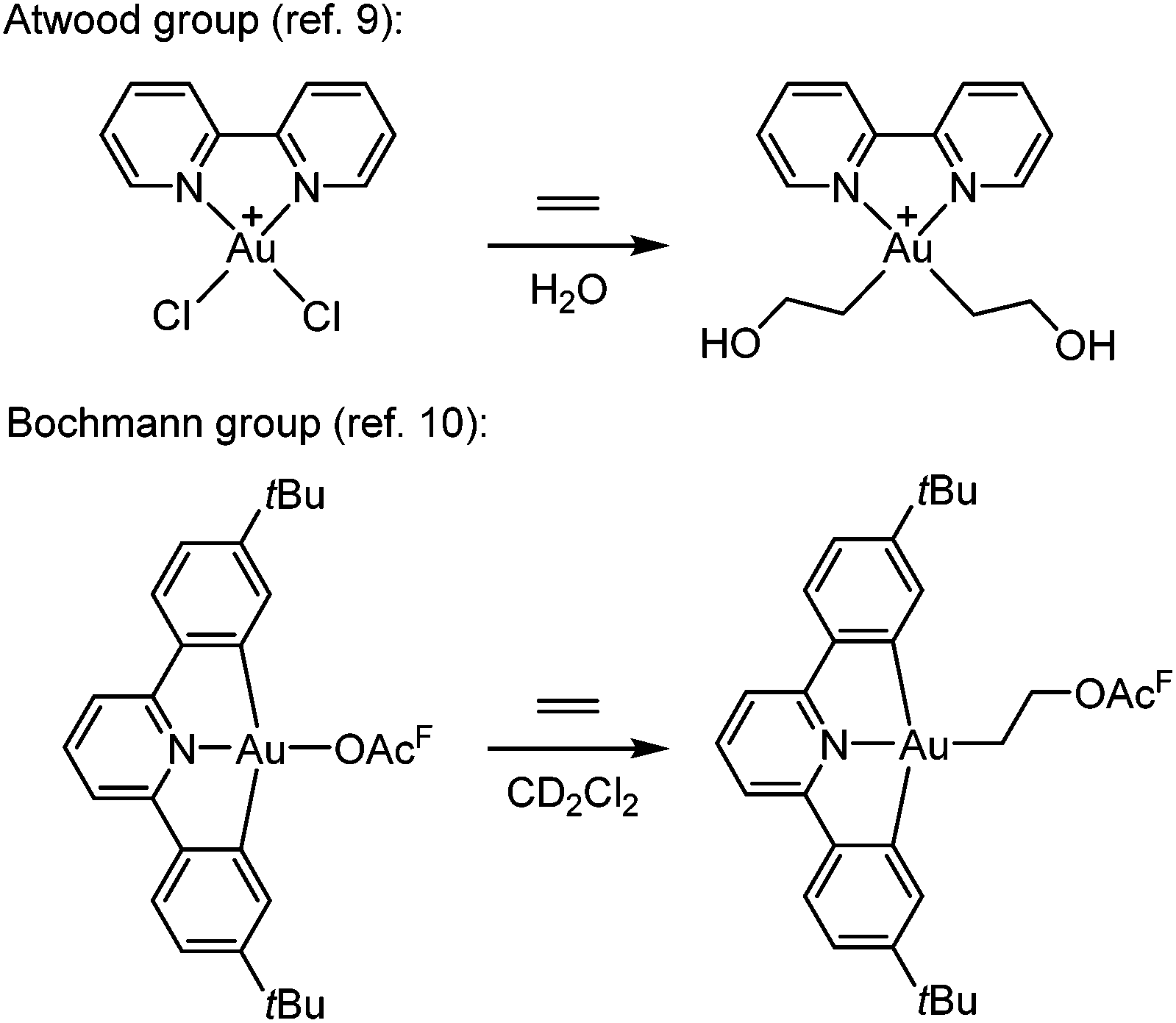 | ||
| Scheme 1 Reactions of ethylene at Au(III) complexes (OAcF = OCOCF3). | ||
Our group recently reported a combined experimental and computational study of the formal insertion of ethylene into an Au–O bond of Au(tpy)(OAcF)2 (1; tpy = 2-(p-tolyl)pyridine; Scheme 2, top).11 Selective insertion into the Au–O bond trans to N of the chelating tpy ligand furnished Au(tpy)(CH2CH2OAcF)(OAcF) (2). Although the trans to C coordination site is kinetically more accessible, the trans to N insertion product is thermodynamically favoured and is formed by nucleophilic attack by free −OAcF at the coordinated ethylene. This discovery led to further investigations, and herein we report that the coordinated and inserted ethylene molecule can undergo further functionalization due to the availability of the trans to C coordination site at Au.
