
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning the photochemical properties of the fulvalene-tetracarbonyl-diruthenium system†
Anders
Lennartson
a,
Angelica
Lundin
a,
Karl
Börjesson
 ab,
Victor
Gray
a and
Kasper
Moth-Poulsen
*a
ab,
Victor
Gray
a and
Kasper
Moth-Poulsen
*a
aDepartment of Chemistry and Chemical Engineering, Chalmers University of Technology, Kemigården 4, SE-412 96 Gothenburg, Sweden. E-mail: asper.moth-poulsen@chalmers.se
bPresent address: Department of Chemistry and Molecular Biology, University of Gothenburg, Kemigården 4, SE-412 96 Gothenburg, Sweden
First published on 11th April 2016
Abstract
In a Molecular Solar–Thermal Energy Storage (MOST) system, solar energy is converted to chemical energy using a compound that undergoes reversible endothermic photoisomerization. The high-energy photoisomer can later be converted back to the parent compound and the excess energy is released as heat. One of the most studied MOST systems is based on fulvalene-tetracarbonyl-diruthenium, and this paper demonstrates, for the first time, the possibility to tune the photochemical properties of this system by positive steric hindrance working on the fulvalene unit.
Introduction
The energy received by planet Earth in the form of sunlight exceeds our energy consumption by orders of magnitude, yet techniques to harvest and store this energy are still at a rather rudimentary stage, when considering the fact that only a very small fraction of the world's energy demand is covered by solar energy. A possible method to store solar energy is by using compounds undergoing reversible, endothermic photoinduced isomerization.1,2 Important parameters to design in such a system are the extended absorption range within the solar spectrum as well as a high energy storage capacity. For efficient utilization of sunlight, the photoisomer must be essentially transparent at the wavelengths of interest. Furthermore, the barrier of back-conversion must be high enough to allow energy to be stored over extended periods of time; back-conversion should preferably be facilitated by catalysis. A number of molecular systems have been proposed for solar energy storage, the most widely studied compounds being small organic molecules, such as norbornadiene, which undergo a photochemical intramolecular [2 + 2] cycloaddition to quadricyclane,3–5trans-stilbene and trans-azobenzene derivatives that undergo photoinduced isomerization to the corresponding cis-forms.6–12 For a detailed account of previous work in the field, we refer to reviews.13–16 A general challenge for several of the systems exemplified above is that they absorb light in the UV-region of the spectrum, where solar emission is low. In this respect, a promising compound is fulvalene-tetracarbonyl-diruthenium (1, Scheme 1) introduced by Vollhardt and co-workers;17–20 irradiation with visible light causes cleavage of both the fulvalene ligand and the metal–metal bond leading to two bridging cyclopentadiene dianions binding in a σ1, η5 mode. The use of ruthenium in these compounds appears to be essential; recently, iron analogues of 1 were prepared, but showed no tendency to photoisomerize, an observation reflecting the differences in electronic structures of iron and ruthenium.21 Since the discovery of 1, efforts have been made in order to modify its properties, e.g. its solubility,22–24 but the photochemical properties have so-far not been subjected to rational design. Tuning the photochemical properties of ruthenium complexes is of importance to several areas of chemistry; ruthenium complexes have been used as photosensitizers since the 1970s25 and have found applications such as in dye-sensitized solar cells,26 organic photoredox reactions,27 water photoreduction,28 water photooxidation.29,30 and carbon dioxide photoreduction.31 Although 1 has an absorption spectrum considerably red shifted compared to e.g. norbornadiene, this type of compounds suffer from relatively low photoisomerization quantum yields.32 In this work we set out to find a strategy to modify the structure of 1 in order to increase the quantum yield and thus achieve a more efficient solar harvesting system.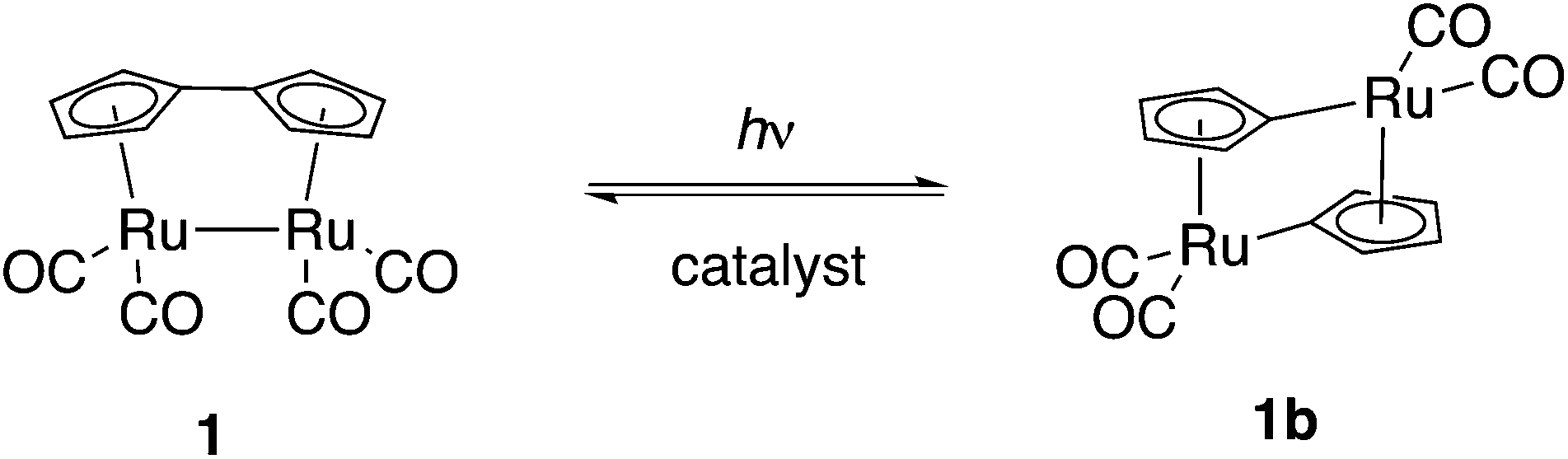 | ||
| Scheme 1 Structure of fulvalene-tetracarbonyl-diruthenium 1 and its photoisomer 1b. | ||
Results and discussion
The mechanism of the photoisomerization of 1 was recently elucidated using a combination of X-ray transient absorption spectroscopy, picosecond IR spectroscopy and computational methods.33,34 This study stated that upon radiation the Ru–Ru bond is broken and a Ru-centered biradical is formed. Due to the high spin–orbit coupling expected for a Ru complex the interconversion between spin states should be facilitated and a stepwise mechanism featuring a biradical intermediate adopting an anti-conformation of the Ru complex is formed, the rate limiting step being rotation about the central C–C bond.34 Based on the proposed mechanism, we worked with a hypothesis that substitutions on the Cp rings would induce a steric constraint on the triplet syn-biradical and the intermediate (the two states having the Cp rings in plane), while leaving the transition states more or less unaffected, thus, facilitating the isomerization. To evaluate this hypothesis, two new compounds with three (2) and four (3) methyl groups were prepared and their properties were compared both experimentally and theoretically with the unsubstituted compound (1).Compounds 2 and 3 were synthesized from triruthenium dodecacarbonyl and the corresponding dihydrofulvalenes in refluxing xylenes (Scheme 2), inspired by the synthesis of (3-tert-butyl-fulvalene)-tetracarbonyl-diruthenium,19cf. ESI.