
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNear infrared light-mediated photoactivation of cytotoxic Re(I) complexes by using lanthanide-doped upconversion nanoparticles†
Ming
Hu
a,
Jixian
Zhao
b,
Xiangzhao
Ai
a,
Maja
Budanovic
a,
Jing
Mu
a,
Richard D.
Webster
a,
Qian
Cao
*b,
Zongwan
Mao
*b and
Bengang
Xing
*ac
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 637371 Singapore
bMOE Key Laboratory of Bioinorganic and Synthetic Chemistry, School of Chemistry and Chemical Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: cesmzw@mail.sysu.edu.cn; caoqian3@mail.sysu.edu.cn
cInstitute of Materials Research and Engineering (IMRE); Agency for Science, Technology and Research (A*STAR); Singapore, 117602 Singapore. E-mail: bengang@ntu.edu.sg
First published on 11th August 2016
Abstract
Platinum-based chemotherapy, although it has been well proven to be effective in the battle against cancer, suffers from limited specificity, severe side effects and drug resistance. The development of new alternatives with potent anticancer effects and improved specificity is therefore urgently needed. Recently, there are some new chemotherapy reagents based on photoactive Re(I) complexes which have been reported as promising alternatives to improve specificity mainly attributed to the spatial and temporal activation process by light irradiation. However, most of them respond to short-wavelength light (e.g. UV, blue or green light), which may cause unwanted photo damage to cells. Herein, we demonstrate a system for near-infrared (NIR) light controlled activation of Re(I) complex cytotoxicity by integration of photoactivatable Re(I) complexes and lanthanide-doped upconversion nanoparticles (UCNPs). Upon NIR irradiation at 980 nm, the Re(I) complex can be locally activated by upconverted UV light emitted from UCNPs and subsequently leads to enhanced cell lethality. Cytotoxicity studies showed effective inactivation of both drug susceptible human ovarian carcinoma A2780 cells and cisplatin resistant subline A2780cis cells by our UCNP based system with NIR irradiation, and there was minimum light toxicity observed in the whole process, suggesting that such a system could provide a promising strategy to control localized activation of Re(I) complexes and therefore minimize potential side effects.
1 Introduction
Transition metal based antitumor complexes, especially platinum drugs such as cisplatin, oxaliplatin and carboplatin have been studied for their abilities in cancer treatment for almost half a century.1 Some of these complexes have gained worldwide applications in chemotherapy against various types of cancers. However, the use of these metal complexes in treatment processes has been mostly associated with severe side effects such as limited tumor specificity, and intrinsic or acquired multi-drug resistance.2 Therefore, rational design through the development of new alternatives with potent anticancer effects and improved specificity is thus the main concern, and extensive studies have been carried out so far.3–16 Recently, some photoactivatable Re(I) complexes which demonstrated great potential to inactivate tumor growth based on increased tumor recognition have been investigated as possible candidates in chemotherapy.17–22 In these cases, light irradiation, which has impressively high spatiotemporal resolution, is required to activate the complexes for enhanced cytotoxicity within the targeted tumor area. Despite their initial success, most of these photoactivatable Re(I) complexes are triggered by short-wavelength light, such as UV, blue and green light. The application of short-wavelength light may cause undesired damage to the tissue, protein and DNA.23 In addition, a limited penetration depth in tissues makes it difficult to reach tumors which are located beneath the skin surface.24 Therefore the application of short-wavelength light in activating Re(I) complexes for a better antitumor effect could be largely restricted. In comparison, long-wavelength light irradiation has been considered to cause less photo-damage and has a deeper tissue penetration, making it more suitable for applications in biological systems. Therefore, considering the potential significance of photoactive Re(I) complexes in antitumor treatment, the rational design of a simple yet specific strategy which allows the activation of Re(I) complexes by long-wavelength light selectively in the tumor region with minimum biological damage is highly desirable, and relevant investigations toward this direction still need to be conducted.25 Recently, lanthanide-doped upconversion nanoparticles (UCNPs) as a promising tool to gain precise control of light irradiation have drawn much attention for their application in sensing,26–28 imaging,29–33 drug delivery,34–41 phototherapy42–47 and theranostics48–51 because of their unique optical properties. Generally, UCNPs can absorb long-wavelength near infrared (NIR) light and exhibit multiple emission ranging from UV to visible and even the NIR region.52–58 Such appealing photo-physical properties enable the local activation of photosensitive metal complexes for therapeutic purposes by remotely controlling long-wavelength light irradiation which is suitable for living systems.16,43,44,49,59–71 For example, the successful NIR photoactivation of Pt metal drug complexes has been reported as being effective in suppressing the viability of cancer cells and inhibiting tumor growth in living animals.16,44,49,71 Similarly, UCNPs have also been utilized for NIR photoactivation of Ru complexes to control the release of antitumor drugs.61,63,65,67,69 Moreover, some other transition metal complexes such as Zn, Fe, Cr and Mn have also been reported for the purpose of photodynamic therapy or the light-mediated delivery of gaseous molecules such as NO and CO.43,59,62,64,66,68,70 Inspired by these pioneering studies, herein, we demonstrate a system for NIR light controlled activation of Re(I) complex cytotoxicity by combining photoactivatable Re(I) complexes with lanthanide-doped upconversion nanoparticles. Upon NIR irradiation, the Re(I) complex can be locally activated by upconverted UV light emitted from UCNPs and subsequently leads to enhanced cell lethality, therefore minimizing potential side effects.2 Experimental section