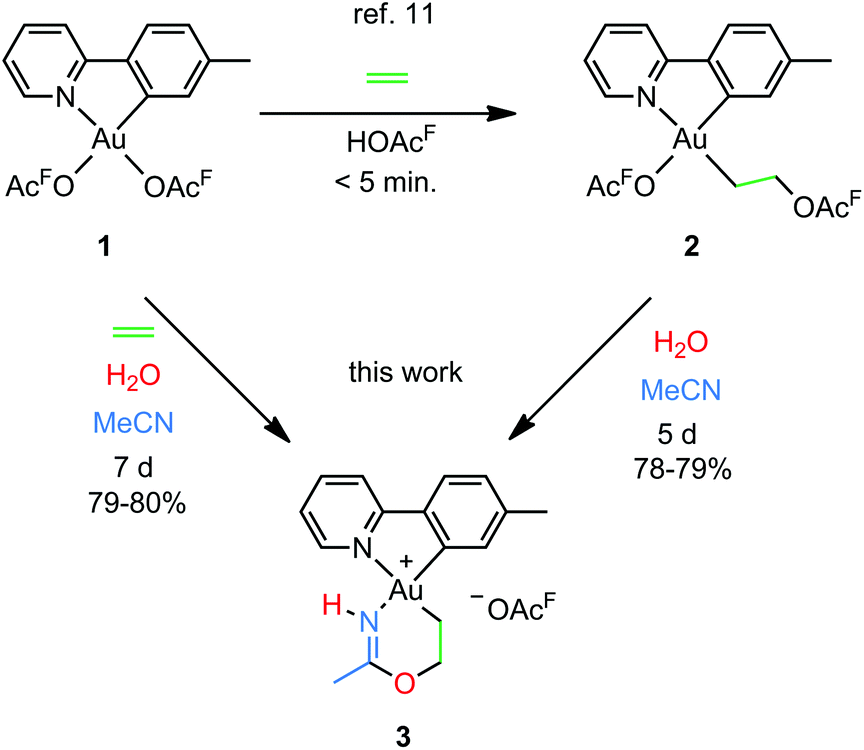 | ||
| Scheme 2 Formal insertion of ethylene into an Au–O bond of Au(tpy)(OAcF)2 (1)11 and preparation of the metallacyclic Au(III) complex 3 from 1 and 2 (this work); (OAcF = OCOCF3). | ||
Results and discussion
Synthesis and characterization
An attempt to crystallize a product mixture containing 2 from acetonitrile led instead to the isolation of small quantities of a new crystalline complex. Examination of these crystals by single-crystal X-ray diffraction revealed that the new complex was a Au(III) metallacycle 3 (Scheme 2, lower part). Synthesis of larger quantities of 3 in about 80% yield was possible simply by stirring 2 in wet acetonitrile for 5 days at ambient temperature. Alternatively, 3 can be obtained in comparable yields by dissolving 1 in acetonitrile and adding water and ethylene. Thus, an efficient synthetic protocol has been developed wherein the three small, readily available building blocks ethylene, water, and acetonitrile are incorporated into the Au(III) metallacycle complex 3 in just one step.The molecular structure, based on the single-crystal X-ray analysis, clearly indicated a complex which contained a new 6-membered chelate ring. Although the X-ray data and the refined structure were of excellent quality, we were concerned about the even slightest possibility of a NH vs. O misassignment,12,13 and sought additional and independent verification of the connectivity seen in the structure. Thus, the three alternatives 3, 4, and 5 in Scheme 3 were initially considered. The 6-ring chelates in these alternatives are formally derived from Au, ethylene, acetonitrile, and one (3, 4) or two (5) equiv. of water. However, involvement of reagents arising from a (possibly gold) catalyzed hydrolysis of acetonitrile, or from impurities inadvertently present in this solvent, must also be considered.
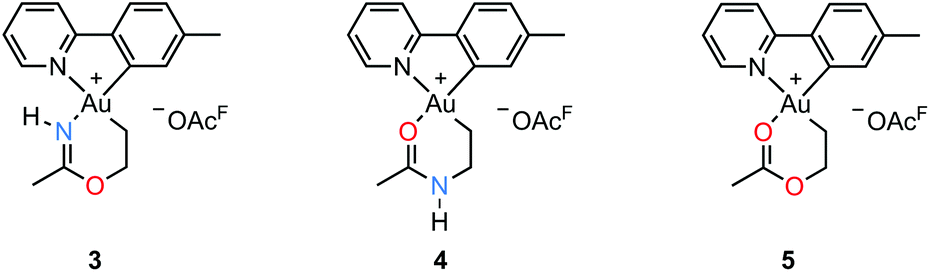 | ||
| Scheme 3 Possible structures of the metallacyclic Au(III) complex. | ||
By positive-ion high resolution mass spectrometry (HRMS), the parent ion at m/z = 451.1068 established the elemental composition C16H18AuN2O for the cation (calcd m/z = 451.1084), in accord with the presence of one N and one O atom in the chelate ring. This composition, which eliminates alternative 5, was corroborated by elemental analysis. Support for the formulation of the N,O-containing chelate 3 was obtained by 1H–15N HMBC NMR spectroscopy. In the spectrum,§ signals arising from two different N atoms are clearly observed. One signal at δ(15N{1H}) = −212 arises from the chelate NH and correlates with the broadened 1H signal of the NH group at δ(1H) = 10.28 and with the signal from the metallacycle methyl group at δ(1H) = 2.51, but not with the other protons of the metallacycle. The other 15N signal at δ(15N{1H}) = −129 arises from the N atom in the tpy ligand and has a correlation with the signal arising from the CH proton α to the pyridyl-N atom at δ(1H) = 9.33. The 19F NMR spectrum shows only one signal, arising from the −OAcF anion. So as to further unambiguously distinguish between alternatives 3 and 4, a NOE experiment was performed. The 1H–1H NOESY spectrum§ shows a clear NOE interaction between the NH proton at δ 10.28 and the CH proton at δ 9.33. These H atoms are therefore in close proximity, supporting the crystallographic analysis and verifying that 3 does indeed represent the structure of the metallacycle.