† The use of tri- and tetramethylcyclopentadiene rather than mono- and dimethylcyclopentadiene, respectively, eliminates the possibility of isomerism. Reports on unsymmetrically substituted fulvalene-tetracarbonyl-diruthenium compounds are otherwise scarce, another example being an indenyl-analogue,35 where the corresponding “dihydrofulvalene” is stable enough to be prepared by Negishi-coupling.36 Like 1, compounds 2 and 3 are bright yellow solids, sparingly soluble in most organic solvents. Prolonged exposure to air leads to slow degradation and discoloration.
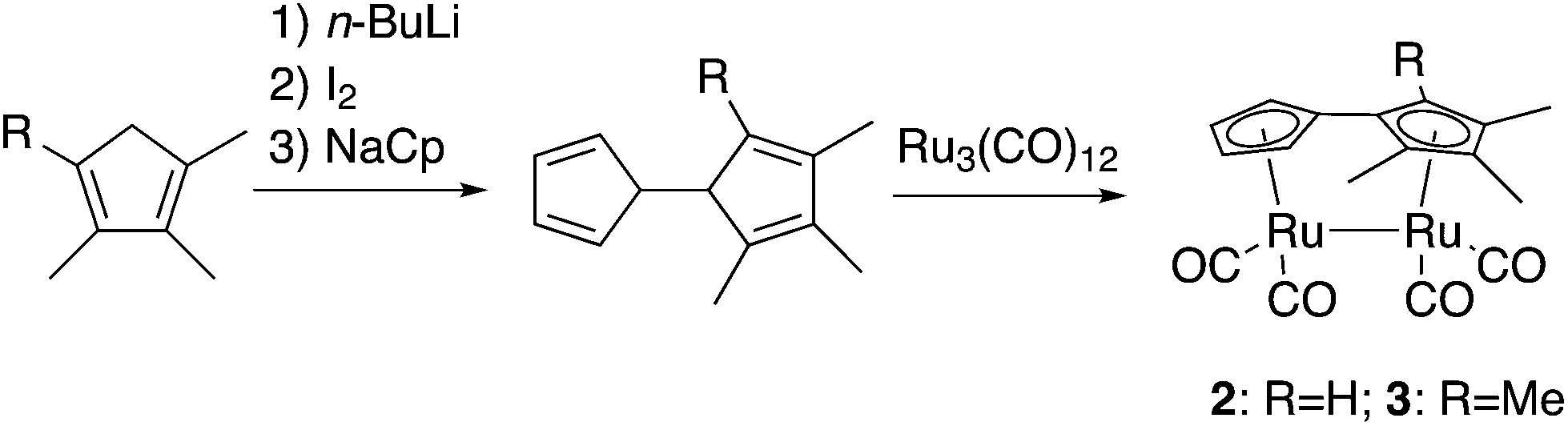 | ||
| Scheme 2 Synthesis of compounds 2 and 3. Tri- and tetramethylcyclo-pentadienes were prepared from the corresponding cyclopentenones via Shapiro reactions, see the ESI† for experimental details. | ||
The absorption spectra (Fig. 1) of 2 and 3 are similar to the spectrum of 1, having absorption on-sets at approx. 460 nm, well within the visible region of the solar spectrum. When irradiating solutions of 1–3 the absorbance decreases until a complete conversion to the photoisomer has occurred. Importantly, the photoisomers do not display any significant absorption of visible light, which satisfies a crucial requirement for MOST systems. Also the calculated absorption follows the same trend, Table 2. One of the most important parameters for a photoswitchable molecule is the photoisomerization quantum yield. This was determined at an irradiation wavelength of 400 ± 5.4 nm using potassium ferrioxalate as a chemical actinometer. Optically thick solutions (abs >2) were used for all samples and NMR spectroscopy was used to determine the isomerization ratio. The quantum yield of 1 (Table 1) was found to be 1.9%. As we used monochromatic light this value is not directly comparable to that previously reported.19 For compounds 2 and 3, the quantum yields were found to be 3.3% and 6.5%, respectively, representing an increase in the quantum yield of up to 3.4 times compared to 1.
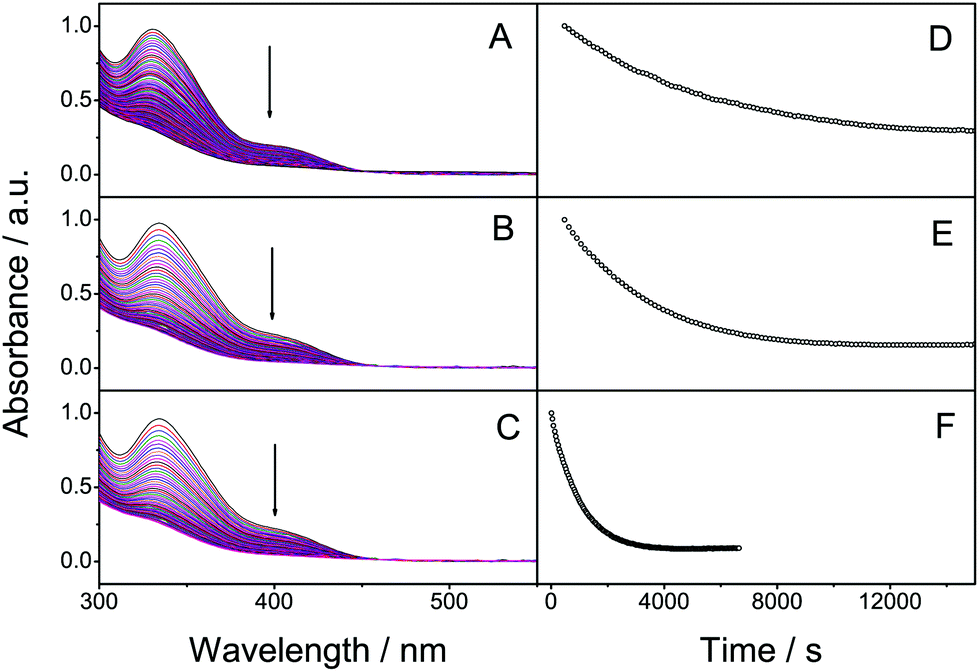 | ||
| Fig. 1 Superimposed absorption spectra of 1 (A), 2 (B) and 3 (C) during photoisomerisation. The decrease in absorbance at 400 nm is shown in D (for 1), E (for 2) and F (for 3). The molar extinction coefficients in toluene at 400 nm for 2 and 3 are 1.6 × 103 M−1 cm−1 which is comparable to 1.2 × 103 M−1 cm−1 for 1. | ||
| Compound | ε max [M−1 cm−1] |
A
onset![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a [nm] a [nm] |
ΔH‡ [kJ mol−1] | ΔS‡ [J mol−1 K−1] |
t
½b [days] |
ϕ [%] |
|---|---|---|---|---|---|---|
| a Absorption onset defined as lg(ε) = 2. b Determined from Eyring parameters at 25 °C. c Measured in toluene-d8 at 400 ± 5.4 nm. | ||||||
