
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Upconverting nanoparticles for the near infrared photoactivation of transition metal complexes: new opportunities and challenges in medicinal inorganic photochemistry
Emmanuel
Ruggiero
a,
Silvia
Alonso-de Castro
a,
Abraha
Habtemariam
b and
Luca
Salassa
*acd
aCIC biomaGUNE, Paseo de Miramón 182, Donostia-San Sebastián, 20009, Spain. E-mail: lsalassa@cicbiomagune.es; lsalassa@dipc.org
bDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK
cKimika Fakultatea, Euskal Herriko Unibertsitatea and Donostia International Physics Center (DIPC), P.K. 1072, Donostia-San Sebastián, 20080, Spain
dIkerbasque, Basque Foundation for Science, Bilbao, 48011, Spain
First published on 20th July 2016
Abstract
The article highlights the emergent use of upconverting nanoparticles as tools for the near infrared photoactivation of transition metal complexes, identifying opportunities and challenges of this approach in the context of medicinal inorganic chemistry.
 Emmanuel Ruggiero (left) and Silvia Alonso-de Castro (right) | Emmanuel Ruggiero was born in Naples (Italy) in 1987. He received his BSc (2009) and MSc (2011) degrees in Chemistry from the University of Naples Federico II. He then moved to Pisa as a research fellow of NEST-Scuola Normale Superiore where he worked on the synthesis of non-natural peptides. In 2013, he started his PhD under the supervision of Dr Luca Salassa at CIC biomaGUNE (Spain), where he is exploring the use of upconverting nanoparticles as tools for the near infrared photoactivation of metal-based prodrugs. Silvia Alonso de Castro obtained her BSc (2011) and MSc (2013) degrees in Applied Chemistry and Pharmacology at the Jaume I University (Castellón de la Plana, Spain). In her undergraduate thesis, she developed new tetrapeptidic hydrogels under the supervision of Dr Juan F. Miravet and studied their rheology in a joint project with P&G. Silvia joined Salassa's group in mid-2014 for her PhD on the development of upconverting nanomaterials for theranostics. |
 Abraha Habtemariam (right) and Luca Salassa (left) | Dr Abraha Habtemariam obtained his BSc degree from the University of East Anglia and his PhD from the University of Sussex. After a post-doctoral fellowship at the University of Toronto (Canada), he joined Peter Sadler's group at Birkbeck College, University of London and in 1996 moved to the University of Edinburgh. In 2007 he moved to the University of Warwick as a Senior Research Fellow. He was Ikerbasque Visiting Professor at CIC biomaGUNE, San Sebastián, Spain (2013–2014). Dr Luca Salassa obtained his MSc degree and PhD from the University of Turin (Italy). After a postdoctoral position in Missoula (USA) at the University of Montana, he was awarded a Marie Curie IEF fellowship (2008) and joined Sadler's group at the University of Warwick (UK). In 2012, he moved to CIC biomaGUNE in San Sebastián (Spain) where he established his research group. Luca has recently been awarded an Ikerbasque Professorship. |
Introduction
The portion of the electromagnetic spectrum that spans from UV to near infrared (NIR) has wavelength-dependent effects on biomolecules and displays different tissue penetration capabilities, as a result of the distinct absorption properties that tissue constituents have.1 As shown in Fig. 1 for skin, NIR light is the most appropriate for maximum tissue penetration2 and for limiting photodamage. In fact, prolonged exposure to short-wavelength light (e.g. UVC-A) can seriously harm cells by inducing photochemical reactions. On the other hand, water absorption is predominant at wavelengths higher than 1000 nm, reducing the penetration of light and causing overheating of biological components.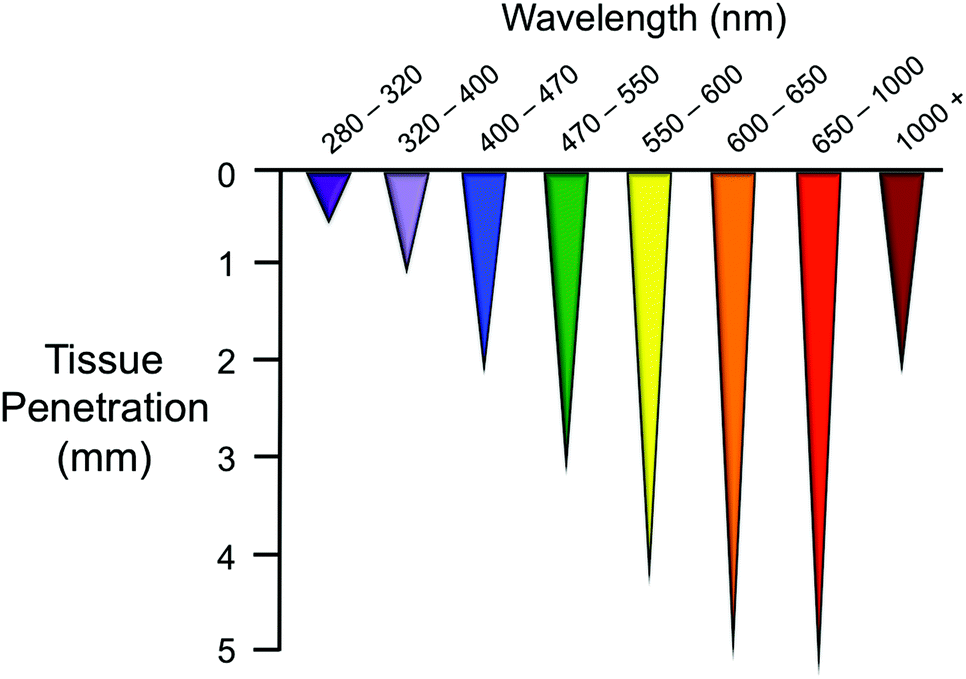 | ||
| Fig. 1 Penetration depth of light of different wavelengths in skin tissue. The image is adapted from ref. 1. | ||
For this reason, the so-called “phototherapeutic window” is considered to be in the range 650–1000 nm. Light of approx. 800 nm is ideal for use in biological systems since it does minimize the excitation of biomolecules and water heating.1
As well documented by several review articles,3 the success of photodynamic therapy (PDT)4 has prompted medicinal inorganic chemists to look at the unique photophysics and photochemistry of metal complexes and to devise new light-activatable prodrugs which improve the selectivity of metal-based anticancer agents. Various design concepts have been adopted in the development of photoactivatable metal complexes. Apart from their use as alternative PDT photosensitizers, metal-based prodrugs have also been designed to exert their antineoplastic action through mechanisms that are different from PDT, i.e. oxygen independent, such as for example DNA-photobinding or photouncaging of bioactive molecules. However, oxygen sensitization is often concurrent with photochemical transformation of the prodrug agent.
