
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
SnPhPc phthalocyanines with dianion Pc2− and radical trianion Pc˙3− macrocycles: syntheses, structures, and properties†
Dmitri V.
Konarev
*a,
Alexey V.
Kuzmin
b,
Yoshiaki
Nakano
c,
Salavat S.
Khasanov
b,
Manabu
Ishikawa
c,
Akihiro
Otsuka
c,
Hideki
Yamochi
c,
Gunzi
Saito
de and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow region 142432, Russia. E-mail: konarev3@yandex.ru
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow region 142432, Russia
cDepartment of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502, Japan
dFaculty of Agriculture, Meijo University, 1-501 Shiogamaguchi, Tempaku-ku, Nagoya 468-8502, Japan
eToyota Physical and Chemical Research Institute, 41-1, Yokomichi, Nagakute, Aichi 480-1192, Japan
First published on 31st May 2016
Abstract
The interaction of SnIVCl2Pc with an excess of NaBPh4 in the presence of fullerenes C60 and C70 provides complete dissolution of SnIVCl2Pc and the formation of blue solutions from which the crystals of [SnPhPc2−]+(BPh4)−·C6H14 (1) or [SnPhPc˙3−]·C6H4Cl2 (2) were selectively isolated. According to the optical spectra, salt 1 contains dianionic Pc2− macrocycles, whereas macrocycles are reduced to form the Pc˙3− radical trianions in 2. As a result, the phthalocyanine macrocycle is dianionic in 1, and the positive charge of SnIV is compensated by the Ph−, Pc2−, and BPh4− anions in this compound. The formally neutral compound 2 contains two anionic species of Ph− and Pc˙3− and the SnIV ion as the counter cation. Phenyl substituents are linked to the SnIV atoms by the Sn–C(Ph) bonds of 2.098(2) (1) and 2.105(2) Å (2) length. The dianionic Pc2− macrocycle significantly deviates from planarity in 1 while Pc˙3− is planar in 2. Salt 1 manifests only a weak impurity EPR signal. Compound 2 manifests an intense EPR signal with g = 2.0046 and a linewidth of 0.5 mT at 298 K due to the presence of Pc˙3−. Spins are weakly antiferromagnetically coupled in the π-stacking [SnPhPc˙3−]2 dimers of 2 with a Weiss temperature of −3 K and the estimated magnetic exchange interaction J/kB = −0.23 K.
Introduction
Promising optical, magnetic and conducting properties of metal phthalocyanines (Pc's) and porphyrins (P's) have evoked great interest of researchers.1–4 Their properties can be modified by adding nitrogen-, oxygen-, or carbon-containing ligands which can coordinate to the metal centers of these macroheterocycles.5–7 Alkyl and phenyl ligands mainly coordinate to the porphyrins and phthalocyanines of indium, gallium, thallium and tin8–17 as well as some transition metals (Fe, Co, Ir).18–21 Organometallic chemistry of these metal macroheterocycles has been developed mainly for metalloporphyrins due to their solubility in organic solvents that allows one to obtain crystals of indium, gallium and tin porphyrins with alkyl, vinyl, phenyl or phenylacetylenyl substituents at the metal centers.8–11,14–17 Compounds with tin porphyrin (SnP) can contain one and two alkyl or phenyl ligands at the metal center, and can be obtained by oxidative addition of CH3I to SnIIP,10 direct synthesis from Li2P and SnCl2R2, or the interaction of the R2Mg and Grignard reagents with SnIVCl2P.11 At the same time, only a few such compounds are known for metal phthalocyanines. For example, the structure of phenylindium octakis(hexyl)phthalocyanine can be found in the CCDC database only for metal phthalocyanines with phenyl substituents at the metal atoms.13 This is surprising particularly in view of the potential biological interest in these compounds as well as their promising optical and magnetic properties.In this work, we developed a new approach to the synthesis of compounds with phenyltin phthalocyanine (SnPhPc). Due to their ionic nature, these compounds are well soluble in organic solvents allowing one to obtain crystalline products. The reaction involves NaBPh4 as a source of phenyl groups and fullerenes C60 and C70. Depending on the fullerene two different complexes are selectively isolated as crystals, namely [SnPhPc]+(BPh4)−·C6H14 (1) and [SnPhPc]·C6H4Cl2 (2). Their crystal structures were determined, optical and magnetic properties were analyzed, and DFT calculations were carried out for SnPhPc to elucidate the electronic structure and magnetic properties of this material. Formally, the neutral compound SnPhPc shows unusual properties due to the existence of paramagnetic Pc˙3− macrocycles which manifest absorption in the NIR range, and have unpaired spins magnetically coupled in the π-stacking [SnPhPc]2 dimers of 2. DFT calculations support the existence of paramagnetic Pc˙3− in 2 and antiferromagnetic coupling between the π-radical spins of the Pc˙3− ligands. The developed approach opens a way for new types of phenyl-substituted metal macroheterocycles.
