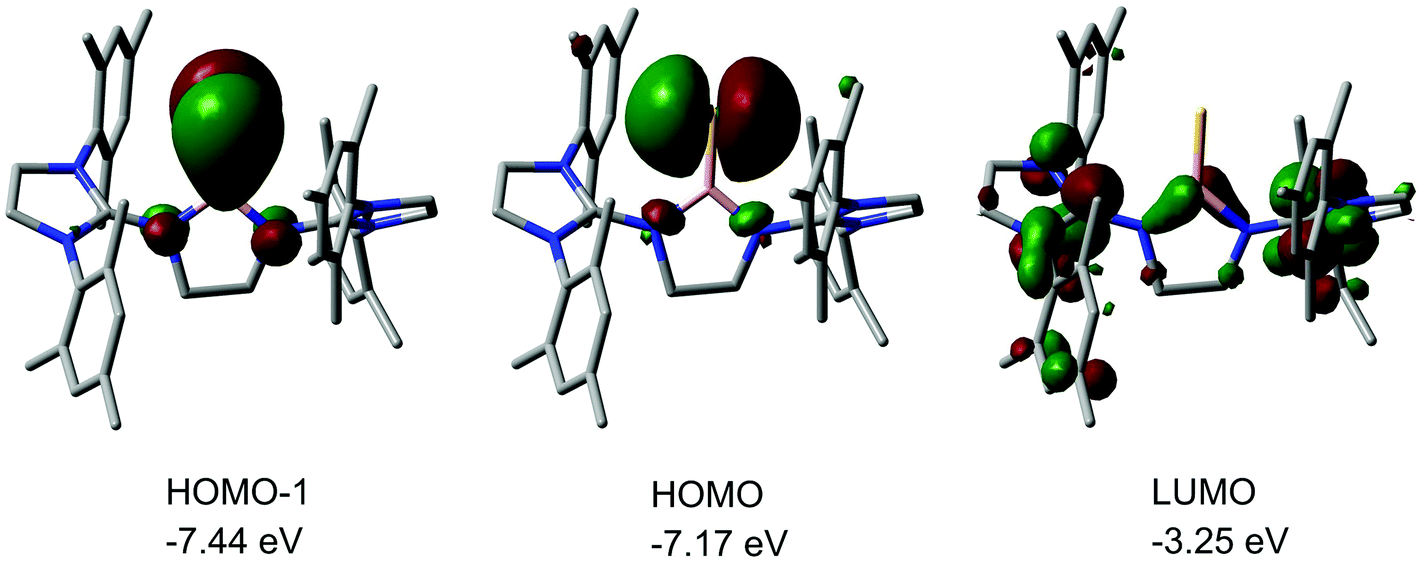DOI:
10.1039/C6DT01413E
(Perspective)
Dalton Trans., 2016,
45, 9385-9397
Advances in the development of complexes that contain a group 13 element chalcogen multiple bond
Received
12th April 2016
, Accepted 16th May 2016
First published on 17th May 2016
Abstract
Inorganic group 13 element (M) chalcogenides (E) based on the general formular M2E3 are ubiquitous in synthesis, catalysis and material science. The parent ME fragment which aggregates to form three dimensional networks in the condensed phase can be expected to exhibit multiple bond character between the elements. Low temperature matrix isolation techniques are required to investigate the nature of this elusive species. An alternate approach for respective studies is the synthesis of electron-precise molecular complexes that contain the ME entity and for which isolation at ambient temperature is possible. This is realized by kinetic stabilization with bulky ligands and thermodynamic stabilization using electron donor, as well as acceptor groups attached to the ME functionality (i.e. donor–acceptor stabilization). In this article we revise the literature on complex compounds that exhibit a bonding interaction between a group 13 element atom and a chalcogen atom that is reasonably to be interpreted in terms of a double- or triple bond.
 Daniel Franz | Daniel Franz was born in Ludwigshafen, Germany, in 1981. He studied chemistry at the Goethe University Frankfurt where he received his Ph.D. degree under the guidance of Prof. Matthias Wagner in 2012. Following this he moved to the Technische Universität Berlin to carry out postdoctoral studies in the group of Prof. Shigeyoshi Inoue. Today he is pursuing his research interests in coordination chemistry of group 13 and 14 elements at the Technische Universität München under the supervision of Prof. Inoue. |
 Shigeyoshi Inoue | Shigeyoshi Inoue received his B.S. (2003), M.Sc. (2005), and Ph.D. (2008) from the University of Tsukuba under the supervision of Prof. Akira Sekiguchi. After a Humboldt postdoctoral fellowship and a JSPS postdoctoral fellowship with Prof. Matthias Drieß at the Technische Universität Berlin, in 2010 he began his independent career at the TU Berlin as a Sofia Kovalevskaja Professor. Since 2015 he has been a W2-Tenure Track Professor for Silicon Chemistry at the Technische Universität München. His research interests focus on the investigation of the synthesis and reactivity of low-valent main group compounds with the goal of finding novel applications. |
Introduction
Background and scope of this Perspective article
The combination of elements of the group 13 (M) and group 16 (E) mostly results in compounds with the typical stoichiometry M2E3 in which the oxidation state +3 is assigned to M and −2 to E. Due to the rather large differences in the electronegativities of M and E, the bonding interaction between the atoms exhibits high ionic character and they form aggregates of high lattice energy in the solid state. A variety of applications in chemical transformation, catalysis, as well as material science have been found for the compound class of group 13 element chalcogenides.1 Low-temperature matrix isolation techniques and theoretical studies are required to investigate the lower-aggregated intermediates and the process of how the bulk materials form from these.2
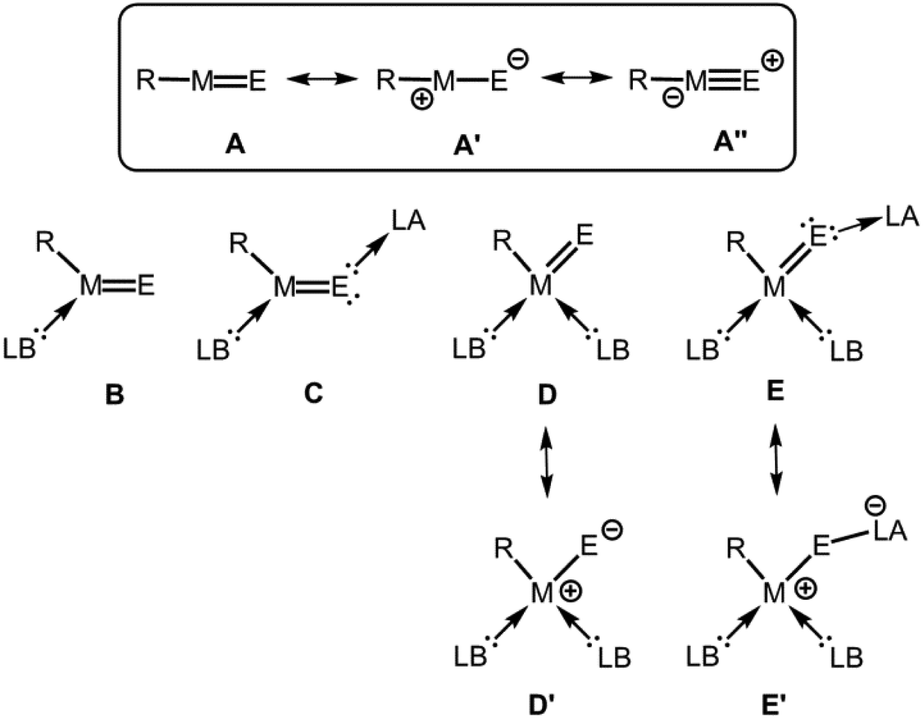
The parent entity of these aggregates is the monotopic ME fragment for which it is reasonable to assume a high degree of bonding interaction between the M and the E atom. As an alternate method to matrix isolation techniques, the stabilization of elusive species via sophisticated ligand systems (e.g. bulky substituents, electron-donor- and electron-acceptor groups) has allowed for the isolation and investigation of well-defined molecular complexes that contain reactive species with pronounced multiple bond character between the elements.3 Fundamental concepts for the isolation of electron-precise complexes that exhibit a group 13 metal chalcogen interaction with high multiple bond character are illustrated in Fig. 1. By use of a very bulky anionic ligand (R−) to the metal centre, kinetic stabilization of the ME fragment might be accomplished and the bond order of the metal–chalcogen interaction should approach a value significantly larger than 1 and, thus, correspond to a metal chalcogen double bond (A, Fig. 1). As a complement to theoretical investigations (e.g. Bond Index, Molecular Orbital Analysis, Natural Resonance Theory) structural information (i.e. M–E bond length) and spectroscopic studies (e.g. NMR, IR, UV/Vis) may elucidate the multiple-bond properties. In fact, the definition of a multiple bond remains subject to controversy and the transition from a single bond to a double bond and to a triple bond is rather fluent. This is particularly true for strongly polarized bonds because the extend of ionic or covalent contribution to the atom–atom interaction is difficult to assess. However, we presume that the comparison of theoretical and structural parameters between compounds for which the bonding situation is undisputed and those for which a new bonding type may apply is conclusive. Generally, the bonding situation of the metal–chalcogen interaction in complexes of the type R–ME can be described as a composite of the resonance structure R–M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E (A) and the zwitterionic forms R–+M–E− (A′), as well as R–−M
E (A) and the zwitterionic forms R–+M–E− (A′), as well as R–−M![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) E+ (A′′, Fig. 1).
E+ (A′′, Fig. 1).
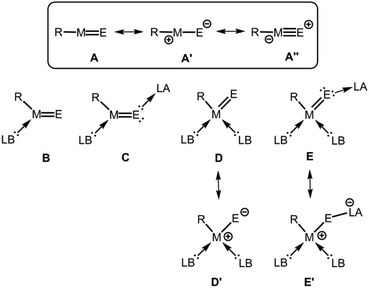 |
| | Fig. 1 The parent group 13 element (M) chalcogen (E) double bond species (A) and conceivable concepts for its thermodynamic (electronic) stabilization (B–E), as well as selected resonance structures (A′, A′′, D′, E′; R = anionic substituent, LB = Lewis base, LA = Lewis acid). | |
Since the parent R–ME group is a Lewis acid base pair that is prone to self-quenching by head-to-tail aggregation thermodynamic (i.e. electronic) stabilization may be required which is achieved by the introduction of an electron-pair donor ligand to the metal centre (B, Fig. 1) and capping the chalcogen atom with an electron-pair acceptor (C, Fig. 1). Instead of implementing the latter, aggregation could also be hampered by an additional Lewis base attached to the group 13 metal centre in order to decrease the intrinsic electron-deficiency of the otherwise only three-coordinate group 13 element atom (D, Fig. 1). Furthermore, the higher coordination of the metal centre might be complemented by tethering a Lewis acid to the group 16 atom for increased stabilization of the ME fragment (E, Fig. 1). It is feasible that the contribution of zwitterionic canonical structures such as D′ and E′ may rise due to the higher coordination number of the group 13 metal centre (Fig. 1).
In this Perspective article, we summarize and assess the present literature on molecular complexes that comprise a group 13 element chalcogen bond with pronounced multiple bond character which allows for the interpretation in terms of a double- or triple bond between the atoms. We focus on classical chemical synthesis in the condensed phase and, hence, gas-phase formation experiments and low temperature matrix isolation techniques are not considered in this paper. Within the subsections of this article the compounds are ordered by chronological appearance in the literature to reflect the evolution of the field in time. Basically, only the non-radioactive elements of group 13 and 16 are considered. A review article from 2010 by Fischer and Power on element–element multiple bonds also includes an outline of group 13 element chalcogen multiple bonds.3
Complexes with a boron chalcogen multiple bond
Background.