The great majority of light-activatable metal-based prodrugs are inactive upon NIR light excitation, since they typically require high-energy UV-visible light to trigger a photochemical response. Such an intrinsic feature of metal complexes hampers somewhat their current progress towards more advanced preclinical and clinical stages. In principle, NIR light sources would favour the use of photoactivatable metal-based prodrugs in a wider range of cancers and allow tuning light doses during treatment. For example, the maximum permissible exposure (MPE) of human skin for visible light (400–700 nm) is 0.20 W cm−2, while MPE for NIR light can reach 0.73 W cm−2.5 Furthermore, the higher penetration depth of NIR light would enable the treatment of thicker tumours.6
Nevertheless, it is fundamental to point out that visible light can be effective in the application of metal complexes in photochemotherapy of more superficial tumours, as recently demonstrated by the approval for clinical trials of a PDT ruthenium-polypyridyl photosensitizer.7
Excluding from the discussion inert metal-based systems that work as PDT photosensitizers, the synthetic design of metal complexes that can be light-activated in the phototherapeutic window has been extremely challenging. Very few exceptions have been reported due to the trade-off that exists between achieving good absorption in the red part of the spectrum and maintaining high photoreactivity.8 This requires an appropriate choice of ligands (e.g. dye-like) and optimal tuning of ligand and metal orbital energies, which in practice is not always feasible. Indeed, photoactivation of metal complexes in the phototherapeutic window or nearby has been reported; however, generally such systems exploit weak absorption bands, whose intensity is hard to improve.9
As an alternative photoactivation approach, inorganic nanomaterials offer outstanding optical features which can serve to shift excitation wavelengths for metal complexes into the red and near infrared regions. Moreover, the versatile surface chemistry of nanomaterials provides a myriad of strategies to develop innovative metal-based prodrug carriers for photochemotherapy.
In this Frontier article we discuss recent research efforts aimed at combining a specific class of nanomaterials, that is, upconverting nanoparticles, with photoactivatable metal complexes of interest in medicinal inorganic chemistry and carry out near infrared photoactivation. For the sake of brevity, we have omitted from this summary the early pioneering work on quantum dots and the promising results obtained by triplet–triplet annihilation upconversion approaches, which have been reviewed elsewhere.10
Upconverting nanoparticles
In recent years, upconverting nanoparticles (UCNPs) have been intensely investigated for application as imaging probes and drug carriers in biology and medicine, due to their exceptional optical and chemical properties. UCNPs are lanthanide-doped crystals which absorb and convert low-energy NIR photons (980 or 808 nm) into high energy UV-visible light.11 In UCNPs, upconversion is a non-linear optical luminescence phenomenon where two or more photons are absorbed by a material and a single photon of shorter wavelength (anti-Stokes shift) is subsequently emitted. Various mechanisms can contribute to upconversion emission, including excited state absorption (ESA), energy transfer upconversion (ETU) and photon avalanche (PA), ETU being the dominating pathway in UCNPs.12 Typically, the brightest UCNPs are composed of a fluorine inert matrix (NaREF4, RE = Y, Gd, Lu), which offers doping sites for optically active lanthanide ions. Ions employed as dopants are divided into two classes: donors (typically Yb3+ca. 20 mol%) and acceptors (Er3+, Tm3+, Ho3+, Eu3+, Tb3+ca. <5 mol%). Fig. 2 shows the emission spectra and energy level diagrams for typical NaYF4:Yb,Tm and NaYF4:Yb,Er UCNPs. Upconversion emission in these UCNPs profit from the good absorption cross-sections of Yb3+ at 980 nm and can be obtained by excitation with relatively low power (0.1–102 W cm−2).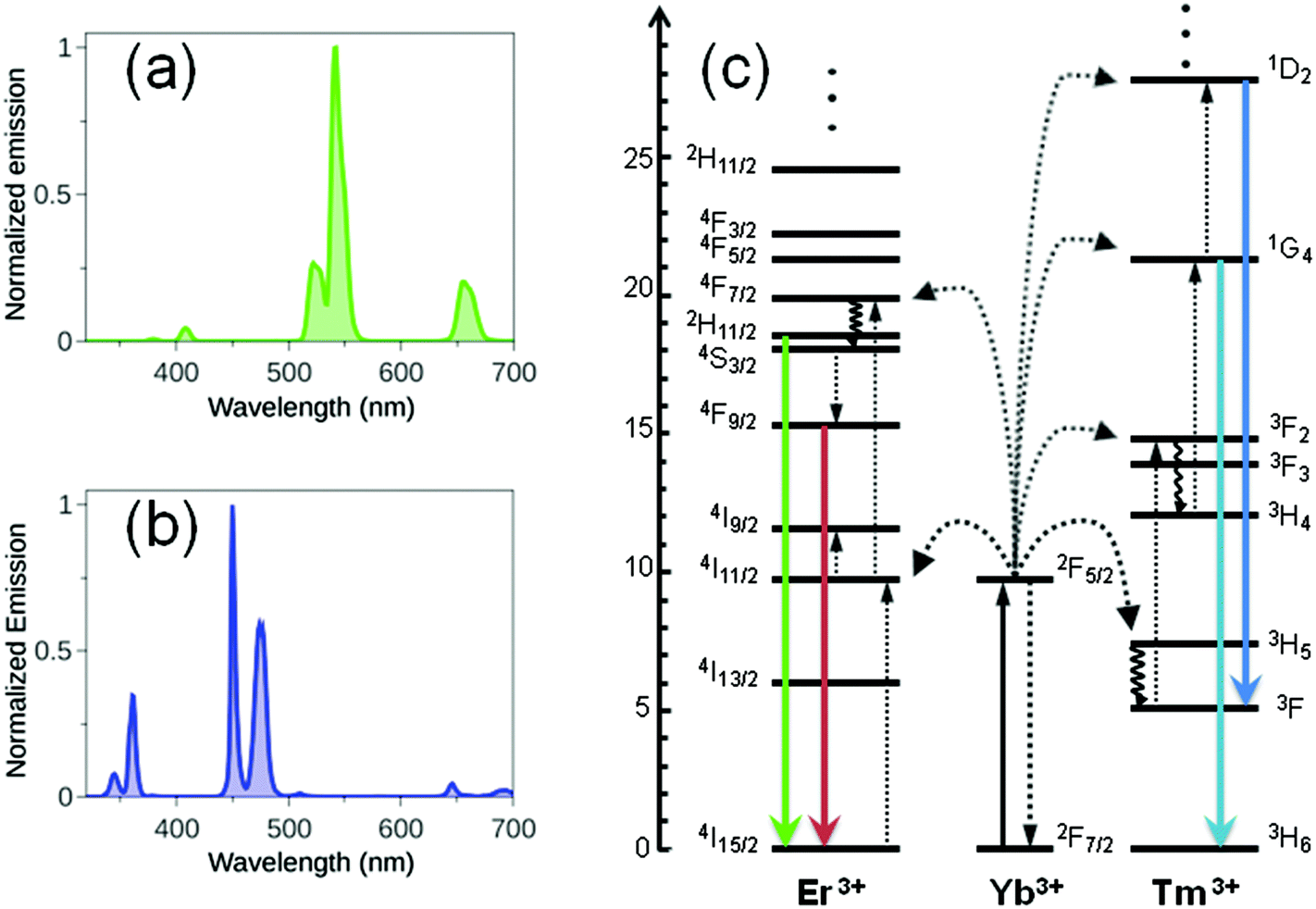 | ||
| Fig. 2 (a) Emission spectra (λexc = 980 nm) of a THF solution of NaYF4:Yb/Er (20%/2%) and NaYF4:Yb/Tm (30%/0.5%) UCNPs and their corresponding (b) energy-level diagrams (adapted from ref. 11b). Full, dotted, and curly arrows indicate radiative and nonradiative energy transfer and multiphonon relaxation processes, respectively. | ||