Results and discussion
Synthesis
Previously, we developed a synthetic method for coordination complexes of zero-valent nickel with fullerenes C60 and C70 by the reduction of NiIIdpppCl2 (dppe: 1,3-bid(diphenylphosphino)propane) and fullerenes with two equivalents of sodium tetraphenylborate (NaBPh4). As a result, crystalline (Ni0dppp)(η2-C60(70))·solvent complexes were obtained.22 Since potentially tin phthalocyanine can also coordinate to fullerenes, we applied the same procedure to obtain its coordination complexes with fullerenes. The reaction of SnIVCl2Pc2− and fullerenes C60 or C70 with an excess of NaBPh4 in o-dichlorobenzene provided complete dissolution of SnIVCl2Pc2− to form deep blue and blue-violet solutions, respectively. Slow diffusion of hexane for 1 month yielded crystals on the walls of the diffusion tube. Examination under a microscope showed that different phases formed during the syntheses with different fullerenes although fullerenes were not involved in the obtained crystals. Synthesis with C60 yielded black blocks of [SnPhPc]+(BPh4)−·C6H14 (1) whereas synthesis with C70 yielded [SnPhPc]·C6H4Cl2 (2) as black prism with copper luster characteristic of phthalocyanines. We found no admixture of phase 1 in the synthesis of 2 and vice versa. Therefore, both phases formed selectively. It is interesting that the reaction of SnIVCl2Pc2− with an excess of NaBPh4 in the absence of fullerenes prevents from complete dissolution of SnIVCl2Pc2− and no crystals were obtained in this synthesis with slow diffusion of n-hexane. We can suppose that NaBPh4 can be a source of phenyl anions to substitute chloride anions at the tin(IV) atom. In this case the second chloride anion can be lost and its negative charge is compensated by the (BPh4)− anions to form [SnPhPc]+(BPh4)−·C6H14 (1). The role of C60, which cannot be reduced with NaBPh4, is to assist the formation of high quality crystals of 1. Fullerene C70 shows slightly stronger acceptor properties, and can be reduced with NaBPh4 in o-dichlorobenzene in the presence of organic cations. In this case, the reaction with C70 provides a different reaction route. The substitution of Cl− by the phenyl anion originated from NaBPh4 is also realized. At the same time, the C70˙− radical anions generated by NaBPh4 can reduce the Pc2− macrocycle to the Pc˙3− radical trianion. Previously, the radical anion salt (PPN)+[SnCl2Pc˙3−]− was obtained by the reduction of SnCl2Pc2− with the fullerene radical anion (PPN+)(C60˙−) salt (PPN: bis(triphenylphosphine)iminium cation). In this case, formally neutral [SnPhPc˙3−]0 is formed, which, however, contains reduced and paramagnetic Pc˙3− macrocycles. That is a preliminary supposition and additional investigations are necessary to elucidate the exact reaction mechanisms for the formation of 1 and 2.Optical properties
The spectra of the starting SnIVCl2Pc2−, the reference compound (PPN)+[SnCl2Pc˙3−]− and compounds 1 and 2 are shown in Fig. 1. SnIVCl2Pc2− manifests the Soret band at 381 nm and the split Q-band at 670, 740 (maximum) and 848 nm (Fig. 1, a). The formation of (PPN)+[SnCl2Pc˙3−]− is accompanied by the appearance of an intense band in the NIR range with the maximum at 1006 nm and a weaker band at 834 nm as well as a strong blue shift of both Soret and Q-bands which are positioned in the spectrum of the salt at 335 nm and 594, 627 (maximum) and 710 nm (Fig. 1, c).23 Similar effects were observed in the formation of the MIIPc˙3− radical anions, unpaired electrons of which are delocalized on the Pc macrocycles (M = NiII, CuII, H2, PbII, SnII, InIIIBr).24–26 The spectrum of salt 1 is similar to that of neutral SnIVCl2Pc2− since the band in the NIR range is nearly absent. The weakly blue-shifted Soret band is located at 360 nm whereas the Q-band retains its position with the maximum at 740 nm (Fig. 1, b). These data allow one to suppose that the Pc macrocycle in 1 has the charge of −2 as in the starting SnIVCl2Pc2−. In contrast to 1, the negatively charged Pc macrocycle in 2 forms the Pc˙3− radical trianions. The spectrum of 2 has an intense band in the NIR range at 1075 nm and a weaker band at 927 nm (Fig. 1, d). The