2.1 Instruments and general methods
The 1H NMR spectrum was recorded on a Bruker AVANCE AV 300 MHz spectrometer. The ESI-MS spectrum was acquired on a Thermo LCQ DECA XP liquid chromatography-mass spectrometer. UV-vis spectra were recorded on a SHIMAZU UV-1800 UV spectrophotometer. Fluorescence spectra were obtained on a SHIMAZU RF-5301PC spectro-fluorophotometer. The fluorescence emission spectra of UCNPs were recorded at an angle of 90° to the excitation laser (980 nm) using an optical SEC-2000 spectrometer coupled with a 2048 pixel CCD array (ALS Co., Ltd). Transmission electron microscopy (TEM) images were recorded using a FEI EM208S TEM (Philips) operating at 100 kV. Dynamic light scattering (DLS) measurements were performed by using a Brookhaven 90 plus Nanoparticle Size analyzer. Rhenium content was measured on an Agilent 7700 Inductively Coupled Plasma Mass Spectrometer (ICP-MS). Cell imaging was carried out on a fluorescence microscope (Nikon, Eclipse TE2000-E).2.2 Synthetic procedure of [Re(DIP)(CO)3(cpy)](PF6)
Typically, the starting material, [Re(DIP)(CO)3Cl] was prepared according to a literature procedure.72 The prepared molecule was then converted to [Re(DIP)(CO)3](OTf). Basically, AgOTf (0.235 mmol) was added to a suspension of [Re(DIP)(CO)3Cl] (0.157 mmol) in 50 mL of CH3CN. The mixture was refluxed under nitrogen overnight in the dark. After removal of the off-white AgCl precipitate, CH3CN was evaporated under reduced pressure. The resulting solid was dissolved in 50 mL of THF containing 3-(hydroxymethyl)pyridine (1.5 mmol) and refluxed under a nitrogen atmosphere for 20 h in the dark. The mixture was then evaporated to dryness and the solid obtained was further dissolved in dichloromethane with 5 mL of thionyl chloride and heated to reflux under a nitrogen atmosphere for 5 h. After removal of dichloromethane and the excess amount of thionyl chloride by nitrogen flushing, the resulting yellow solid was dissolved in CH3CN and added dropwise into a saturated NH4PF6 solution. The yellow precipitate was collected and washed with distilled water and diethylether followed by recrystallization from diethylether and dried under vacuum to afford the final product. (Yield: 77%) 1H NMR (300 MHz, DMSO) δ 9.85 (d, J = 5.4 Hz, 2H; H of Ph2-phen), 8.68 (s, 1H; H of pyridine), 8.52 (d, J = 4.9 Hz, 1H; H of pyridine), 8.22 (d, J = 5.4 Hz, 2H; H of Ph2-phen), 8.14 (s, 2H; H of Ph2-phen), 7.99 (d, J = 8.1 Hz, 1H; H of pyridine), 7.69 (s, 10H; C6H5 at Ph2-phen), 7.42 (t, J = 7.9, 5.7 Hz, 1H; H of pyridine), 4.70 (s, 2H; CH2). ESI-MS: m/z 729.9 [M − PF6−]+ Elemental Analysis (%) Calcd: C33H22ClF6N3O3PRe·5H2O: C, 41.06; H, 3.34; N, 4.35; C/N, 9.41. Found: C, 39.79; H, 2.97; N, 4.11; C/N, 9.68.2.3 Preparation of NaYF4:Yb3+/Tm3+ core nanoparticles
NaYF4:Yb3+/Tm3+ core nanoparticles were synthesized according to the method reported previously.73 In general, 1 mmol of RE(CH3 CO2)3 (RE = 59.5% Y + 40% Yb + 0.5% Tm), 12 mmol of NaF, and 20 mL of an oleic acid (OA)/1-octadecene (ODE) (v/v = 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixed solvent were added to the round bottom flask and heated to 110 °C under vacuum with magnetic stirring for 30 min to remove the residual water and oxygen. Then, the temperature was further increased to 320 °C and maintained for 1.5 h under a nitrogen atmosphere. The resulting UCNPs were obtained by centrifugation for the following application.
:1) mixed solvent were added to the round bottom flask and heated to 110 °C under vacuum with magnetic stirring for 30 min to remove the residual water and oxygen. Then, the temperature was further increased to 320 °C and maintained for 1.5 h under a nitrogen atmosphere. The resulting UCNPs were obtained by centrifugation for the following application.
2.4 Fabrication of NaYF4:Yb3+/Tm3+@NaYF4 core–shell nanoparticles
A cyclohexane solution of the as-prepared NaYF4:Yb3+/Tm3+ core nanoparticles (200 mg) and a 20 mL mixture of OA and ODE (OA/ODE = v/v = 1:1) were added to a round bottom flask and heated to 120 °C under vacuum with magnetic stirring for 1 h and flushed with nitrogen. Then, the temperature was further heated to 310 °C. A mixture of 3 mL of OA and ODE (OA/ODE = v/v = 1:1), Y(CH3CO2)3 (1 mmol) and Na(CH3CO2) (1 mmol) was added to the solution immediately. The reaction was maintained at 310 °C for 1 h under a nitrogen atmosphere. The resulting core–shell UCNPs were isolated by centrifugation for further surface modification.
2.5 Synthesis of PAA-functionalized UCNPs (UCNP-PAA)
A ligand exchange process was performed using poly(acrylic acid) (PAA) to replace the original hydrophobic ligands on the nanoparticle surface.74 PAA (50 mg) was dissolved in ethanol (5 mL) and mixed with 2 mL of the as-prepared core–shell UCNP dispersion in chloroform (20 mg). The resulting mixture was kept overnight with magnetic stirring. The solution was then centrifuged at 2 × 104 rpm for 10 minutes. The precipitate was washed 3 times with ethanol to obtain the UCNP-PAA particles.2.6 Surface coating of chitosan
The reagents EDC (28.7 mg) and NHS (11.6 mg) were added to an aqueous dispersion (10 mg, 2.5 mL) of the as-prepared UCNP-PAA particles to activate the carboxyl group on UCNP-PAA for 30 min. Then an aqueous solution of chitosan (4 mg mL−1, 2.5 mL) was added and kept overnight with magnetic stirring. The solution was then centrifuged at 2 × 104 rpm for 10 minutes. The precipitate was washed 3 times with deionized water to obtain the UCNP-PAA-chitosan (UPC) particles.2.7 Loading of Re(I) complex on UPC
The previously prepared Re(I) complex (0.8 mg) was dissolved in 20 μL of CH3CN to give a yellow solution. The resulting solution was added into PBS (1 mL, 10 mM, pH 7.4) containing UCNP-PAA-chitosan particles (5.2 mg). The mixture was then stirred overnight. The resulting Re(I) loaded UCNPs (Re-UPC) was obtained by centrifugation at 2 × 104 rpm for 10 minutes and washed with PBS/CH3CN (20:1) three times to remove the excess amount of the Re(I) complex. Loading of the Re(I) complex was determined by both fluorescence spectroscopy and the ICP-MS method.