As a consequence of the differences in electronegativities, the bonds between the metalloid boron and chalcogen atoms are strongly polarized and the negative charge density accumulates at the group 16 element (Table 1). In contrast to the heavier homologues of the group 13 family, boron is inclined to be the centre of a trigonal-planar structure motif. This tendency of the boron atom towards three-coordination increases if the ligand atoms compensate the metalloid's electron deficiency via π-donation into the unoccupied p-orbital at the boron centre. Thus, one could assume that synthetic access to a BO double bond should be more simple than for the heavier ME analogues because π-interaction benefits from the comparably small atomic radii of boron and oxygen (Table 1). However, as apparent from the literature, this is not the case. Presumably, the tendency for aggregation inflicted by the strong polarization of the boron–oxygen bond cannot be easily compensated by π-bonding interaction.
Table 1 Covalent radii and values for electronegativities of group 13 and (non-radioactive) group 16 elements7
| |
Covalent radius [Å] |
EN (Mulliken) |
EN (Pauling) |
EN (Allred-Rochow) |
| B |
0.84 |
1.83 |
1.88 |
2.01 |
| Al |
1.21 |
1.37 |
1.62 |
1.47 |
| Ga |
1.22 |
1.34 |
1.77 |
1.82 |
| In |
1.42 |
n.a. |
1.63 |
1.49 |
| Tl |
1.45 |
n.a. |
n.a. |
1.44 |
| O |
0.66 |
3.21 |
3.61 |
3.50 |
| S |
1.05 |
2.65 |
2.64 |
2.44 |
| Se |
1.20 |
2.51 |
2.46 |
2.48 |
| Te |
1.38 |
2.34 |
2.29 |
2.01 |
Boron oxygen double bond.
A pioneering study of Pachaly and West demonstrated the remarkable reactivity of the oxoborane functionality. Irradiation of dioxaboretane F affords the transient oxoborane 1 as concluded from its trapping products with oxadisilolane and ketone, respectively (G, H, Scheme 1).4 As apparent from the elusive nature of 1 the very bulky supermesityl group does not suffice to kinetically stabilize this highly elusive oxoborane species at ambient temperature. Notably, one would expect partial boron oxygen double- or triple bond character for the three- or two-coordinate boron species, respectively (F–H, 1). In the context of discussing 1, it is of interest that the related oxoborane (2,4,6-(CH3)3-C6H2)BO was presumed to be produced as an intermediate species in the reaction of the two-coordinate dimesitylborinium ion (2,4,6-(CH3)3-C6H2)2B+ with carbon dioxide.5
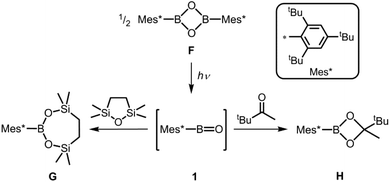 |
| | Scheme 1 Irradiation of the dioxaboretane F to yield the intermediate oxoborane 1. Trapping reactions of 1 to form G and H. | |
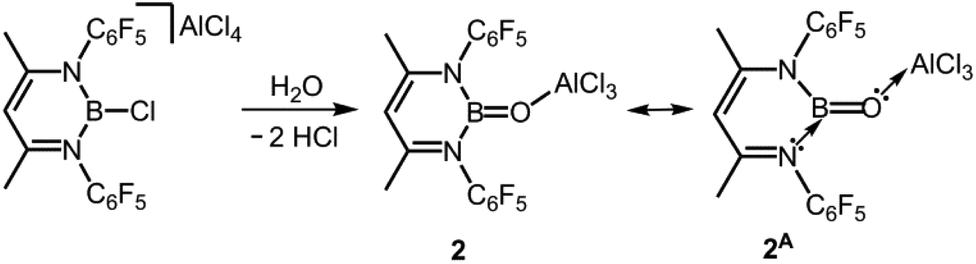
In seminal work from 2005, Cowley and coworkers reported the oxoborane complex 2 with a very short B–O distance of 1.304(2) Å (Scheme 2, Fig. 2).6 For comparison, the sum of the covalent radii of boron and oxygen amounts to 1.50 Å (Table 1).7 The B–O single bond lengths in representative organoborates (four-coordinate boron centre) range from 1.46 Å to 1.54 Å and tend to decrease with the number of oxygen atoms attached to the metalloid centre.8 For trigonal-planar organoboranes typical values for B–O bond lengths are found between 1.36 Å and 1.37 Å and the interaction between these atoms often possesses partial double-bond character.9 A 1,3-diketimino group was used as an ancillary ligand to the boron centre in 2. The oxygen atom is protected by a capping AlCl3 moiety (Al–O = 1.720(1) Å). Accordingly, the concept of donor–acceptor stabilization of type C of the BO fragment is employed as illustrated by resonance structure 2A (Fig. 1, Scheme 2). Synthetic access to the oxoborane 2 is granted by controlled hydrolysis of the boron halide salt HC{C(CH3)N(C6F5)}2BCl[AlCl4] (Scheme 2). The authors carried out DFT (Density Functional Theory) calculations on 2, as well as the model compound 2′ which has no AlCl3 group attached to the oxygen atom and found that the B–O distance decreased only marginally upon removal of the Lewis acid (1.292 Å). However, the molecular orbital analysis revealed significant differences between 2 and 2′ as one would expect. For example, the HOMO (highest occupied molecular orbital) of 2 mainly consists of orbital lobes at the Cl atoms while the HOMO in 2′ principally possesses oxygen lone pair character. The 11B NMR spectroscopic analysis of 2 exhibited a signal at 40.1 ppm.
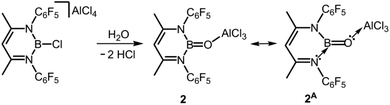 |
| | Scheme 2 Synthesis of the oxoborane 2 and the selected resonance structure 2A. | |
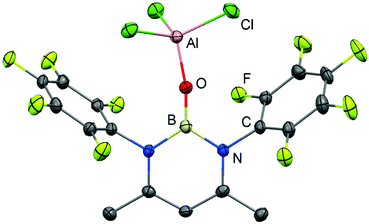 |
| | Fig. 2 Ellipsoid plot (50% level) of the molecular structure of 2 in the solid state as derived from XRD analysis. Hydrogen atoms have been omitted. | |
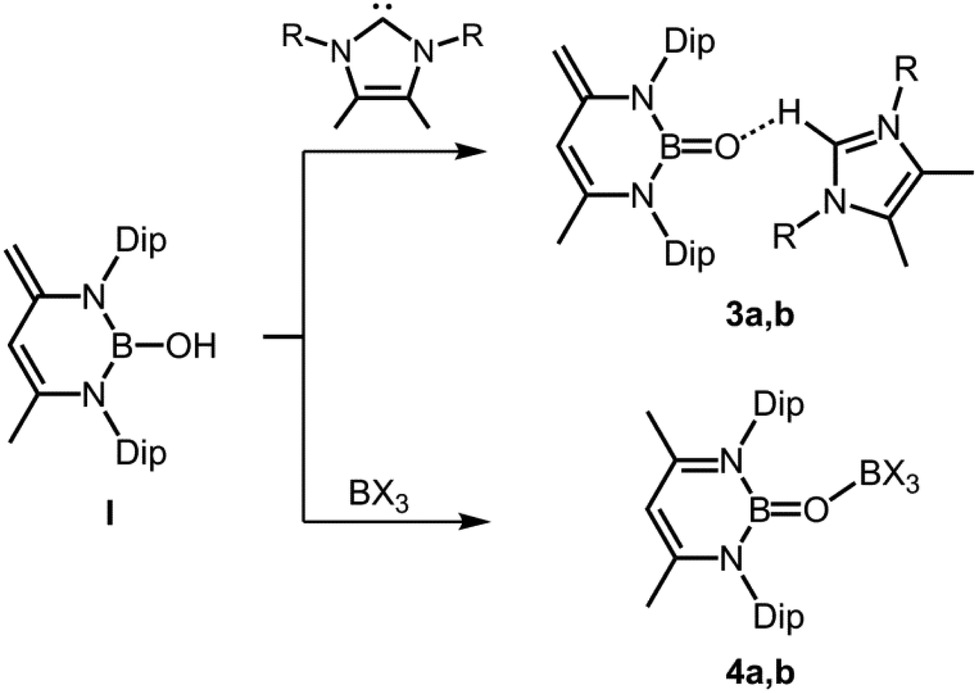
In 2011 Cui and coworkers enriched the field of oxoborane chemistry by successful isolation of 3 and 4 derived from the hydroxoborane precursor I in which a 1,3-diketimino group is employed as a stabilizing ligand to the boron centre (Scheme 3).10 The Lewis basicity of the oxygen atom is quenched by coordination to a weak Brønsted acid ([H–NHC]+, NHC = N-heterocyclic carbene) or a strong Lewis acid (B(C6F5)3, BCl3), respectively (stabilization concept: C, Fig. 1). The bulkiness of the proton donor was varied (3a, 3b) and two different boron-based Lewis acids were implemented (4a, 4b), however, only 3a and 4a were structurally characterized via XRD (X-ray diffraction) analysis. The B–O distances observed for 3a amount to 1.287(4) Å and 1.296(3) Å (the compound contains two crystallographically independent molecules in the asymmetric unit), which is slightly shorter than in 2 (vide supra). The respective bond lengths for 4a have values of 1.311(3) Å and 1.314(3) Å (here also: two independent molecules found in the asymmetric unit). The authors reasoned that the marginally longer B–O distances for 4a are caused by the increased steric congestion of the BO entity inflicted by the B(C6F5)3 moiety as compared to the more remote [H–NHC]+ group in 3a. DFT calculations were carried out on a sterically reduced model compound to 4b because this complex had not been crystallized and the B–O distance was computed to 1.30308 Å. Interestingly, the BO double bond length of a model compound to 3 for which the protecting weak Brønsted acid had been removed was calculated to 1.28240 Å which suggests only moderate influence of the O⋯H interaction in 3 on the degree of its BO double bond character. Moreover, the application of DFT methods on a sterically reduced model compound to 3 revealed a boron–oxygen π-bonding interaction in the HOMO−7. The 11B NMR chemical shifts of 3a and 4a were found at 19.8 ppm and 22.8 ppm, respectively.
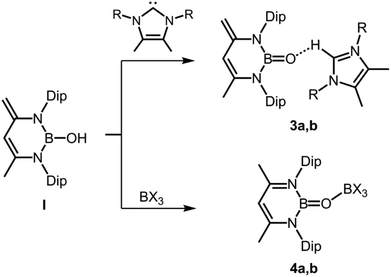 |
| | Scheme 3 Syntheses of oxoboranes via proton association (3a,b) and via proton shift (4a,b) from hydroxoborane I (R = iPr for 3a, Me for 3b; X = C6F5 for 4a, Cl for 4b, Dip = 2,6-diisopropylphenyl). | |
Curran and coworkers described the NHC-stabilized dihydroxoborenium salt 5[OTf] (Scheme 4, Tf = SO3CF3) in 2012.11 The compound was isolated after stepwise treatment of the borane–NHC adduct with an excess of triflic acid and structurally studied by XRD analysis (Scheme 4).11 The short B–O distances of 1.307(4) Å and 1.310(6) Å suggest that a considerable degree of double bond character can be attributed to the boron–oxygens bonds in 5+. This is illustrated by the canonical forms 5A+ and 5B+ (Scheme 4) and as apparent from 5A+ the bonding situation corresponds to the stabilization concept of type C (Fig. 1). A resonance at 22.5 ppm in the 11B NMR spectrum is correlated to this three-coordinate boron cation.
