
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Investigation of reactivity and structure formation in a K–Te–U oxo-system under high-temperature/high-pressure conditions†
Bin
Xiao
ab,
Philip
Kegler
a,
Dirk
Bosbach
a and
Evgeny V.
Alekseev
*ab
aInstitute for Energy and Climate Research (IEK-6), Forschungszentrum Jülich GmbH, 52428 Jülich, Germany. E-mail: e.alekseev@fz-juelich.de
bInstitut für Kristallographie, RWTH Aachen University, 52066 Aachen, Germany
First published on 19th August 2016
Abstract
The high-temperature/high-pressure treatment of the K–Te–U oxo-family at 1100 °C and 3.5 GPa results in the crystallization of a series of novel uranyl tellurium compounds, K2[(UO2)3(TeIVO3)4], K2[(UO2)TeIV6O14], α-K2[(UO2)TeVIO5] and β-K2[(UO2)TeVIO5]. In contrast to most of the reported uranyl compounds which are favorable in layered structures, we found that under extreme conditions, the potassium uranyl oxo-tellurium compounds preferably crystallized in three-dimensional (3D) framework structures with complex topologies. Anion topology analysis indicates that the 3D uranyl tellurite anionic framework observed in K2[(UO2)3(TeIVO3)4] is attributable to the additional linkages of TeO3 polyhedra connecting with TeO4 disphenoids from the neighboring U–Te layers. The structure of K2[(UO2)TeIV6O14] can be described based on [UTe6O26]22− clusters, where six TeO5 polyhedra enclose a hexagonal cavity in which a UO8 polyhedron is located. The [UTe6O26]22− clusters are further linked by TeO5 square pyramids to form the 3D network. Similar to uranyl tellurates, both α-K2[(UO2)TeVIO5] and β-K2[(UO2)TeVIO5] contain TeO6 octahedra which share a common face to form a dimeric Te2O10 unit. However, in α-K2[(UO2)TeVIO5], these Te2O10 units connect with UO6 tetragonal bipyramids to form a 3D structural framework, while in β-K2[(UO2)TeVIO5], the same Te2O10 dimers are observed to link with UO7 pentagonal bipyramids, forming 2D layers. Raman measurements were carried out and the vibration bands related to TeIV–O, TeVI–O and UVI–O bonds are discussed.
Introduction
First known from a handful of minerals including cliffordite (UO2(Te3O7)),1 moctezumite (PbUO2(TeO3)2)2 and schmitterite (UO2(TeO3)),3 the chemistry of the uranium tellurium family has attracted increasing attention in recent years. This is, in particular, with regard to its diverse structural chemistry as well as the key roles it plays in environmental issues. Tellurium is one of the highly corrosive fission products that forms in different chemical states in spent nuclear fuel.4,5 The reaction of tellurium with actinides such as Th, U and Pu, as well as other fission products can produce currently unknown metallic and oxide compounds.6,7 The phase formation and structural chemistry of these tellurium-bearing compounds are the prerequisites for understanding and modelling the complicated processes in a reactor.Under oxidizing conditions, uranium is usually found in a hexavalent state in solutions and in the solid-state. The UVI is dominated by the configuration of the so-called uranyl cation (UO22+) formed via connecting two oxo oxygen atoms with one uranium center. It exhibits a linear geometry that can further coordinate with four to six ligands perpendicular to its linear direction resulting in tetragonal, pentagonal and hexagonal bipyramidal polyhedra.8–12
Tellurium, typically occurring in either a tetravalent or a hexavalent oxidation state, also bears fascinating structural chemistry. TeIV has a very rich structural chemistry coordinating three, four, or even five O atoms to form trigonal pyramidal (TeIVO3), disphenoidal (TeIVO4) and square pyramidal (TeIVO5) geometries. These oxo-anions can be further interlinked to form condensed oligomeric and polymeric structures.13 Unlike the versatile TeIV, TeVI normally occurs in a distorted octahedral arrangement by coordinating with six O atoms. Thus, one could expect that uranyl tellurates would be structurally much simpler. In fact, since TeVI is easily reduced to TeIV, the uranyl tellurates are extremely sparse, and only three uranyl tellurate compounds have been reported in the literature. These include one recently discovered mineral Pb[(UO2)(TeO6)] (Markcooperite14) and two synthetic Ag[(UO2)(HTeO5)] and Pb2[(UO2)(TeO6)].15
One of the current interests in uranium chemistry is to find suitable synthetic methods that permit the isolation of stable three-dimensional (3D) framework structures. These framework materials attract significant interest because they are able to accommodate guest atoms, ions or molecules in their cavities. This feature enables them to demonstrate extraordinary physicochemical performance, including selective-oxidation catalysis,16,17 ionic exchange properties,18,19 and storage metrics for radionuclides.20 In recent years, due to the improvement of chemical technology and the arduous efforts of research, uranium compounds have undergone a respectable rate of expansion with a great quantity of products being isolated and chemically elucidated almost on a weekly basis. However, most of these uranium compounds, encompassing ubiquitous linear uranyl (UO22+) units, are based on two-dimensional layered structures. This phenomenon can be attributed to the terminal feature of the trans dioxo oxygen from the UO22+ units that are normally arranged in a parallel fashion. However, this trend has not always followed the expectations. One of the most remarkable exceptions is the uranyl tellurite family.15,21,22 Equipped with a stereochemically active lone-pair of electrons, the uranyl tellurites have the ability to form unusual structural building units which are not restricted to layered structures. Some examples of 1D uranyl tellurites are Pb[(UO2)(TeO3)2],23 Sr3[UO2(TeO3)2](TeO3)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 and β-Tl2[(UO2)(TeO3)2],21 whereas Na8[(UO2)6(TeO3)10]24 and UO2(Te3O7)1 are crystallized in 3D framework structures. Due to the 5s25p0 electronic configuration, the p-block cation Te4+ has lone-pairs with an effective volume nearly the same as that of an O2−, which plays the role of a terminating ligand to cause one-sided coordination.12,25,26 As a consequence, the presence of a non-bonding volume on the Te4+ centers can either reduce overall dimensionality from 2D to 1D through blocking the propagation of structural linkages or cause pore volumes through opening up the crystal structure.27,28
21 and β-Tl2[(UO2)(TeO3)2],21 whereas Na8[(UO2)6(TeO3)10]24 and UO2(Te3O7)1 are crystallized in 3D framework structures. Due to the 5s25p0 electronic configuration, the p-block cation Te4+ has lone-pairs with an effective volume nearly the same as that of an O2−, which plays the role of a terminating ligand to cause one-sided coordination.12,25,26 As a consequence, the presence of a non-bonding volume on the Te4+ centers can either reduce overall dimensionality from 2D to 1D through blocking the propagation of structural linkages or cause pore volumes through opening up the crystal structure.27,28
In spite of the promising application potential for open framework materials, detailed studies associated with a targeted design have been barely reported. Recently, we addressed that the formation of a 3D framework can be directly affected by the reaction conditions such as temperature and pressure.29 Following this, we discussed the influence of high-temperature/high-pressure conditions on the formation of atypical structural features present in the solid-state uranium compounds.30 The extreme synthesis conditions play an important role in enhancing and improving the structural diversity and complexity that uranium can adopt. Under high-temperature (up to 1200 °C) and high-pressure (near 2.5 GPa) conditions, a totally novel family of uranyl borates where UO6, UO7, and UO8 tetragonal, pentagonal, and hexagonal bipyramids are all present within one compound were prepared.31 In addition, this method is also helpful for yielding structural features that have not been observed with traditional synthesis conditions, as exemplified by the isolation of the first thorium compound with mixed-valent oxoarsenic(III)/arsenic(V),32 as well as the first actinide aluminoborate.29 Continuing in this direction, here, we substantially expand the uranyl tellurium family through the application of a high-temperature/high-pressure route that yields four new potassium uranyl tellurium compounds, K2[(UO2)3(TeIVO3)4], K2[(UO2)TeIV6O14], α-K2[(UO2)TeVIO5] and β-K2[(UO2)TeVIO5].