Upconversion luminescence increases with the size of UCNPs. For instance, quantum yield values for NaYF4:Yb,Er (20%/2%) with different diameters (from 10 to 100 nm) span from 0.005% to 0.3%. Growing a protective shell with different thicknesses and compositions around an active core serves to modulate and/or improve the optical properties of UCNPs. In fact, shell passivation reduces surface defects in nanocrystals and protects optically active lanthanide ions from non-radiative deactivation by solvents and organic capping ligands which cover the UCNP surface. Core@shell nanoparticles are generally brighter than core-only nanoparticles, with an increase in upconverted luminescence up to 10–20 folds.13 Importantly, co-doping of UCNPs with Yb3+ and Nd3+ donor ions affords nanoparticles that can be excited at 808 nm (reduced water heating) where Nd3+ has a large absorption cross section.12
The principal approach employed to fabricate bright UCNPs is the thermal decomposition method, which produces nanoparticles with controlled composition, crystalline phase and size (detailed studies on the crystalline properties and growth mechanism of UCNPs have been reported for example by Haase and co-workers).14
NIR activation of metal complexes by UCNPs
The interest of inorganic medicinal chemists in UCNPs began to show an upsurge recently (around 2012), motivated by the prospect that upconverted UV-visible emissions might activate transition metal complexes adsorbed or anchored onto the surface of nanoparticles by means of NIR-light excitation. Few, yet encouraging, systems have demonstrated that Er- and Tm-doped UCNPs indeed provide a novel route for near infrared photochemistry in metal complexes. These early examples involve so far four transition metals, Mn, Fe, Ru and Pt, all of which undergo chemical transformation upon UV-visible light excitation. The primary and basic objectives of these studies were to explore different anchoring strategies for loading photoactivatable metal complexes onto UCNPs and evaluate the photochemical behaviour of the resulting nanomaterials under NIR light irradiation. Evidently, several of the systems reported here should be improved both in terms of photoreactivity and applicability in biological environments for use in photochemotherapy. However, they represent a benchmark which may serve to develop new light-activation approaches and to improve the design of UCNP-based nanomaterials for more advanced applications in biology and medicine.In 2012 Ford and Zhang first reported, in a joint article, the NIR-light activation of the photoactive Roussin's black salt anion, Na[Fe4S3(NO)7] (RBS).15 This inorganic salt has a broad absorption band in the visible region (400–600 nm) and has been extensively investigated as a nitric oxide (NO) photoreleasing moiety (Fig. 3a). NO plays a key role in vasodilatation, immune response, tumour growth and suppression.16 Core@shell NaYF4:Yb,Er@NaYF4 UCNPs (ca. 60 nm) were selected for attempting NIR photo-stimulation of the RBS anion. Silica coating of UCNPs served to load RBS via impregnation of the mesoporous SiO2 shell, whose pores were eventually covered with polyallylamine hydrochloride (PAA HCl) to afford nanoconstructs capable of photoreleasing NO. The use of an NO sensitive electrode confirmed that the nanoparticles successfully generated NO upon NIR-light remote activation in a power-dependent manner (1–4.5 W) and using sequential excitation times of 30, 60, 120, and 240 s.
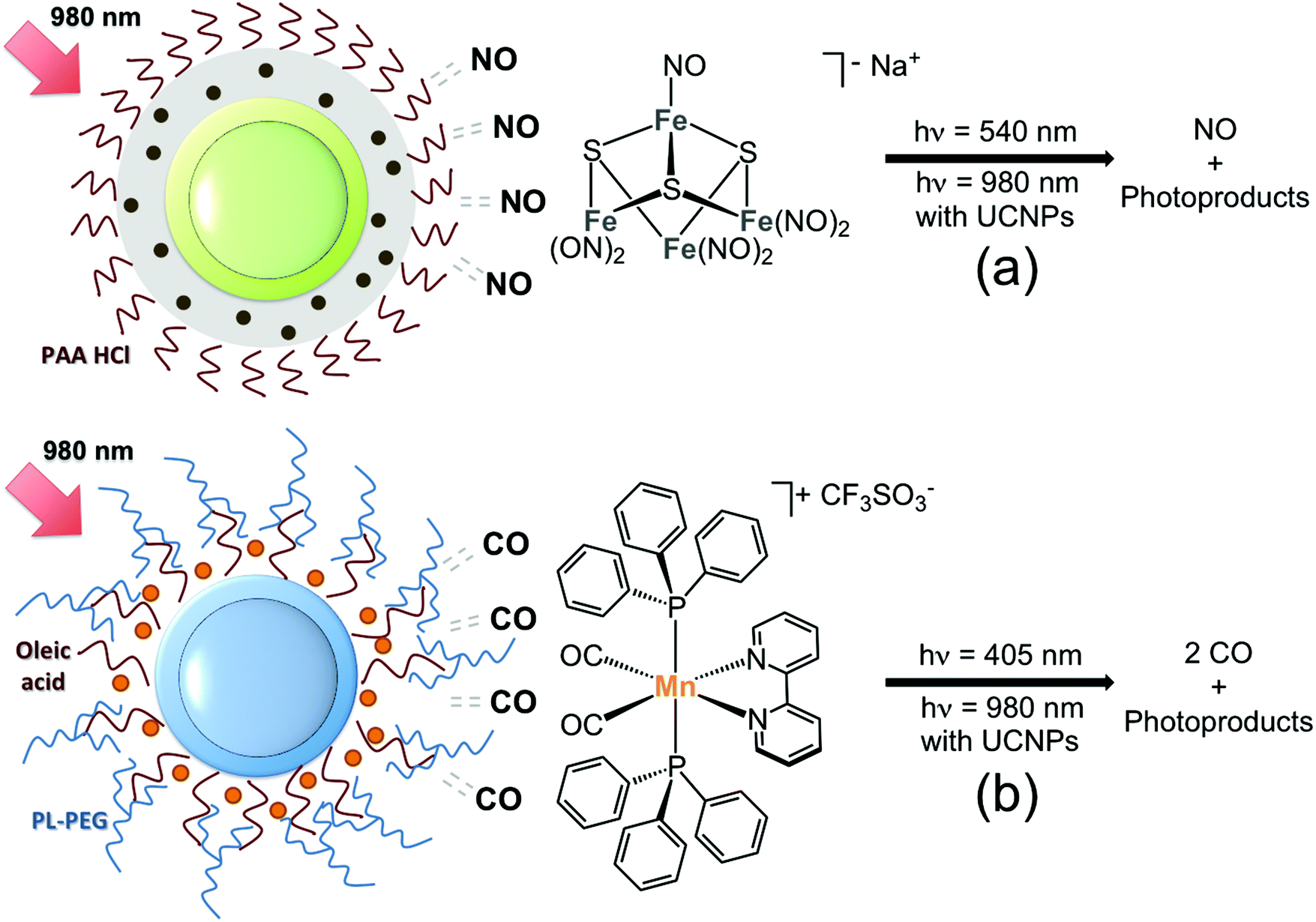 | ||
Fig. 3 Schematic representation of UCNP nanocarriers for the NIR-light photorelease of (a) NO from RBS ( ) and (b) CO from the complex cis,trans-[Mn(bpy)(CO)2(PPh3)2][CF3SO3] ( ) and (b) CO from the complex cis,trans-[Mn(bpy)(CO)2(PPh3)2][CF3SO3] ( ). Adapted from ref. 15 and 18. ). Adapted from ref. 15 and 18. | ||
Shortly afterwards, the Ford group encapsulated UCNPs and the RBS anion in a NIR transparent biocompatible polymer disk, as an implant model for the controlled therapeutic release of NO.17 The disk was made of a poly(dimethylsiloxane) matrix where RBS and Er- or Tm-doped core@shell NaGdF4@NaYF4 particles were successfully trapped due to their affinity for the hydrophobic polymer. UCNPs with different acceptor ions (Er3+, Tm3+) were capable of photo-stimulating NO release from RBS upon 980 nm irradiation (2.5 W cm−2). When different porcine tissue samples (skin, muscle, fat) were employed as a barrier in front of the polymer disk, irradiation experiments demonstrated that NIR light could penetrate the tissue protective layer, and generate NO from the RBS precursor, even if in lower amounts. An oscillating laser beam photolysis apparatus was designed to deliver heterogeneously high laser power densities (300 W cm−2) to the polymeric disk in these experiments and avoid tissue damage.