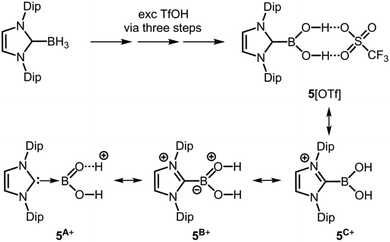 |
| | Scheme 4 Conversion of borane–NHC adduct to NHC-stabilized hydroxoborenium salt 5[OTf] with very short B–O bond lengths. Selected resonance structures of the cation (5A+, 5B+ and 5C+; NHC = N-heterocyclic carbene, Dip = 2,6-diisopropylphenyl, Tf = triflyl). | |
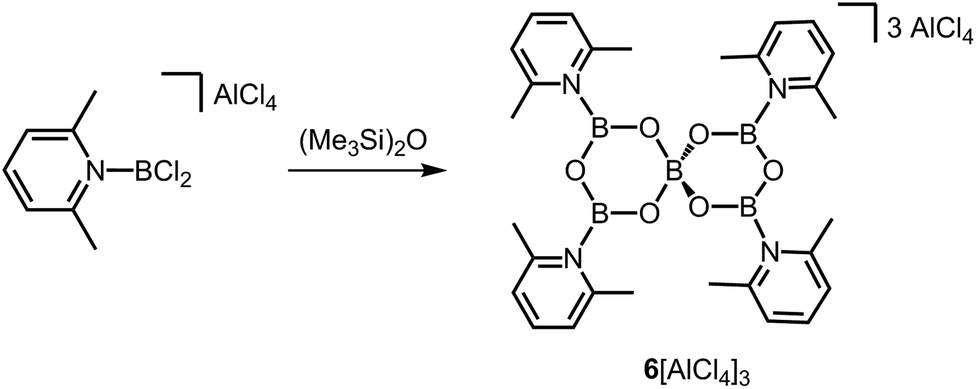
Very short B–O bond lengths (1.296(4) Å and 1.308(4) Å) are observed in the crystal structure of the ionic compound 6[AlCl4]3 which has been described by Ingleson and coworkers as a tricationic boroxine analogue (Scheme 5).12 Considering only the crystallographically independent B–O distances of the three-coordinate boron centres in 63+ each B-atom possesses one shorter (vide supra) and one longer (1.374(4) Å and 1.378(4) Å) B–O bond length. Presumably, the short B–O bond lengths correlate to the high cationic charge of the complex 63+. In the 11B NMR spectroscopic analysis signals at 25 ppm and 5.2 ppm were attributed to 63+.
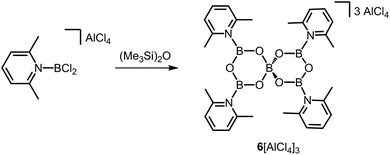 |
| | Scheme 5 Conversion of a 2,6-lutidine-stabilized chloroborenium salt to the boroxine salt 6[AlCl4]3. | |
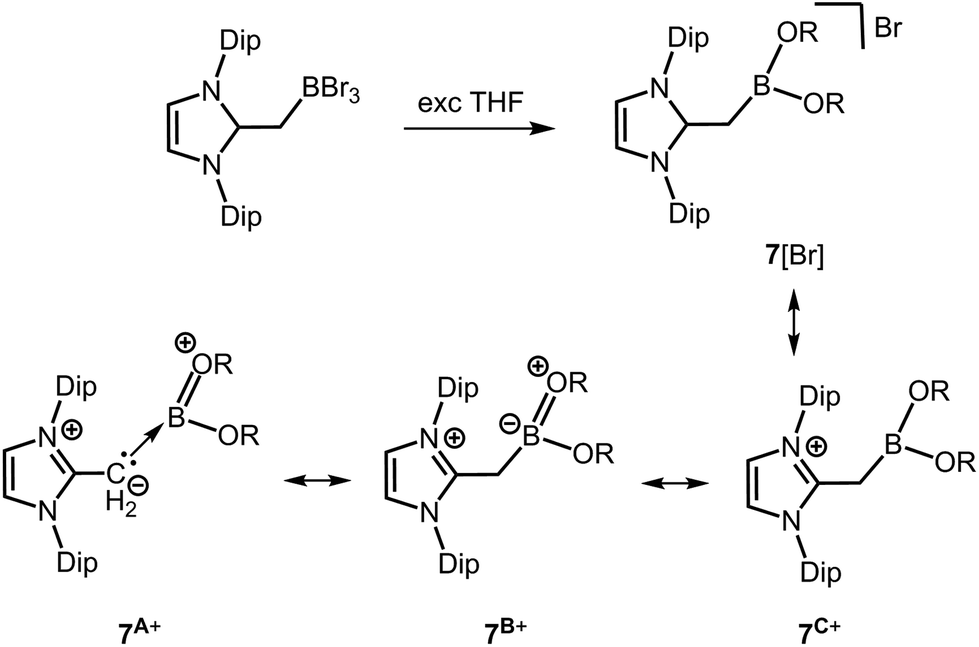
In 2013 Robinson and coworkers reported compound 7[Br] which is a borenium salt reminiscent of 5[OTf] (Scheme 4, Scheme 6).13 Though the B–O distances in 7+ (1.339(11) Å and 1.345(11) Å) clearly reside below the typical length of a B–O single bond (vide supra) they are longer than in 5+. Accordingly, the double bond character as represented by the canonical forms 7A+ and 7B+ is probably lower than in 5+ (Scheme 6). However, the structural features of 7+ support the assumption that the introduction of cationic charge to a boron complex may generally strengthen the boron–oxygen interaction as in 5+ and 6+. The 11B NMR chemical shift of 7+ was observed at 26.60 ppm.
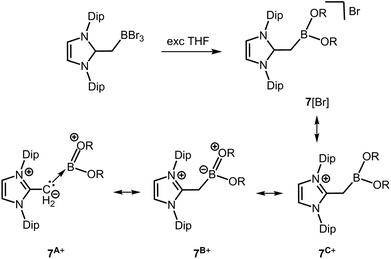 |
| | Scheme 6 Reaction of bromoborane–NHO adduct to the alkoxy borenium salt 7[Br] via ether cleavage (NHO = N-heterocyclic olefin, Dip = 2,6-diisopropylphenyl, R = (CH2)4Br). Selected resonance structures 7A+, 7B+ and 7C+. | |
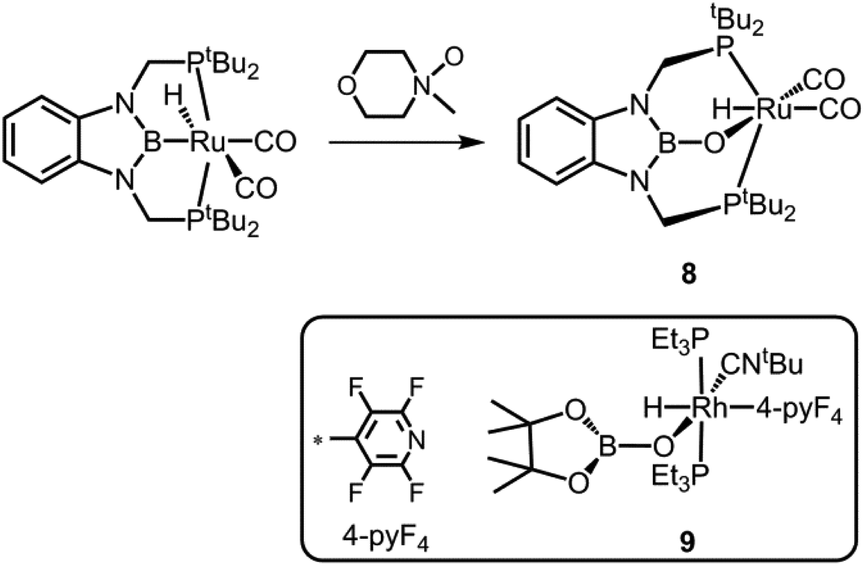
Also in 2013 Miyada and Yamashita described the oxoborane ruthenium compound 8 which is derived from its boryl pincer complex precursor by insertion of oxygen into the B–Ru bond (Scheme 7).14 As a source of oxygen they implemented N-methylmorpholine N-oxide. The B–O distance in 8 amounts to 1.329(6) Å and, thus, it is shorter than in 7+ but elongated in comparison to the uncharged oxoborane 2. Interestingly, a WBI (Wiberg Bond Index) of 1.04 was calculated for the boron–oxygen interaction in 8 and this value is considerably higher than expected for a boron oxygen single bond if one takes the strong polarization along the B–O vector into account. As apparent from the direct comparison of the B–N distances in 8 (1.457(7) Å, 1.450(6) Å) with its boryl precursor (1.432(9) Å, 1.436(9) Å) the introduction of the oxygen atom as an additional π-donor substituent weakens the π-interaction between the boron atom and the nitrogen ligands. Notably, compound 8 is related to the oxoborane rhodium complex 9 for which the distance between the boron centre and the rhodium-bonded oxygen atom was observed at 1.328(11) Å (Scheme 7).15 Compound 8 gave rise to a peak at 22.7 ppm in the 11B NMR spectrum.
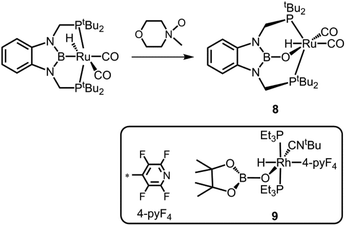 |
| | Scheme 7 Synthesis of the oxoborane ruthenium complex 8. The rhodium congener 9 (boxed). | |
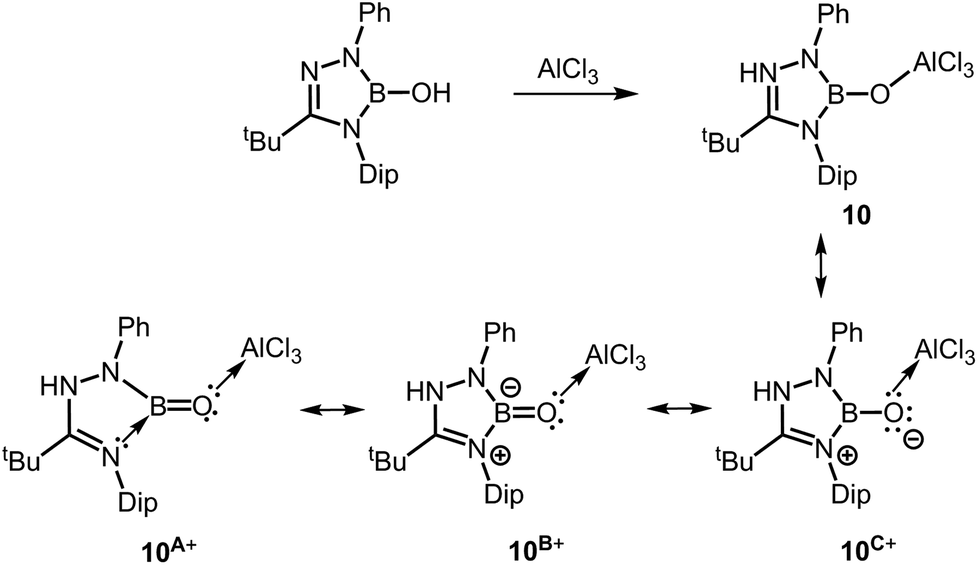
In a report from the year 2014 Kinjo and coworkers described the 1,2,3,4-triazaborole-based oxoborane 10 (Scheme 8) in which the oxygen atom is attached to a protecting AlCl3 group, similar to complex 2.16 The B–O distance amounts to 1.304(3) Å and the Al–O bond length was determined to 1.7073(16) Å. These values are very similar to 2 for which relevant BO double bond character has been acknowledged. As apparent from resonance form 10A+ concept C may apply for the stabilization of the BO fragment (Fig. 1, Scheme 8). For comparison, the hydroxoborane precursor of 10 exhibits a B–O distance of 1.351(7) Å (Scheme 8). Remarkably, a BO bond length of 1.264 Å was calculated for a model compound of 10 that is free of the AlCl3 moiety. Furthermore, a WBI of 1.07 was reported for the strongly polarized boron–oxygen bond in the optimized structure of 10 and that value is marginally larger than in compound 8. In the infrared spectroscopic study of 10 a band at 1636 cm−1 was assigned to the boron oxygen stretch vibration. Notably, in the 11B NMR spectrum a resonance at 18.9 ppm was attributed to 10.
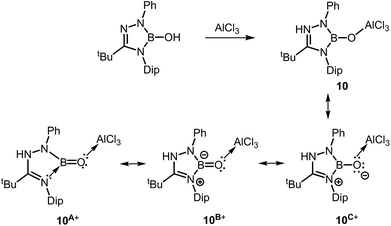 |
| | Scheme 8 Synthesis of the oxoborane 10 and selected resonance structures 10A+, 10B+ and 10C+ (Dip = 2,6-diisopropylphenyl). | |
Boron oxygen triple bond.