Experimental section
Crystal syntheses
Note: U used in this study belongs to an α emitting radioisotope and thus standard precautions for handling radioactive materials should be strictly obeyed at all times.The titled four uranyl tellurium crystals were prepared by using high-temperature/high-pressure experiments using the piston cylinder module of a Voggenreiter LP 1000-540/50. The raw chemicals used were A. R. grade UO3, KNO3 (Alfa-Aesar), TeO2 (Alfa-Aesar) and H6TeO6 (Alfa-Aesar) and used without further purification. The UO3 was obtained by heating (UO2)(NO3)2·6H2O at 400 °C for 5 h. All the syntheses were performed using similar experimental procedures; the only differences were the ratios of starting chemicals, applied temperature and pressure. First, the starting materials of UO3, KNO3, TeO2 and H6TeO6 were weighed with the desired ratio, then were mixed and finely ground. Second, the obtained mixture was filled into a platinum capsule (outer diameter: 4 mm, wall thickness: 0.2 mm, length: 7 mm). Third, the capsule was sealed on both sides with an impulse micro welding device (Lampert PUK U4) and placed into the center of a ½-inch piston cylinder talc-Pyrex assembly. The calibration procedure of the piston cylinder module is described in ref. 32. After this, the corresponding pressure and temperature were applied on the capsule to perform the crystal synthesis. A detailed description of the chemical ratio as well as the pressure and temperature profile for each crystal synthesis is as below.
:K:Te ratio of 1:2:4. A pressure of 3.5 GPa was applied within 30 minutes and this pressure was kept constant for the whole experimental run. The temperature program was started only after the desired pressure was reached, and the associated temperature is as follows: first, the temperature was increased to 1100 °C within 30 minutes, and then held at this temperature for 180 minutes. After this, the temperature was decreased to 900 °C within 60 minutes. Then, the temperature was slowly decreased to 350 °C at a rate of 0.14 °C min−1 followed by quenching to room temperature. The quenching time of the sample is about 2 to 3 seconds. After quenching, the pressure was released in a period of 30 minutes. Finally, the platinum capsule was crushed in order to extract the reaction products out of the high pressure assembly. The resulting products with dark yellow color were obtained for further investigations. The powder X-ray diffraction patterns of the obtained products show that the mineral cliffordite (UO2(Te3O7)) is the major phase. K2[(UO2)3(TeIVO3)4] presents as a minor phase (see the ESI†).
:K:Te ratio of 1:4:7. After filling the mixture into the capsule and sealing it, a pressure of 3.5 GPa was applied within 30 minutes and this pressure was kept constant for the whole experimental running time. The temperature program was started only after the desired pressure was reached, and the temperature profile is the same as that of K2[(UO2)3(TeIVO3)4]. The resulting products are light brown crystals. The powder X-ray diffraction patterns of the synthesized products indicate that the yield of K2[(UO2)3(TeIVO3)4] is more than 95% (see the ESI†).
:K:Te ratio of 1:2:1. A pressure of 3.5 GPa was applied within 30 minutes and this pressure was kept constant for the whole experiment. The temperature program was started only after the desired pressure was reached, and the associated temperature is very similar to that of K2[(UO2)3(TeIVO3)4]. But here we used a rate of 0.10 °C min−1 to decrease the temperature from 900 °C to 350 °C. The resulting products with dark yellow color were obtained for further investigations. Powder X-ray diffraction patterns indicate that the UO3 is the main phase. α-K2[(UO2)TeIV6O14] can only be found as a minor product (see the ESI†).
:K:Te ratio of 1:2:3. A pressure of 3.5 GPa was applied within 30 minutes and this pressure was kept constant for the whole experimental run. The temperature program was started shortly after the desired pressure was reached, and the temperature program is the same as that of α-K2[(UO2)TeVIO5]. After the experiment, the products with dark yellow color were found. Because of the difficulty to discern the crystals from broken glass pieces, the yield cannot be obtained. Powder XRD shows that β-K2[(UO2)TeVIO5] is the minor product. The main phase is α-K2[(UO2)TeVIO5]. Similar to α-K2[(UO2)TeVIO5], powder diffraction shows that β-K2[(UO2)TeVIO5] presents as the minor phase with UO3 presenting as the major one (see the ESI†).
Crystallographic studies
The as-obtained uranyl tellurium crystals were selected for data collection. The crystals were mounted on glass fibers and optically aligned on an Agilent single crystal diffractometer (SuperNova, Dual Source). The data collection was done using a monochromatic Mo-Kα tube which has an incident wavelength of 0.71073 Å and runs at 50 kV and 0.8 mA providing a beam size of approximately 30 μm. A scan width of 0.75°/ω and an exposure time of 35 s per frame were used for data collection, respectively. The unit-cell dimensions for these crystals were refined using least-squares techniques against the positions of all measured reflections. More than a hemisphere of data were collected for each crystal and the three-dimensional data were reduced and filtered for statistical outliers using the standard CrysAlisPro program. Data were corrected for Lorentz, polarization, absorption and background effects. The crystal structure determination and refinement were carried out using the SHELXL-97 program.33 The data and crystallographic information are given in Table 1. The structures were solved by direct methods and refined to R1 = 0.0377 for K2[(UO2)3(TeIVO3)4], R1 = 0.0298 for K2[(UO2)TeIV6O14], R1 = 0.0344 for α-K2[(UO2)TeVIO5] and R1 = 0.0225 for β-K2[(UO2)TeVIO5], respectively.| Compound | K2[(UO2)3(TeIVO3)4] | K2[(UO2)TeIV6O14] | α-K2[(UO2)TeVIO5] | β-K2[(UO2)TeVIO5] |
|---|---|---|---|---|
| Dimension | 3D | 3D | 3D | 2D |
| Formula weight | 1590.69 | 1337.82 | 555.83 | 555.83 |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) |
P21/n | C2/c |
| a (Å) | 6.8463(6) | 11.394(3) | 7.9021(6) | 14.201(3) |
| b (Å) | 7.0274(7) | 11.394(3) | 10.1355(9) | 13.8632(12) |
| c (Å) | 9.4044(9) | 11.394(3) | 8.5671(7) | 7.1186(6) |
| α | 73.579(9)° | 90 | 90 | 90 |
| β | 81.098(8)° | 90 | 95.591(7) | 113.761(14) |
| γ | 81.761(8)° | 90 | 90 | 90 |
| V (Å3) | 426.41(7) | 1479.2(7) | 682.89(10) | 1282.7(3) |
| Z | 1 | 1 | 4 | 8 |
| λ (Å) | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| F(000) | 666 | 2290 | 952 | 1943 |
| ρ calcd (g cm−3) | 6.195 | 6.031 | 5.406 | 5.416 |
| R 1 | 0.0377 | 0.0298 | 0.0344 | 0.0225 |
| wR2 (Fo2) | 0.0886 | 0.0680 | 0.0832 | 0.0533 |
Raman studies