An ORTEP plot of the molecular structure of 3 is shown in Fig. 1, with selected bond distances and angles. The Au(III) centre has the expected square planar geometry. The deviation of the C9–Au–N1 bond angle in the tpy chelate from 90 to 80.60(10)° is typical, and the Au–ligand distances Au1–N1, Au1–C9, and Au1–C4 (sp3) are within the ranges that have previously been observed for the related complexes Au(tpy)Me2,14 Au(tpy)(CH2CH2OAcF)(OAcF),11 and Au(tpy)(CH2CH2OCH2CF3)(OAcF).11 The N5–C3 (1.284(4) Å) and C3–O2 (1.332(3) Å) bond distances are consistent with the corresponding average N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (1.282 Å) and C–O (1.325 Å) in Pt(II) imino ether complexes.15–22¶ These distances are, however, distinctly different from the distances in a [(tpa)RhIII]2+ (tpa = N,N,N-tri(2-pyridylmethyl)amine) analogue to 3, in which the corresponding N–C and C–O bond distances are 1.245(19) and 1.374(17) Å, respectively.23 The directionality of the N5⋯O1A interaction between the NH of the 6-membered ring and the nearest O (O1A) of the trifluoroacetate anion suggests the presence of a hydrogen bond; in fact, the final refinement shows a donor–acceptor distance (N5⋯O1A) of 2.8708(2) Å. This is in excellent agreement with the proposed structure 3. The crystal structure of 3 is monoclinic, but with β = 90°. In addition, it is twinned via a twofold rotation about the crystallographic a axis. The structure displays parallel displaced π–π stacking along the a direction, and the distance between the intersecting planes of the tpy ligands alternates between 3.42 and 3.44 Å (Fig. 2).
C (1.282 Å) and C–O (1.325 Å) in Pt(II) imino ether complexes.15–22¶ These distances are, however, distinctly different from the distances in a [(tpa)RhIII]2+ (tpa = N,N,N-tri(2-pyridylmethyl)amine) analogue to 3, in which the corresponding N–C and C–O bond distances are 1.245(19) and 1.374(17) Å, respectively.23 The directionality of the N5⋯O1A interaction between the NH of the 6-membered ring and the nearest O (O1A) of the trifluoroacetate anion suggests the presence of a hydrogen bond; in fact, the final refinement shows a donor–acceptor distance (N5⋯O1A) of 2.8708(2) Å. This is in excellent agreement with the proposed structure 3. The crystal structure of 3 is monoclinic, but with β = 90°. In addition, it is twinned via a twofold rotation about the crystallographic a axis. The structure displays parallel displaced π–π stacking along the a direction, and the distance between the intersecting planes of the tpy ligands alternates between 3.42 and 3.44 Å (Fig. 2).