2.8 Stability evaluation of Re-UPC
The Re-UPC particles (50 μM) were incubated with PBS (10 mM, pH 5.0 or 7.4) at 37 °C for different time intervals. The released Re(I) complex from Re-UPC was obtained by centrifugation at 2 × 104 rpm for 10 minutes. The amount of the released Re(I) complex in the resulting supernatant was measured by fluorescence spectroscopy.2.9 Cellular uptake
Cellular uptake was investigated by using a fluorescence microscope. Drug susceptible human ovarian carcinoma A2780 cells or cisplatin resistant human ovarian carcinoma A2780cis cells were seeded at a density of 105 cells per well in a 35 mm u-dish (ibidi) containing RPMI 1640 medium (Gibco) supplemented with 10% fetal bovine serum (FBS, HyClone) and antibiotics (100 units per mL penicillin and 100 μg mL−1 streptomycin) at 5% CO2 and 37 °C for 24 hours prior to treatment with Re-UPC or the Re(I) complex. After 2 hours of incubation, the media were removed and the cells were washed. The images were acquired on a fluorescence microscope (Nikon Eclipse TE2000) with the excitation filter at 364 nm.2.10 Cell viability assay
In the cell viability assay, the cells were seeded in 96-well plates with a density of 104 cells per well. After 24 hours of incubation at 5% CO2 and 37 °C, the medium was replaced with fresh medium containing the Re(I) complex or UPC and Re-UPC. After another 24-hour incubation at 37 °C, culture media were removed, the cells were washed and then incubated with 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) containing culture media. After 5 hours of incubation, the media were removed and 100 μL of DMSO was added. The absorbance at 570 nm was measured by using a Tecan's Infinite M200 microplate reader. For the NIR light irradiation experiment, the cells were incubated with Re-UPC for 2 hours. Subsequently, the drug-containing medium was replaced with fresh medium and the cells were irradiated with NIR (980 nm, 1.5 W cm−2, 5 min break after 5 min irradiation). After 24 hours of incubation cell viability was evaluated by standard MTT assays. In phototoxicity tests, the cells were exposed to NIR for different periods of time followed by 24 hours of incubation and MTT assays to investigate cell viability.3 Results and discussion
3.1 Synthesis and characterization of Re-UPC
Scheme 1 illustrates the design of NIR-mediated activation of the Re(I) complex on chitosan-coated UCNPs (UCNP-PAA-chitosan). The Re(I) complex was obtained through a facile synthesis by refluxing a mixture of [Re(DIP)(CO)3(CH3CN)](PF6) and 4-(chloromethyl)pyridine in THF under a nitrogen atmosphere. The compound was characterized by 1H NMR and ESI-MS (Fig. S1 and S2†). To establish our platform, first, NaYF4:Yb3+/Tm3+ core nanoparticles were prepared using a thermal decomposition method, followed by coating of the NaYF4 shell on the surface to afford the core–shell UCNPs. TEM images (Fig. S3†) demonstrate that the as-prepared core nanoparticles have a narrow size distribution of around 25 nm. After coating with NaYF4, the size of particles increased to about 35 nm. The as-prepared core–shell particles are terminated with oleic ligands which is hydrophobic. To improve the solubility of the particles in water, the core–shell UCNPs were transferred from a nonpolar solvent to an aqueous environment by using poly(acrylic acid) (PAA) as functional ligands to replace the hydrophobic oleic ligands. | ||
| Scheme 1 Schematic representation for the synthesis of the Re(I) complex loaded UCNPs (Re-UPC). | ||
Moreover, to facilitate the loading of the Re(I) complex, as well as biocompatibility of the nanoparticles, the carboxyl groups of PAA on the PAA-coated UCNPs (UCNP-PAA) were activated using the EDC/NHS method, and followed by further conjugation with amine groups on amphiphilic chitosan structures. The size of the obtained PAA-coated UCNPs and chitosan-coated UCNPs was determined by dynamic light scattering (DLS) and transmission electron microscopy (TEM). As given in Fig. 1, the obtained UCNPs (UCNP-PAA and UCNP-PAA-chitosan) have good mono-dispersity and uniform morphology. The inner core–shell structures of both UCNP-PAA and UCNP-PAA-chitosan were ∼35 nm. The hydrodynamic diameter distribution, measured by DLS is ∼76 nm for UCNP-PAA and ∼118 nm for UCNP-PAA-chitosan. In addition, the successful coating of PAA and chitosan was further confirmed by Fourier transform infrared (FTIR) spectroscopy. As shown in Fig. 2A the disappearance of the vibration of –CH3 at 2927 cm−1 and 2855 cm−1 suggests the successful removal of oleic acid from the particle surface. Further conjugation of chitosan on the particle surface can be confirmed by the peaks situated at 1080 cm−1 and 1643 cm−1 corresponding to the C–O stretching and the formation of amide between the carboxyl group on PAA and the amine group on chitosan. Moreover, spectroscopy studies indicated that the absorption of the Re(I) complex overlaps the emission of UCNP-PAA-chitosan (Fig. 2B and S4†) at 291 nm (1I6–3H6), 350 nm (1I6–3F4) and 365 nm (1D2–3H6), suggesting that the as-prepared UCNP-PAA-chitosan can be utilized for activation of the Re(I) complex. Next, to afford the final product Re-UPC (Re(I) complex loaded UCNP-PAA-chitosan), the Re(I) complex was dissolved in aqueous solution (PBS/CH3CN = 50:1) in which UCNP-PAA-chitosan (UPC) was suspended to facilitate the incorporation of the complex onto the particles surface (UPC). Loading of the Re(I) complex was confirmed (Fig. S5†) by fluorescence emission of the Re complex on Re-UPC. The loading amount of the complex was quantified by fluorescence spectroscopy and ICP-MS analysis. The amount of the complex on Re-UPC was determined to be around 6.14 wt%, which is comparable to the value in the nano-platforms reported previously.75 Furthermore, in order to examine the stability of Re-UPC under biologically relevant conditions, the possible release of the Re(I) complex was evaluated. In general, the aqueous suspensions containing Re-UPC were incubated at 37 °C (pH 5.0 and 7.4) for different time durations within 24 hours. The release of the Re(I) complex was quantified by measuring the fluorescence intensity of the supernatant after centrifugation.17 As shown in Fig. 2c, there was no obvious release of the Re complex observed under different pH conditions (e.g. pH 5.0 and pH 7.4) in PBS buffer at 37 °C, suggesting that loading of the Re complex is stable under both conditions.