Utilizing a Peltier cooled multi-channel CCD detector, the unpolarized Raman spectra were recorded with a Horiba LabRAM HR spectrometer. All the samples were in the form of single crystals. An objective with a 50× magnification was linked to the spectrometer, allowing the analysis of samples as small as 2 μm in diameter. The incident radiation was produced by a He–Ne laser at a power of 17 mW (λ = 632.81 nm). The focal length of the spectrometer was 800 mm and a 1800 gr mm−1 grating was used. The spectral resolution was around 1 cm−1 with a slit of 100 μm. The Raman spectroscopic investigation for all the samples was executed at room temperature in the range of 100–1050 cm−1.Bond-valence analysis
Bond-valence sums (BVS) for all atom positions in the four uranium tellurium compounds were calculated. The bond-valence parameters for U(VI)–O are obtained by Burns,34 and the bond-valence parameters for Te(IV)–O and Te(VI)–O are used according to Brese and O'Keeffe.35 The BVS for all atoms are consonant with their expected formal valences.Scanning electron microscopy/energy-dispersive spectroscopy (SEM/EDS)
Scanning electron microscopy images and energy-dispersive spectroscopy (SEM/EDS) data were collected using a FEI Quanta 200F environment scanning electron microscope. The EDS results are in good agreement with the proposed chemical compositions for all the four tellurium uranium compounds.Results and discussion
Synthesis
Attributable to the weak thermal stability of tellurium compounds,36–38 their synthesis needs to be performed either at relatively low temperature or under sealed conditions. In fact, most reported uranyl tellurium compounds were either from the low-temperature (180–220 °C) hydrothermal method24,39 or a high-temperature (around 800 °C) flux method40 which allows the crystal growth at a temperature far below the melting point of the solute. In our recent work,41 we described the preparation of a series of novel sodium uranium tellurium compounds using the high-temperature/high-pressure technique. The advantages of the high-temperature/high-pressure method include the ability to stabilize crystalline phases which are difficult to obtain under normal conditions. Besides, the sealed conditions prevent the sublimation of tellurium oxide and evaporation of H2O molecules.The formation of the four uranyl tellurium compounds is strongly associated with the molar ratio of the starting materials (KNO3, TeO2, H6TeO6 and uranium). We found that the appearance of TeO2 is necessary for the isolation of uranyl tellurites (K2[(UO2)3(TeIVO3)4] and K2[(UO2)TeIV6O14]). Our initial effort to synthesize the uranyl mixed-valent Te4+/Te6+ compounds by adding tetravalent TeO2 and hexavalent H6TeO6 as reacting sources was not successful. By comparison, thorium Te4+/Te6+ compounds can be prepared under similar conditions, but these are the subject of ongoing work and will be published in the near future. It is noted that α-K2[(UO2)TeVIO5] was favored in the reactions with low Te:U ratios, while higher Te:U ratios can lead to β-K2[(UO2)TeVIO5].
Structure descriptions
, and forms a 3D U–Te anionic framework which is charge balanced by K+ cations. It contains two symmetrically unique U6+ sites (U(1) and U(2)) and two different Te4+ sites (Te(1) and Te(2)) in the asymmetric unit. Both uranium sites are coordinated with two nearly linear trans oxygen atoms at the axial positions, forming a uranyl (UO22+) unit. U(1) forms in a highly distorted UO6 tetragonal bipyramidal geometry by binding with four equatorial O atoms, a less common coordination environment for uranium. The U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond distance of U(1) is 1.812 Å (×2), which falls in the typical range of uranyl coordination.42 Its equatorial U–O distances, with an average value of 2.310 Å, are obviously distorted in the O(4) direction, this being the shortest U–O distance of 2.198(9) Å. U(2) is found in a UO7 pentagonal bipyramidal configuration with the UO bond distances from 1.817(9) to 1.820(9) Å. Compared to U(1), U(2) experiences less perturbation within its equatorial plane, but still shows a slight variation, which ranges from 2.268(8) to 2.383(9) Å.
O bond distance of U(1) is 1.812 Å (×2), which falls in the typical range of uranyl coordination.42 Its equatorial U–O distances, with an average value of 2.310 Å, are obviously distorted in the O(4) direction, this being the shortest U–O distance of 2.198(9) Å. U(2) is found in a UO7 pentagonal bipyramidal configuration with the UO bond distances from 1.817(9) to 1.820(9) Å. Compared to U(1), U(2) experiences less perturbation within its equatorial plane, but still shows a slight variation, which ranges from 2.268(8) to 2.383(9) Å.
Te(1) is in the form of a disphenoidal geometry (TeO4). It is coordinated with four oxygen atoms, and with its electron lone pairs pointing towards the cross corner (see Fig. 1(c)), whereas Te(2) is surrounded with three oxygen atoms in a trigonal pyramidal configuration (TeO3). The Te(2) atom is off-centered from the triangular base (see Fig. 1(d)). It is noted that the fourfold TeIVO4 or threefold TeIVO3 coordinations are quite common among the tellurite structures.43–45 The Te(1)–O bond lengths vary within a considerably wide range from 1.845(9) to 2.171(9) Å, and the mean value is around 1.999 Å. In contrast, the Te(2)O3 trigonal pyramid is less distorted, but still shows substantial variations in Te–O bond lengths and O–Te–O angles, which range from 1.853(9) to 1.890(9) Å and from 87.8(4)° to 103.0(4)°, respectively.
 | ||
| Fig. 1 Representation of the structure of K2[(UO2)3(TeIVO3)4]. (a) Illustration of the anionic uranyl tellurite framework which is charge balanced by K+. (b) View of the [TeO3]2− chain comprised of alternating connections of TeO3 and TeO4 polyhedra. (c) The local coordination environment of Te(1). (d) The local coordination environment of Te(2). Legends: UO7 pentagonal bipyramids are shown in yellow. TeO3 polyhedra are in light blue and their long pairs are shown in purple. K+ cations are in blue nodes. | ||
In K2[(UO2)3(TeIVO3)4], two U(2) pentagonal bipyramids share a common edge, leading to a U2O12 dimer. The neighboring U2O12 dimers are fused together via sharing four vertexes with four equatorial corners from UO6 polyhedra, resulting in a zigzag uranyl ribbon running in the [010] direction (Fig. 1(a)). The Te(1)O4 disphenoids and Te(2)O3 trigonal pyramids are connected alternately by sharing common corners to form infinite [TeO3]2− chains propagating along the a-axis direction (Fig. 1(b)). The lone electron pairs of Te(1) and Te(1) are arranged exclusively vertical to the chain direction. Finally, these [TeO3]2− chains further link with the above-mentioned uranyl ribbons in a corner-sharing manner, completing the [(UO2)3Te4O12]2− framework. Bond valence calculation results in 4.05 and 3.97 v.u. for Te(1) and Te(2), respectively, both of which are consistent with Te in tetravalent oxidation.