In 2015, Ford, Zheng and their co-workers have directed the use of UCNPs towards the phototriggered release of carbon monoxide (CO) from the complex cis,trans-[Mn(bpy)(CO)2(PPh3)2][CF3SO3] (where bpy = 2,2′-bipyridine and PPh3 = triphenylphosphine, Fig. 2b).18 CO is an attractive small molecule for phototherapeutic applications due to its cytoprotective and anti-inflammatory properties.19 The Mn complex was loaded on core@shell NaGdF4:Yb,Tm@NaGdF4 UCNPs (20–30 nm) functionalized with an amphiphilic PEG polymer, which provided the appropriate hydrophobic environment to entrap securely the water insoluble cis,trans-[Mn(bpy)(CO)2(PPh3)2]+. Loading efficiency was determined by ICP-AES, indicating that at best 80% of the Mn complex used in the preparation protocol could be incorporated onto the UCNPs. In addition, this nanocarrier could elicit photochemical uncaging of CO under NIR-light irradiation (2 W, t = 0–5 min) as evidenced by gas chromatography and myoglobin binding studies.
In 2013, our laboratory started a research programme on upconverting nanomaterials for NIR-light photoactivation of coordination and organometallic compounds. Our first study was concerned with the UCNP-mediated photoactivation of cis-[Ru(bpy)2(py)2][Cl]2 (where py = pyridine),20 chosen as a model for biologically active ruthenium polypyridyl complexes, which have found application in neuronal stimulation,21 protein inhibition22 and cancer therapy.8a,23cis-[Ru(bpy)2(py)2][Cl]2 has a well-understood photochemical behaviour24 and photodissociates one pyridine ligand with high efficiency (Fig. 4a) upon excitation of its metal-to-ligand charge transfer band. In this study, 980 nm photoactivation (25 W cm−2, t = 0.5–4 h) of the complex was achieved by employing large NaYF4:Yb,Er nanoparticles (80 nm), selected for their brightness. Photocleavage of one coordinated pyridine ligand and subsequent formation of the cis-[Ru(bpy)2(py)(H2O)]2+ complex were monitored over time by UV-visible (red-shift of the lowest-energy absorption band) and by 1H NMR (appearance of diagnostic signals for the photoproducts), analyzing aliquots of the supernatant solution obtained upon centrifugation of the reaction mixture. After 4 h of NIR light irradiation, 70% of cis-[Ru(bpy)2(py)2][Cl]2 underwent photoconversion as determined by integration of 1H NMR signals.
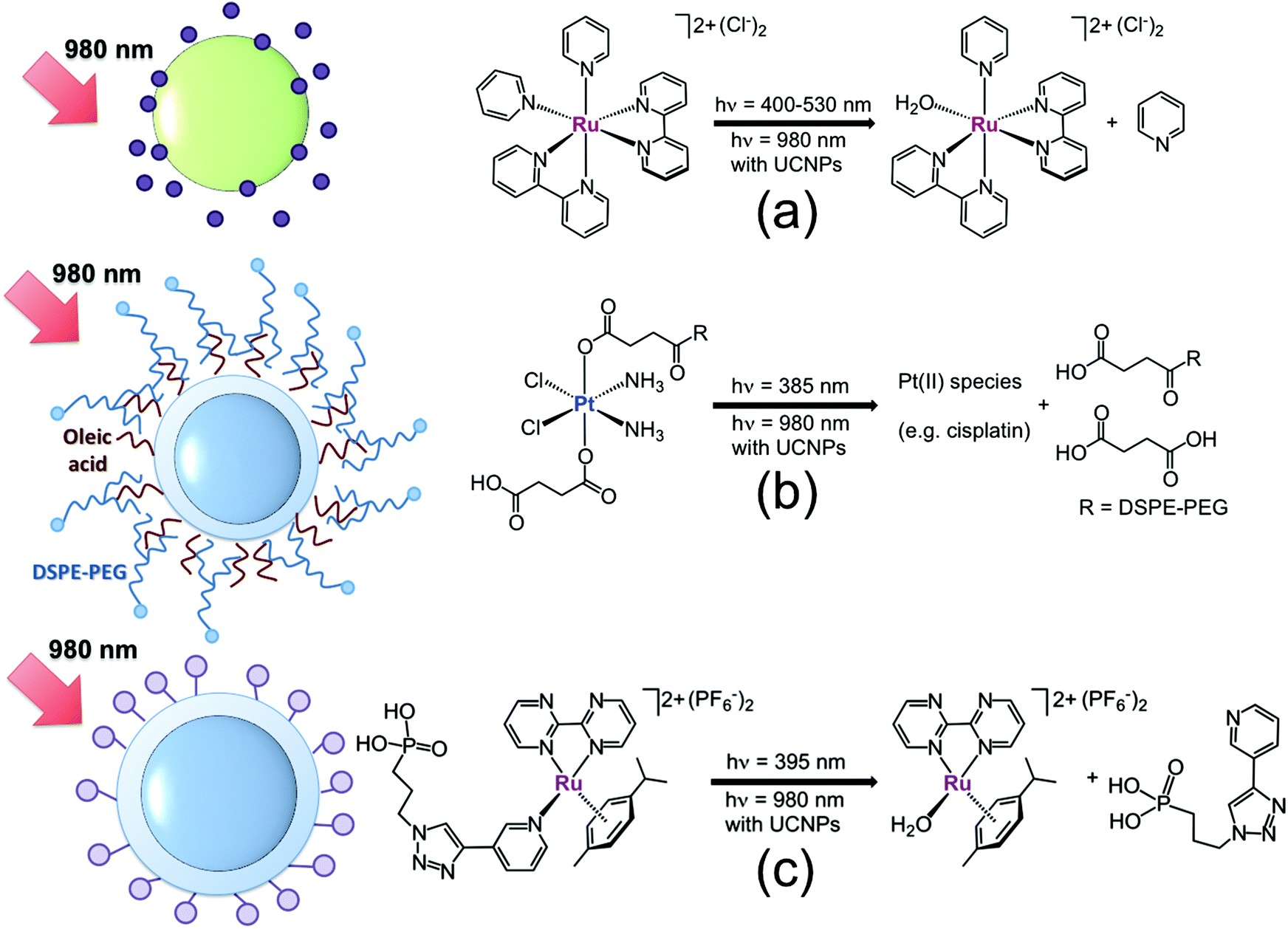 | ||
Fig. 4 Schematic representation of UCNP systems for the NIR-light activation of (a) cis-[Ru(bpy)2(py)2][Cl]2 ( ), (b) cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2H)2] functionalized with DSPE-PEG ( ), (b) cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2H)2] functionalized with DSPE-PEG ( ) and (c) [( η6-p-cym)Ru(bpm)(P-Trz-Py)][PF6]2 ( ) and (c) [( η6-p-cym)Ru(bpm)(P-Trz-Py)][PF6]2 ( ). Adapted from ref. 20, 26 and 28. ). Adapted from ref. 20, 26 and 28. | ||
UCNPs were previously treated with HCl to remove oleic acid ligands from their surface, improve their aqueous solubility and facilitate surface adsorption of the charged Ru complexes. XPS analysis revealed that a fraction of cis-[Ru(bpy)2(py)2][Cl]2 was still adsorbed onto the surface of UCNPs even after extensive washing, indicating electrostatic interactions between the negatively charged naked UCNPs and the positively charged complexes were sufficient to provide enough confinement for the NIR photochemistry to occur.