In 2010 Braunschweig and coworkers published their seminal report on boron–oxygen triple bonds stabilized in the coordination sphere of platinum (11, 12, Scheme 9).17 The B–O distance of 11 was computed to 1.226 Å (XRD data did not suffice discussion of structural parameters) and the NRT (Natural Resonance Theory) bond order for the boron–oxygen interaction was determined to 2.83 which is a high value if one considers the presumably strong polarization along the B–O vector. Though the presence of the low-coordinate boron species in 11 is not verified by the 11B NMR signal observed at 17 ppm (strong inductive effects will be produced by the adjacent transition metal) the infrared spectroscopic analysis confirmed the strength of the BO bond (bands at 1797 cm−1 and 1853 cm−1). Notably, these vibrations indicate higher bond energy than observed for the boron oxygen double bond in 10 (band at 1636 cm−1, vide supra). DFT methods revealed that the π-MOs in 11 possess considerable overlap between the BO fragment and d-type orbitals at the platinum centre. Interestingly, via stepwise addition of BrSiMe3 to 11 it was verified that the terminal oxoborane is in dynamic equilibrium with its bromoboryl precursor (Scheme 9).17 The conversion of 11 with Bu4N[SPh] afforded 12 for which a B–O distance of 1.210(3) Å was found via XRD analysis (Scheme 9, Fig. 3).17 The 11B NMR analysis of 12 revealed a signal at 24 ppm. Further derivatization experiments on 11 resulted in the syntheses of complexes 13–15 (Scheme 9).18 In addition, a variety of adducts between the platinum oxoboryl system and group 13 metal-based Lewis acids has been reported very recently.19
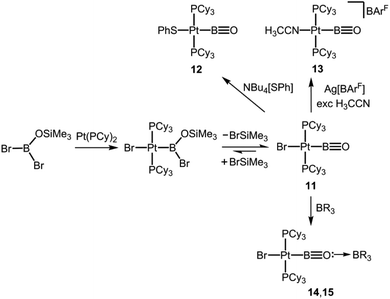 |
| | Scheme 9 Oxidative addition of siloxy bromoborane to form a platinum phosphine complex and its conversion to oxoborane 11. Reactions of 11 to phenylsulfide 12 nitrile complex 13, as well as the oxoborane adducts 14 and 15 (Cy = cyclohexyl, ArF = 3,5-(CF3)2-C6H3, R = ArF or C6F5). | |
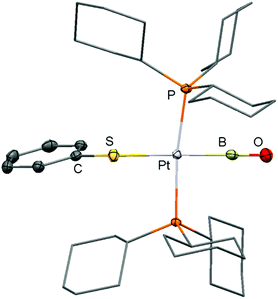 |
| | Fig. 3 Ellipsoid plot (50% level) of the molecular structure of 12 in the solid state as derived from XRD analysis. Hydrogen atoms have been omitted. Cyclohexyl groups are depicted as stick model. | |
Boron sulfur- and boron selenium double bond.
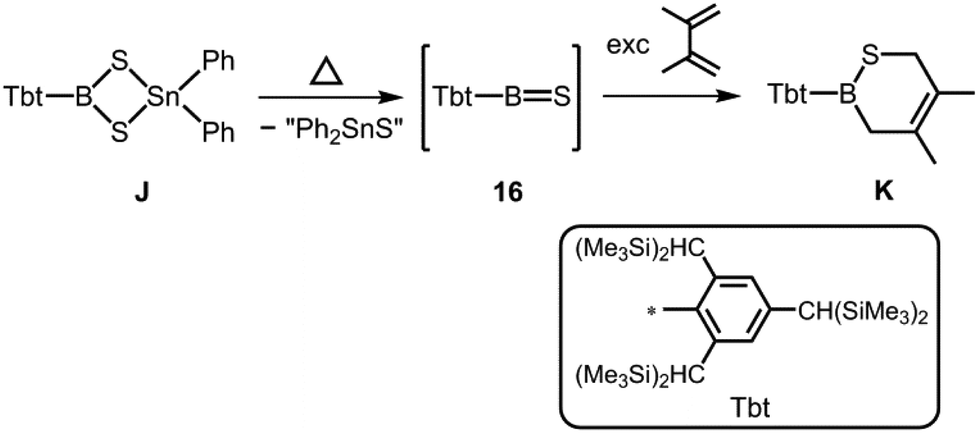
Tokitoh et al., in their seminal work, described the reaction of a dithiastannaboretane (J) with 1,3-dienes at elevated temperature which afforded boron–sulfur heterocycles (K, Scheme 10).20 The formation of K was reasoned by the intermediate presence of the thioxoborane 16 with a BS double bond that engages in cycloaddition reactions with the olefin. However, the characterization and isolation of this highly elusive species (16) could not be accomplished.
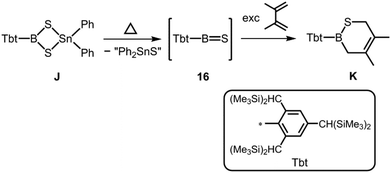 |
| | Scheme 10 Formation of thioxoborane intermediate 16 from dithiastannaboretane J and its representative trapping product with 2,3-dimethyl-butadiene (K). | |
In 2010 Cui and coworkers presented their study on the 1,3-diketiminato-stabilized thioxoborane 17, as well as its heavier selenoxoborane congener 18 (Scheme 11).21 As apparent from the structural formulation the BE entity (E = S or Se) bears two ligands to the metal centre but lacks an additional stabilizing Lewis acid at the chalcogen atom as implemented for the BO moiety in respective oxygen congeners (vide supra). Hence, the system corresponds to the concept of type B for thermodynamic stabilization as illustrated by the canonical forms 17A and 18A (Fig. 1, Scheme 11). Access to these compounds (17, 18) is granted by conversion of the hydridoborane precursor with elemental sulfur or selenium. Interestingly, the reaction runs via hydrogen shift from the putative hydrogenchalcogenide intermediate to the backbone of the diketiminato ligand (Scheme 11). The XRD study of the final products showed B–E distances that amount to 1.741(2) Å in 17 and 1.896(4) Å in 18 and for the former the authors pointed out the decrease in the B–S distance as compared to typical three-coordinate boron compounds with a B–SR moiety. The difference in the bond lengths between the analogues (0.155 Å) corresponds to the deviation in the covalent radii (r) of sulfur and selenium (rS = 1.05 Å, rSe = 1.20 Å).7 Moreover, the structural parameters of the NBN moiety in 17 and 18 are very similar. Consequently, it is reasonable to assume strong resemblance in the nature of the boron–chalcogen bonds of both compounds. The B–N distances are in a narrow range of 1.48 Å to 1.49 Å. This is increased with respect to 1,3-diketimino boranes with a single bond to a chalcogen atom as the hydroxoborane I (Scheme 3, 1.433(5) Å and 1.436(5) Å). This accounts for the higher boron nitrogen dative bond character in 17 and 18 which complies with the stabilization concept B (Fig. 1). For 17 the BS double bond was verified by Natural Bond Order analysis and Kohn–Sham depictions of the frontier orbitals showed that the HOMO−1 comprises significant contribution from a boron sulfur π bond. In the infrared spectra of 17 and 18 the BE stretching vibration modes are observed at 1161 cm−1 and 1136 cm−1, respectively. The 11B NMR spectrum of 17, as well as 18 showed a signal at 36.79 ppm and at 40.88 ppm, respectively.
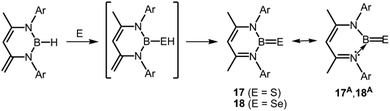 |
| | Scheme 11 Reaction of diketimino borane to intermediate hydrogenchalcogenide and its conversion to the thioxoborane 17, as well as the selenoxoborane 18. Selected resonance structures 17A and 18A (Ar = 2,6-(CH3)2-C6H3). | |
A rare example for the stabilization of a putative boron–sulfur double bond in the coordination sphere of a transition metal was given by the group of Braunschweig in 2013.22 The metallacycle intermediate 19 was observed via NMR spectroscopy in the reaction of a manganese borylene system with triphenylphosphorane sulfide to afford a manganese phosphine complex (in the 11B NMR analysis a broad signal at 88.6 ppm was attributed to 19; Scheme 12). The resonance structure 19A, that is an η2-bonded thioxoborane with a BS double bond stabilized in the coordination sphere of the transition metal, may have relevant contribution to the bonding situation (Scheme 12). The existence of 19 was supported by DFT calculations, but the compound was not characterized by X-ray diffraction analysis.
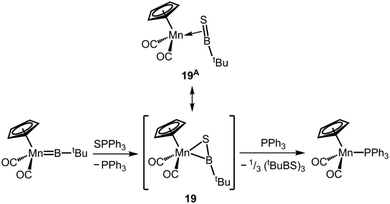 |
| | Scheme 12 Reaction of a manganese borylene complex to a manganese phosphine complex via the metallacycle intermediate 19 and its resonance structure representing thioxoborane character (19A). | |
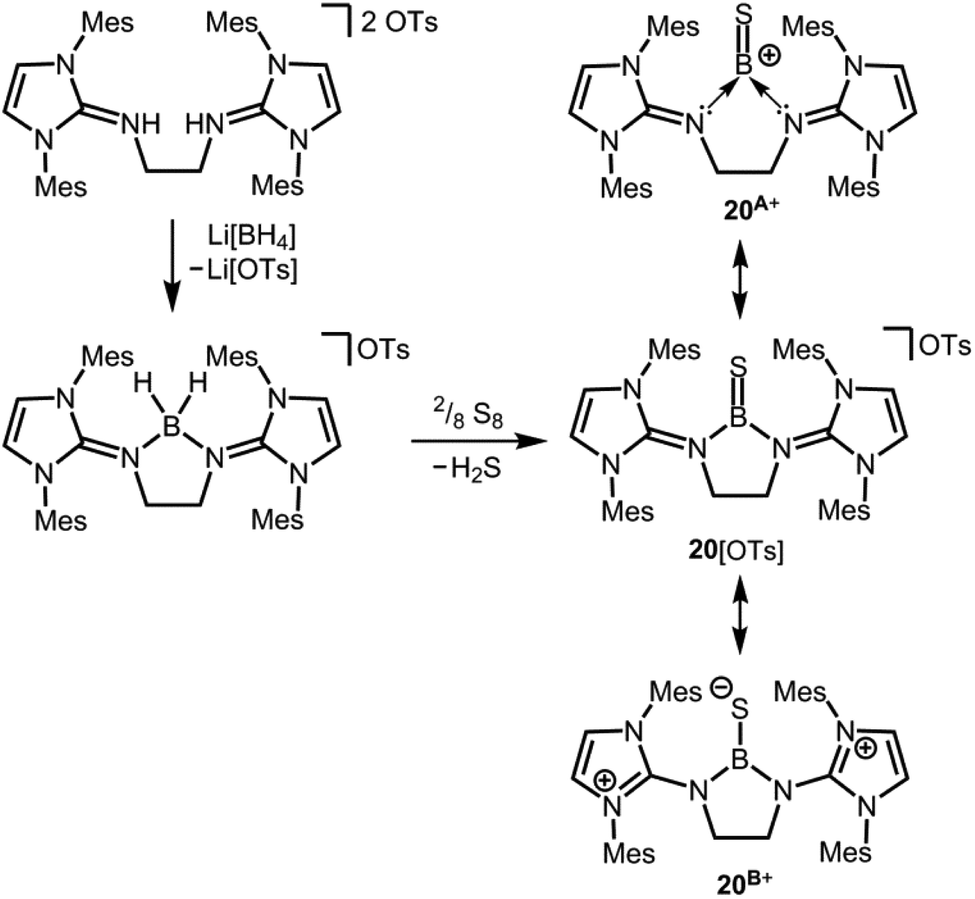
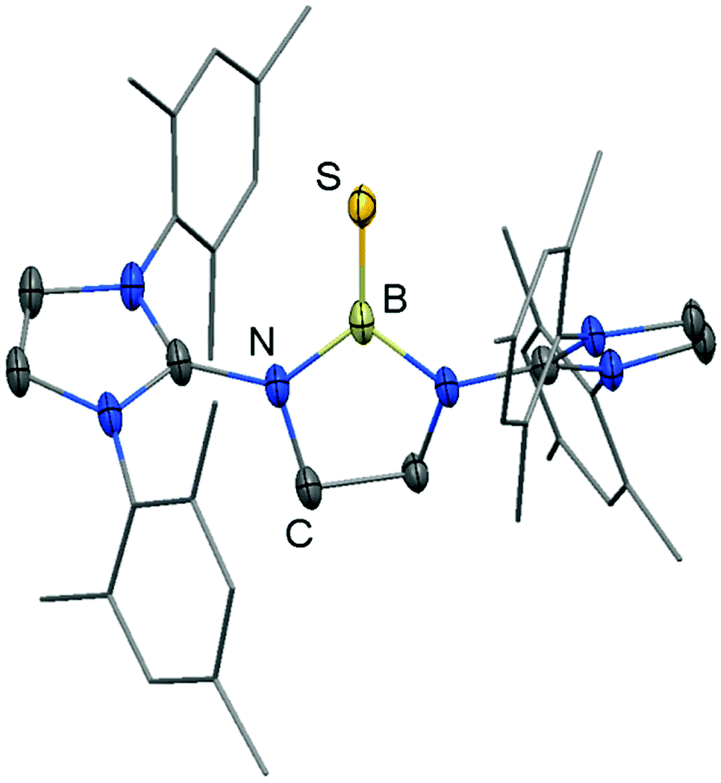
We made a contribution to the field by our report on the cationic thioxoborane 20+ in 2014 (Scheme 13, Fig. 4).23 The XRD analysis revealed a B–S distance of 1.710(5) Å which is even shorter than in 17 (vide supra). The calculated Natural Bond Orbital (NBO) charge of +0.634 at the boron atom indicates that 20+ possesses boron-centred cation character as represented by the canonical structure 20A+ (Scheme 13). Moreover, the calculated WBI of 1.739 verifies the BS double bond nature particularly if one considers the strong polarization along the BS vector (NBO charge at S = −0.578). Accordingly, the HOMO−1 in 20+ is marked by the π bonding interaction between the boron and the sulfur atom whereas HOMO and LUMO comprise the lone pair at the sulfur atom and ligand-centred orbital contributions, respectively (Fig. 5). Notably, the B–N distances in 20+ amount to 1.483(5) Å and 1.493(5) Å and, thus, partial boron nitrogen double bond character is concluded (Fig. 4). Vice versa the C–Nimino bonds are longer than typically found for CN double bonds. This accounts for the partial delocalization of positive charge density into the imidazoline rings as represented by the resonance structure 20B+ (Scheme 13). The 11B NMR chemical shift of 20+ was determined to 33.9 ppm.