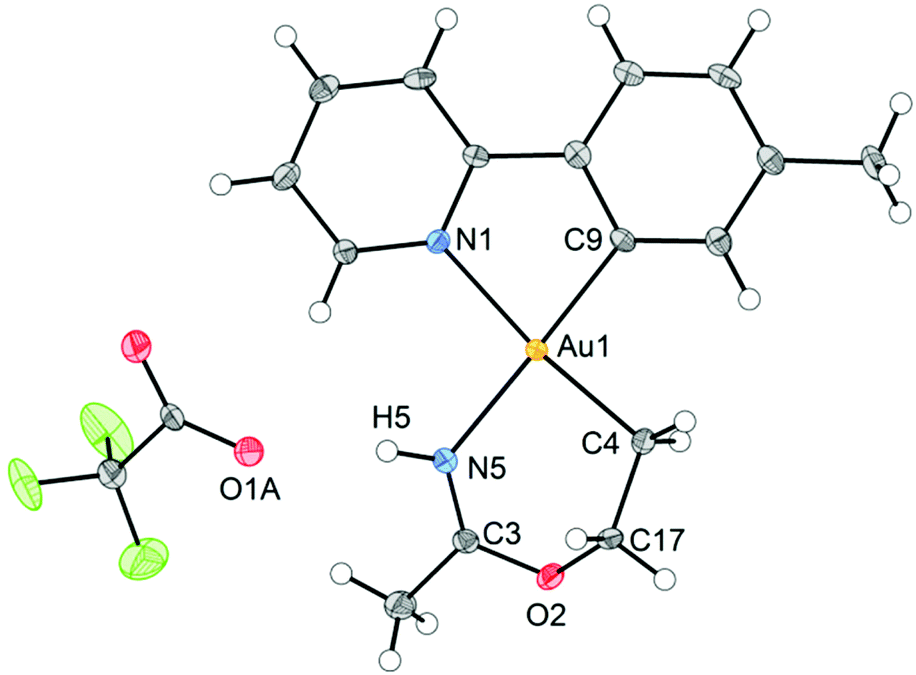 | ||
| Fig. 1 ORTEP drawing of Au(III) complex 3, with 50% probability ellipsoids. Selected bond distances (Å), angles, and torsional angles (°): Au1–C4, 2.042(3); Au1–C9, 2.019(3); Au1–N1, 2.126(2); Au1–N5, 2.092(3); C3–N5, 1.284(4); C3–O2, 1.332(3); C17–O2, 1.449(4); C4–C17, 1.510(4); O1A...N5, 2.8708(2); C4–Au1–C9, 92.39(12); C4–Au1–N5, 89.54(11); C9–Au1–N1, 80.60(10); N1–Au1–N5, 97.43(9); C4–Au1–N1, 172.99(11); C9–Au1–N5, 174.24(12); C3–O2–C17–C4, −70.8(4); Au1–C4–C17–O2, 60.2(3); Au1–N5–C3–O2, 26.5(5); C17–O2–C3–N5, 21.8(5). | ||
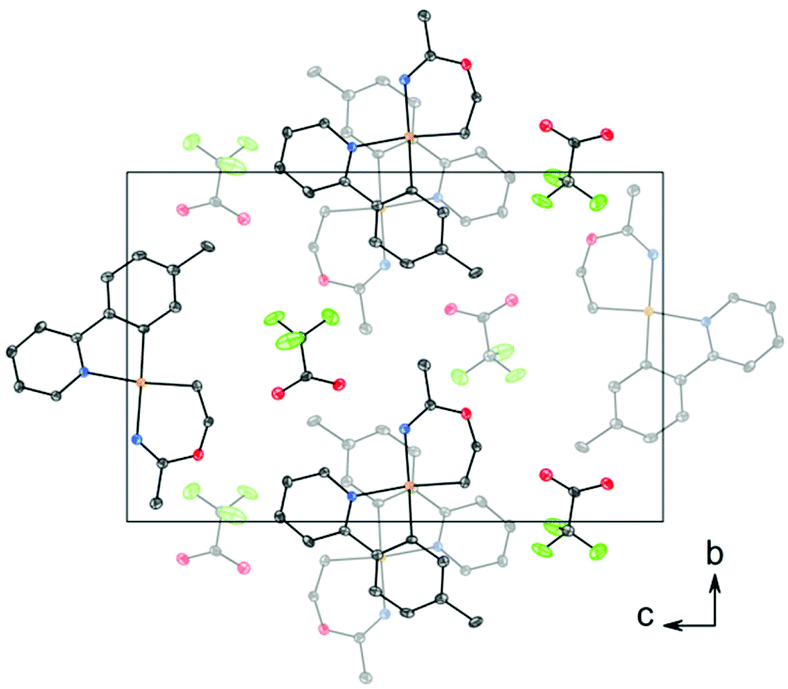 | ||
| Fig. 2 Packing diagram of structure 3 viewed along the a axis, showing stacking between the tpy ligands. The thermal ellipsoids are displayed at 50% probability, and H atoms are omitted for clarity. | ||
DFT calculations were performed on the cations of 3, 4, and 5 in order to further assist with the verification of the structure. Gratifyingly, comparisons of the crystallographic data with the optimized structures showed that the average r.m.s. deviations of the atomic positions were considerably smaller for 3 (average deviation 0.015 Å) than for 4 (0.022 Å) and 5 (0.032 Å), lending further support for the structural assignment.