| 1 | 6000 (330 nm) | 458 | 133.6 ± 6.4 | 63.5 ± 18.1 | 165 | 1.9 |
| 2 | 6950 (333 nm) | 460 | 127.4 ± 21.9 | 47.2 ± 61.5 | 94 | 3.3 |
| 3 | 6800 (335 nm) | 461 | 137.9 ± 12.5 | 78.6 ± 35.2 | 149 | 6.5 |
| P S0–S1 (1st excit) | P S0–B S0 | B S0–C S0 | P S0–PI S0 | P S0–A T1 | A T1–B T1 | B S0–B T1 | B T1–C T1 | C S0–C T1 | PI T1–C T1 | PI S0–PI T1 | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 277.5 | 127.4 | 41.5 | 90.7 | 139.2 | −4.2 | 7.6 | 237.9 | 204.0 | 43.0 | 78.2 |
| 2 | 279.5 | 120.7 | 44.9 | 88.6 | 134.7 | 0.5 | 14.5 | 232.3 | 202.0 | 79.3 | 76.9 |
| 3 | 279.6 | 118.9 | 45.5 | 81.4 | 123.1 | 1.3 | 5.5 | 240.0 | 200.1 | 83.9 | 83.0 |
A rationalization of the observed increase in the quantum yield going from 1 to 3 was sought using Density Functional Theory (DFT) calculations. Geometry optimizations where done in a vacuum using the 6-311+G(d,p)37 basis set for all elements except for Ru where the effective core potential LANL2DZ38,39 was used in combination with the hybrid B3LYP40,41 functional. Singlet and triplet calculations were done for the parent compounds (P), the anti-biradical intermediate (B) and the photoisomers (PI) of the three compounds 1, 2 and 3, respectively (Fig. 2 and Table 2). In addition, transition state searches were done between A and B; B and PI, respectively. Earlier computational studies have used DFT to calculate a relatively high barrier of the first transition state between A and B (38 kJ mol−1).42 However, due to the high biradical character of the first transition state (A to B), DFT is not suitable to describe this multi-reference problem. We are therefore using more conventional arguments to rationalize the difference in the photoisomerization efficiency between 1, 2 and 3, with respect to the initial rotation to form B from P. The transition state between B and PI is found and well characterized.
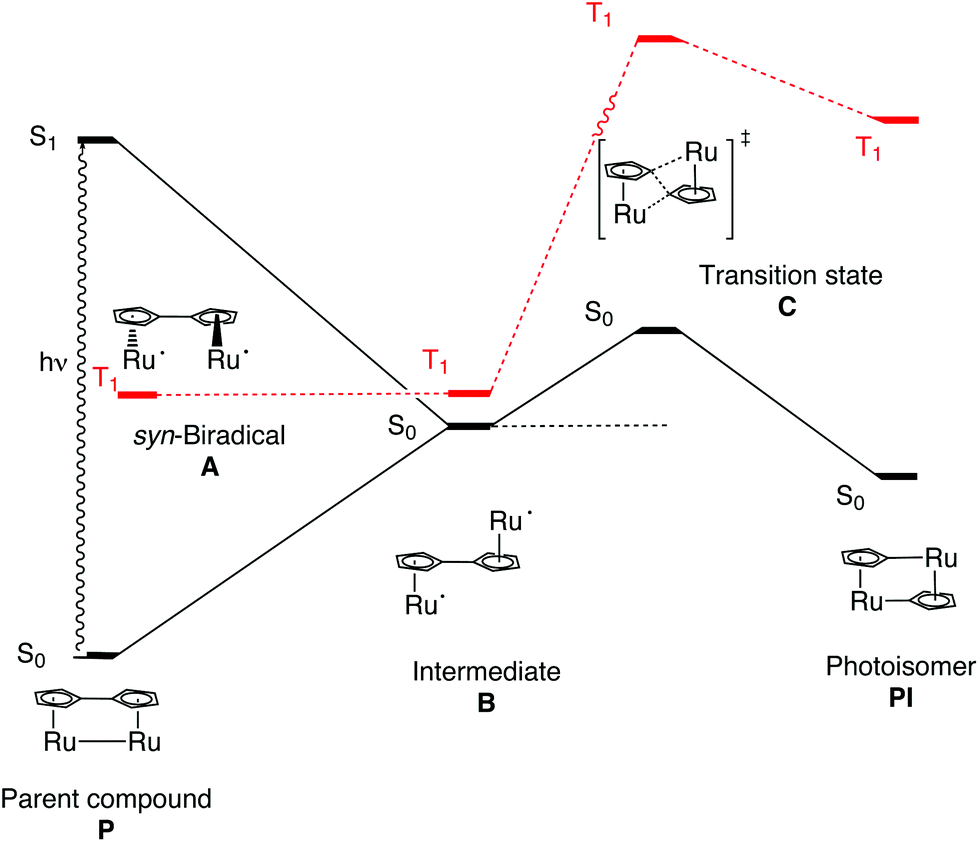 | ||
| Fig. 2 Schematic energy profile for the photoisomerization of 1 on the singlet surface (solid line), and triplet surface (red, dotted line). Note that representations of molecular structures are only schematic. | ||
As P is excited, the Ru–Ru bond is cleaved and a syn-biradical (A) is formed, the relaxed triplet lying well below the excited singlet and the reaction will cross-over to the triplet surface. As can be seen in Fig. 3 and ESI Table S1,† the relaxed triplet A T1 structures are already twisted by 38.8–60.8°, where the largest twist is for the substituted compounds 2 and 3. When compared with the relaxed structure of B T1 we see that these are also twisted by 131.8–172.4° where the least twist is for 2 and 3. Thus, the degree of rotation required for converting A to B decreases in the order 1 >2 >3.