 | ||
| Fig. 1 Characterization of the prepared nanoparticles. (A) and (B) TEM images of PAA-coated UCNPs (UCNP-PAA) and chitosan-coated nanoparticles (UCNP-PAA-chitosan, UPC), respectively. Scale bar = 50 nm. (C) Size distributions of the prepared UCNP-PAA and UCNP-PAA-chitosan (UPC) determined by DLS. | ||
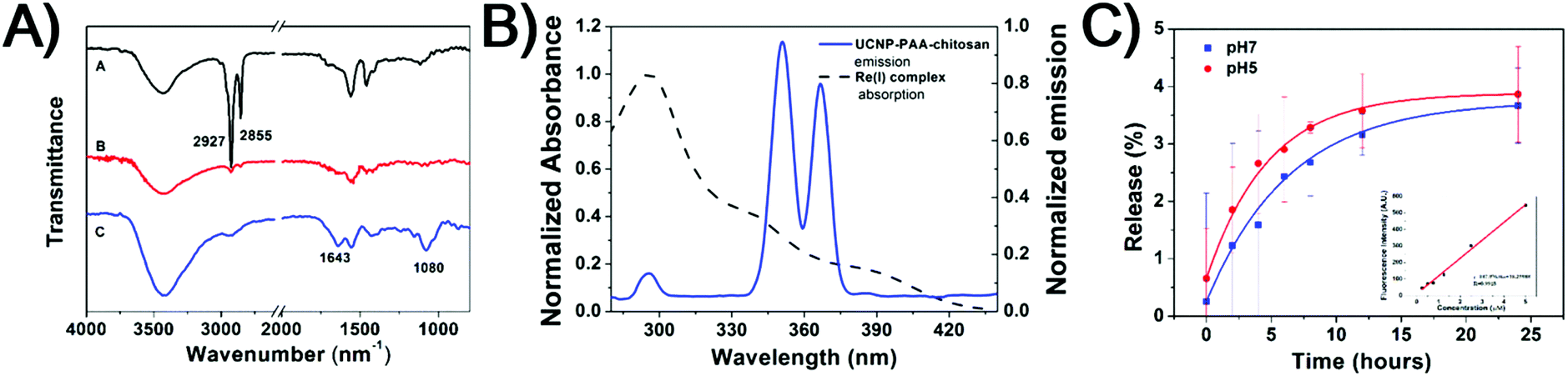 | ||
| Fig. 2 (A) FT-IR spectra of core–shell UCNPs (black), UCNP-PAA (red) and UCNP-PAA-chitosan (UPC) (blue). (B) Absorbance spectra of the Re(I) complex (black dash line) and emission spectra of UPC (blue line, λex = 980 nm). (C) Cumulative release of the Re(I) complex from Re-UPC at 37 °C in PBS buffer. pH 5.0 (red) and pH 7.4 (blue). Inset: the calibration curve used to determine the concentration of the released Re(I) complex. | ||
3.2 Cellular uptake monitored by fluorescence microscopy
To evaluate the cellular uptake of Re-UPC, a fluorescence imaging study was carried out. In this typical study, A2780 cells, a well characterized cisplatin susceptible human ovarian carcinoma cell line, and its cisplatin resistant subline, A2780cis were chosen as the target cells. Both cells were incubated with Re-UPC (10 μM) in the dark for 2 hours before fluorescence imaging. As shown in Fig. 3 and S6† the green fluorescence signal of the Re(I) complex loaded on Re-UPC was observed within both cell lines, indicating the successful internalization of Re-UPC into the cells. To further investigate the possible mechanism of cellular uptake of Re-UPC, both types of cells were treated with Re-UPC (10 μM) at 4 °C in the dark for 2 hours. It can be observed that (Fig. 3 and S6†) at a lower temperature (∼4 °C), there was little green fluorescence inside the cells, suggesting the inhibited internalization of Re-UPC. These results indicated that the uptake of Re-UPC followed a temperature dependent endocytic pathway.76 Moreover, the uptake of the free Re(I) complex was investigated as a control. It can be observed that the fluorescence intensity of Re-UPC inside the cells is much higher than that of the free complex (Fig. S6 and S7†), demonstrating more cell uptake via Re-UPC. Moreover, it suggested that UPC can serve as a reliable platform for effective delivery of the Re(I) complex, which can potentially lead to a better antitumor effect in vitro and in living cells.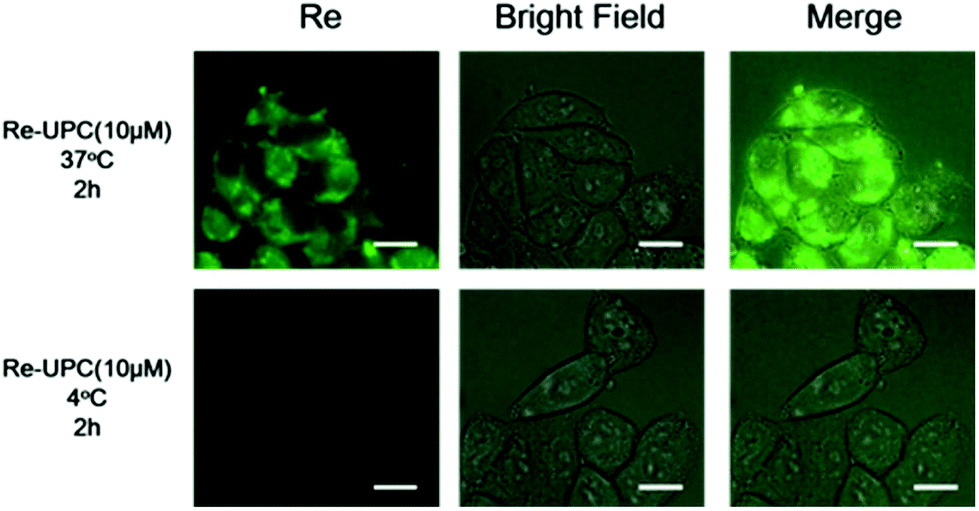 | ||
| Fig. 3 Fluorescence microscopy images of A2780 cells incubated with Re-UPC (10 μM) at 37 °C and 4 °C for 2 h, λex = 364 nm. All scale bars are 10 μm. | ||
3.3 Cytotoxicity assay upon light irradiation
Moreover, we evaluated the cytotoxicity of our system in living cells. Both drug susceptible human ovarian carcinoma A2780 cells and its cisplatin resistant subline A2780cis were chosen as the targeted cell lines. Basically, the two types of cells were treated with Re-UPC and 1 hour of NIR irradiation (980 nm) followed by 24 hours of incubation before cell viability assays. Similar cellular incubation in the absence of NIR irradiation was used as a control.Tumor cells alone and the cells treated with UPC were irradiated under NIR to assess the potential phototoxicity. As indicated in Fig. 4A and B, no significant toxicity was detected in the Re-UPC treated A2780 cells and the A2780cis cells without NIR irradiation. However, when the cells were treated with Re-UPC and 1 h of NIR irradiation (at 980 nm) more potent cytotoxicity could be achieved. Cell viability of the A2780 cells dramatically decreased from 89% to around 55% (0.5 μM) exhibiting the enhanced cytotoxicity of Re-UPC upon NIR irradiation (Fig. 4A). Similarly, improved cell lethality was also achieved in the cisplatin resistant cell line A2780cis. There was a 47% cell viability observed in the A2780cis cells treated with Re-UPC (1 μM) and NIR irradiation, which was much lower than (∼74%) the similarly treated cells without NIR activation (Fig. 4B). These results clearly suggested that NIR irradiation of Re-UPC is essential for activated toxicity.