Inert PtIV complexes have attracted a lot of attention in the last decade as light-activatable prodrug candidates. They often combine little or no dark toxicity in cells and high stability in cellular environments with the capacity for being photoconverted into the anticancer-active square planar PtII species under UVA-visible light irradiation.25 However, PtIV complexes suffer from extremely poor absorptions in the visible region, which rarely extend above 400 nm. In order to demonstrate that this limitation could be overcome, we employed NIR light (7.3 W cm−2, t = 0.5–3.5 h) and Tm-doped NaYF4:Yb,Tm@NaYF4 UCNPs (40 nm) to promote the photoreduction of cis,cis,trans-[Pt(NH3)2(Cl)2(O2CCH2CH2CO2H)2] to cisplatin-like PtII species (Fig. 4b).26 A biocompatible (FDA-approved) PEGylated phospholipid was coupled with the PtIV complex to decorate UCNPs with prodrug molecules. In the nanocarrier, the oleate chains capping UCNPs hydrophobically interact with the tails of the PEGylated phospholipid, while the hydrophilic PEG units of the polymer are exposed to the solvent, conferring further stabilization and water solubility on the system. After 3.5 h of NIR-light irradiation, the PtIV prodrug agent loaded onto the nanocarrier was fully converted into PtII species, as determined by XPS and NMR analyses of the supernatant solution (separated from the UCNP-containing pellet by centrifugation).
Earlier this year, we showed that UCNPs were also suitable for NIR-light photoactivation of pyridinato RuII–arene complexes. Previous studies27 showed that such a class of compounds is stable in the dark and selectively dissociates the monodentate pyridinato ligand coordinated to the Ru centre upon visible light excitation, hence generating highly reactive aqua species with a potential to exert anticancer activity. In order to control, by NIR light, this key reaction in the mechanism of action of anticancer Ru–arene piano-stool complexes, we prepared two novel pyridinato RuII–arene complexes and functionalized via click chemistry the photolabile pyridinato ligand with a phosphonate moiety. The derivative [(η6-p-cym)Ru(bpm)(P-Trz-Py)][PF6]2 (where bpm = 2,2′-bipyrimidine and P-Trz-Py = [3-(1-pyridin-3-yl-[1,2,3]triazol-4-yl)-propyl]phosphonic acid diethyl ester, Fig. 4c) was directly loaded onto NaYF4:Yb,Tm@NaYF4 UCNPs (40 nm) exploiting the high affinity of phosphonates for the UCNP surface. A rough estimate by UV-visible provided a complex/UCNP ratio of approximately 3000![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (3.5 wt%). Once conjugated to UCNPs, the Ru complex selectively generated the aqua derivative [(η6-p-cym)Ru(bpm)(H2O)]2+ upon 980 nm excitation. Approximately 55% of the starting Ru complex was photoconverted after 4.5 h at a power density of 8.1 W cm−2. NMR and UPLC-MS demonstrated that the aqua photoproduct was able to bind the DNA model base guanosine 5′-monophosphate when the two were co-incubated.28
:1 (3.5 wt%). Once conjugated to UCNPs, the Ru complex selectively generated the aqua derivative [(η6-p-cym)Ru(bpm)(H2O)]2+ upon 980 nm excitation. Approximately 55% of the starting Ru complex was photoconverted after 4.5 h at a power density of 8.1 W cm−2. NMR and UPLC-MS demonstrated that the aqua photoproduct was able to bind the DNA model base guanosine 5′-monophosphate when the two were co-incubated.28
Later on in 2016, Wang's group used Tm-doped UCNPs to activate another Ru polypyridyl complex by NIR light (Fig. 5). [Ru(bpy)2(C18H37CN)2][Cl]2 was loaded onto diamond-shaped LiYF4:Yb,Tm UCNPs (70 nm along the long axis), taking advantage of the aliphatic chain of the C18H37CN ligands to promote strong hydrophobic interactions with the oleic acid ligands of UCNPs. Afterwards, UCNPs were capped with PEG to improve aqueous solubility. ICP-AES indicated that a loading capability of 0.01 g of complex per gram of particle was achieved. The resulting nanocarriers released the two nitrile monodentate ligands upon 980 nm excitation (3 W cm−2, t = 10–70 min), generating the bio-active species [Ru(bpy)2(H2O)2]2+. UV-visible measurements of supernatant solutions after centrifugation showed that ca. 80% of photoconversion is reached upon 40 minutes of NIR-light irradiation. As reported earlier,29 such an aqua product was able to bind plasmid DNA whilst no change in electrophoresis mobility was noted under dark conditions.30