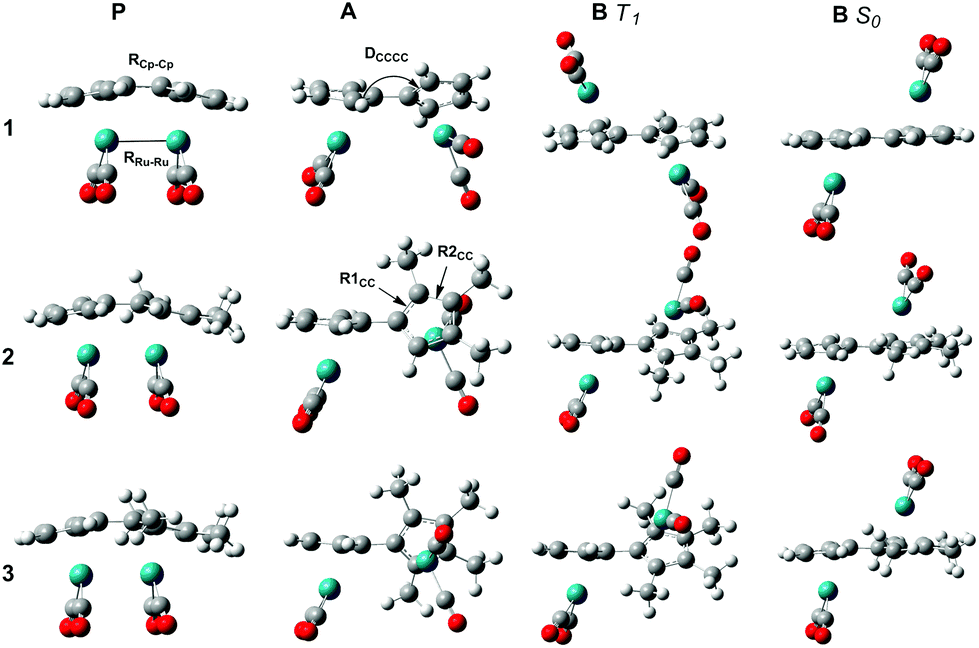 | ||
| Fig. 3 Optimized structures of P S0, A T1, B T1 and B S0 at B3LYP/6-311+G(d,p)/LANL2DZ. | ||
The C–C bond lengths (RCp–Cp; ESI Table S1†) between the Cp rings are in the order of 1.416–1.473 Å indicating that these bonds have a single bond character between two sp2 hybridized C atoms. When comparing with C–C bond lengths (R1CC and R2CC) in the Cp rings we see that they only differ by a maximum of 0.039 Å (B S0) showing the full conjugation of the rings. Taken together, this indicates delocalized Cp-rings connected with a single bond rather than a fulvalene ligand with localized double bonds in the five-membered rings and a double bond between the rings. For 1, A is 4.2 kJ mol−1 lower in energy than B T1, as opposed to 2 and 3 where it is 0.5 and 1.3 kJ mol−1 higher respectively, see Table 2. Rotation around a carbon–carbon single bond is typically low (12 kJ mol−1 for ethane and about 25 kJ mol−1 for butane).43 Therefore we conclude that, as the reaction takes place on the triplet surface after the excitation, and that the relaxed A T1 and B T1 already are rotated around the single bond between the Cp rings, there should only be a small barrier connected with this rotation. The higher quantum yields of 2 and 3 compared to 1 can thus be rationalized as; (1) A and B T1 structures are most alike (less difference in rotational angle DCCCC) (2) B T1 is lower in energy than A T1. The S–T splitting of B is 5.5–14.6 kJ mol−1 allowing the compounds to relax and the reaction to proceed on the singlet surface. As can be seen in Fig. 2 and Table 2 the enthalpies on the triplet surface for transition states C T1 and PI T1 lie significantly higher than the corresponding S0 states.