Mechanistic studies by experiment and DFT calculations
Initial mechanistic experiments probed whether a gold-mediated or gold-catalyzed hydrolysis of acetonitrile to form acetamide might be involved in the process (Scheme 4). First, no formation of 3 was seen when 2 was treated with only acetamide in CH2Cl2 (Scheme 4A). Second, the reaction of 2 with wet acetonitrile-d3 in the presence of acetamide led to the generation of 3-d3 and a minor amount of 3 as inferred by integration of the metallacycle–methyl resonance at δ 2.51 in the 1H NMR spectrum (Scheme 4B). These experiments suggest that acetonitrile, and not acetamide, is the main reactant in the formation of the metallacycle.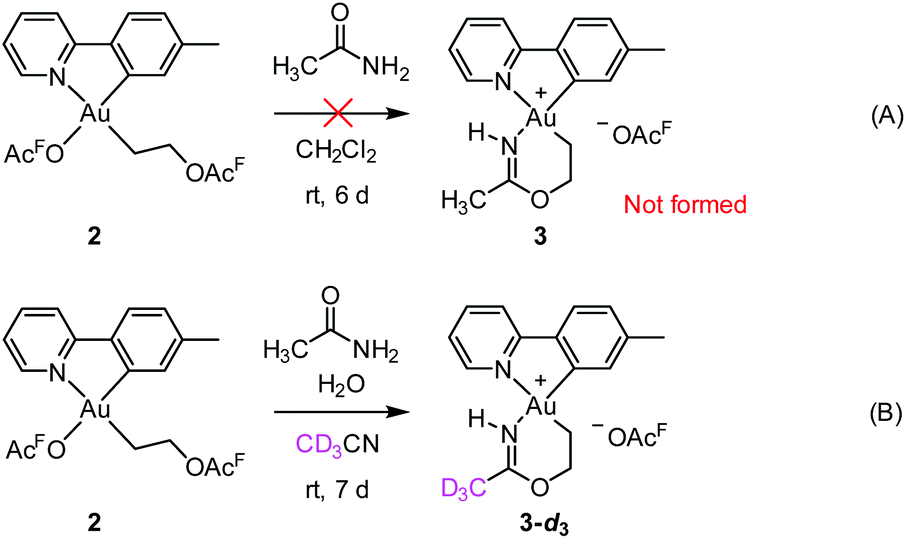 | ||
| Scheme 4 Potential role of acetamide in the formation of 3. | ||
Scheme 5 shows a possible mechanism for the formation of 3. The unobserved cationic ethylene complex 6 is generated from either 1 or 2, in agreement with our previous findings.11 The hydroxyethyl complex 7 is formed from 2 after nucleophilic attack by water, by analogy with Atwood's study,9 and parallel to reaction of trifluoroethanol with 2, which proceeds via6 yielding 10.11 Acetonitrile substitution for the −OAcF anion of 7 at the kinetically most accessible position11trans to tpy-C furnishes a crucial intermediate 8, which undergoes cyclization by an intramolecular nucleophilic attack of the hydroxyethyl-O atom on the nitrile-C atom of acetonitrile, analogous to the alcoholysis of Pt(II)-coordinated nitriles to provide Pt(II) imino ethers.20–22 An −OAcF anion assisted proton transfer from O to N finally provides 3. Close analogues of the putative intermediates 7 and 8 exist and support their mechanistic viability. The hydroxyethyl complex 7 is an analogue of Au(tpy)Me(OTf) and Au(tpy)Me(OAc); both have been structurally characterized as the pertinent stereoisomers with Me trans to N.24 Complex 8 is closely related to [Au(tpy)Me(NCMe)][BF4] (9) which has been prepared with the relevant stereochemistry at Au by bromide abstraction from AuBrMe(tpy) with AgBF4 in acetonitrile.25
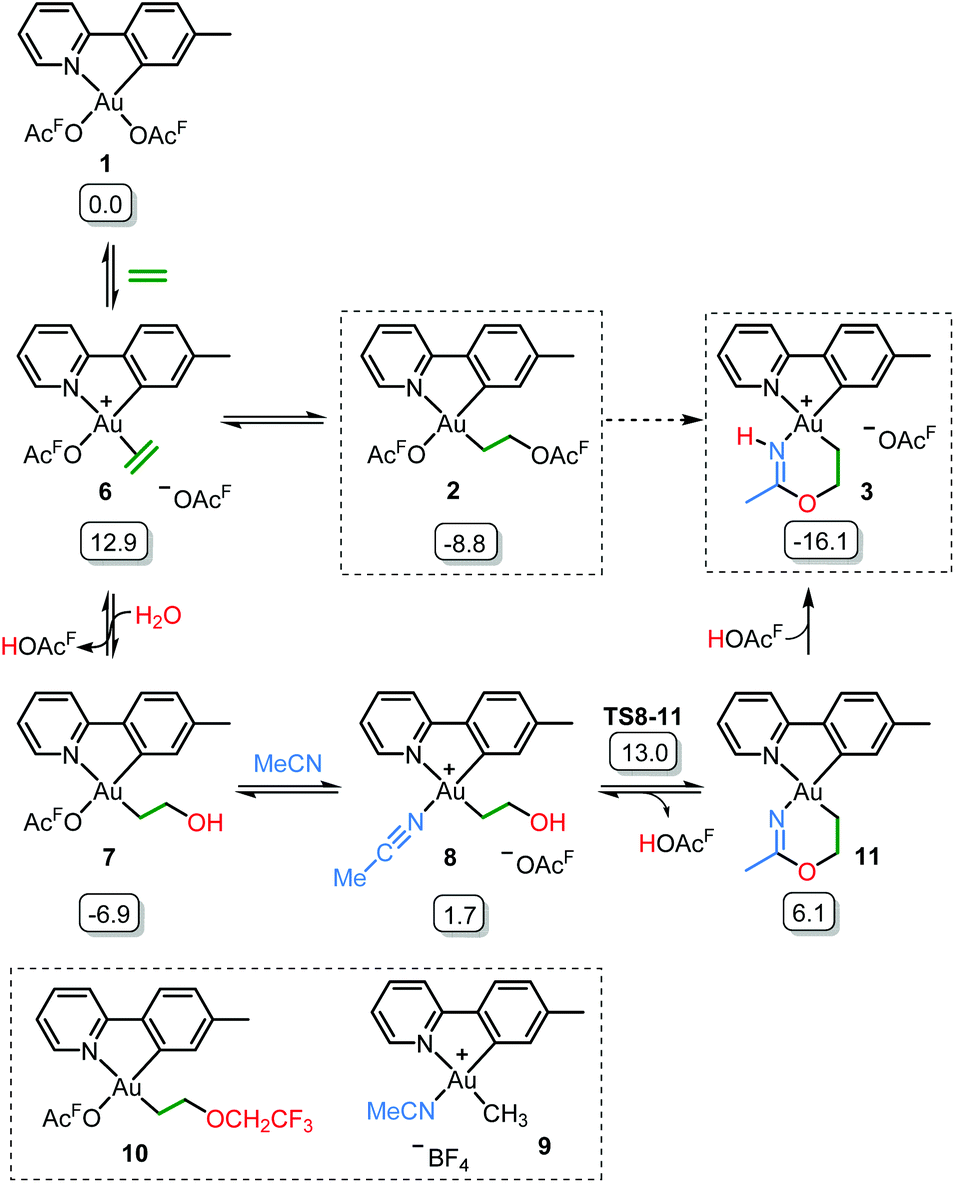 | ||
| Scheme 5 Postulated reaction mechanism for the formation of 3 (OAcF = OCOCF3). Framed species have been characterized by X-ray diffraction in either this or previous (ref. 11) work. Free energies obtained from DFT calculations in acetonitrile are given in kcal mol−1. | ||