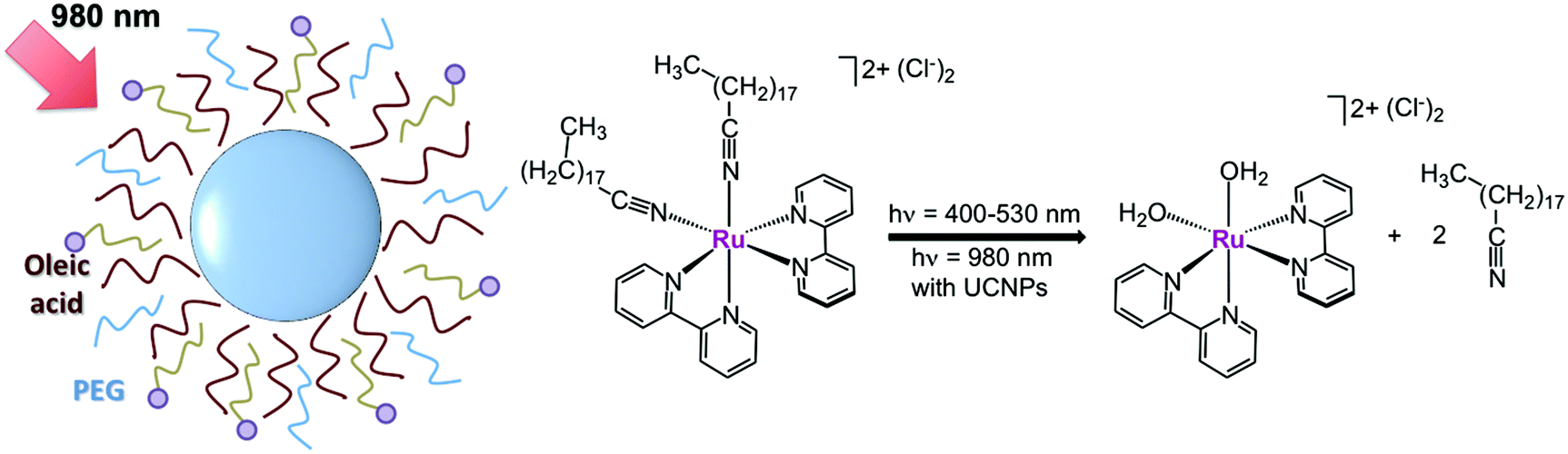 | ||
Fig. 5 Schematic representation of the NIR-light activation of the prodrug candidate [Ru(bpy)2(C18H37CN)2][Cl]2 ( ) mediated by UCNPs. Adapted from ref. 30. ) mediated by UCNPs. Adapted from ref. 30. | ||
Wu's group has exploited Ru-polypyridyl photochemistry to achieve NIR-light controlled release of the clinically approved anticancer drug doxorubicin. They prepared NaYF4:Yb,Tm@NaYF4 particles coated with mesoporous silica to encapsulate doxorubicin and the complex [Ru(bpy)2(PMe3)((3-aminopropyl)(EtO)3Si)][PF6]2. The silyl group of the aminopropyl ligand was key for covalently anchoring the Ru compound onto UCNPs. As schematized in Fig. 6, the Ru complex acted as a photocleavable molecular valve, which trapped doxorubicin molecules inside silica pores.31 Under NIR-light irradiation as low as 0.35 W cm−2, the complex photodissociated the silyl ligand and liberated doxorubicin from the UCNPs. The authors performed control experiments on nanoparticles grafted with azobenzene, which was selected as a photoactive but non-cleavable valve. Doxorubicin release profiles of these nanomaterials proved that the delivery of the drug was efficiently controlled by the photochemistry of the ruthenium complex. A laser power density of 0.64 W cm−2 could induce doxorubicin release from the system through a thick pork tissue. Upon 10–30 min of NIR-light exposure (0.35 W cm−2), the nanocarrier (300 μg ml−1) incubated in HeLa cells decreased the cell viability to 30–40%, while the system displayed no toxicity under dark conditions. Control experiments under NIR light irradiation also demonstrate that UCNPs loaded with the Ru complex without doxorubicin did not affect cell viability. Remarkably, the maximum light intensity used to photoactivate the Wu nanoconstruct was lower than the maximum permissible exposure of skin.
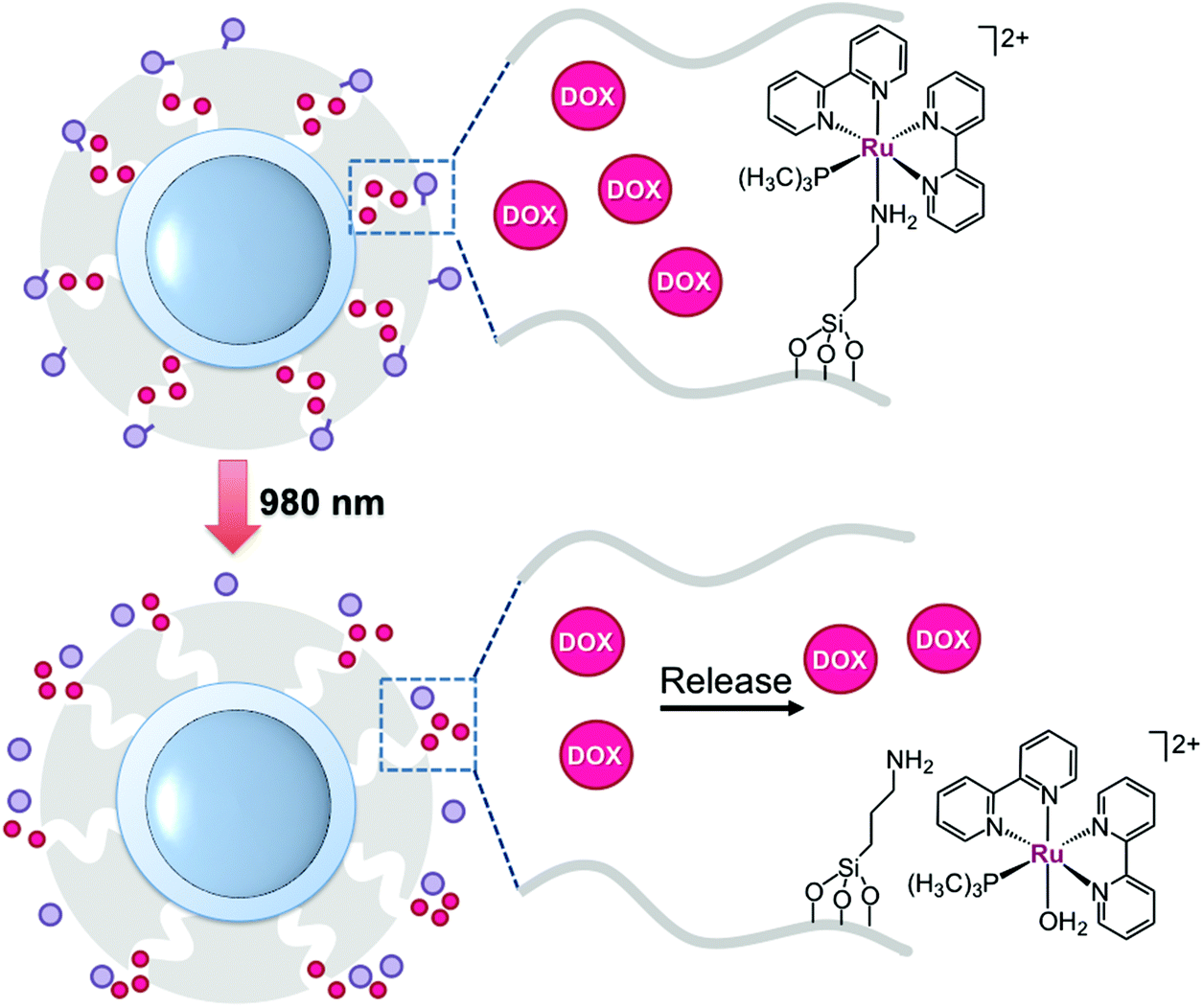 | ||
Fig. 6 Schematic representation of the nanocarrier developed by Wu and coworkers. Doxorubicin (DOX,  ) is released by NIR-light activation of a Ru polypyridyl complex ( ) is released by NIR-light activation of a Ru polypyridyl complex ( ) acting as a valve. The figure is adapted from ref. 31. ) acting as a valve. The figure is adapted from ref. 31. | ||
Conclusions and future outlook
NIR-light photochemical activation of biologically relevant metal complexes is still in its infancy. UCNPs hold great promise as NIR-light sensitizers and the examples reported indeed show that UCNPs can shift activation wavelengths of metal-based prodrugs into the so-called phototherapeutic window. However, systems based on UCNPs and metal complexes work so far at a proof-of-concept level and many more obstacles need to be overcome to advance forward. In the first instance, lowering the laser power density employed during light administration and the light exposure time are fundamental requirements. In this context, it will be crucial to improve the upconversion efficiency of UCNPs and optimize the photochemical response of nanoconstructs for maximal efficiency. The ultimate goal is to achieve excitation intensities that are lower than 0.73 W cm−2. Although much needs to be improved, Wu's group has shown that the choice of the photosensitive compounds is critical and that activation of ruthenium metal complexes can be achieved with such intensities or lower and through tissues.31,32In addition, NIR-sensitive hybrid materials based on UCNP and metal complexes have to face and tackle challenges that are typically encountered in the development of nanodelivery systems and nanodrugs. These include aspects of the chemical design such as optimizing the solubility of nanoplatforms and their loading with metal-based drugs. Moreover, systematic biological studies are fundamental to obtain a thorough knowledge of the interactions between UCNPs and the healthy and tumour cell microenvironments, understanding how to improve materials circulation and distribution in organisms, and tailoring their toxicity mechanism of action to specific tumours.33
Therefore, many more years of investigation are required to eventually translate the use of UCNPs into a real clinical application. Yet, NIR-activated photochemistry of metal complexes may lead, in the process, to new discoveries and also provide innovative solutions for other applications.