It is noteworthy that the structure of K2[(UO2)3(TeIVO3)4] is closely related to that of A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs). Both structures can be seen as based on similar U–Te building fragments, shown in Fig. 2(b and c). The local linkages inside such building fragments can be described using the method of anion topology46 and the result is shown in Fig. 2(a). In this anion topology, the triangles are filled with Te polyhedra (TeO3 or TeO4) while the squares and pentagons are occupied by UO6 and UO7, respectively. In the former structure, the observed U–Te building fragments are fused together, with the linkage of additional TeO3 polyhedra, resulting in a terraced configuration (Fig. 2(d)), whereas the adjacent U–Te building fragments in the latter compounds are fused side-by-side within a plane (Fig. 2(e)). As a result, K2[(UO2)3(TeIVO3)4] forms a 3D framework while A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs) crystallizes in a 2D layered structural type. It is obvious that this dimensionality difference is due to the coordination geometries of the Te polyhedra within the respective U–Te building fragment. As mentioned above, the anion topology of both structural types contains triangles that represent the Te polyhedra. In K2[(UO2)3(TeIVO3)4], the corresponding triangular area in the anion topology graph cannot be exactly occupied by a four-coordinated TeO4 disphenoid. In this case, the remaining two oxygen corners of this TeO4 which do not participate in triangle composition can be further linked “up” and “down” relative to the U–Te building plane with additional TeO3 units (highlighted in Fig. 2(d) in pink color). In contrast, for A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs), the corresponding triangle in anion topology can be exactly filled by three oxygen atoms of a TeO3 trigonal pyramid.
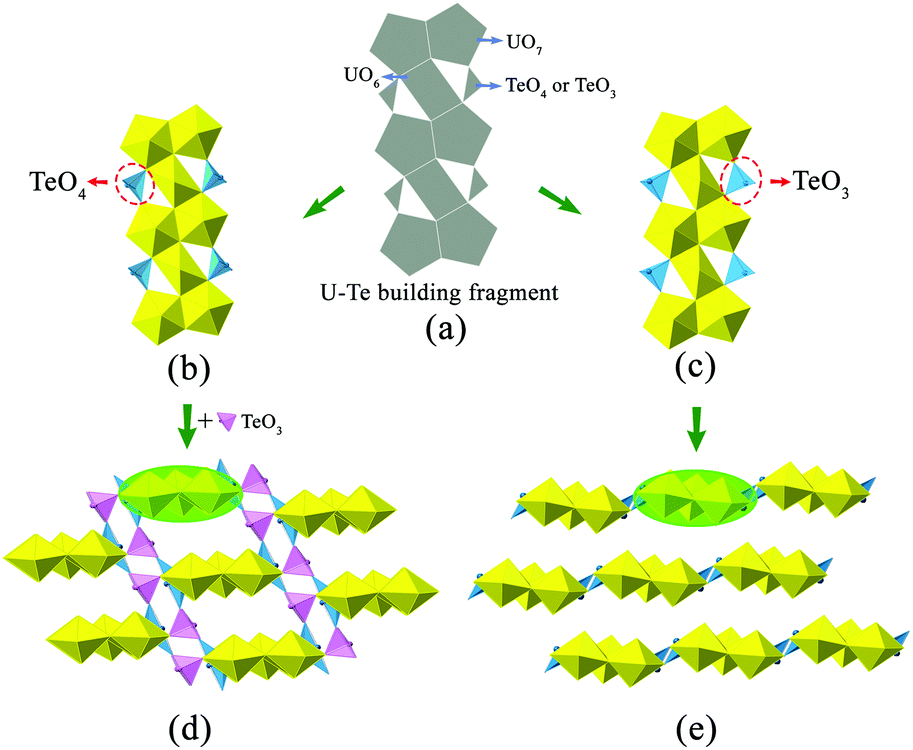 | ||
| Fig. 2 Structural comparison between 3D K2[(UO2)3(TeIVO3)4] and 2D A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs). (a) The anion topology of the uranyl tellurite building block. (b) The uranyl tellurite building block observed in K2[(UO2)3(TeIVO3)4]. (c) The uranyl tellurite building block observed in A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs). (d) 3D framework in K2[(UO2)3(TeIVO3)4] formed by connecting uranyl tellurite building blocks with additional TeO3 linkers (the TeO3 linkers are highlighted in pink). (e) 2D layered structure in A2[(UO2)3(TeO3)2O2] (A = K, Rb, Cs) composed of linking uranyl tellurite building blocks side-by-side in a plane. Legends: UO7 pentagonal bipyramids are shown in yellow. TeOx (x = 3 or 4) polyhedra are in light blue and pink, respectively. | ||
O bond distances of 1.788(10) Å (×2), and the OUO angle of 180.0(0)°. The equatorial U–O bond distances are all equal to 2.445(6) Å. As given in Fig. 3(b), each U shares all six edges with six TeO5 polyhedra in the equatorial plane, giving rise to a [UTe6O26]22− cluster. Each Te(1) site is coordinated by five oxygen atoms with the electron lone pairs pointing towards the square base, creating a distorted square pyramidal coordination geometry. The resulting Te(1)O5 polyhedron has one μ2–O atom linked with another Te(1), and four μ3–O atoms, two of which are linked with one Te(1) and one U(1) and another two are linked with two Te(1) atoms. The topology of oxo-tellurium is shown in Fig. 3(c). The Te–O bond distances varying from 1.873(6) to 2.363(6) Å are within the range seen in other tellurites containing TeO5 polyhedra.47,48 Finally, the TeO5 square pyramids further bridge with [UTe6O26]22− clusters to complete the 3D network structure of K2[(UO2)TeIV6O14]. Bond valence sum calculations are consistent with UVI and TeIV, providing the values of 6.0 and 3.9 v.u., respectively.
 | ||
| Fig. 3 (a) A view of the three-dimensional K2[(UO2)TeIV6O14] extending along the c-axis. (b) The local coordination geometry of [UTe6O26]22− clusters in K2[(UO2)TeIV6O14]. (c) The topology of oxo-tellurium. UO8 hexagonal bipyramids are shown in yellow. TeO5 polyhedra are in light blue. K+ cations are blue nodes. | ||
The structure of K2[(UO2)TeIV6O14] is highly related to that of cliffordite (UO2(Te2O7)). Both are constructed from the same structural building units of UO8 hexagonal bipyramids and TeO5 square pyramids. Their structural difference comes from the number and arrangement of the UO8 polyhedra in each structural unit. As shown in Fig. 4, only one U site is observed in K2[(UO2)TeIV6O14]. However, two symmetrically independent U sites are found in the structure of cliffordite, one of which is partially occupied and the other is fully occupied. For cliffordite, 27 UO8 hexagonal bipyramids reside in each unit cell, forming a regular pcu (primitive cubic lattice) net. In contrast, only 13 U atoms are found in one unit cell for K2[(UO2)TeIV6O14], and they are arranged into a cuboctahedral conformation with a fcu-a (augmented face-centered cubic lattice) topology, much like the uranium arrangement in the Na-based counterpart of Na[(UO2)Te6O13(OH)].41 In fact, the U arrangement in K2[(UO2)TeIV6O14] and cliffordite can be transformed from one to another by removing the 8 corners and 6 face-centers from the cliffordite-based topology.
 | ||
| Fig. 4 Arrangement of UO8 hexagonal pyramids in one unit cell for (a) cliffordite (UO2(Te3O7)) and (b) K2[(UO2)TeIV6O14], respectively. The corresponding topologies of the pcu and fcu-a net for UO2(Te3O7) and K2[(UO2)TeIV6O14], respectively. Topological transformation of U arrangement from pcu observed in K2[(UO2)TeIV6O14] to fcu-a observed in UO2(Te3O7) can be achieved by removing the 8 corners and 6 face-centers. UO8 hexagonal bipyramids are shown in yellow nodes. | ||
O, and 2.221(10)–2.246(9) Å are observed for equatorial U–Oeq. One symmetrically distinct Te6+ cation, with the BVS result of 5.97 v.u., is present in TeO6 octahedral coordination. The bond angles are 78.9(4)–99.9(4)° and 169.3(4)–179.5(4)° Å for Te–O in cis- and trans-configurations, respectively.