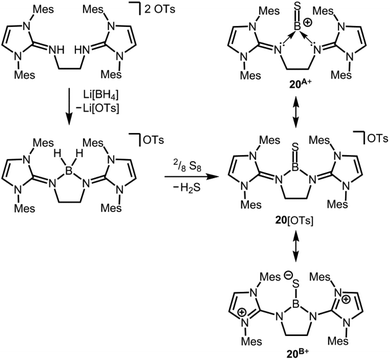 |
| | Scheme 13 Synthesis of a dihydridoboronium salt and its conversion to the tosylate salt of the cationic thioxoborane (20[OTs]). Selected resonance structures 20A+, as well as 20B+ (Mes = mesityl, Ts = tosyl). | |
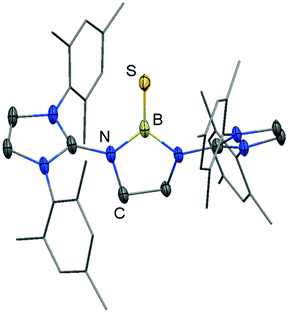 |
| | Fig. 4 Ellipsoid plot (30% level) of the molecular structure of 20+ in the solid state as derived from XRD analysis. Hydrogen atoms have been omitted. Mesityl groups are depicted as a stick model. | |
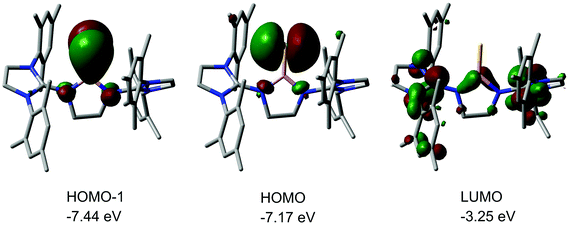 |
| | Fig. 5 Kohn–Sham depictions and relative energy levels with occupancy of selected molecular orbitals of 20+ (LUMO, HOMO, HOMO−1). Colours: Pink (boron), yellow (sulfur), blue (nitrogen), gray (carbon). Hydrogen atoms omitted. | |
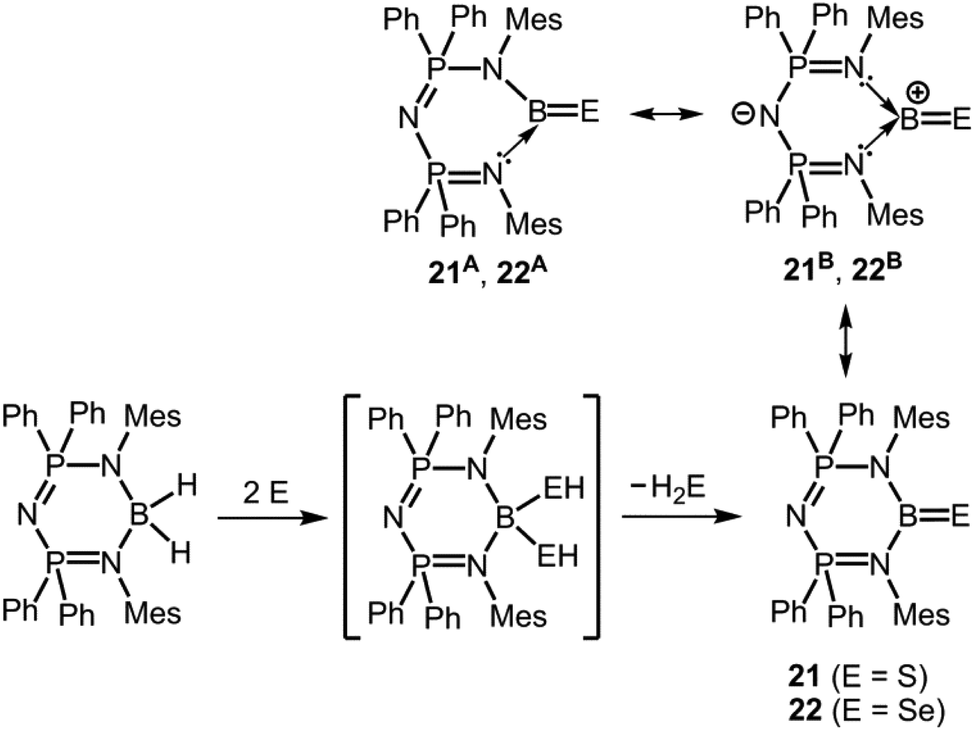
Singh and coworkers converted a boron dihydride complex with two equivalents of elemental sulfur or selenium to obtain the thioxoborane 21 and the selenoxoborane 22, respectively (Scheme 14).24 The bis(phosphoranimino)aminato ligand is a monoanionic system which is less inclined to switch to a dianionic group by proton transfer as observed for the 1,3-diketiminato ligand in 17 and 18. In structural resemblance to these the boron centre in 21 and in 22 is attached to two nitrogen atoms incorporated into a six-membered ring system. The B–S distance of 1.752(5) Å in 21 is slightly elongated in comparison to the BS double bond in 17, however, the B–Se bond length of 1.871(5) Å in 22 is decreased with respect to its ketimine congener 18 (1.896(4) Å). Similar to the formation of 17, 18 and 20+ a hydrogenchalcogenide intermediate was presumed for the reaction pathway that leads to 21 and to 22 (Scheme 14; monohydrogensulfide species for 17 and 18; bis(hydrogensulfide) species for 20+, 21 and bis(hydrogenselenide) species for 22). The three-coordinate borane (21, 22) results from elimination of EH2 from the four-coordinate species (as in 20+) which contrasts the hydrogen shift mechanism involved in the formation of the congeners 17 and 18 (vide supra, Schemes 11 and 14). The infrared spectroscopic study of 21 and 22 revealed the stretching vibration modes of the boron chalcogen bonds at 1096 cm−1 and 1076 cm−1, respectively. These values are at lower wave number than observed for 17 and 18 (vide supra). In the 11B NMR spectroscopic study 21 and 22 produced resonances at 41 ppm and 45.2 ppm, respectively.
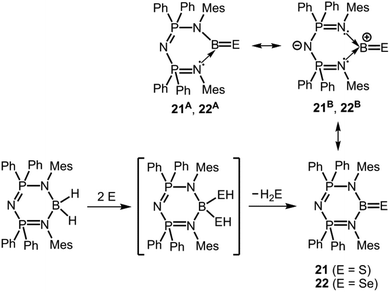 |
| | Scheme 14 Conversion of bis(phosphoranimino)amino boron dihydride to the thioxoborane 21, as well as selenoxoborane 22via a postulated bis(hydrogenchalcogenide) intermediate. Selected resonance structures 21A, 22A and 21B, 22B (Mes = mesityl). | |
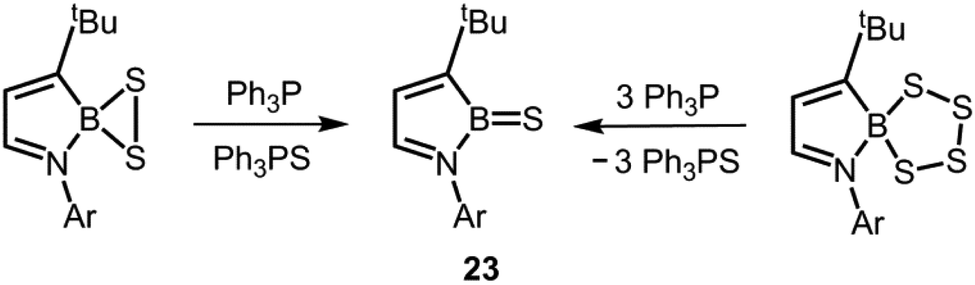
Very recently, Cui's group described the desulfination of boron polysulfides to the thioxoborane 23 by means of implementing triphenylphosphine as a chalcogen scavenger (Scheme 15).25 No structural data was reported for 23. The compound 23 gave rise to a peak at 50.3 ppm in the 11B NMR spectrum. If one considers that the number of competing π-donor ligands in 23 is decreased with respect to 17, 20+ and 21, it is reasonable to expect an increased boron sulfur bond order for the compound due to stronger electron donation from a lone pair at the sulfur atom to the boron-centred p-orbital.
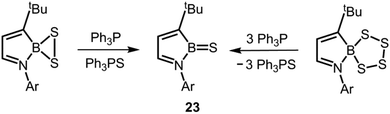 |
| | Scheme 15 Synthesis of the thioxoborane 23via desulfination of boron persulfide (left) or boron tetrasulfide (right; Ar = 2,6-(CH3)2-C6H3). | |
Related boron tellurium compounds.
There are a number of borane cluster compounds with a boron tellurium interaction.26 In sharp contrast, molecular electron-precise complexes with a boron–tellurium bond of any type are surprisingly rare. In fact, we found only the (9-BBN)2Te and {(9-BBN)Te}2 as related examples.27 Due to the unoccupied p-orbital at the boron centre, one would expect partial boron–tellurium double bond character for these compounds to a minor degree, though, the covalent radius of Te is comparably large (Table 1). However, the nature of the interaction was not conclusively elucidated by means of structural or theoretical investigation. Of course, the formation of aggregates in the condensed phase in which the electron deficiency of the boron atom is compensated by intermolecular interaction between the boron- and tellurium centres is feasible, as well. The 11B NMR resonances produced by (9-BBN)2Te and {(9-BBN)Te}2 were observed at 93.5 ppm and 87.0 ppm, respectively.