Soret (334 nm) and Q-bands (565, 612 (maximum) and 705 nm) are noticeably blue-shifted in the spectrum of 2, and became closer to the positions in the spectrum of (PPN)+[SnCl2Pc˙3−]− containing unpaired electron in the Pc˙3− macrocycle. It is seen that the addition of the phenyl group to SnPc in 2 noticeably shifts the absorption bands of Pc˙3− in the NIR range to lower energies from 1006 and 834 nm in the spectrum of (PPN)+[SnCl2Pc˙3−]− to 1075 and 927 nm in the spectrum of 2. The spectrum of 2 also contains a broad low-energy band with the maximum at 1560 nm (0.79 eV), which can be attributed to charge transfer (CT) between negatively charged Pc macrocycles within the [SnPhPc˙3−]2 pairs. The IR-spectra of the starting and reference compounds and salts 1 and 2 are listed in Table S1† and shown in Fig. S1 and S2.† It is seen that some absorption bands of SnIVCl2Pc2− strongly shifting at 409 cm−1 increases in intensity (Table S1†). Positions of the absorption bands in the IR spectrum of 1 are close to those in the spectrum of SnIVCl2Pc2− whereas the positions of the bands in the spectrum of 2 are close to those in the spectrum of (PPN)+[SnCl2Pc˙3−]−. Thus, the IR spectra also justify the formation of −2 and −3 charged Pc macrocycles in 1 and 2, respectively. | ||
| Fig. 1 UV-visible-NIR spectra of: (a) starting SnIVCl2Pc2−; (b) salt [SnPhPc]+(BPh4)−·C6H14 (1); (c) radical anion (PPN)+[SnCl2Pc˙3−]− salt containing Pc˙3−; (d) [SnPhPc˙3−]·C6H4Cl2 (2) measured in KBr pellets. Pellets for spectra b–d were prepared under anaerobic conditions. | ||
Crystal structures
Crystal structures of 1 and 2 were solved at 150 and 120 K, respectively. Since these compounds contain differently charged Pc macrocycles, we can compare how the reduction affects the geometry of SnPhPc. There are two types of C–N bonds in the Pc macrocycle. Shorter bonds are formed with imine (Im) nitrogen atoms and longer bonds are formed with pyrrole (Py) nitrogen atoms. The bond lengths in the dianionic Pc2− macrocycle of 1 are 1.325(2) and 1.387(2) Å being close to those in SnIVCl2Pc2− of 1.334(2) and 1.375(3) Å, respectively.28 In both cases no alternation of the C–N(Im) or C–N(Py) bonds is observed. The average length of the C–N(Py) bonds in negatively charged Pc macrocycles is 1.393(2) Å in 2. The C–N(Im) bonds alternate between shorter (1.308(2) Å) and longer bonds (1.350(2) Å). These bonds are located in such a way that shorter or longer bonds belong to two oppositely located isoindole units. Alternation of the bonds can be attributed to partial disruption of the aromaticity of the Pc macrocycle in the reduction. Similar alternation of the C–N(Im) bonds is observed for different MIIPc˙3− radical anions, in which the Pc macrocycle is reduced. Phenyl substituents are linked to tin atoms by the Sn–C bonds of 2.098(2) and 2.105(2) Å length for 1 and 2, respectively. In SnIVCl2Pc2−, tin atoms are positioned exactly in the 24-atom Pc plane with an average Sn–N(Pc) bond length of 2.054(2) Å. Due to the formation of the Sn–C bonds with phenyl groups tin atoms displace out of the 24-atom Pc plane in 1 and 2 by 0.853 and 0.625 Å. As a result, the Sn–N(Pc) bonds elongated to 2.103(2) and 2.089(2) Å, respectively. The dianionic Pc2− macrocycle deviates essentially stronger from the planarity in 1 in comparison with the nearly planar Pc˙3− macrocycle in 2. The Pc macrocycle in 1 is distorted to show the butterfly shape with the dihedral angle of 16.15° between the two nearly planar halves of the Pc macrocycle.The SnPhPc molecules in 1 form the [SnPhPc]2 dimers with π–π stacking between two halves of the Pc macrocycles (Fig. 2a). The interplanar distance in the dimers is short (3.207 Å) and provides the formation of multiple van der Waals C,N⋯C,N contacts in the 3.21–3.37 Å range. The dimers form chains along the a-axis and separated from the neighboring chains by BPh4− anions and solvent C6H14 molecules (Fig. 2a). The interplanar distance between the Pc macrocycles belonging to two neighboring [SnPhPc]2 dimers is essentially longer (3.527 Å), and no van der Waals C,N⋯C,N contacts are formed, indicating that phthalocyanine dimers are isolated in 1.