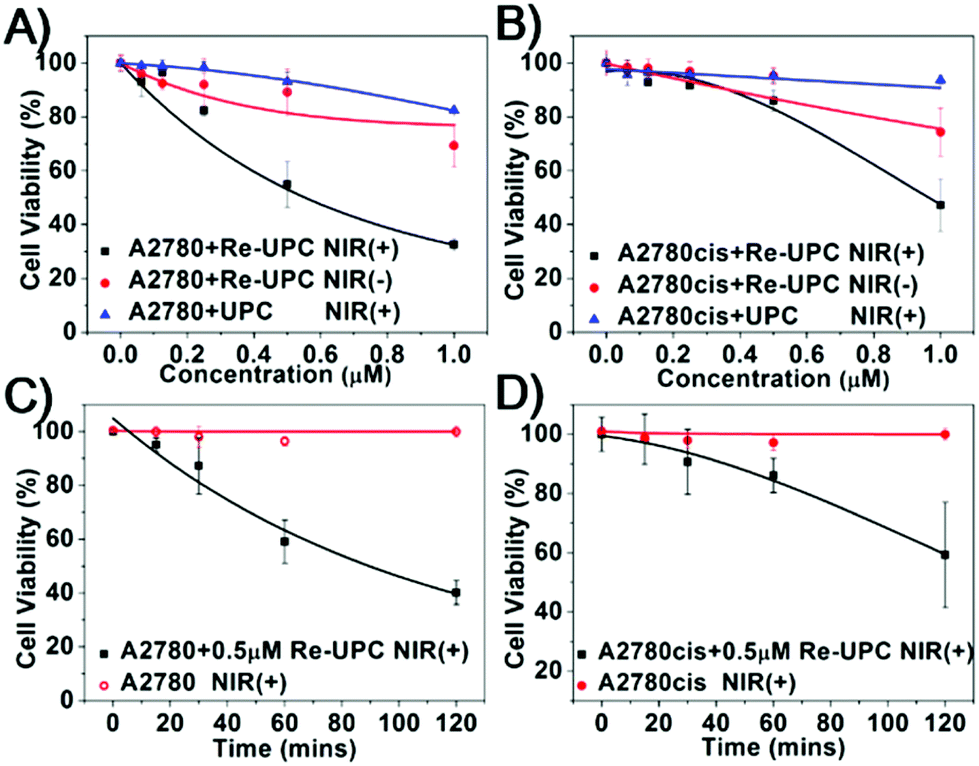 | ||
| Fig. 4 The cytotoxicity assays of the Re-UPC (black) and UPC (blue) at different concentrations with 1 h of NIR irradiation (1.5 W cm−2) in (A) A2780 and (B) cisplatin-resistant A2780cis cells. The cells treated with Re-UPC but without light illumination were used as controls (red). (C) A2780 cells and (D) A2780cis cells under different exposure times of NIR with treatment of Re-UPC (0.5 μM, black) or without Re-UPC (red). | ||
In addition, the light-mediated cytotoxicity in both A2780 and A2780cis cell lines exhibited dependence on the concentration of Re-UPC and the duration of NIR irradiation. In particular, the increased cytotoxicity could be achieved when a higher concentration of Re-UPC was applied. As indicated in Fig. 4A and B, under 1 h NIR light irradiation, Re-UPC (0.5 μM) was found to exhibit obvious cytotoxicity against both A2780 cells and A2780cis cells, and there were 55% and 86% cell viability detected respectively. Moreover, the more potent cell lethality (e.g. 32% and 47% cell viability) of A2780 and A2780cis cells was observed when a higher concentration of Re-UPC (1 μM) was used. Similarly, the more significant cell lethality could be also observed when the prolonged NIR irradiation was applied. It can be observed that (Fig. 4C and D) the cells treated with 0.5 μM of Re-UPC under 2 h NIR irradiation exhibited more distinct toxicity towards A2780 and A2780cis cells with a cell viability of 40% and 59% respectively than that under 1 h NIR illumination. The potent NIR-triggered cytotoxicity in both A2780 and A2780cis cells demonstrated that Re-UPC can be successfully used in activating the Re(I) complex for effective inactivation of both drug susceptible and cisplatin-resistant tumor cells. As a control, a similar free Re(I) complex under UV irradiation also exhibited photoactivatable cytotoxicity which was found to be dependent upon the concentration of the complex and the duration of UV irradiation (Fig. S8A and S8B†). Although photoactivated cytotoxicity can be achieved by both NIR and UV irradiation, NIR activated Re-UPC showed a slightly better antitumor effect in comparison with the free Re(I) complex irradiated with UV light. For instance, the A2780 cells incubated with 1 μM of Re-UPC followed by 1 h NIR irradiation result in more obvious toxicity (32% cell viability, Fig. 4A) than the cells treated with 1 μM of the Re(I) complex and 5 min UV illumination (43% cell viability, Fig. S8A†). Similarly in the A2780cis cell line, 1 μM of Re-UPC with 1 h NIR irradiation caused more cell death (47% cell viability, Fig. 4B) than 1 μM of the Re(I) complex and 5 min UV illumination (60% cell viability, Fig. S8B†). Despite the fact that it is possible to obtain an improved cytotoxicity of the Re(I) complex with prolonged UV irradiation, overexposure to UV light may cause drastic photo-damage to the cells (Fig. S8C and S8D†). In contrast, more potent cytotoxicity of Re-UPC can be achieved by prolonged NIR irradiation (Fig. 4C and D) without obvious cell lethality. Therefore, these results suggested the combination of UCNPs and photoactivatable Re(I) complexes of cytotoxicity for inhibition of the targeted tumor cells in both drug susceptible cells and cisplatin resistant cells. Moreover, the use of NIR illumination, compared to UV light, can largely reduce photo-damage to cells.