Since the step from A to B involves a considerable molecular rearrangement (in contrast to the B to PI step), the viscosity of the reaction medium may influence the quantum yield: lower viscosity would assist rotation. To investigate this, we measured the photoisomerization quantum yield in a series of alkane solvents (pentane to decane) of increasing viscosity (Fig. 4). The study was made using bis(1,1-dimethyl-tridecyl)fulvalene-tetracarbonyl-diruthenium (4; Fig. 5),23 which has a higher solubility in alkanes than does 1. The viscosity was found to have a significant effect on the quantum yield, which was more than twice as high in pentane, the least viscous solvent, compared to decane. This is similar to the photoisomerization of trans-stilbene, where the quantum yield also decreases with increasing viscosity,44 and gives further experimental support for a mechanism where the A to B rearrangement is the rate-limiting step. It has previously been observed that the quantum yield is dependent on temperature,34 and this is also the case here. From measurements of the photoisomerization quantum yield in heptane at different temperatures it is clear that the change in viscosity as a function of temperature does not fully account for the temperature dependence of the quantum yield.
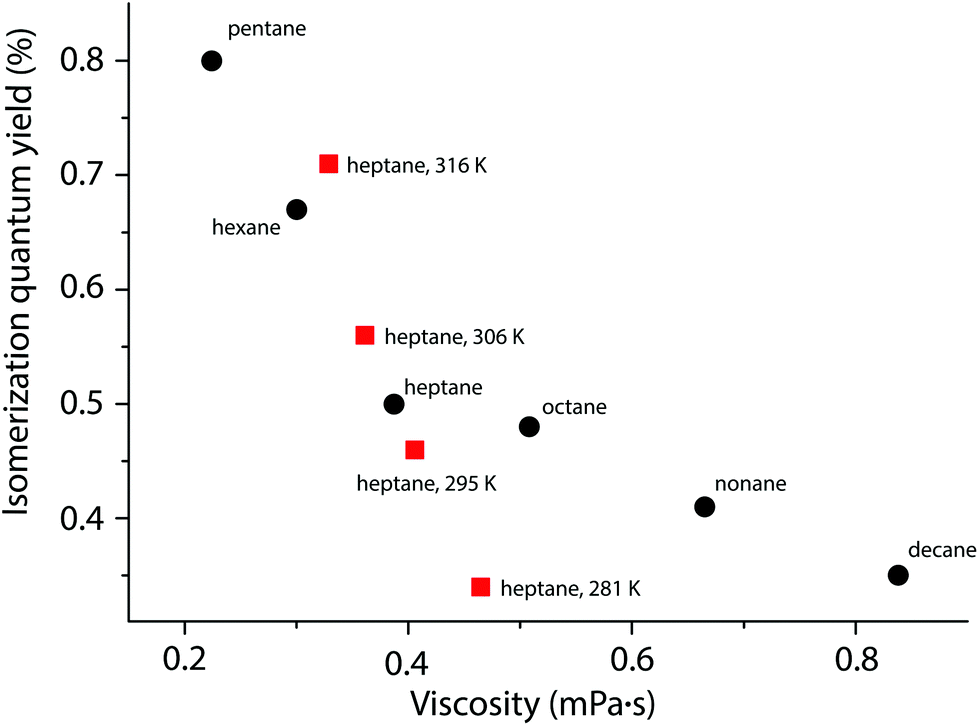 | ||
| Fig. 4 Quantum yield of 4 as a function of viscosity. The viscosity was modulated using different linear aliphatic solvents at ambient temperatures (circles: pentane to decane), or by changing the temperature (squares: in heptane). | ||
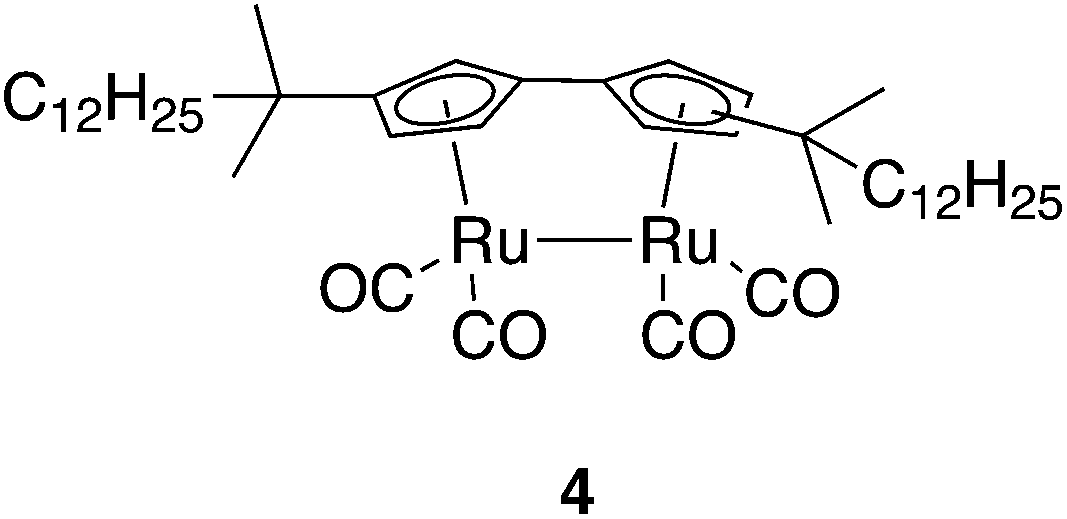 | ||
| Fig. 5 Compound 4 is obtained as a mixture of two isomers with indistinguishable photochemical properties. | ||
For fulvalene-tetracarbonyl-diruthenium to be used as a MOST system, the photoisomer needs to be stable over an extensive period of time. To determine the thermal activation barrier for back conversion from e.g. compound 1b to 1, the first order rate constants of the reaction were determined at different temperatures by both UV-vis spectroscopy and NMR. Eyring plots of the rate constants as a function of T are shown in ESI Fig. S4–6† and the associated thermodynamic parameters are summarized in Table 1. The compounds 1–3 all show similar values of ΔG‡, indicating that the thermal back isomerisation is not affected in the same way as the photo-induced isomerization.
Conclusions