The relative stability of all intermediates shown in Scheme 5 has been assessed by means of DFT calculations, which show that they are all accessible under the experimental conditions.|| The TS from 8 to 11 has also been computed (TS8-11, Fig. 3) and it shows that cyclization is triggered by the deprotonation of the hydroxyl group by an external −OAcF. This transition state is 13.0 kcal mol−1 higher than 1 and involves an effective energy barrier of 21.8 kcal mol−1 relative to 2. The long experimental reaction time required for this transformation may be due to the existence of many equilibria, which are dependent on the different reactant concentrations.
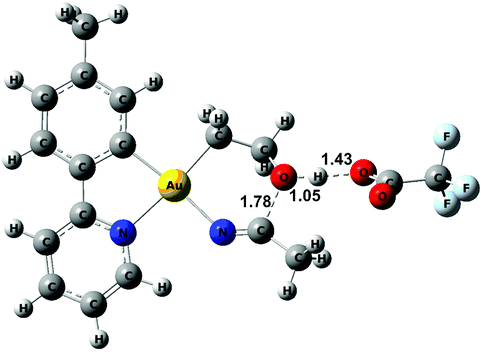 | ||
| Fig. 3 Optimized geometry for TS8-11 with selected bond distances (Å). | ||
Concluding remarks
An eventual reductive elimination from 3 would generate 2-methyl-2-oxazoline, with an important heterocyclic ring structure. Previously, 2-oxazolines have been prepared by Sawamura and coworkers26–28 by Au(I) catalyzed asymmetric aldol reactions of isocyanides and aldehydes. Hashmi and coworkers more recently29,30 prepared oxazolines by Au(I)-catalyzed cycloisomerizations of propargyl amides. Interestingly, it has been recently reported that cyclometallated Au(III) aryl–pyridine complexes may act as efficient catalysts for the three-component synthesis of propargylic amines from aldehydes, secondary amines, and alkynes, and of substituted oxazoles from N-benzyl imines, alkynes, and acyl chlorides.31,32 The [(tpa)RhIII]2+ analogue to 3 was synthesized in a two-step process by H2O2 oxidation of a coordinated ethylene, followed by reaction with acetonitrile and NH4PF6.23 In this context, our one-step assembly of an oxazoline from an alkene, a nitrile, and water represents a new strategy with obvious possibilities in organic synthesis.33In conclusion, the convenient and high-yield one-pot, four-component assembly of a new cationic metallacyclic Au(III) complex has been described. In the process, the three small, readily available building blocks ethylene, water, and acetonitrile have been incorporated into one product. The transformation demonstrates that both of the potentially labile coordination sites in Au(tpy)(OAcF)2 can be utilized – a fact that may be of importance for catalytic applications. The metallacycle is constructed in such a fashion that the high trans-effect (alkyl) end of the assembled chelate ligand occupies the position that is trans to the low trans-effect (N donor) of the tpy supporting ligand, and vice versa.