 | ||
| Fig. 2 Crystal structure of: (a) salt [SnPhPc]+(BPh4)−·C6H14 (1), view on the chains from the π-stacking {[SnPhPc]+}2 dimers; (b) compound [SnPhPc]·C6H4Cl2 (2) formed by closely packed π-stacking [SnPhPc]2 dimers. van der Waals C,N⋯C,N contacts are shown by green dashed lines. Solvent C6H4Cl2 molecules are not shown for 2 for clarity. | ||
Compound 2 contains no cations, and the SnPhPc units are closely packed in a crystal (Fig. 2b). They also form the π-stacking [SnPhPc]2 dimers, but in contrast to 1, the Pc macrocycles are planar in 2, and the whole SnPhPc molecule is involved in the π–π stacking within the dimer. The interplanar distance between the Pc macrocycles is only 3.266 Å, and multiple van der Waals contacts are formed between the Pc macrocycles in the dimers. The [SnPhPc]2 dimers are closely packed (Fig. 2b). However, the interplanar distance between the Pc macrocycles from the neighboring dimers is essentially longer (3.594 Å), and only one van der Waals C⋯C contact between Pc macrocycles is formed in this case (Fig. 2b). Phenyl substituents of SnPhPc form channels arranged along the b-axis to accommodate C6H4Cl2 molecules (not shown in Fig. 2b).
To elucidate the peculiarities of the SnPhPc packing in 2, overlap integrals between Pc macrocycles were calculated by the extended Hückel method29 from the X-ray diffraction data. Since the Pc macrocycle is negatively charged, SnPhPc has an occupied HOMO and a singly occupied LUMO which can be regarded as the SOMO. There are face to face stacks between Pc macrocycles in the dimers of 2 (overlap integral s1). In this case large SOMO–SOMO (0.014) and HOMO–HOMO (0.0106) overlap integrals are observed (Fig. 3, s1). There are also stacks between the Pc macrocycles from the neighboring dimers: slipped stacks (Fig. 3, s2) with the SOMO–SOMO and HOMO–HOMO overlap integrals of 0.0001 and 0.0008 and quasi stacks (Fig. 3, s3) with the SOMO–SOMO and HOMO–HOMO overlap integrals of 0.0003 and 0.0002. These data indicate strong dimerization of the SnPhPc units in 2. The interdimer interactions are rather weak to realize, for example, conductivity through the Pc˙3− macrocycles in 2.
 | ||
| Fig. 3 (a) Three types of overlapping integrals (s1, s2 and s3) between the SnPhPc units in the phthalocyanine layer of 2; (b) overlapping modes for the s1, s2 and s3 integrals. One of two molecules for the s1 and s2 integrals is shown by red and blue colors. | ||