In conclusion, we have re-investigated the mechanism of photoisomerization of fulvalene-tetracarbonyl-diruthenium by DFT calculations, a study suggesting that increased steric hindrance around the central carbon–carbon bond would facilitate isomerization. By synthesizing a series of compounds, it was shown that the isomerization quantum yield indeed is sensitive to steric hindrance. Introducing methyl groups in the 2 and 5 positions increases the quantum yield from 1.9% for compound 1 to 3.3% and 6.5%, for compounds with one or two methyl groups, respectively. This constitutes the first example of tuning the photochemical properties in this important class of compounds.Acknowledgements
The authors would like to thank the following funding agencies for financial support: Swedish Research Council (young researcher grant), Swedish Strategic Research Council (Future Research Leader 5), K. & A. Wallenberg Foundation (Wallenberg Academy Fellow 2014), Chalmers Materials and Energy Areas of Advance.Notes and references
- F. Weigert, Jahrbuch für Photographie, Kinematographie und Reproduktionsverfahren, 1909, p. 109 Search PubMed.
- Chem. Abstr., 1910, 4, 3170 Search PubMed.
- S. J. Cristol and R. L. Snell, J. Am. Chem. Soc., 1958, 80, 1950–1952 CrossRef CAS.
- W. G. Dauben and R. L. Cargill, Tetrahedron, 1961, 15, 197–201 CrossRef CAS.
- G. S. Hammond, N. J. Turro and A. Fischer, J. Am. Chem. Soc., 1961, 83, 4674–4675 CrossRef CAS.
- J. Wislicenus and M. Jahrmarkt, Chem. Zentralbl., 1901, I, 463–464 Search PubMed.
- R. Stoermer, Ber. Dtsch. Chem. Ges., 1909, 42, 4865–4871 CrossRef.
- J. Saltiel, J. D'Agostino, E. D. Megarity, L. Metts, K. R. Neuberger, M. Wrighton and O. C. Zafiriou, Org. Photochem., 1973, 3, 1–113 CAS.
- H. Goerner and H. J. Kuhn, Adv. Photochem., 1995, 19, 1–117 CAS.
- Y. Feng, H. Liu, W. Luo, E. Liu, N. Zhao, K. Yoshino and W. Feng, Sci. Rep., 2013, 3, 3260 Search PubMed.
- T. J. Kucharski, N. Ferralis, A. M. Kolpak, J. O. Zheng, D. G. Nocera and J. C. Grossman, Nat. Chem., 2014, 6, 441–447 CrossRef CAS PubMed.
- K. Masutani, M.-a. Morikawa and N. Kimizuka, Chem. Commun., 2014, 50, 15803–15806 RSC.
- V. A. Bren, A. D. Dubonosov, V. I. Minkin and V. A. Chernoivanov, Russ. Chem. Rev., 1991, 60, 451–469 CrossRef.
- A. D. Dubonosov, V. A. Bren and V. A. Chernoivanov, Russ. Chem. Rev., 2002, 71, 917–927 CrossRef CAS.
- T. J. Kucharski, Y. Tian, S. Akbulatov and R. Boulatov, Energy Environ. Sci., 2011, 4, 4449–4472 CAS.
- A. Lennartson, A. Roffey and K. Moth-Poulsen, Tetrahedron Lett., 2015, 56, 1457–1465 CrossRef CAS.