In addition to their use as phototriggers and luminescent probes, UCNP nanomaterials also offer outstanding opportunities for other imaging modalities. Their composition and structural features make UCNPs suitable for magnetic resonance imaging (MRI), computed tomography (CT), positron emission tomography (PET) and single-photon emission computed tomography (SPECT). Often these modalities have been successfully exploited simultaneously in one UCNP.11a
Together with the good biocompatibility and relatively low toxicity profile in vitro and in vivo,34 these features make UCNPs worth investigating for application in theranostics.
Acknowledgements
We gratefully acknowledge the Spanish Ministry of Economy and Competitiveness (grant CTQ2012-39315 and BES-2013-065642) and the Department of Industry of the Basque Country (grant ETORTEK). L. S. and E. R. were supported by the MICINN of Spain with the Ramón y Cajal Fellowship RYC-2011-07787 and by the MC CIG fellowship UCnanomat4iPACT (grant no. 321791). We thank Ikerbasque for the Visiting Professor Fellowship to A. H. (2013–2014) and members of the European COST Actions CM1105 and CM1403 for stimulating discussions.References
- D. Barolet, Semin. Cutaneous Med. Surg., 2008, 27, 227 CrossRef CAS PubMed.
- S. Stolik, J. A. Delgado, A. Perez and L. Anasagasti, J. Photochem. Photobiol., B, 2000, 57, 90 CrossRef CAS.
- (a) N. J. Farrer, L. Salassa and P. J. Sadler, Dalton Trans., 2009, 10690 RSC; (b) U. Schatzschneider, Eur. J. Inorg. Chem., 2010, 1451 CrossRef CAS; (c) E. C. Glazer, Isr. J. Chem., 2013, 53, 391 CrossRef CAS.
- J. M. Dabrowski and L. G. Arnaut, Photochem. Photobiol. Sci., 2015, 14, 1765 CAS.
- Laser safety handbook, Northwestern University, Evanston, 2011 Search PubMed.
- K. Plaetzer, B. Krammer, J. Berlanda, F. Berr and T. Kiesslich, Lasers Med. Sci., 2009, 24, 259 CrossRef CAS PubMed.
- (a) G. Shi, S. Monro, R. Hennigar, J. Colpitts, J. Fong, K. Kasimova, H. Yin, R. DeCoste, C. Spencer, L. Chamberlain, A. Mandel, L. Lilge and S. McFarland, Coord. Chem. Rev., 2015, 282–283, 127 CrossRef CAS; (b) http://mcfarland.acadiau.ca/news-reader/items/our-licensed-pdt-agent-nears-human-clinical-trials.html .
- (a) E. Wachter, D. K. Heidary, B. S. Howerton, S. Parkin and E. C. Glazer, Chem. Commun., 2012, 48, 9649 RSC; (b) M. J. Rose and P. K. Mascharak, Inorg. Chem., 2009, 48, 6904 CrossRef CAS PubMed; (c) M. J. Rose, N. L. Fry, L. Hinck and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 8834 CrossRef CAS PubMed.
- (a) B. Banik, K. Somyajit, G. Nagaraju and A. R. Chakravarty, RSC Adv., 2014, 4, 40120 RSC; (b) S. L. H. Higgins and K. J. Brewer, Angew. Chem., Int. Ed., 2012, 51, 11420 CrossRef CAS PubMed; (c) M. A. Sgambellone, A. David, R. N. Garner, K. R. Dunbar and C. Turro, J. Am. Chem. Soc., 2013, 135, 11274 CrossRef CAS PubMed; (d) J. Wang, J. Newman Jr., S. L. H. Higgins, K. M. Brewer, B. S. J. Winkel and K. J. Brewer, Angew. Chem., Int. Ed., 2013, 52, 1262 CrossRef CAS PubMed; (e) B. Maity, M. Roy and A. R. Chakravarty, J. Organomet. Chem., 2008, 693, 1395 CrossRef CAS; (f) J. D. Knoll, B. A. Albani and C. Turro, Acc. Chem. Res., 2015, 48, 2280 CrossRef CAS PubMed; (g) U. Basu, I. Pant, P. Kondaiah and A. R. Chakravarty, Eur. J. Inorg. Chem., 2016, 1002 CrossRef CAS; (h) Y. Sun, L. E. Joyce, N. M. Dickson and C. Turro, Chem. Commun., 2010, 46, 6759 RSC; (i) T. Gianferrara, C. Spagnul, R. Alberto, G. Gasser, S. Ferrari, V. Pierroz, A. Bergamo and E. Alessio, ChemMedChem, 2014, 9, 1231 CrossRef CAS PubMed; (j) H. Yin, M. Stephenson, J. Gibson, E. Sampson, G. Shi, T. Sainuddin, S. Monro and S. A. McFarland, Inorg. Chem., 2014, 53, 4548 CrossRef CAS PubMed.
- (a) C. R. Maldonado, L. Salassa, N. Gomez-Blanco and J. C. Mareque-Rivas, Coord. Chem. Rev., 2013, 257, 2668 CrossRef CAS; (b) P. T. Burks and P. C. Ford, Dalton Trans., 2012, 41, 13030 RSC; (c) S. Bonnet, Comments Inorg. Chem., 2015, 35, 179 CrossRef CAS.