Magnetic properties
Salt 1 shows a single weak EPR signal. It has a Lorentzian shape at room temperature (RT) with a g-factor of 2.0036 and the line halfwidth (ΔH) of 0.57 mT. The signal becomes asymmetric below 160 K, and can be fitted by two Lorentzian lines with g1 = 2.0034 (ΔH = 0.58 mT) and g2 = 1.9927 (ΔH = 7.34 mT) at 160 K. The signal is even more asymmetric below 6 K and can be fitted by three Lorentzian lines (Fig. S3 in the ESI†). The parameters of these lines are g1 = 2.0027 (ΔH = 0.39 mT), g2 = 1.9989 (ΔH = 0.86 mT) and g3 = 1.9871 (ΔH = 2.77 mT) at 4.2 K (Fig. S3†). The intensity of this signal is very weak, and corresponds to the contribution of less than 1.6% of spins from the total amount of SnPhPc. That is only the admixture of some products with the reduced Pc˙3− macrocycle.Formally neutral compound 2 contains a negatively charged paramagnetic Pc˙3− macrocycle. As a result, 2 shows an intense EPR signal whose integral intensity corresponds to the contribution of about one S = 1/2 spin per formula unit. The signal has a Lorentzian shape at 298 K with a g-factor of 2.0046 and a ΔH of 0.49 mT (Fig. 4, spectrum at 298 K). The signal splits into two lines below 80 K with g1 = 2.0062 (ΔH = 0.33 mT) and g2 = 2.0045 (ΔH = 0.23 mT) at 80 K. The two-component signal preserves down to 4.2 K (g1 = 2.0060, ΔH = 0.40 mT and g2 = 2.0044, ΔH = 0.22 mT) (Fig. 4, spectrum at 4.2 K). The temperature decrease results in the shift of g-factor to higher values (Fig. 5a) and noticeable narrowing of the main component down to 30 K (Fig. 5b). However, after splitting of the signal into two lines, the additional component with a g-factor of 2.0045 and the main component with a g-factor of 2.0060 begin to broaden below 80 and 30 K, respectively (Fig. 5b). Most probably low-temperatures broadening of both components is due to the antiferromagnetic interaction between spins which can be realized in closely packed π-stacking [SnPhPc]2 dimers. Indeed, fitting of the temperature dependence of the integral intensity of both components by the Curie–Weiss law in the 10–300 K range yields a Weiss temperature of −3 K (red curve in Fig. 5c) indicating weak antiferromagnetic coupling between spins. The temperature dependence of the integral intensity of the EPR signal can also be approximated well by the Heisenberg model for isolated pairs of antiferromagnetically interacting spins30 with magnetic exchange interaction J/kB = −0.23 K (red curve in Fig. 5d). This value indicates only weak antiferromagnetic coupling between spins in the [SnPhPc]2 dimers.