Furthermore, to study the mechanism of photoactivated cytotoxicity, the N,N-dimethyl-4-nitrosoaniline (RNO) imidazole assay was carried out. In general, PBS solution containing RNO, imidazole and the Re(I) complex or Re-UPC was irradiated in fluorescence quartz cuvettes. The absorbance of N,N-dimethyl-4-nitrosoaniline at 440 nm was monitored by using a UV-vis spectrometer. As indicated in Fig. S9,† compared to the sample without light irradiation, an obvious decrease in the absorbance of RNO was observed when the Re(I) complex was activated by UV irradiation. Similarly, Re-UPC under NIR irradiation also caused a decreased absorbance of RNO, suggesting that the photo-activated cytotoxicity observed was probably associated with ROS generated during photoactivation.19,77 In addition, to better understand the feasibility concerning photoactivation of the Re(I) complex, further studies have also been carried out to investigate the possible ligand dissociation of the complex before and after light treatment.17,78,79 Typically, the myoglobin assay77 and NMR analysis were performed to study the photochemistry of the rhenium(I) complex. By following the standard myoglobin assay to determine CO release, a solution containing 50 μM myoglobin (Mb), 10 mM dithionite and 15 μM of the Re(I) complex in PBS solution was prepared and irradiated with UV light (8.9 mW cm−2). The possible CO dissociation was tracked by monitoring the absorbance of Mb at 557 nm.80,81 Compared to the sample without photoactivation (in Fig. S11†), the decreased absorbance band at 557 nm of Mb was observed upon light irradiation. Meanwhile, two bands of the MbCO adduct also appeared at around 540 nm and 577 nm. Similar spectral changes were also observed when the Re-UPC conjugate was irradiated under NIR light (Fig. S12†). These results indicated the conversion of Mb to MbCO, suggesting the possible dissociation of CO from the complex upon light irradiation.80 Moreover, although the detailed mechanism remains under further investigation, further NMR analysis demonstrated that there are no observable peaks found corresponding to the control molecule (e.g. free pyridine) after light irradiation (Fig. S13†), suggesting that the pyridine ligand was most likely kept in the Re complex and there was no obvious dissociation of pyridine from the whole molecular structure.63,82
4 Conclusions
In summary we have demonstrated an upconversion nanoparticle-based system for NIR activated cytotoxicity of a Re(I) complex. Typically, UCNPs can be used to activate the complex with upconversion luminescence triggered by NIR. The system can be effectively taken up by tumor cells via endocytosis. In addition, upon NIR irradiation, the locally activated drug exhibited enhanced cytotoxicity against both drug susceptible A2780 cells demonstrating that NIR irradiation combined with our system can achieve potent photoactivated cytotoxicity while minimizing unwanted photodamage to cells. Therefore, such a combination of UCNPs and a photoactivatable drug can provide a promising strategy to remotely control the activation of cytotoxicity to inhibit targeted tumor cell growth in both drug susceptible cells and cisplatin resistant cells and thus minimize potential side effects.Acknowledgements
We acknowledge the Start-Up Grant (SUG), Tier 1 RG64/10, RG11/13, RG35/15 awarded by Nanyang Technological University, Singapore and the National Natural Science Foundation of China (No. 21401217).Notes and references
- T. C. Johnstone, K. Suntharalingam and S. J. Lippard, Chem. Rev., 2016, 116, 3436–3486 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265–7279 CrossRef CAS PubMed.
- P. C. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- J. S. Butler and P. J. Sadler, Curr. Opin. Chem. Biol., 2013, 17, 175–188 CrossRef CAS PubMed.
- N. Graf and S. J. Lippard, Adv. Drug Delivery Rev., 2012, 64, 993–1004 CrossRef CAS PubMed.
- W. Han Ang and P. J. Dyson, Eur. J. Inorg. Chem., 2006, 2006, 4003–4018 CrossRef.
- S. K. Singh and D. S. Pandey, RSC Adv., 2014, 4, 1819–1840 RSC.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- L. He, Y. Li, C.-P. Tan, R.-R. Ye, M.-H. Chen, J.-J. Cao, L.-N. Ji and Z.-W. Mao, Chem. Sci., 2015, 6, 5409–5418 RSC.
- Y. Li, C. P. Tan, W. Zhang, L. He, L. N. Ji and Z. W. Mao, Biomaterials, 2015, 39, 95–104 CrossRef CAS PubMed.
- J. N. Liu, W. B. Bu and J. L. Shi, Acc. Chem. Res., 2015, 48, 1797–1805 CrossRef CAS PubMed.
- H. Chen, J. Tian, W. He and Z. Guo, J. Am. Chem. Soc., 2015, 137, 1539–1547 CrossRef CAS PubMed.
- P. Zhang, Y. Chen, Y. Zeng, C. Shen, R. Li, Z. Guo, S. Li, Q. Zheng, C. Chu, Z. Wang, Z. Zheng, R. Tian, S. Ge, X. Zhang, N. S. Xia, G. Liu and X. Chen, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6129–E6138 CrossRef CAS PubMed.
- S. Imstepf, V. Pierroz, R. Rubbiani, M. Felber, T. Fox, G. Gasser and R. Alberto, Angew. Chem., Int. Ed., 2016, 55, 2792–2795 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, L. A. Adams, N. Barlow, S. Ferrari, B. Graham and G. Gasser, ACS Med. Chem. Lett., 2014, 5, 809–814 CrossRef CAS PubMed.
- S. Perfahl, M. M. Natile, H. S. Mohamad, C. A. Helm, C. Schulzke, G. Natile and P. J. Bednarski, Mol. Pharm., 2016, 13, 2346–2362 CrossRef CAS PubMed.
- I. Chakraborty, S. J. Carrington, J. Hauser, S. R. J. Oliver and P. K. Mascharak, Chem. Mater., 2015, 27, 8387–8397 CrossRef CAS.