Experimental and computational section
General experimental methods
Gold(III) complexes 1 and 2 were prepared by previously reported procedures.11,14 Distilled water was used. CH3CN and CH2Cl2 were purified using a MB SPS-800 solvent purifying system from MBraun. The gold(III) complexes studied here are not sensitive to air, so inert atmosphere was not utilized, except for the synthesis of 9 which is described in the ESI.† NMR spectra were recorded on Bruker Avance DPX200, AVII400, DRX500, AVII600 and AV600 instruments at ambient temperature. 1H and 13C spectra have been referenced relative to the residual solvent signals. 19F has been referenced to CFCl3 by using C6F6 (−164.9 ppm with respect to CFCl3 at 0 ppm) as an internal standard. The 15N chemical shifts have been calibrated using MeNO2 as an external standard at 0 ppm. The peaks in the 1H NMR and 13C NMR spectra were assigned by the aid of 2D NMR techniques such as HSQC, HMBC, COSY, NOESY, and 1H–15N HMBC according to the numbering scheme shown below. Mass spectra (ESI) were obtained on a Micromass QTOF II spectrometer and a Bruker Daltronics maXisII spectrometer. Elemental analysis was performed by Microanalytisches Laboratorium Kolbe, Mülheim an der Ruhr, Germany.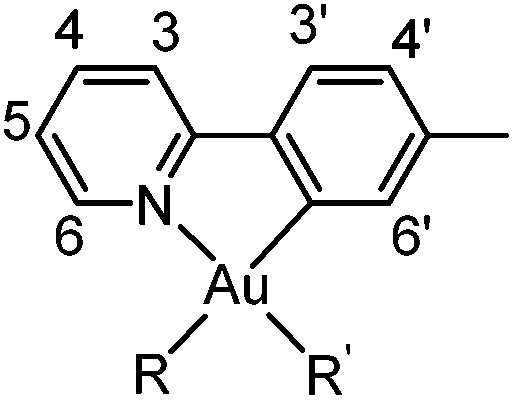
Computational details
Calculations were carried out at the DFT level as implemented in the Gaussian09 software package.34 The hybrid PBE0+GD3 functional35,36 including Grimme's model for dispersion forces was used to optimize all geometries. This methodology was selected because previous studies have proven its solid performance in the modeling of Au(III) alkene complexes.8,11,37 C, H, N and O were described with the all-electron triple-ζ 6-311+G** basis set,38,39 whereas Au was described with the Stuttgart–Köln basis set including a small-core quasi-relativistic pseudopotential.40,41 Geometries were fully optimized without any constraint. Vibrational frequencies were computed analytically to verify that the stationary points found were energy minima or transition states. All optimizations needed for the mechanism proposal were carried out in solvent (acetonitrile) using the SMD solvation model.42 Complexes 3, 4 and 5 were optimized in gas phase with the aim of comparing their geometries with an X-ray crystal structure. Gibbs energies were obtained for T = 298.15 K and p = 1 atm. In the bimolecular steps, these energies were corrected for the 1 M standard state (T = 298.15 K and p = 24.465 atm). Further computational details are given in the ESI.†Acknowledgements
We are grateful for financial support from the Research Council of Norway for funding provided through the Centre of Excellence for Theoretical and Computational Chemistry (CTCC; Grant 179568/V30) and for stipends to AN, EL and MSMH (Grants 185513/I30 and 221801/F20), and the Norwegian Metacenter for Computational Science (NOTUR; Grant nn4654k). DB also thanks the EU REA for a Marie Curie Fellowship (Grant CompuWOC/618303). This work was also supported by COST Action CM1205 CARISMA (Catalytic Routines for Small Molecule Activation).Notes and references
- M. Chiarucci and M. Bandini, Beilstein J. Org. Chem., 2013, 9, 2586–2614 CrossRef PubMed.
- A. S. K. Hashmi, S. Schäfer, M. Wölfle, C. Diez Gil, P. Fischer, A. Laguna, M. C. Blanco and M. C. Gimeno, Angew. Chem., Int. Ed., 2007, 46, 6184–6187 CrossRef CAS PubMed.
- Modern Gold Catalyzed Synthesis, ed. A. S. K. Hashmi and F. D. Toste, Wiley-VCH, Weinheim, 2012 Search PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- R. E. M. Brooner and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2013, 52, 11714–11724 CrossRef CAS PubMed.
- R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef CAS PubMed.
- H. Schmidbaur and A. Schier, Organometallics, 2010, 29, 2–23 CrossRef CAS.
- D. Balcells, O. Eisenstein, M. Tilset and A. Nova, Dalton Trans., 2016, 45, 5504–5513 RSC.
- C. E. Rezsnyak, J. Autschbach, J. D. Atwood and S. Moncho, J. Coord. Chem., 2013, 66, 1153–1165 CrossRef CAS.
- N. Savjani, D.-A. Roşca, M. Schormann and M. Bochmann, Angew. Chem., Int. Ed., 2013, 52, 874–877 CrossRef CAS PubMed.
- E. Langseth, A. Nova, E. A. Tråseth, F. Rise, S. Øien, R. H. Heyn and M. Tilset, J. Am. Chem. Soc., 2014, 136, 10104–10115 CrossRef CAS PubMed.