 | ||
| Fig. 5 (a) View of the structure of α-K2[(UO2)TeVIO5] extending along the bc plane. The fundamental building block (FBB) is highlighted by a red line. (b) Projection of the structure of α-K2[(UO2)TeVIO5] along the b-axis. (c) The FBB is constructed from two UO6 tetragonal bipyramids which share corners with a Te2O10 dimer. (d) The corresponding ball-and-stick presentation of the FBB. UO6 tetragonal bipyramids are shown in yellow. TeO6 octahedra are in light blue. K+ cations are blue spheres. | ||
The fundamental building block (FBB) in the 3D structural framework of α-K2[(UO2)TeVIO5] is composed of two TeO6 octahedra and two UO6 tetragonal bipyramids, shown in Fig. 5(c and d). Within it, the [Te2O10]4− anions, formed by two edge-sharing TeO6 octahedra, share four corners linking with two UO6 tetragonal bipyramids. The bridging Te–O(4), with a bond length of 1.995(9) Å, shows significant lengthening with respect to the remaining Te–O bonds, which range from 1.869(10) to 1.911(9). The Te–O–Te angles in [Te2O10]4− anions are around 101°, and the Te–O–O–Te dihedral angle is nearly 180°. It is to be noted that analogues of the [Te2O10]4− anions can also be found in several inorganic tellurates.15,49
The FBBs link together via a corner-sharing manner to form a complex 3D system of channels. When viewing from the c-axis, one can find that the largest channel runs though the U–Te lattice (highlighted in Fig. 6(a)). This channel, with an eight-ring pore opening, is occupied by charge compensating K+ cations. It has a cross-section of an elliptic shape and the dimensions of around 5.4 Å × 3.2 Å. Besides, additional smaller interlacing channels can also be detected propagating in the [101], [10] and [110] directions. In order to describe the connection behavior of uranium and tellurium polyhedra along the channel direction, here we use the black-and-white nodal representation where the UO6 tetragonal bipyramids and TeO6 octahedra are simplified as black and white nodes, respectively. The nodes are connected by single or double lines if the corresponding polyhedra share a corner or an edge with each other. This method permits one to elucidate the local topological feature inside the channel structure and has been widely used to describe the open-framework actinide compounds containing tubular units, such as uranyl selenate [(UO2)3(SeO4)5]4−,50 uranyl molybdate [(UO2)5(MoO4)]4− (ref. 51) and thorium molybdate [Th3(MoO4)7]2−.52 The largest channels propagating along the c-axis is shown in Fig. 6(b and c). The corresponding topological net is given in Fig. 6(d). From the idealized unfold topology (see Fig. 6(e)), one can note that the infinite U–Te channel is assembled from seven- and three-membered rings. Due to the existence of a twofold rotational axis, the topological net is achiral, which is consistent with the space group P21/n. Each uranium polyhedron (black node) is four-connected while each tellurium polyhedron (white node) is three-connected. It is noted that this channel structure can be re-constructed using a folding and gluing procedure. First, the equivalent points on the sides of the tape were labelled by letters a, b and c. Then the tape was folded by joining the corresponding opposite sides (aa′, bb′ and cc) to make a cylinder. The idealized topological structure for the tubular unit in α-K2[(UO2)TeVIO5] is demonstrated in Fig. 6(f).
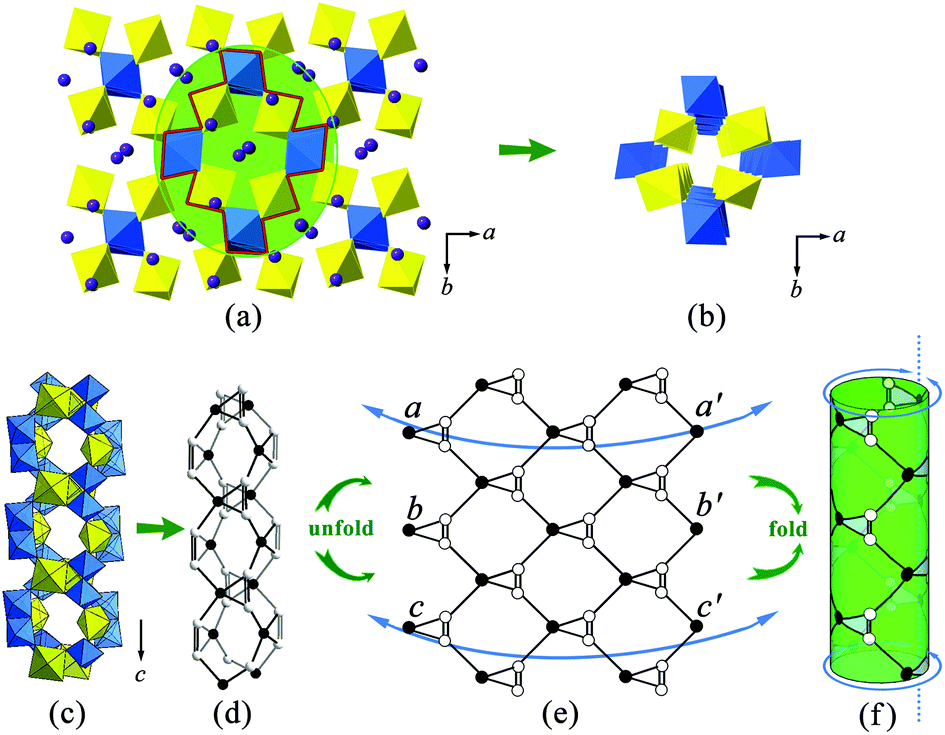 | ||
| Fig. 6 Channels in the structure of α-K2[(UO2)TeVIO5]. The structure can be considered as consisting of a cylinder unit. (a) The polyhedral presentation viewed along the b-axis. (b) Extraction of one of the channels from the structure of α-K2[(UO2)TeVIO5]. (c) Viewing this channel along the direction that is perpendicular to the c-axis. (d) The black-and-white presentation of the corresponding channel topology. (e) The ideal unfold vision of the channel topology, which shows that the channel is composed of seven- and three-membered rings. (f) The ideal vision of the channel structure obtained through simple folding and gluing procedures. | ||
O bonds of the uranyl unit, and from 2.241(5) to 2.541(5) Å for the U–O in the equatorial plane. These UO7 polyhedra share common edges with each other to result in infinite chains, with one UO7 polyhedron wide, running along the c-axis. In fact, these U chains have been commonly observed among several minerals and synthetic uranium tellurites/tellurates, such as moctezumite Pb[(UO2)(TeO3)2]2 and UTeO53. As for the Te site, it exhibits a slightly distorted octahedral environment. The distortion can be best shown from the cis O–Te–O angles in the range of 80.0(2)° to 95.4(2)°. The Te–O bond distances vary within an appreciable range of 1.886(4) to 1.983(5) Å, the average distance being 1.929 Å. The bond valence sum calculations give the values of 5.9 v.u. for the U site, and 5.8 v.u. for the Te site, respectively, confirming that all the U and Te are all in the +6 oxidation state.
 | ||
| Fig. 7 View of the layered structure of β-K2[(UO2)TeVIO5] along the b-axis. (b) One of the [UTeO7]2− anionic layers projected along the a-axis. (c) The local coordination environment of Te2O10 dimers and UO7 polyhedra shown in ball-and-stick presentation. UO7 pentagonal bipyramids are shown in yellow. TeO6 octahedra are in light blue. K+ cations are blue spheres. | ||
As shown in Fig. 7(c), two TeO6 octahedra share a common edge, O(6)–O(6)′, leading to a Te2O10 dimer, similar to the one reported in a series of compounds with the formula A2[Te3O8(OH)4] (A = Na, K, Rb, Cs).53 However, the Te2O10 dimers have different coordination environments in both structures. In β-K2[(UO2)TeVIO5], these Te2O10 dimers are embedded into the above-mentioned [UTeO7]2− anionic layers by sharing one edge (O(3) and O(5)) and one corner (O(7)) from each TeO6 octahedron. In contrast, the corresponding Te2O10 dimers in A2[Te3O8(OH)4] are further fused together through corners resulting in infinite chains.