- K. P. C. Vollhardt and T. W. Weidman, J. Am. Chem. Soc., 1983, 105, 1676–1677 CrossRef CAS.
- K. P. C. Vollhardt and T. W. Weidman, Organometallics, 1984, 3, 82–86 CrossRef CAS.
- R. Boese, J. K. Cammack, A. J. Matzger, K. Pflug, W. B. Tolman, K. P. C. Vollhardt and T. W. Weidman, J. Am. Chem. Soc., 1997, 119, 6757–6773 CrossRef CAS.
- K. Börjesson, D. Coso, V. Gray, C. Grossman Jeffrey, J. Guan, B. Harris Charles, N. Hertkorn, Z. Hou, Y. Kanai, D. Lee, P. Lomont Justin, A. Majumdar, K. Meier Steven, K. Moth-Poulsen, L. Myrabo Randy, C. Nguyen Son, A. Segalman Rachel, V. Srinivasan, B. Tolman Willam, N. Vinokurov, K. P. C. Vollhardt and W. Weidman Timothy, Chem. – Eur. J., 2014, 20, 15587–15604 CrossRef PubMed.
- Z. Hou, S. C. Nguyen, J. P. Lomont, C. B. Harris, N. Vinokurov and K. P. C. Vollhardt, Phys. Chem. Chem. Phys., 2013, 15, 7466–7469 RSC.
- B. Zhu, O. S. Miljanic, K. P. C. Vollhardt and M. J. West, Synthesis, 2005, 3373–3379 CAS.
- K. Moth-Poulsen, D. Coso, K. Börjesson, N. Vinokurov, S. K. Meier, A. Majumdar, K. P. C. Vollhardt and R. A. Segalman, Energy Environ. Sci., 2012, 5, 8534–8537 CAS.
- K. Börjesson, A. Lennartson and K. Moth-Poulsen, J. Fluorine Chem., 2014, 161, 24–28 CrossRef.
- A. W. Adamson and J. N. Demas, J. Am. Chem. Soc., 1971, 93, 1800–1801 CrossRef CAS.
- B. O'Regan and M. Graetzel, Nature, 1991, 353, 737–740 CrossRef.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- J. Kiwi and M. Gratzel, Nature, 1979, 281, 657–658 CrossRef CAS.
- V. Y. Shafirovich, N. K. Khannanov and V. V. Strelets, Nouv. J. Chim., 1980, 4, 81–84 CAS.
- M. D. Kaerkaes, O. Verho, E. V. Johnston and B. Åkermark, Chem. Rev., 2014, 114, 11863–12001 CrossRef CAS PubMed.
- J. Hawecker, J. M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1985, 56–58 RSC.
- The quantum yield of 1 was originally reported to be 15%, however, our measurements have indicated a quantum yield of 1.9%.
- Y. Kanai, V. Srinivasan, S. K. Meier, K. P. C. Vollhardt and J. C. Grossman, Angew. Chem., Int. Ed., 2010, 49, 8926–8929 CrossRef CAS PubMed.
- M. R. Harpham, S. C. Nguyen, Z. Hou, J. C. Grossman, C. B. Harris, M. W. Mara, A. B. Stickrath, Y. Kanai, A. M. Kolpak, D. Lee, D.-J. Liu, J. P. Lomont, K. Moth-Poulsen, N. Vinokurov, L. X. Chen and K. P. C. Vollhardt, Angew. Chem., Int. Ed., 2012, 51, 7692–7696 CrossRef CAS PubMed.
- B. Li, X. Tan, S. Xu, H. Song and B. Wang, J. Organomet. Chem., 2009, 694, 1503–1508 CrossRef CAS.
- D. Tews and P. Escarpa Gaede, Tetrahedron Lett., 2004, 45, 9029–9031 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- M. R. Harpham, S. C. Nguyen, Z. Hou, J. C. Grossman, C. B. Harris, M. W. Mara, A. B. Stickrath, Y. Kanai, A. M. Kolpak, D. Lee, D.-J. Liu, J. P. Lomont, K. Moth-Poulsen, N. Vinokurov, L. X. Chen and K. P. C. Vollhardt, Angew. Chem., Int. Ed., 2012, 51, 7692–7696 CrossRef CAS PubMed.
- M. B. Smith and J. March, March's Advanced Organic Chemistry Reactions, Mechanisms and Structure, John Wily & Sons, Inc, New York, 5th edn, 2001 Search PubMed.
- D. Gegiou, K. A. Muszkat and E. Fischer, J. Am. Chem. Soc., 1968, 90, 12–18 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details of synthesis, quantum yield determination, determination of Eyring parameters and quantum mechanical calculations. See DOI: 10.1039/c6dt01343k |
| This journal is © The Royal Society of Chemistry 2016 |