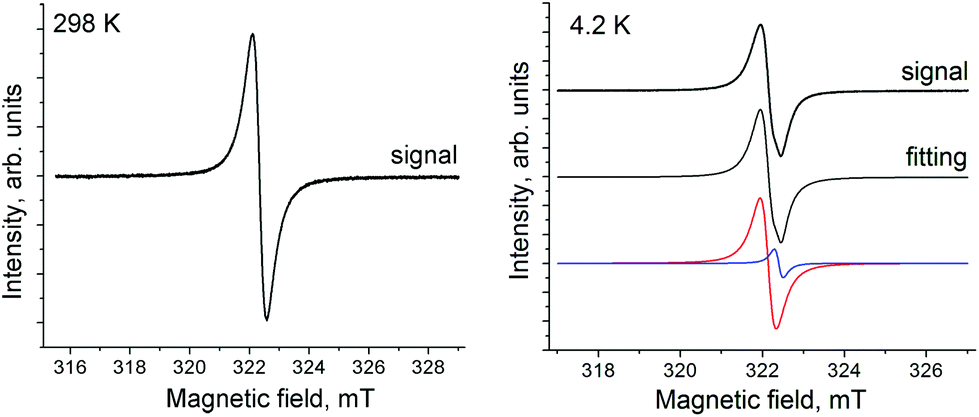 | ||
| Fig. 4 The EPR signal of polycrystalline compound 2 at 298 K (sweep width 15 mT, modulation width 0.1 mT, microwave power 1 mW, frequency 9.04355 GHz, ampl. 10) and 4.2 K (sweep width 10 mT, modulation width 0.1 mT, microwave power 1 mW, frequency 9.03844 GHz, ampl. 1.6). | ||
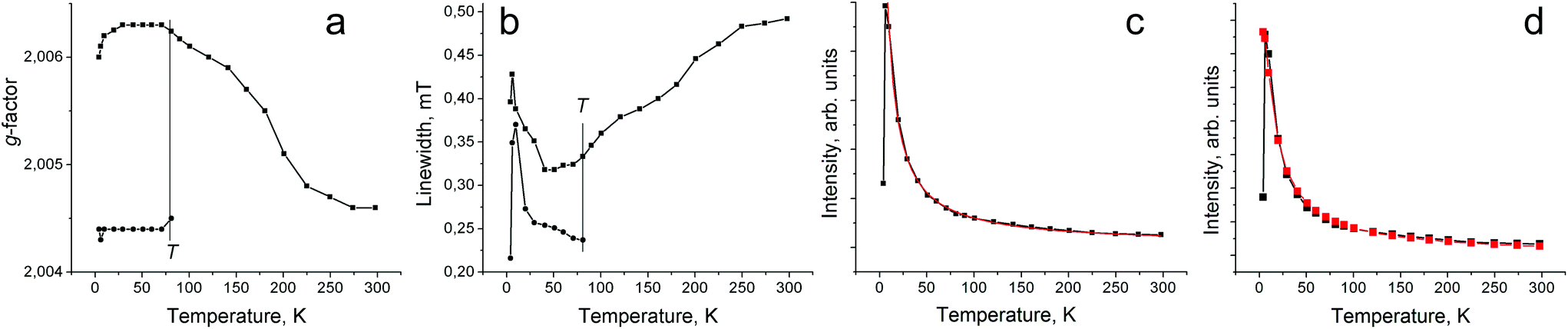 | ||
| Fig. 5 Temperature dependence of g-factor (a), linewidth (b) and integral intensity (c) of EPR signal from polycrystalline 2. Fitting of the temperature dependence in (c) by the Curie–Weiss law with a Weiss temperature of −3 K is shown by a red curve. (d) Temperature dependence of integral intensity of EPR signal approximated by the Heisenberg model for isolated pairs of antiferromagnetically interacting spins30 with J/kB = −0.23 K (red curve). | ||
Theoretical analysis
To investigate the electronic structure of [SnPhPc]·C6H4Cl2 (2), theoretical analyses were performed at the UCAM-B3LYP-D3 and UM11/cc-pVTZ-PP/cc-pVDZ levels of theory. Only the coordinates of hydrogen atoms in the [SnPhPc]0 monomer and the [SnPhPc]20 dimer were geometry-optimized. The total and relative energies, 〈S2〉 values, and charge and spin densities of the doublet state in the [SnPhPc]0 monomer and the broken-symmetry singlet and triplet states of the [SnPhPc]20 dimer are summarized in Tables S2–S4.† The UCAM-B3LYP-D3 and UM11 functionals afforded similar results, and it is mainly described based on UCAM-B3LYP-D3 below.The energy diagram for the frontier Kohn–Sham orbitals of the 2A state in the [SnPhPc]0 monomer is shown in Fig. 6a. The 164th and 165th orbitals are singly occupied (SO) and the lowest unoccupied (LU) orbitals, respectively, which are derived from the doubly degenerate LUMO of the D4h-symmetric phthalocyaninate dianion (Pc2−), and the 163rd orbital is the highest occupied (HO) orbital from the HOMO of D4h-symmetric Pc2−. The calculated charge and spin densities on the Sn atom and the Pc ligand in the [SnPhPc]0 monomer are similar to those in the [SnCl2Pc˙3−]˙− (Tables S3 and S4†). Therefore, the Pc ligand and the Sn ion in the [SnPhPc]0 monomer can be regarded as an open-shell Pc˙3− radical trianion and Sn4+ cation, respectively.