- (a) J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323 RSC; (b) M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808 CrossRef CAS PubMed; (c) J. Shen, G. Chen, A.-M. Vu, W. Fan, O. S. Bilsel, C.-C. Chang and G. Han, Adv. Opt. Mater., 2013, 1, 644 CrossRef.
- L. Tu, X. Liu, F. Wu and H. Zhang, Chem. Soc. Rev., 2015, 44, 1331 RSC.
- (a) F. Vetrone, R. Naccache, V. Mahalingam, C. G. Morgan and J. A. Capobianco, Adv. Funct. Mater., 2009, 19, 2924 CrossRef CAS; (b) D. Chen and P. Huang, Dalton Trans., 2014, 43, 11299 RSC.
- (a) R. Komban, J. P. Klare, B. Voss, J. Nordmann, H. J. Steinhoff and M. Haase, Angew. Chem., Int. Ed., 2012, 51, 6506 CrossRef CAS PubMed; (b) B. Voss and M. Haase, ACS Nano, 2013, 7, 11242 CrossRef CAS PubMed; (c) T. Rinkel, J. Nordmann, A. N. Raj and M. Haase, Nanoscale, 2014, 6, 14523 RSC.
- J. V. Garcia, J. P. Yang, D. K. Shen, C. Yao, X. M. Li, R. Wang, G. D. Stucky, D. Y. Zhao, P. C. Ford and F. Zhang, Small, 2012, 8, 3800 CrossRef CAS PubMed.
- (a) L. J. Ignarro, Nitric Oxide: Biology and Pathobiology, Elsevier Inc., Burlington, 2nd edn, 2010 Search PubMed; (b) K. D. Bloch, F. Ichinose, J. D. Roberts Jr. and W. M. Zapol, Cardiovasc. Res., 2007, 75, 339 CrossRef CAS PubMed.
- P. T. Burks, J. V. Garcia, R. GonzalezIrias, J. T. Tillman, M. T. Niu, A. A. Mikhailovsky, J. P. Zhang, F. Zhang and P. C. Ford, J. Am. Chem. Soc., 2013, 135, 18145 CrossRef CAS PubMed.
- A. E. Pierri, P. J. Huang, J. V. Garcia, J. G. Stanfill, M. Chui, G. Wu, N. Zheng and P. C. Ford, Chem. Commun., 2015, 51, 2072 RSC.
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug. Discovery, 2010, 9, 728 CrossRef CAS PubMed.
- E. Ruggiero, A. Habtemariam, L. Yate, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2014, 50, 1715 RSC.
- (a) E. Fino, R. Araya, D. S. Peterka, M. Salierno, R. Etchenique and R. Yuste, Front. Neural Circuits, 2009, 3, 1 Search PubMed; (b) R. Araya, V. Andino-Pavlovsky, R. Yuste and R. Etchenique, ACS Chem. Neurosci., 2013, 4, 1163 CrossRef CAS PubMed.
- (a) T. Respondek, R. N. Garner, M. K. Herroon, I. Podgorski, C. Turro and J. J. Kodanko, J. Am. Chem. Soc., 2011, 133, 17164 CrossRef CAS PubMed; (b) R. Sharma, J. D. Knoll, P. D. Martin, I. Podgorski, C. Turro and J. J. Kodanko, Inorg. Chem., 2014, 53, 3272 CrossRef CAS PubMed; (c) T. Respondek, R. Sharma, M. K. Herroon, R. N. Garner, J. D. Knoll, E. Cueny, C. Turro, I. Podgorski and J. J. Kodanko, ChemMedChem, 2014, 9, 1306 CrossRef CAS PubMed; (d) S. D. Ramalho, R. Sharma, J. K. White, N. Aggarwal, A. Chalasani, M. Sameni, K. Moin, P. C. Vieira, C. Turro, J. J. Kodanko and B. F. Sloane, PLoS One, 2015, 10, e0142527 Search PubMed.
- (a) B. S. Howerton, D. K. Heidary and E. C. Glazer, J. Am. Chem. Soc., 2012, 134, 8324 CrossRef CAS PubMed; (b) M. Dickerson, B. Howerton, Y. Bae and E. C. Glazer, J. Mater. Chem. B, 2016, 4, 394 RSC.
- (a) D. V. Pinnick and B. Durham, Inorg. Chem., 1984, 23, 1440 CrossRef CAS; (b) E. Borfecchia, C. Garino, L. Salassa, T. Ruiu, D. Gianolio, X. Zhang, K. Attenkofer, L. X. Chen, R. Gobetto, P. J. Sadler and C. Lamberti, Dalton Trans., 2013, 42, 6564 RSC.
- E. Ruggiero, S. Alonso-de Castro, A. Habtemariam and L. Salassa, Struct. Bonding, 2014, 165, 69 CrossRef.
- E. Ruggiero, J. Hernandez-Gil, J. C. Mareque-Rivas and L. Salassa, Chem. Commun., 2015, 51, 2091 RSC.
- (a) S. Betanzos-Lara, L. Salassa, A. Habtemariam, O. Novakova, A. M. Pizarro, G. J. Clarkson, B. Liskova, V. Brabec and P. J. Sadler, Organometallics, 2012, 31, 3466 CrossRef CAS; (b) S. Betanzos-Lara, L. Salassa, A. Habtemariam and P. J. Sadler, Chem. Commun., 2009, 6622 RSC.
- E. Ruggiero, C. Garino, J. C. Mareque-Rivas, A. Habtemariam and L. Salassa, Chem. – Eur. J., 2016, 22, 2801 CrossRef CAS PubMed.
- T. N. Singh and C. Turro, Inorg. Chem., 2004, 43, 7260 CrossRef CAS PubMed.
- Y. Chen, G. Jiang, Q. Zhou, Y. Zhang, K. Li, Y. Zheng, B. Zhang and X. Wang, RSC Adv., 2016, 6, 23804 RSC.
- S. He, K. Krippes, S. Ritz, Z. Chen, A. Best, H.-J. Butt, V. Mailänder and S. Wu, Chem. Commun., 2015, 51, 431 RSC.
- Z. Chen, W. Sun, H.-J. Butt and S. Wu, Chem. – Eur. J., 2015, 21, 9165 CrossRef CAS PubMed.
- (a) Z. Cheng, A. Al Zaki, J. Z. Hui, V. R. Muzykantov and A. Tsourka, Science, 2012, 338, 903 CrossRef CAS PubMed; (b) V. P. Chauhan and R. K. Jain, Nat. Mater., 2012, 12, 958 CrossRef PubMed; (c) V. R. Devadasu, V. Bhardwaj and M. N. V. R. Kumar, Chem. Rev., 2013, 113, 1686 CrossRef CAS PubMed; (d) A. Gnach, T. Lipinski, A. Bednarkiewicz, J. Rybka and J. A. Capobianco, Chem. Soc. Rev., 2015, 44, 1561 RSC.
- Y. Sun, W. Feng, P. Yang, C. Huang and F. Li, Chem. Soc. Rev., 2015, 44, 1509 RSC.
| This journal is © The Royal Society of Chemistry 2016 |