Raman spectral analysis
The Raman spectra for all the four uranyl tellurium compounds are presented in Fig. 8. It is noteworthy that the vibrational data on tellurites/tellurates are well-established in the literature,54 however, very little research has been undertaken on the vibrational spectroscopy of actinide tellurates/tellurites. The infrared spectrum studies of schmitterite (UO2TeO3)55 and cliffordite (UTe3O9)56 were reported by Botto et al., and Raman vibrational studies of schmitterite (UO2TeO3)57 and moctezumite (Pb(UO2)(TeO3)2)58 have been recently discussed by Frost et al.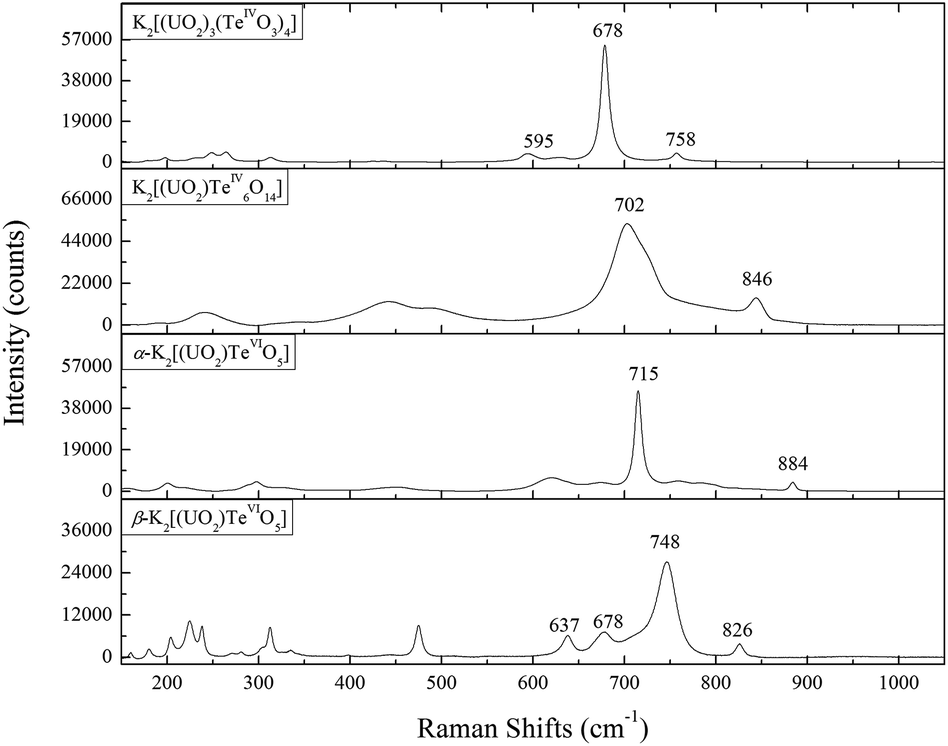 | ||
| Fig. 8 Raman shifts from 150 to 1050 cm−1 for K2[(UO2)3(TeIVO3)4], K2[(UO2)TeIV6O14], α-K2[(UO2)TeVIO5], and β-K2[(UO2)TeVIO5]. | ||
The Raman spectra for all the above-discussed uranyl tellurium compounds are characterized by motions from uranyl (UO22+) linearity and the tellurite (TeIVO3) trigonal pyramid or tellurate (TeVIO6) octahedron. The ideal uranyl (UO22+) unit with D∞h symmetry has three normal modes in aqueous solution, that is, v1 symmetric stretching mode (approximately from 860 to 880 cm−1), v3 anti-symmetrical stretching (approximately from 930 to 960 cm−1), and v2 bending mode (approximately from 199 to 210 cm−1).59,60 The free TeIVO3 trigonal pyramid has the C3v symmetry, with motions being assigned as A1(ν1), A1(ν2), E(ν3) and E(ν4),61 where ν1, ν2, ν3 and ν4 modes are denoted as non-degenerate symmetric stretching, non-degenerate symmetric bending, doubly degenerate asymmetric stretching and asymmetric bending vibrations, respectively. In aqueous solution, these modes are observed to be located at 758 cm−1 for ν1, 703 cm−1 for ν3, 364 cm−1 for ν2 and 326 cm−1 for ν4, respectively.62 The ideal TeVIO6 octahedron has three kinds of Raman active vibrational bands, ν1(A1g), ν2(Eg) and ν5(F2g), and the stretching and bending vibrations for most tellurate compounds are distributed in the range of 550–750 cm−1 and 350–450 cm−1, respectively.63
For all the four uranyl compounds, the high frequency bands between 600 and 800 cm−1 are mainly attributed to the stretching vibrations of U–O and Te–O, whereas the low frequency part below 250 cm−1 corresponds to the motions of the crystal lattice. Due to the large number of atoms in the unit cell and to the relatively overlapping vibrational regions of U–O and Te–O, here we only assign the signature peaks for our uranyl tellurium compounds. The assignment of the vibrational bands was mainly based on the comparison of two uranyl mineral schmitterite (UO2TeO3)57 and moctezumite (Pb(UO2)(TeO3)2).58 Moreover, the identification of the stretching modes (ν1 and ν3) of UO22+ are also attempted from crystallographic parameters using the empirical equations64 from Bartlett and Cooney:
| ν1 = [(dU–O − 0.575)/106.5]−3/2 | (1) |
| ν3 = [(dU–O − 0.804)/91.42]−3/2 | (2) |
| Compound | Uranyl sites | Bond length (Å) | Calculated | Observed | ||
|---|---|---|---|---|---|---|
| ν 1 (cm−1) | ν 3 (cm−1) | ν 1 (cm−1) | ν 3 (cm−1) | |||
| K2[(UO2)3(TeIVO3)4] |

|
U(1)–O(3): 1.812(9) | 799 | 864 | 758 | |
|
U(2)–O(1): 1.817(9) | 794 | 858 | 760 | ||
| U(2)–O(2): 1.820(9) | 792 | 854 | ||||
| K2[(UO2)TeIV6O14] |
|
U(1)–O(1): 1.788(10) | 822 | 895 | 846 | 920 |
| α-K2[(UO2)TeVIO5] |
|
U(1)–O(1): 1.824(9) | 788 | 849 | 884 | |
| U(1)–O(6): 1.829(9) | 783 | 842 | ||||
| β-K2[(UO2)TeVIO5] |

|
U(1)–O(1): 1.807(5) | 804 | 870 | 826 | 898 |
| U(1)–O(2): 1.799(5) | 811 | 880 | ||||
Specifically for K2[(UO2)3(TeIVO3)4], the very strong peak at 678 cm−1 may be assigned to the symmetric stretching of ν1 TeIVO3. The corresponding band is shifted to a higher frequency for K2[(UO2)TeIV6O14] (at 702 cm−1), but still agrees well with that for schmitterite where it is also observed at 724 cm−1 as the strongest peak in the spectrum.57 The symmetric stretching ν1 of UO22+ presents a slightly broad structure with subcomponents composed of two subpeaks (758 and 760 cm−1). This is due to the multiple crystallographic sites of U in K2[(UO2)3(TeIVO3)4] (see Table 2), which gives rise to multiple ν1 bands. The triply-degenerate asymmetric stretching ν1 of UO22+ is not clearly shown from the spectrum but can be calculated as 854, 858 and 864 cm−1. The Raman spectrum of K2[(UO2)TeIV6O14] is very similar to that of Na[(UO2)Te6O13(OH)].41 This is due to the fact that both K2[(UO2)TeIV6O14] and Na[(UO2)Te6O13(OH)] can be considered as the derivatives of cliffordite (UO2(Te3O7)). The main feature of the K2[(UO2)TeIV6O14] spectrum is the presence of broad, partially overlapping bands, which exhibit a high degree of symmetry because of the cubic crystal system. The band located at 846 cm−1 may be assigned to the ν1 (UO2)2+ symmetric stretching vibration, which can be compared with 823 cm−1 in calcurmolite (Ca(UO2)3(MoO4)3(OH)2·H2O)65 and 826 cm−1 in moctezumite,58 respectively. The presence of different peaks in α- and β-K2[(UO2)(TeVIO5)] reflects the structural differences in both modifications. The largest peak which can be assigned as ν1 of TeVIO6 is lower for α-K2[(UO2)(TeVIO5)] (at 715 cm−1) than that for β modification (at 748 cm−1), but still comparable well with most tellurate compounds.66,67