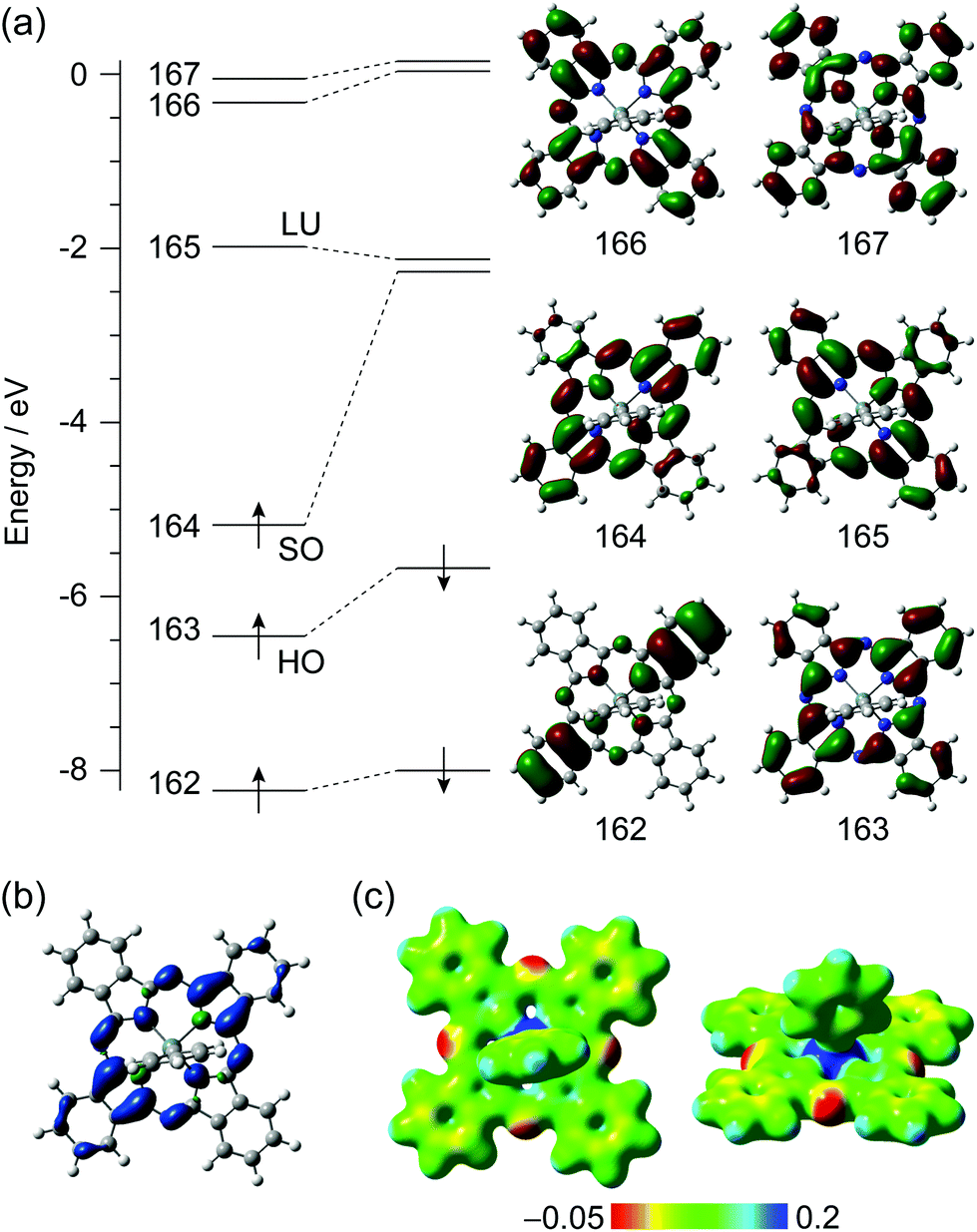 | ||
| Fig. 6 (a) Energy diagram for the frontier Kohn–Sham orbitals of the 2A state in [SnPhPc]0 monomer calculated at the UCAM-B3LYP-D3/cc-pVTZ-PP/cc-pVDZ level of theory. HO, SO, and LU denote the highest occupied, singly occupied, and the lowest unoccupied orbitals, respectively. The α-orbitals are shown on the right side. (b) The isosurface plot for spin density distribution, where the isosurface value is 0.0016 electron per au3. The isosurfaces in blue and green denote positive and negative spin density, respectively. (c) Electrostatic potential maps on the 0.02 electron per au3 of electron density surface. | ||
Considering the electroneutrality, the electronic structure of the [SnPhPc]0 monomer can be concluded as [Sn4+Ph−Pc˙3−]0 on the formal charge basis. The spin density distribution also indicates that the magnetic properties stem from the π-radical spin on the Pc moiety (Fig. 6b). It is seen from the electrostatic potential map that the isoindole moieties, the SO orbital of which spreads over, are more negatively charged than the others (Fig. 6c).
The broken-symmetry singlet state in the [SnPhPc]20 dimer was found to be more stable in energy than the 3Au state (Table S2†), and the intermolecular antiferromagnetic interaction was calculated to be J = −1105 K, which supports the observed antiferromagnetic behavior. The energy diagram for the frontier Kohn–Sham orbitals of the broken-symmetry singlet state in the [SnPhPc]20 dimer is shown in Fig. S6.† The 327th α- and β-HO orbitals are the SO orbitals, related to the magnetic properties of the [SnPhPc]20 dimer. The electronic structure of the [SnPhPc]20 dimer inherits that of the 2A state in the [SnPhPc]0 monomer. The spin density distribution in the [SnPhPc]20 dimer spreads over the Pc ligands, and the π-radical spins on the Pc ligands show the intermolecular antiferromagnetic interaction (Fig. S6b†). The charge and spin density are also similar to each other between the [SnPhPc]0 monomer and the [SnPhPc]20 dimer calculations (Tables S3 and S4†).
To obtain further insight into the optical properties of [SnPhPc]·C6H4Cl2 (2), the excited states of the [SnPhPc]0 monomer and the [SnPhPc]20 dimer were examined based on time-dependent density functional theory (TD-DFT) at the same level of theory.