Complexes with an aluminium chalcogen multiple bond
Background.
Aluminium is marked by electronegativity values significantly smaller than that of boron (Table 1). As a result, the polarization of aluminium chalcogen bonds is usually stronger in comparison to boron chalcogen bonds and therefore the Lewis acid base pair character of the AlE fragment should be more pronounced, as well (vide supra). Moreover, the increased atomic radius of aluminium with respect to boron increases its tendency to form complexes with a higher coordinate metal centre (Table 1). In fact, three-coordinate aluminium complexes with chalcogen substituents at the metal centre have been described in the literature,28 but they are remarkably rare as compared to trigonal planar boron compounds. As another consequence of aluminium's larger atomic radius, the π-interaction with attached chalcogen atoms is generally weaker than that for boron. Accordingly, related examples with aluminium chalcogen double bond character rather follow the concepts D and E for thermodynamic stabilization via a four-coordinate metal centre (vide infra, Fig. 1). In this context, it has to be pointed out that isolated complexes that contain multiple bonds between aluminium atoms or between aluminium and a group 14 or 15 element are surprisingly rare in comparison to its neighbours in the periodic table. A monomeric iminoalane (AlN double bond) was described by Cui and coworkers.29 Extensive studies that aimed at the isolation of a dialumene (AlAl double bond) had been carried out by Power's and by Tokitoh's group, however, only trapping products of this elusive species were isolated.30 Maybe closest to complexes with AlAl double bonds are related examples of Power, as well as Pörschke and coworkers who isolated the lithium salts of radical compounds which were described to comprise a one-π-electron bond in addition to the Al–Al single bond.31
Aluminium oxygen double bond.
In 2002 Roesky and coworkers reported their outstanding study of the monotopic alumoxane 24 (Scheme 16).32 The metal centre is incorporated into a 1,3-diketimino scaffold and the bonding situation corresponds to the Lewis acid base-stabilization concept E as represented by the canonical structure 24A (Fig. 1, Scheme 16). The remarkably low Al–O distance of 1.659(3) Å is shorter than in typical ditopic aluminium oxygen complexes with a diketiminate ligand and four-coordinate aluminium centres (1.68–1.91 Å).28e,32,33 It is in the range of aluminium compounds with the very bulky 2,6-ditertbutyl-4-methyl-phenoxide ligand (1.64–1.69 Å) which mark a class of aluminum complexes that often comprise the metal centre in the coordination number three that is scarcely-found for aluminium.28 Notably, one Al–O distance in a tellurium-bridged ditopic 1,3-diketimino aluminium oxide was determined to 1.588(3) Å.33c However, this very short bond length does not imply aluminium oxygen double bond character as obvious from the bonding situation.
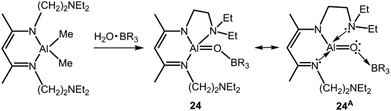 |
| | Scheme 16 Conversion of the diketimino dimethylalane to the alumoxane 24 (R = C6F5). Selected resonance structure 24A illustrates the donor–acceptor stabilization mode. | |
Related aluminium sulfur or -selenium compounds.
Interestingly, to the best of our knowledge, there are no examples reported for complexes that contain an aluminium and a sulfur or selenium atom that engage in an interaction of considerable double bond character. Several aluminium bis(hydrogensulfide)- and bis(hydrogenselenide) complexes, as well as related hydride hydrogenchalcogenides were described.33b,34 However, the elimination of H2S or dihydrogen, respectively, to yield monotopic thioxoalane similar to the formation of thioxoboranes from their boron hydrogensulfide precursors (vide supra) has not been reported to date.
Aluminium tellurium double bond.
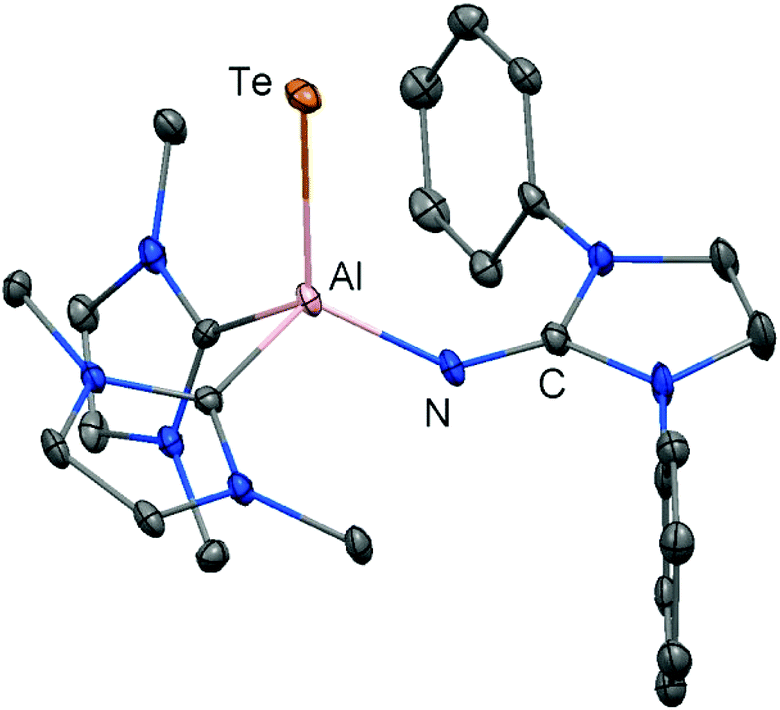
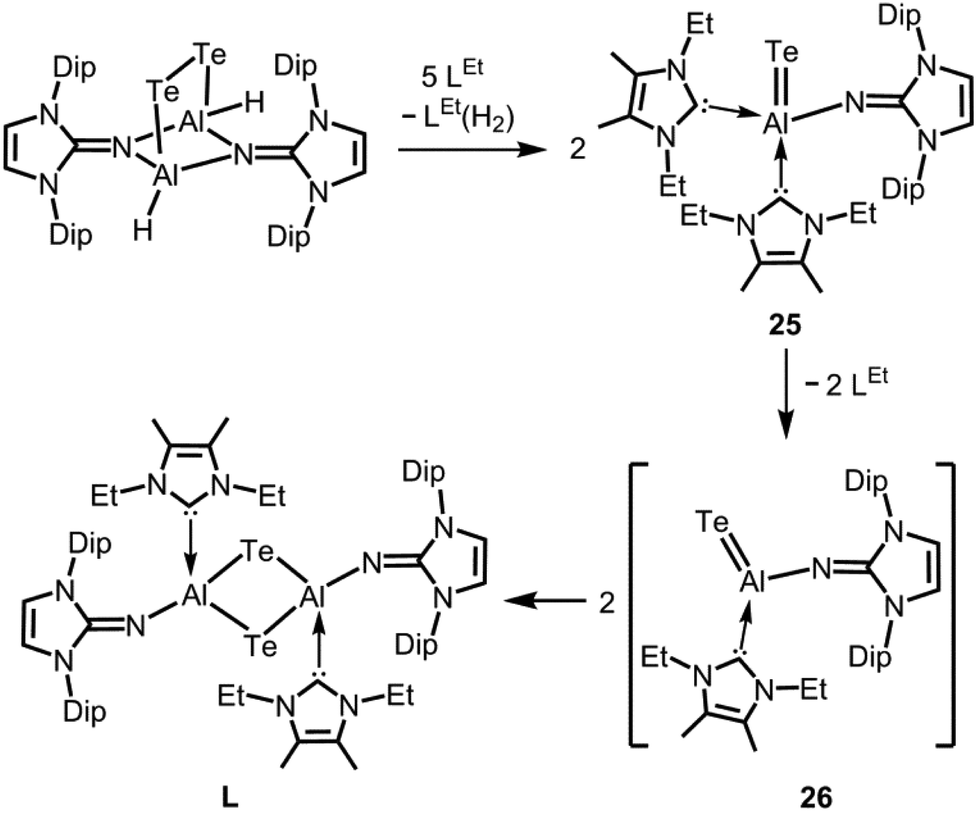
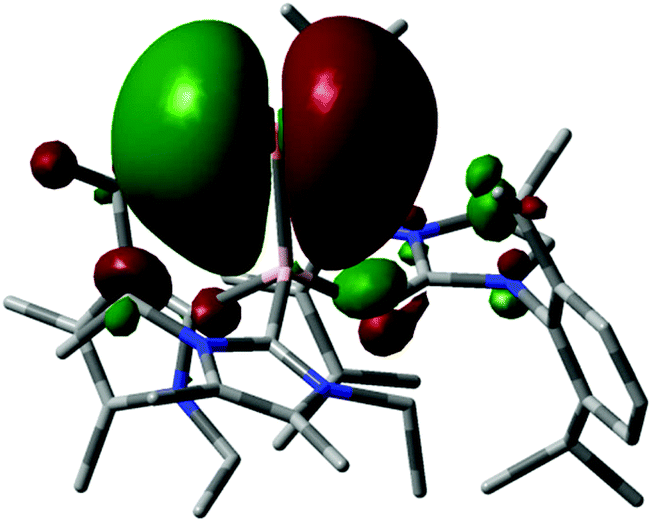
In 2015 we found a ditopic aluminium ditelluride with the chalcogen in the oxidation state −1 that reacts with NHC (LEt, 5 equiv., LEt = 1,3-diethyl-4,5-dimethyl-imidazolin-2-ylidene) in a dehydrogenative redox process.35 In this process the monotopic aluminium telluride 25 with the chalcogen in the formal oxidation state −2 is furnished along with dihydrogenated NHC (LEt(H2); Fig. 6, Scheme 17; concept D, Fig. 1).35 The structural study of 25 revealed a remarkably short Al–Te distance of 2.5130(14) Å and DFT calculations determined an enhanced aluminium–tellurium interaction (WBIAlTe = 1.20; NPA charges: Al = +1.24, Te = −0.95; NPA = Natural Population Analysis). Interestingly, the terminal position of the tellurium atom is a very scant structure motif as group 16 atoms commonly assume bridging positions in aluminium chalcogenides. Accordingly, it does not come as a surprise that upon heating a benzene solution of 25 to 80 °C one of the two LEt ligands is released. The intermediate LDipN(AlTe)LEt (26, Al–Te = 2.428 Å; LDip = 1,3-bis(2,6-diisopropylphenyl)-imidazolin-2-ylidene) aggregates to tellurium-bridged L and this process was computed by DFT methods (Scheme 17). The XRD analysis of L revealed significantly elongated Al–Te distances (2.6143(14) Å, 2.6211(15) Å) and a decreased bond order for the aluminium–tellurium interaction (WBIAlTe = 0.75; NPA charges: Al = +1.21, Te = −0.79) with respect to 25.35 Taking into account the salient changes in the Al–Te distances and the values for the WBIAlTe upon transformation of 25 into L, the nature of the aluminium–tellurium interaction in 25 was presumed to possess high AlTe double bond character. This is supported by a pronounced π-symmetric orbital lobe in the HOMO−1 which expands between the aluminium centre and the tellurium atom (Fig. 7). Notably, the aluminium species in 25 is isoelectronic to a sila-acylium ion for which significant SiO double bond character had been verified by NBO- and NRT analysis, as well as Mayer Bond Index and infrared spectroscopy.36
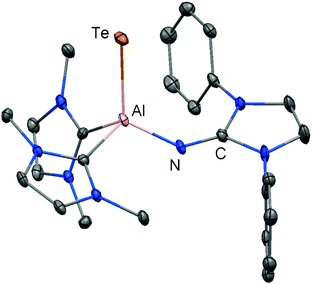 |
| | Fig. 6 Ellipsoid plot (30% level) of the molecular structure of aluminium telluride 25 in the solid state as derived from XRD analysis. Hydrogen atoms isopropyl groups and non nitrogen-bonded methyl groups have been omitted. | |
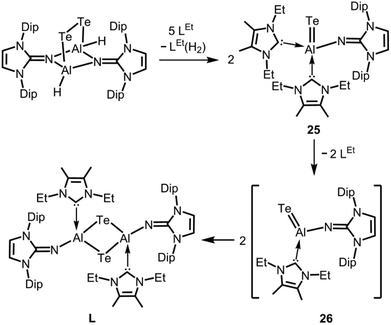 |
| | Scheme 17 Reaction of a ditopic aluminium ditelluride to the monotopic aluminium telluride 25 and its thermal transformation to ditopic Lvia the calculated intermediate 26 (Dip = 2,6-diisopropylphenyl, LEt = 1,3-diethyl-4,5-dimethyl-imidazolin-2-ylidene). | |
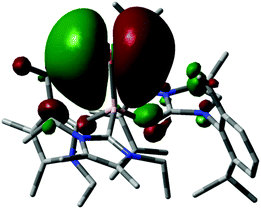 |
| | Fig. 7 Depiction of the HOMO−1 of 25. Colours: Pink (aluminium), yellow (tellurium), blue (nitrogen), gray (carbon). Hydrogen atoms omitted. | |
Complexes with a heavier group 13 metal chalcogen multiple bond
Background.