Conclusion
This work enhances the poorly investigated area of the actinide chemistry under extreme conditions of pressures and temperatures. Amid all the synthetic uranyl tellurium compounds in the literature, most were isolated via employing mild hydrothermal (around 200 °C) conditions in a Teflon-lined autoclave, and a few were synthesized at high-temperature (around 800 °C) using alkaline chloride as the flux.22,40 The obtained novel uranyl tellurium compounds in this work are structurally more complex than those produced under ambient pressure or mild hydrothermal conditions.The structures of the title compounds demonstrate the considerable flexibility of tellurium polyhedral geometries (TeIVO3 trigonal pyramid, TeIVO4 disphenoid, TeIVO5 square pyramid and TeVIO6 octahedron) in forming diverse structural topologies. Specifically, TeIVO3 trigonal pyramids and TeIVO4 disphenoids are connected in [TeO3]2− chains extending along the a-axis in K2[(UO2)3(TeIVO3)4]. TeIVO5 square pyramids play the role of structural linkers to connect [UTe6O26]22− clusters into K2[(UO2)TeIV6O14], and TeVIO6 octahedra are found to form Te2O10 dimers among both and α- and β-K2[(UO2)TeVIO5]. Moreover, three out of four uranyl compounds, K2[(UO2)3(TeIVO3)4], K2[(UO2)TeIV6O14] and α-K2[(UO2)TeVIO5] are isolated in the 3D framework, which is a sharp contrast to the dominant 2D layered structures among uranyl compounds synthesized under regular conditions. Thus, it is clear that the extreme conditions allow for the formation of atypical structure types with structures more complex and dimensionalities more variable than expected.
The chemical behavior of uranyl tellurium compounds under extreme conditions is very sensitive to the nature of counter cations. Small changes in the composition can give rise to dramatic different chemical compositions and structural properties. Compared to the sodium uranyl tellurium family41 where the high probability of noncentrosymmetric and polar structures are observed, the larger cations of the K+-based family in this work are centrosymmetric and nonpolar. The ionic size of the present cations is of major influence to control the macroscopic polarity (eight-coordinated Na+, 1.18 Å; eight-coordinated K+, 1.51 Å),68 which should therefore be taken into consideration for the further investigations aiming at elucidation of the pressure influence on the structural and chemical formation of the actinide inorganic materials.
Acknowledgements
The authors are grateful to Dr. Martina Klinkenberg (IEK-6, Forschungszentrum Jülich) for the kind help in electron microscopy and EDX experiments. We are grateful to the Helmholtz Association for funding within the VH-NG-815 grant.Notes and references
- J. Galy and G. Meunier, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1971, 27, 608–616 CrossRef CAS.
- G. H. Swihart, P. K. Sen Gupta, E. O. Schlemper, M. E. Back and R. V. Gaines, Am. Mineral., 1993, 78, 835–839 CAS.
- G. Meunier and J. Galy, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1973, 29, 1251–1255 CrossRef CAS.
- M. S. Samant, S. R. Bharadwaj, A. S. Kerkar, S. N. Tripathi and S. R. Dharwadkar, J. Nucl. Mater., 1994, 211, 181–185 CrossRef CAS.
- H. Kleykamp, J. Nucl. Mater., 1985, 131, 221–246 CrossRef CAS.
- B. R. Bowsher, Prog. Nucl. Energy, 1987, 20, 199–233 CrossRef CAS.
- R. de Boer and E. H. P. Cordfunke, J. Nucl. Mater., 1997, 240, 124–130 CrossRef CAS.
- B. Xiao, T. M. Gesing, P. Kegler, G. Modolo, D. Bosbach, H. Schlenz, E. V. Suleimanov and E. V. Alekseev, Inorg. Chem., 2014, 53, 3088–3098 CrossRef CAS PubMed.
- B. Xiao, T. M. Gesing, L. Robben, D. Bosbach and E. V. Alekseev, Chem. – Eur. J., 2015, 21, 7629–7629 CrossRef CAS PubMed.
- B. Xiao, T. M. Gesing, L. Robben, D. Bosbach and E. V. Alekseev, Chem. – Eur. J., 2015, 21, 7746–7754 CrossRef CAS PubMed.
- B. Xiao, M. Klinkenberg, D. Bosbach, E. V. Suleimanov and E. V. Alekseev, Inorg. Chem., 2015, 54, 5981–5990 CrossRef CAS PubMed.
- P. C. Burns, Can. Mineral., 2005, 43, 1839–1894 CrossRef CAS.
- M. G. Johnston and W. T. A. Harrison, J. Am. Chem. Soc., 2002, 124, 4576–4577 CrossRef CAS PubMed.
- A. R. Kampf, S. J. Mills, R. M. Housley, J. Marty and B. Thorne, Am. Mineral., 2010, 95, 1554–1559 CrossRef CAS.
- J. Ling, M. Ward and P. C. Burns, J. Solid State Chem., 2011, 184, 401–404 CrossRef CAS.
- G. J. Hutchings, C. S. Heneghan, I. D. Hudson and S. H. Taylor, Nature, 1996, 384, 341–343 CrossRef CAS.
- S. D. Pollington, A. F. Lee, T. L. Overton, P. J. Sears, P. B. Wells, S. E. Hawley, I. D. Hudson, D. F. Lee and V. Ruddock, Chem. Commun., 1999, 725–726, 10.1039/A900802K.
- P. O. Adelani and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2010, 49, 8909–8911 CrossRef CAS PubMed.
- T. Y. Shvareva, S. Skanthakumar, L. Soderholm, A. Clearfield and T. E. Albrecht-Schmitt, Chem. Mater., 2007, 19, 132–134 CrossRef CAS.
- E. Pichot, N. Dacheux, V. Brandel and M. Genet, New J. Chem., 2000, 24, 1017–1023 RSC.