- R. L. Harlow, J. Res. Natl. Inst. Stand. Technol., 1996, 101, 327–339 CrossRef CAS.
- A. Spek, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- E. Langseth, C. H. Görbitz, R. H. Heyn and M. Tilset, Organometallics, 2012, 31, 6567–6571 CrossRef CAS.
- D. Giardina-Papa, F. P. Intini, C. Pacifico and G. Natile, Inorg. Chem., 2013, 52, 13058–13067 CrossRef CAS PubMed.
- D. Giardina-Papa, F. P. Intini, G. Natile and C. Pacifico, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2012, 68, m300–m302 CAS.
- T. G. Chulkova, P. V. Gushchin, M. Haukka and V. Y. Kukushkin, Inorg. Chem. Commun., 2010, 13, 580–583 CrossRef CAS.
- S. M. Sbovata, F. Bettio, C. Marzano, A. Tassan, M. Mozzon, R. Bertani, F. Benetollo and R. A. Michelin, J. Inorg. Biochem., 2008, 102, 882–891 CrossRef PubMed.
- S. M. Sbovata, F. Bettio, M. Mozzon, R. Bertani, A. Venzo, F. Benetollo, R. A. Michelin, V. Gandin and C. Marzano, J. Med. Chem., 2007, 50, 4775–4784 CrossRef PubMed.
- P. D. Prenzler, D. C. R. Hockless and G. A. Heath, Inorg. Chem., 1997, 36, 5845–5849 CrossRef CAS PubMed.
- R. Cini, P. A. Caputo, F. P. Intini and G. Natile, Inorg. Chem., 1995, 34, 1130–1137 CrossRef CAS.
- J. M. Casas, M. H. Chisholm, M. V. Sicilia and W. E. Streib, Polyhedron, 1991, 10, 1573–1578 CrossRef CAS.
- B. de Bruin, M. J. Boerakker, R. de Gelder, J. M. M. Smits and A. W. Gal, Angew. Chem., Int. Ed., 1999, 38, 219–222 CrossRef CAS.
- A. Venugopal, A. P. Shaw, K. W. Törnroos, R. H. Heyn and M. Tilset, Organometallics, 2011, 30, 3250–3253 CrossRef CAS.
- The synthesis, characterization, and X-ray structure of Au(tpy)(Me)(NCMe)+BF4− is given in the ESI.†.
- Y. Ito, M. Sawamura and T. Hayashi, J. Am. Chem. Soc., 1986, 108, 6405–6406 CrossRef CAS.
- Y. Ito, M. Sawamura, M. Kobayashi and T. Hayashi, Tetrahedron Lett., 1988, 29, 6321–6324 CrossRef CAS.
- Y. Ito, M. Sawamura, E. Shirakawa, K. Hayashizaki and T. Hayashi, Tetrahedron Lett., 1988, 29, 235–238 CrossRef CAS.
- A. S. K. Hashmi, J. P. Weyrauch, W. Frey and J. W. Bats, Org. Lett., 2004, 6, 4391–4394 CrossRef CAS PubMed.
- S. Doherty, J. G. Knight, A. S. K. Hashmi, C. H. Smyth, N. A. B. Ward, K. J. Robson, S. Tweedley, R. W. Harrington and W. Clegg, Organometallics, 2010, 29, 4139–4147 CrossRef CAS.
- H. von Wachenfeldt, A. V. Polukeev, N. Loganathan, F. Paulsen, P. Rose, M. Garreau, O. F. Wendt and D. Strand, Dalton Trans., 2015, 44, 5347–5353 RSC.
- K. K.-Y. Kung, V. K.-Y. Lo, H.-M. Ko, G.-L. Li, P.-Y. Chan, K.-C. Leung, Z. Zhou, M.-Z. Wang, C.-M. Che and M.-K. Wong, Adv. Synth. Catal., 2013, 355, 2055–2070 CrossRef CAS.
- Oxazoles: Synthesis, Reactions, and Spectroscopy, Part B, ed. D. C. Palmer, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2004 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Rev. D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. Langseth, M. L. Scheuermann, D. Balcells, W. Kaminsky, K. I. Goldberg, O. Eisenstein, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2013, 52, 1660–1663 CrossRef CAS PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- D. Figgen, G. Rauhut, M. Dolg and H. Stoll, Chem. Phys., 2005, 311, 227–244 CrossRef CAS.
- D. Figgen, K. A. Peterson, M. Dolg and H. Stoll, J. Chem. Phys., 2009, 130, 164108 CrossRef PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experimental and computational details, characterization data, and spectra. CCDC 1443527 and 1443528. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01648k |
| ‡ Current address: SINTEF Materials and Chemistry, P.O. Box 124 Blindern, N-0314 Oslo, Norway. |
| § See ESI pages S5, S13, and S14† for details. |
| ¶ No structural examples of Au imino ethers were found via SciFinder. |
| || For an estimation of the transition state energies involved in the transformation of 1 to 7, see ref. 11. |
| This journal is © The Royal Society of Chemistry 2016 |