The increase in atomic number of the group 13 elements gallium, indium and thallium corresponds to electronegativity values that are mostly higher than found for aluminium (Table 1). Hence, the polarization of the M–E (M = Ga, In, Tl) bonds is generally weaker than for aluminium. Albeit the covalent radius of gallium is very similar to aluminium's the radii of indium and thallium are considerably increased and respective impact on the preferred coordination number (i.e. larger) and the π-interaction with chalcogen atoms (i.e. weaker) is to be expected (Table 1). Consequently, the tendency to form double-bonded systems is likely to be diminished for indium and thallium. In fact, we found no report for a thallium chalcogen multiple bond in the literature.
Gallium chalcogen double bond.
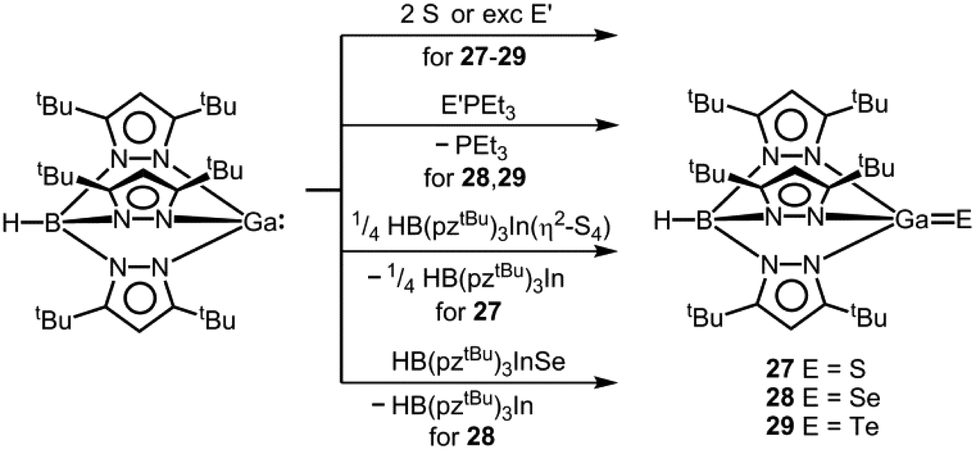
As the only examples for electron-precise complexes that contain a potential gallium chalcogen multiple bond, we found the work of Kuchta and Parkin on the complexes 27–29 with the tris(3,5-ditertbutyl)pyrazolylhydridoborato ligand ([HB(pztBu)3]−, Scheme 18).37,38 With regard to the ligand system concept D for stabilization of the GaE species would apply. However, if one considers that the electronic nature of the trispyrazolylborato ligand – its complexes are often referred to as “scorpionates” – has been compared to the cyclopentadienide anion unambiguous assignment according to the concepts for thermodynamic stabilization as outlined in this introduction is difficult.39 Compounds 27–29 derive from direct conversion of the trigonal pyramidal gallium(I) scorpionate HB(pztBu)3Ga with the respective chalcogen in elemental form (Scheme 18).37,38,40
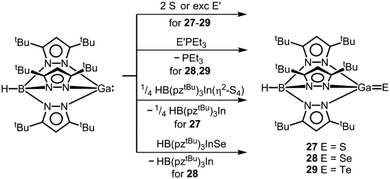 |
| | Scheme 18 Conversion of a trispyrazolylboryl gallium(I) compound to the gallium chalcogenides 27–29 (pztBu = 3,5-ditertbutyl-pyrazolyl, E′ = Se or Te). | |
The Ga–S distance in 27 amounts to 2.093(2) Å which is significantly shorter than the sum of the covalent radii (2.27 Å, Table 1, Fig. 8) and shorter than typical Ga–S single bonds that the authors retrieved from the database.37 Interestingly, synthetic access to 27 is also granted by conversion of HB(pztBu)3Ga with the indium sulfur complex HB(pztBu)3In(η2-S4) in a chalcogen exchange reaction (Scheme 18).37,38 It is of note that for the indium scorpionate system no InS species with a terminal sulfur atom was described.41 The heavier chalcogenides 28 and 29 exhibit gallium–chalcogen bond lengths of Ga–Se = 2.214(1) Å and Ga–Te = 2.422(1) Å.38 These values are markedly below the sums of the respective covalent radii of 2.42 Å and 2.60 Å (Table 1). For comparison, the authors referenced 2.324(2) Å as the shortest average Ga–Se bond length observed for a complex compound. Similar to the gallium sulfide 27, the gallium selenide 28 can be generated via conversion of HB(pztBu)3Ga with HB(pztBu)3InSe (30, vide infra, Scheme 18).38,42 Furthermore, 28 and 29 can be furnished by implementation of SePEt3 and TePEt3 as a chalcogen source, respectively (Scheme 18). The bonding situation of the gallium–chalcogen interaction in HB(pztBu)3GaE (27–29, E = S, Se, Te) was described as a composite of the resonance structures [GaE], [+Ga–E−], as well as [−GaE+] and a main contribution of the zwitterionic form [+Ga–E−] was pronounced (cf. Fig. 1, A, A′, A′′). However, no theoretical investigation was presented to verify this presumption. In fact, taking into account the theoretical studies on the nature of the AlTe bond in 25, as well as the SiO bond in its sila-acylium ion congener (vide supra),35,36 the weight of the canonical structure [GaE] should not be underrated.
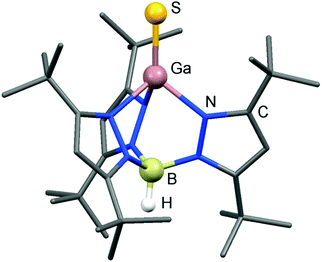 |
| | Fig. 8 Ball&stick model of the molecular structure of the trispyrazolylboryl gallium sulfide 27 in the solid state. Hydrogen atoms except on boron have been omitted. Pyrazolyl groups are depicted as a stick model. | |
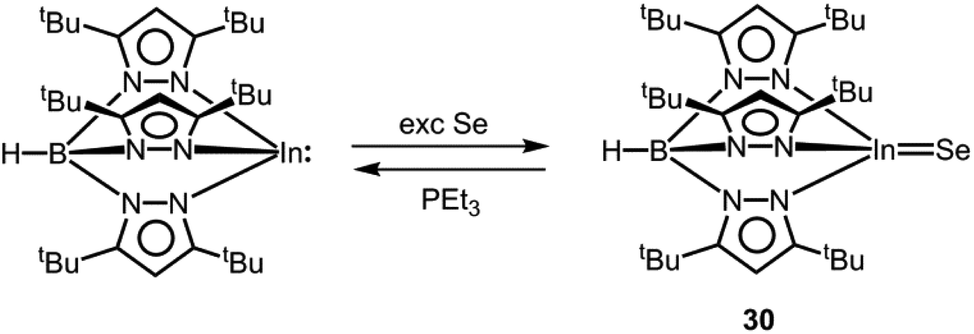
Indium chalcogen double bond.
As a scant example for an electron-precise complex that contains a potential indium chalcogen multiple bond, we found the scorpionate HB(pztBu)3InSe (30) in the literature (vide supra, Scheme 19).42 The compound was synthesized via conversion of HB(pztBu)3In with elemental selenium (Scheme 19). Unlike the gallium congener 28, it cannot be accessed via use of SePEt3 as a selenium source. On the contrary, it readily transfers the chalcogen atom to PEt3 (Scheme 19). The In–Se distance in 30 amounts to 2.376(1) Å which is considerably shorter than the sum of the covalent radii (2.62 Å, Table 1) and also reduced with respect to the mean In–Se single bond length that the authors determined to 2.65 Å.42 Interestingly, the selenium atom in HB(pztBu)3InSe (30) exchanges the chalcogen atom with HB(pztBu)3Ga to produce HB(pztBu)3In and HB(pztBu)3GaSe (28, Scheme 18). Interestingly, the heavier analogue HB(pztBu)3InTe was not obtained by the reaction of HB(pztBu)3In with tellurium metal.38 As outlined by Kuchta and Parkin, the indium–selenium interaction is composed of the three major resonance structures [InSe], [+In–Se−] and [−InSe+] (cf. Fig. 1, A, A′, A′′).42 With regard to the susceptibility of HB(pztBu)3InSe (30) towards chalcogen scavengers (e.g. HB(pztBu)3Ga, PEt3), the strength of the InSe bond can be assessed as comparably low. In consideration of the higher atomic radius and the lower electronegativity of indium in comparison to gallium the contribution of the zwitterionic form [+In–Se−] is presumed to be significantly higher than for the gallium congener 28. However, profound theoretical investigation (e.g. bond order, bond polarization, NRT study) would be required to shed more light on the bonding situation.
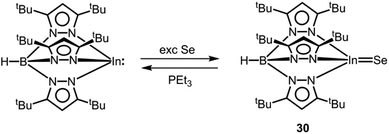 |
| | Scheme 19 Reaction of a trispyrazolylboryl indium(I) complex to the indium selenide 30 and regeneration of its precursor. | |
Conclusions
In summary this article has provided an overview on reports of electron-precise molecular complexes that contain a potential multiple bond between a group 13 element (M) and a chalcogen atom (E). No example for a purely kinetically stabilized multiple-bonded system of the type R–ME or LB→ME (LB = Lewis base) that can be isolated in the condensed phase at ambient temperature is found in the literature (R = anionic substituent). However, various related species have been highlighted which exploit the concepts for thermodynamic stabilization via electron-donating and electron-accepting ancillary groups. Furthermore, isolation of the BO triple bond was achieved via electronic stabilization in the coordination sphere of platinum. The given examples demonstrate that the lability of the ME fragment increases with the polarization of the M–E bond, as well as the atomic radius of the group 13 element which is in accordance with the general expectation. Among the group 13 elements examples for BE double bonds are most abundant and for thallium non-existent. Whether this is due to a bias in common research interests or results from a higher challenge for the isolation of respective substances is unclear. One way or the other, a higher demand for stabilization with rising metal character and larger coordination sphere of the element is to be expected. For the chalcogen group oxygen stands out in that no example has been reported in which it assumes a terminal position. Moreover, tellurium lives up to its maverick position among the group 16 elements with only one double-bonded species reported.
Future studies should generally focus on applying bulkier ligand systems to grasp the intrinsically elusive parent ME species. The application of metal chalcogen multiple bonded systems in the field of Small Molecule Activation remains largely unexplored, but high prospect in this regard is implied by work of Pachaly and West, as well as Tokitoh and coworkers. Additionally, the capabilities of the gallium or indium chalcogen systems reported by Kuchta and Parkin which may function as chalcogen acceptor, as well as transfer reagents illustrate the tremendous potential of this compound class as an activator or catalyst.
Acknowledgements
Financial support of the WACKER Chemie AG, as well as the European Research Council (ERC Starting Grant: SILION 637394) is gratefully acknowledged.