- P. M. Almond and T. E. Albrecht-Schmitt, Inorg. Chem., 2002, 41, 5495–5501 CrossRef CAS PubMed.
- J. D. Woodward and T. E. Albrecht-Schmitt, J. Solid State Chem., 2005, 178, 2922–2926 CrossRef CAS.
- G. H. Swihart, P. Sen Gupta, E. O. Schlemper, M. E. Back and R. V. Gaines, Am. Mineral., 1993, 78, 835–839 CAS.
- P. M. Almond, M. L. McKee and T. E. Albrecht-Schmitt, Angew. Chem., Int. Ed., 2002, 41, 3426–3429 CrossRef CAS.
- I. Zimmermann and M. Johnsson, Cryst. Growth Des., 2014, 14, 5252–5259 CAS.
- R. G. Pearson, J. Am. Chem. Soc., 1969, 91, 4947–4955 CrossRef CAS.
- M. Johnsson, K. W. Törnroos, P. Lemmens and P. Millet, Chem. Mater., 2003, 15, 68–73 CrossRef CAS.
- M. Johnsson, K. W. Törnroos, F. Mila and P. Millet, Chem. Mater., 2000, 12, 2853–2857 CrossRef CAS.
- S. Wu, O. Beermann, S. Wang, A. Holzheid, W. Depmeier, T. Malcherek, G. Modolo, E. V. Alekseev and T. E. Albrecht-Schmitt, Chem. – Eur. J., 2012, 18, 4166–4169 CrossRef CAS PubMed.
- S. Wu, P. Kegler, S. Wang, A. Holzheid, W. Depmeier, T. Malcherek, E. V. Alekseev and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 3941–3943 CrossRef CAS PubMed.
- S. Wu, S. Wang, M. Polinski, O. Beermann, P. Kegler, T. Malcherek, A. Holzheid, W. Depmeier, D. Bosbach, T. E. Abrecht-Schmitt and E. V. Alekseev, Inorg. Chem., 2013, 52, 5110–5118 CrossRef CAS PubMed.
- N. Yu, V. V. Klepov, P. Kegler, D. Bosbach, T. E. Albrecht-Schmitt and E. V. Alekseev, Inorg. Chem., 2014, 53, 8194–8196 CrossRef CAS PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. C. Burns, R. C. Ewing and F. C. Hawthorne, Can. Mineral., 1997, 35, 1551–1570 CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- W. Zhang, J. Sun, X. Wang, G. Shen and D. Shen, CrystEngComm, 2012, 14, 3490–3494 RSC.
- W. Zhang, X. Tao, C. Zhang, H. Zhang and M. Jiang, Cryst. Growth Des., 2009, 9, 2633–2636 CAS.
- T. Zhu, J. Qin and P. S. Halasyamani, Dalton Trans., 2011, 40, 8527–8532 RSC.
- F. Brandstätter, Z. Kristallogr., 1981, 155, 193–200 Search PubMed.
- J. D. Woodward, P. M. Almond and T. E. Albrecht-Schmitt, J. Solid State Chem., 2004, 177, 3971–3976 CrossRef CAS.
- B. Xiao, P. Kegler, D. Bosbach and a. E. V. Alekseev, Inorg. Chem., 2016, 55, 4626–4635 CrossRef CAS PubMed.
- P. C. Burns, Can. Mineral., 2005, 43, 1839–1894 CrossRef CAS.
- V. Balraj and K. Vidyasagar, Inorg. Chem., 1999, 38, 1394–1400 CrossRef CAS.
- A. Guesdon and B. Raveau, Chem. Mater., 2000, 12, 2239–2243 CrossRef CAS.
- S.-e. Bang, Z. Pan, Y. H. Kim, D. W. Lee and K. M. Ok, J. Solid State Chem., 2013, 208, 65–70 CrossRef CAS.
- S. Krivovichev, P. Burns and I. Tananaev, Structural chemistry of inorganic actinide compounds, Elsevier, 2006 Search PubMed.
- M. K. Kim, S. H. Kim, H. Y. Chang, P. S. Halasyamani and K. M. Ok, Inorg. Chem., 2010, 49, 7028–7034 CrossRef CAS PubMed.
- D. Xiao, S. Wang, E. Wang, Y. Hou, Y. Li, C. Hu and L. Xu, J. Solid State Chem., 2003, 176, 159–164 CrossRef CAS.
- M. Weil, Z. Anorg. Allg. Chem., 2007, 633, 1217–1222 CrossRef CAS.
- S. V. Krivovichev, V. Kahlenberg, R. Kaindl, E. Mersdorf, I. G. Tananaev and B. F. Myasoedov, Angew. Chem., Int. Ed., 2005, 117, 1158–1160 CrossRef.
- S. V. Krivovichev, C. L. Cahill and P. C. Burns, Inorg. Chem., 2003, 42, 2459–2464 CrossRef CAS PubMed.
- B. Xiao, J. Dellen, H. Schlenz, D. Bosbach, E. V. Suleimanov and E. V. Alekseev, Cryst. Growth Des., 2014, 14, 2677–2684 CAS.
- M. P. Minimol and K. Vidyasagar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2003, 42, 2244–2249 Search PubMed.
- J. Hanuza, M. Mączka, J. Lorenc, A. A. Kaminskii, L. Bohaty and P. Becker, Spectrochim. Acta, Part A, 2008, 71, 68–72 CrossRef CAS PubMed.
- I. L. Botto, Acta Sud Am. Quim., 1984, 4, 71–74 Search PubMed.
- I. L. Botto and E. J. Baran, Z. Anorg. Allg. Chem., 1982, 484, 210–214 CrossRef CAS.
- R. L. Frost, M. L. Weier, B. J. Reddy and J. Čejka, J. Raman Spectrosc., 2006, 37, 816–821 CrossRef CAS.
- R. L. Frost, J. Čejka and M. J. Dickfos, J. Raman Spectrosc., 2009, 40, 38–41 CrossRef CAS.
- L. H. Jones, Spectrochim. Acta, 1958, 10, 395–403 CrossRef CAS.
- D. D. Schnaars and R. E. Wilson, Inorg. Chem., 2013, 52, 14138–14147 CrossRef CAS PubMed.
- V. Farmer, The infrared spectra of minerals, The Mineralogical Society, London, 1974 Search PubMed.
- K. V. Domoratskii, V. I. Pastukhov, A. Y. Kudzin, L. Y. Sadovskaya, V. M. Rizak and V. A. Stefanovich, Phys. Solid State, 2000, 42, 1443–1446 CrossRef CAS.
- G. Sekar, V. Ramakrishnan and G. Aruldhas, Infrared Phys., 1987, 27, 253–256 CrossRef CAS.
- J. R. Bartlett and R. P. Cooney, J. Mol. Struct., 1989, 193, 295–300 CrossRef CAS.
- R. L. Frost, J. Čejka and M. J. Dickfos, J. Raman Spectrosc., 2008, 39, 779–785 CrossRef CAS.
- J. Llanos, R. Castillo, D. Barrionuevo, D. Espinoza and S. Conejeros, J. Alloys Compd., 2009, 485, 565–568 CrossRef CAS.
- S. Raman, Inorg. Chem., 1964, 3, 634–638 CrossRef CAS.
- R. Shannon, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 751–767 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD patterns, BSE image, EDS measuring points and Raman fitting results. See DOI: 10.1039/c6dt01350c |
| This journal is © The Royal Society of Chemistry 2016 |