- A. Kastl, S. Dieckmann, K. Wahler, T. Volker, L. Kastl, A. L. Merkel, A. Vultur, B. Shannan, K. Harms, M. Ocker, W. J. Parak, M. Herlyn and E. Meggers, ChemMedChem, 2013, 8, 924–927 CrossRef CAS PubMed.
- A. Leonidova, V. Pierroz, R. Rubbiani, J. Heier, S. Ferrari and G. Gasser, Dalton Trans., 2014, 43, 4287–4294 RSC.
- K. Wahler, A. Ludewig, P. Szabo, K. Harms and E. Meggers, Eur. J. Inorg. Chem., 2014, 2014, 807–811 CrossRef PubMed.
- K. Yin Zhang, K. Ka-Shun Tso, M.-W. Louie, H.-W. Liu and K. Kam-Wing Lo, Organometallics, 2013, 32, 5098–5102 CrossRef.
- A. Leonidova and G. Gasser, ACS Chem. Biol., 2014, 9, 2180–2193 CrossRef CAS PubMed.
- K. Szacilowski, W. Macyk, A. Drzewiecka-Matuszek, M. Brindell and G. Stochel, Chem. Rev., 2005, 105, 2647–2694 CrossRef CAS PubMed.
- N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690–10701 RSC.
- G. Gasser and T. Joshi, Synlett, 2015, 275–284 Search PubMed.
- R. Deng, X. Xie, M. Vendrell, Y. T. Chang and X. Liu, J. Am. Chem. Soc., 2011, 133, 20168–20171 CrossRef CAS PubMed.
- P. Huang, W. Zheng, S. Zhou, D. Tu, Z. Chen, H. Zhu, R. Li, E. Ma, M. Huang and X. Chen, Angew. Chem., Int. Ed., 2014, 53, 1252–1257 CrossRef CAS PubMed.
- L. Zhou, R. Wang, C. Yao, X. Li, C. Wang, X. Zhang, C. Xu, A. Zeng, D. Zhao and F. Zhang, Nat. Commun., 2015, 6, 6938 CrossRef CAS PubMed.
- Y. Yang, Q. Shao, R. Deng, C. Wang, X. Teng, K. Cheng, Z. Cheng, L. Huang, Z. Liu, X. Liu and B. Xing, Angew. Chem., Int. Ed., 2012, 51, 3125–3129 CrossRef CAS PubMed.
- L. Cheng, C. Wang, X. Ma, Q. Wang, Y. Cheng, H. Wang, Y. Li and Z. Liu, Adv. Funct. Mater., 2013, 23, 272–280 CrossRef CAS.
- Q. Liu, W. Feng, T. Yang, T. Yi and F. Li, Nat. Protoc., 2013, 8, 2033–2044 CrossRef CAS PubMed.
- H. Dong, S. R. Du, X. Y. Zheng, G. M. Lyu, L. D. Sun, L. D. Li, P. Z. Zhang, C. Zhang and C. H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS PubMed.
- J. Rieffel, F. Chen, J. Kim, G. Chen, W. Shao, S. Shao, U. Chitgupi, R. Hernandez, S. A. Graves, R. J. Nickles, P. N. Prasad, C. Kim, W. Cai and J. F. Lovell, Adv. Mater., 2015, 27, 1785–1790 CrossRef CAS PubMed.
- G. Tian, Z. Gu, L. Zhou, W. Yin, X. Liu, L. Yan, S. Jin, W. Ren, G. Xing, S. Li and Y. Zhao, Adv. Mater., 2012, 24, 1226–1231 CrossRef CAS PubMed.
- P. Ma, H. Xiao, X. Li, C. Li, Y. Dai, Z. Cheng, X. Jing and J. Lin, Adv. Mater., 2013, 25, 4898–4905 CrossRef CAS PubMed.
- Y. Yang, F. Liu, X. Liu and B. Xing, Nanoscale, 2013, 5, 231–238 RSC.
- Y. Yang, B. Velmurugan, X. Liu and B. Xing, Small, 2013, 9, 2937–2944 CrossRef CAS PubMed.
- X. Li, L. Zhou, Y. Wei, A. M. El-Toni, F. Zhang and D. Zhao, J. Am. Chem. Soc., 2014, 136, 15086–15092 CrossRef CAS PubMed.
- W. Fan, W. Bu, Z. Zhang, B. Shen, H. Zhang, Q. He, D. Ni, Z. Cui, K. Zhao, J. Bu, J. Du, J. Liu and J. Shi, Angew. Chem., Int. Ed., 2015, 54, 14026–14030 CrossRef CAS PubMed.
- N. M. Idris, M. K. Jayakumar, A. Bansal and Y. Zhang, Chem. Soc. Rev., 2015, 44, 1449–1478 RSC.
- D. Yang, P. Ma, Z. Hou, Z. Cheng, C. Li and J. Lin, Chem. Soc. Rev., 2015, 44, 1416–1448 RSC.
- L. Cheng, K. Yang, Y. Li, J. Chen, C. Wang, M. Shao, S. T. Lee and Z. Liu, Angew. Chem., Int. Ed., 2011, 50, 7385–7390 CrossRef CAS PubMed.
- N. M. Idris, M. K. Gnanasammandhan, J. Zhang, P. C. Ho, R. Mahendran and Y. Zhang, Nat. Med., 2012, 18, 1580–1585 CrossRef CAS PubMed.
- Y. Dai, H. Xiao, J. Liu, Q. Yuan, P. Ma, D. Yang, C. Li, Z. Cheng, Z. Hou, P. Yang and J. Lin, J. Am. Chem. Soc., 2013, 135, 18920–18929 CrossRef CAS PubMed.
- C. Wang, L. Cheng, Y. Liu, X. Wang, X. Ma, Z. Deng, Y. Li and Z. Liu, Adv. Funct. Mater., 2013, 23, 3077–3086 CrossRef CAS.
- W. Fan, W. Bu, B. Shen, Q. He, Z. Cui, Y. Liu, X. Zheng, K. Zhao and J. Shi, Adv. Mater., 2015, 27, 4155–4161 CrossRef CAS PubMed.
- X. Zhu, W. Feng, J. Chang, Y. W. Tan, J. Li, M. Chen, Y. Sun and F. Li, Nat. Commun., 2016, 7, 10437 CrossRef CAS PubMed.