References
-
(a) N. Kambe, K. Kondo, S. Morita, S. Murai and S. Sonoda, Angew. Chem., Int. Ed. Engl., 1980, 19, 1009 Search PubMed
 ;
(b) I. Levin and D. Brandon, J. Am. Ceram. Soc., 1998, 81, 1995 CrossRef CAS ;
(c) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS PubMed ;
(d) G. Busca, Catal. Today, 2014, 226, 2 CrossRef CAS ;
(e) W. Lee and S.-J. Park, Chem. Rev., 2014, 114, 7487 CrossRef CAS PubMed ;
(f) M. Xia, K. Ding, F. Rao, X. Li, L. Wu and Z. Song, Sci. Rep., 2015, 5, 8548 CrossRef CAS PubMed .
;
(b) I. Levin and D. Brandon, J. Am. Ceram. Soc., 1998, 81, 1995 CrossRef CAS ;
(c) E. Y.-X. Chen and T. J. Marks, Chem. Rev., 2000, 100, 1391 CrossRef CAS PubMed ;
(d) G. Busca, Catal. Today, 2014, 226, 2 CrossRef CAS ;
(e) W. Lee and S.-J. Park, Chem. Rev., 2014, 114, 7487 CrossRef CAS PubMed ;
(f) M. Xia, K. Ding, F. Rao, X. Li, L. Wu and Z. Song, Sci. Rep., 2015, 5, 8548 CrossRef CAS PubMed .
-
(a) H.-J. Himmel, A. J. Downs and T. M. Greene, Chem. Rev., 2002, 102, 4191 CrossRef CAS PubMed ;
(b) A. Y. Timoshkin and H. F. Schaefer III, J. Phys. Chem. A, 2008, 112, 13180 CrossRef CAS PubMed ;
(c) N. A. Young, Coord. Chem. Rev., 2013, 157, 956 CrossRef ;
(d) T. Yang, D. S. N. Parker, B. B. Dangi, R. I. Kaiser, D. Stranges, Y.-H. Su, S.-Y. Chen, A. H. H. Chang and A. M. Mebel, J. Am. Chem. Soc., 2014, 136, 8387 CrossRef CAS PubMed .
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877 CrossRef CAS PubMed .
- B. Pachaly and R. West, J. Am. Chem. Soc., 1985, 107, 2987 CrossRef CAS .
- Y. Shoji, N. Tanaka, K. Mikami, M. Uchiyama and T. Fukushima, Nat. Chem., 2014, 6, 498 CrossRef CAS PubMed .
- D. Vidovic, J. A. Moore, J. N. Jones and A. H. Cowley, J. Am. Chem. Soc., 2005, 127, 4566 CrossRef CAS PubMed .
-
(a) B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832 RSC ;
(b) R. S. Mulliken, J. Chem. Phys., 1934, 2, 782 CrossRef CAS ;
(c) A. L. Allred and E. G. Rochow, J. Inorg. Nucl. Chem., 1958, 5, 264 CrossRef CAS ;
(d) D. Bergmann and J. Hinze, Angew. Chem., Int. Ed. Engl., 1996, 35, 150 CrossRef CAS .
-
(a) A. T. de Sousa, K. E. Bessler, S. S. Lemos, F. B. Gomes, G. A. Casagrande and E. Schulz Lang, Z. Anorg. Allg. Chem., 2007, 633, 771 CrossRef CAS ;
(b) Y. Yamamoto, M. Takizawa, X.-Q. Yu and N. Miyaura, Angew. Chem., Int. Ed., 2008, 47, 928 CrossRef CAS PubMed ;
(c) A. Budanow, E. von Grotthuss, M. Bolte and M. Wagner, Tetrahedron, 2016, 72, 1477 CrossRef CAS .
-
(a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., Int. Ed., 2009, 48, 5350 CrossRef CAS PubMed ;
(b) D. Franz, M. Bolte, H.-W. Lerner and M. Wagner, Dalton Trans., 2011, 40, 2433 RSC ;
(c) J. M. Breunig, A. Hübner, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2013, 32, 6792 CrossRef CAS .
- Y. Wang, H. Hu, J. Zhang and C. Cui, Angew. Chem., Int. Ed., 2011, 50, 2816 CrossRef CAS PubMed .
- A. Solovyev, S. J. Geib, E. Lacôte and D. P. Curran, Organometallics, 2012, 31, 54 CrossRef CAS .
- A. Del Grosso, E. R. Clark, N. Montoute and M. J. Ingleson, Chem. Commun., 2012, 48, 7589 RSC .
- Y. Wang, M. Y. Abraham, R. J. Gilliard Jr., D. R. Sexton, P. Wei and G. H. Robinson, Organometallics, 2013, 32, 6639 CrossRef CAS .
- T. Miyada and M. Yamashita, Organometallics, 2013, 32, 5281 CrossRef CAS .
- M. A. Salomon, T. Braun and A. Penner, Angew. Chem., Int. Ed., 2008, 47, 8867 CrossRef CAS PubMed .
- Y. K. Loh, C. C. Chong, R. Ganguly, Y. Li, D. Vidovic and R. Kinjo, Chem. Commun., 2014, 50, 8561 RSC .
- H. Braunschweig, K. Radacki and A. Schneider, Science, 2010, 328, 345 CrossRef CAS PubMed .
- H. Braunschweig, K. Radacki and A. Schneider, Chem. Commun., 2010, 46, 6473 RSC .
- S. Bertsch, J. Brand, H. Braunschweig, F. Hupp and K. Radacki, Chem. – Eur. J., 2015, 21, 6278 CrossRef CAS PubMed .
- N. Tokitoh, M. Ito and R. Okazaki, Tetrahedron Lett., 1996, 37, 5145 CrossRef CAS .
- H. Wang, J. Zhang, H. Hu and C. Cui, J. Am. Chem. Soc., 2010, 132, 10998 CrossRef CAS PubMed .
- J. Bauer, H. Braunschweig, A. Damme, J. O. C. Jimenez-Halla, T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726 CrossRef CAS PubMed .
- D. Franz, E. Irran and S. Inoue, Angew. Chem., Int. Ed., 2014, 53, 14264 CrossRef CAS PubMed .
- K. Jaiswal, B. Prashanth, S. Ravi, K. R. Shamasundar and S. Singh, Dalton Trans., 2015, 44, 15779 RSC .
- P. Chen and C. Cui, Chem. – Eur. J., 2016, 22, 2902 CrossRef CAS PubMed .
-
(a) G. Ferguson, J. F. Gallagher, M. McGrath, J. P. Sheehan, T. R. Spalding and J. D. Kennedy, J. Chem. Soc., Dalton Trans., 1993, 27 RSC ;
(b) D. K. Roy, S. K. Bose, K. Geetharani, K. K. V. Chakrahari, S. M. Mobin and S. Ghosh, Chem. – Eur. J., 2012, 18, 9983 CrossRef CAS PubMed ;
(c) A. Thakur, S. Sao, V. Ramkumar and S. Ghosh, Inorg. Chem., 2012, 51, 8322 CrossRef CAS PubMed .
- R. Köster, G. Seidel, W. Schüßler and B. Wrackmeyer, Chem. Ber., 1995, 128, 87 CrossRef .
-
(a) A. P. Shreve, R. Mulhaupt, W. Fultz, J. Calabrese, W. Robbins and S. D. Ittel, Organometallics, 1988, 7, 409 CrossRef CAS ;
(b) R. Benn, E. Janssen, H. Lehmkuhl, A. Rufińska, K. Angermund, P. Betz, R. Goddard and C. Krüger, J. Organomet. Chem., 1991, 411, 37 CrossRef CAS ;
(c) M. D. Healy and A. R. Barron, Angew. Chem., Int. Ed. Engl., 1992, 31, 921 CrossRef ;
(d) M. D. Healy, M. R. Mason, P. W. Gravelle, S. G. Bott and A. R. Barron, J. Chem. Soc., Dalton Trans., 1993, 441 RSC ;
(e) W. Clegg, E. Lamb, S. T. Liddle, R. Snaith and A. E. H. Wheatley, J. Organomet. Chem., 1999, 573, 305 CrossRef CAS ;
(f) Y. Yang, H. Li, C. Wang and H. W. Roesky, Inorg. Chem., 2012, 51, 2204 CrossRef CAS PubMed ;
(g) C. Descour, T. J. J. Sciarone, D. Cavallo, T. Macko, M. Kelchtermans, I. Korobkov and R. Duchateau, Polym. Chem., 2013, 4, 4718 RSC ;
(h) L. Wang and L. Yang, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2014, 70, m352 CAS .
- J. Li, X. Li, W. Huang, H. Hu, J. Zhang and C. Cui, Chem. – Eur. J., 2012, 18, 15263 CrossRef CAS PubMed .
-
(a) R. J. Wright, A. D. Phillips and P. P. Power, J. Am. Chem. Soc., 2003, 125, 10784 CrossRef CAS PubMed ;
(b) Y. Zhao, Y. Lei, Q. Dong, B. Wu and X.-J. Yang, Chem. – Eur. J., 2013, 19, 12059 CrossRef CAS PubMed ;
(c) T. Agou, K. Nagata and N. Tokitoh, Angew. Chem., Int. Ed., 2013, 52, 10818 CrossRef CAS ;
(d) K. Nagata, T. Agou and N. Tokitoh, Angew. Chem., Int. Ed., 2014, 53, 3881 CrossRef CAS PubMed .
-
(a) C. Pluta, K.-R. Pörschke, C. Krüger and K. Hildenbrand, Angew. Chem., Int. Ed. Engl., 1993, 32, 388 CrossRef ;
(b) R. J. Wehmschulte, K. Ruhlandt-Senge, M. M. Olmstead, H. Hope, B. E. Sturgeon and P. P. Power, Inorg. Chem., 1993, 32, 2983 CrossRef CAS .
- D. Neculai, H. W. Roesky, A. M. Neculai, J. Magull, B. Walfort and D. Stalke, Angew. Chem., Int. Ed., 2002, 41, 4294 CrossRef CAS .
-
(a) H. Zhu, J. Chai, V. Jancik, H. W. Roesky, W. A. Merrill and P. P. Power, J. Am. Chem. Soc., 2005, 127, 10170 CrossRef CAS PubMed ;
(b) S. González-Gallardo, V. Jancik, R. Cea-Olivares, R. A. Toscano and M. Moya-Cabrera, Angew. Chem., Int. Ed., 2007, 46, 2895 CrossRef PubMed ;
(c) S. González-Gallardo, A. S. Cruz-Zavala, V. Jancik, F. Cortés-Guzmán and M. Moya-Cabrera, Inorg. Chem., 2013, 52, 2793 CrossRef PubMed .
-
(a) V. Jancik, Y. Peng, H. W. Roesky, J. Li, D. Neculai, A. M. Neculai and R. Herbst-Irmer, J. Am. Chem. Soc., 2003, 125, 1452 CrossRef CAS PubMed ;
(b) A. P. Gómora-Figueroa, V. Jancik, R. Cea-Olivares and R. A. Toscano, Inorg. Chem., 2007, 46, 10749 CrossRef PubMed ;
(c) D. Franz, E. Irran and S. Inoue, Dalton Trans., 2014, 43, 4451 RSC ;
(d) D. Franz and S. Inoue, Chem. – Eur. J., 2014, 20, 10645 CrossRef CAS PubMed .
- D. Franz, T. Szilvási, E. Irran and S. Inoue, Nat. Commun., 2015, 6, 10037 CrossRef CAS PubMed .
- S. U. Ahmad, T. Szilvási, E. Irran and S. Inoue, J. Am. Chem. Soc., 2015, 137, 5828 CrossRef CAS PubMed .
- M. C. Kuchta and G. Parkin, J. Chem. Soc., Dalton Trans., 1998, 2279 RSC .
- M. C. Kuchta and G. Parkin, Inorg. Chem., 1997, 36, 2492 CrossRef CAS .
- F. T. Edelmann, Angew. Chem., Int. Ed., 2001, 40, 1656 CrossRef CAS .
- M. C. Kuchta, J. B. Bonanno and G. Parkin, J. Am. Chem. Soc., 1996, 118, 10914 CrossRef CAS .
- M. C. Kuchta and G. Parkin, Main Group Chem., 1996, 1, 291 CrossRef CAS .
- M. C. Kuchta and G. Parkin, J. Am. Chem. Soc., 1995, 117, 12651 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a