- G. Chen, H. Qiu, P. N. Prasad and X. Chen, Chem. Rev., 2014, 114, 5161–5214 CrossRef CAS PubMed.
- Y. Min, J. Li, F. Liu, E. K. Yeow and B. Xing, Angew. Chem., Int. Ed., 2014, 53, 1012–1016 CrossRef CAS PubMed.
- S. Lu, D. Tu, P. Hu, J. Xu, R. Li, M. Wang, Z. Chen, M. Huang and X. Chen, Angew. Chem., Int. Ed., 2015, 54, 7915–7919 CrossRef CAS PubMed.
- X. Ai, C. J. Ho, J. Aw, A. B. Attia, J. Mu, Y. Wang, X. Wang, Y. Wang, X. Liu, H. Chen, M. Gao, X. Chen, E. K. Yeow, G. Liu, M. Olivo and B. Xing, Nat. Commun., 2016, 7, 10432 CrossRef CAS PubMed.
- L. D. Sun, Y. F. Wang and C. H. Yan, Acc. Chem. Res., 2014, 47, 1001–1009 CrossRef CAS PubMed.
- F. Wang, R. Deng and X. Liu, Nat. Protoc., 2014, 9, 1634–1644 CrossRef CAS PubMed.
- E. M. Chan, E. S. Levy and B. E. Cohen, Adv. Mater., 2015, 27, 5753–5761 CrossRef CAS PubMed.
- G. Chen, H. Agren, T. Y. Ohulchanskyy and P. N. Prasad, Chem. Soc. Rev., 2015, 44, 1680–1713 RSC.
- X. Chen, D. Peng, Q. Ju and F. Wang, Chem. Soc. Rev., 2015, 44, 1318–1330 RSC.
- A. Sedlmeier and H. H. Gorris, Chem. Soc. Rev., 2015, 44, 1526–1560 RSC.
- B. Zhou, B. Shi, D. Jin and X. Liu, Nat. Nanotechnol., 2015, 10, 924–936 CrossRef CAS PubMed.
- S. Cui, D. Yin, Y. Chen, Y. Di, H. Chen, Y. Ma, S. Achilefu and Y. Gu, ACS Nano, 2012, 7, 676–688 CrossRef PubMed.
- S. Wu and H. J. Butt, Adv. Mater., 2016, 28, 1208–1226 CrossRef CAS PubMed.
- S. He, K. Krippes, S. Ritz, Z. Chen, A. Best, H. J. Butt, V. Mailander and S. Wu, Chem. Commun., 2015, 51, 431–434 RSC.
- A. E. Pierri, P. J. Huang, J. V. Garcia, J. G. Stanfill, M. Chui, G. Wu, N. Zheng and P. C. Ford, Chem. Commun., 2015, 51, 2072–2075 RSC.
- E. Ruggiero, A. Habtemariam, L. Yate, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2014, 50, 1715–1718 RSC.
- Z. Chen, W. Sun, H. J. Butt and S. Wu, Chem. – Eur. J., 2015, 21, 9165–9170 CrossRef CAS PubMed.
- E. Ruggiero, C. Garino, J. C. Mareque-Rivas, A. Habtemariam and L. Salassa, Chem. – Eur. J., 2016, 22, 2801–2811 CrossRef CAS PubMed.
- P. T. Burks, J. V. Garcia, R. GonzalezIrias, J. T. Tillman, M. Niu, A. A. Mikhailovsky, J. Zhang, F. Zhang and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 18145–18152 CrossRef CAS PubMed.
- H. Shi, T. Fang, Y. Tian, H. Huang and Y. Liu, J. Mater. Chem. B, 2016, 4, 4746–4753 RSC.
- J. D. Mase, A. O. Razgoniaev, M. K. Tschirhart and A. D. Ostrowski, Photochem. Photobiol. Sci., 2015, 14, 775–785 CAS.
- Y. Chen, G. Jiang, Q. Zhou, Y. Zhang, K. Li, Y. Zheng, B. Zhang and X. Wang, RSC Adv., 2016, 6, 23804–23808 RSC.
- J. V. Garcia, J. Yang, D. Shen, C. Yao, X. Li, R. Wang, G. D. Stucky, D. Zhao, P. C. Ford and F. Zhang, Small, 2012, 8, 3800–3805 CrossRef CAS PubMed.
- E. Ruggiero, J. Hernandez-Gil, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2015, 51, 2091–2094 RSC.
- B. Machura, M. Wolff, M. Jaworska, P. Lodowski, E. Benoist, C. Carrayon, N. Saffon, R. Kruszynski and Z. Mazurak, J. Organomet. Chem., 2011, 696, 3068–3075 CrossRef CAS.
- Y. Wei, F. Lu, X. Zhang and D. Chen, Chem. Mater., 2006, 18, 5733–5737 CrossRef CAS.
- H. Zhang, Y. Li, I. A. Ivanov, Y. Qu, Y. Huang and X. Duan, Angew. Chem., Int. Ed., 2010, 49, 2865–2868 CrossRef CAS PubMed.
- K. Baranowska and J. Okal, Appl. Catal., A, 2015, 499, 158–167 CrossRef CAS.
- Y. Cheng, S. Yu, J. Wang, H. Qian, W. Wu and X. Jiang, Macromol. Biosci., 2012, 12, 1326–1335 CrossRef CAS PubMed.
- I. Kraljić and S. E. Mohsni, Photochem. Photobiol., 1978, 28, 577–581 CrossRef.
- S. Sato, A. Sekine, Y. Ohashi, O. Ishitani, A. M. Blanco-Rodríguez, A. Vlček, T. Unno and K. Koike, Inorg. Chem, 2007, 46, 3531–3540 CrossRef CAS PubMed.
- P. V. Simpson, B. W. Skelton, P. Raiteri and M. Massi, New J. Chem., 2016, 40, 5797–5807 RSC.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC.
- H. Tabe, T. Shimoi, M. Boudes, S. Abe, F. Coulibaly, S. Kitagawa, H. Mori and T. Ueno, Chem. Commun., 2016, 52, 4545–4548 RSC.
- Y. Zhao, N. J. Farrer, H. Li, J. S. Butler, R. J. McQuitty, A. Habtemariam, F. Wang and P. J. Sadler, Angew. Chem., Int. Ed., 2013, 52, 13633–13637 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt01569g |
| This journal is © The Royal Society of Chemistry 2016 |