The calculated excitation energies, oscillator strengths, 〈S2〉 values, and assignments on the low-lying excited states of the [SnPhPc]0 monomer and the [SnPhPc]20 dimer are summarized in Tables S5–S7.† The observed UV-vis-NIR spectrum of [SnPhPc]·C6H4Cl2 (2) and the calculated spectra of the [SnPhPc]0 monomer and the [SnPhPc]20 dimer are compared in Fig. 7 and S7.†
 | ||
| Fig. 7 (a) The observed UV-vis-NIR spectrum of SnPhPc·C6H4Cl2 (2) in KBr. Calculated spectra of (b) the broken-symmetry singlet state in [SnPhPc]20 dimer and (c) the 2A state in [SnPhPc]0 monomer at the TD-UCAM-B3LYP-D3/cc-pVTZ-PP/cc-pVDZ level of theory. The charge transfer transitions are indicated as red lines. | ||
The first and second excited states of the [SnPhPc]0 monomer are the SO → LU and HO → LU excitations, respectively (Table S5† and Fig. 7). Although the CAM-B3LYP-D3 and M11 functionals altered the order of the first and second excited states, the main spectral feature is not affected since these oscillator strengths are weak. The third and fourth excited states are the HO → SO and HO → LU excitations, respectively, where these excitations have the Q-band character and are denoted as Qx and Qy. In the case of TD-UCAM-B3LYP-D3 calculation, the second and fourth excited states seem to be derived from the splitting of the Q-band in [SnIVPc2−]2+, and the second one with weak oscillator strength and the fourth one with strong oscillator strength are red-shifted and blue-shifted from the original Q-band, respectively.31 The fifth excited state is the SO → LU+1 excitation.
As for the [SnPhPc]20 dimer, the spectral feature takes over that of the [SnPhPc]0 monomer except for CT transition between [SnPhPc]0 monomers. In the case of TD-UCAM-B3LYP-D3 calculation (Table S7†), the first and second excited states are the SO → LU excitations, the third, fourth, 13th, and 17th excited states are the HO → LU excitations (Qy), the fifth and tenth excited states are the HO → SO excitations (Qx), and the 15th and 18th excited states are the SO → LU+1 excitations, which are the intramolecular electronic transitions in the [SnPhPc]0 monomer. In the calculation of the [SnPhPc]20 dimer (Fig. 7b), the additional bands that do not exist in the [SnPhPc]0 monomer are ascribed to the intermolecular CT transitions. The sixth and seventh excited states are the intermolecular SO → SO excitations, the eighth and ninth are the intermolecular SO → LU excitations, the 11th and 12th are the intermolecular HO → LU excitations, and the 14th and 16th are the intermolecular HO → SO excitations. Comparing the observed UV-vis-NIR spectrum with the calculated ones, the band at 0.79 eV (1560 nm) can be assigned to the CT transition with the character of the intermolecular SO → SO excitation although the CAM-B3LYP-D3 functional overestimated the excitation energy. The band at 1.15 eV (1075 nm) can be regarded as the intramolecular HO → SO excitations with the Q-band character (Qx), and the band at 2.02 eV (612 nm) as the mixed HO → LU (Qy) and SO → LU+1 excitations. Although the bands at 1.34 eV (927 nm) and 1.75 eV (705 nm) may also be due to the intermolecular CT transitions, further investigation would be necessary due to a great number of candidates for the assignments. The M11 functional afforded has a tendency to afford higher excitation energy in the CT transition than the CAM-B3LYP-D3 (Fig. S7 and Table S6†).
Neutral phthalocyanine compounds containing radical trianionic macrocycles
One of the interesting possibilities which can be realized for metal phthalocyanines is the preparation of formally neutral compounds which, however, contain radical trianionic macrocycles. The presence of unpaired spins on these macrocycles can allow the preparation of single-component molecular conductors or magnets. The search for compounds of such type is realized in different classes of materials39 since they show some advantages compared with normal two-component salts containing cations and anions. In the case of single-component phthalocyanine compounds, the advantages are solubility in most of the organic solvents, intense absorption in the visible and even in the NIR range and easy preparation of films and composites. Some other formally neutral metal phthalocyanine compounds, in which the phthalocyanine macrocycle is in radical trianionic or tetraanionic states, are also known. These are (AlIIIPc˙3−)(anisole)240 and (GeIVPc4−)(pyridine)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 41 containing radical trianionic and tetraanionic phthalocyanine macrocycles, respectively. The coordination complex Ph5CpRu(CO)2[SnIIPc˙3−] is also formally neutral but contains the Pc˙3− radical trianions. As a result, very strong magnetic coupling is attained between the spins in this complex with the exchange interaction of J/kB = −187 K.27
41 containing radical trianionic and tetraanionic phthalocyanine macrocycles, respectively. The coordination complex Ph5CpRu(CO)2[SnIIPc˙3−] is also formally neutral but contains the Pc˙3− radical trianions. As a result, very strong magnetic coupling is attained between the spins in this complex with the exchange interaction of J/kB = −187 K.27
Conclusions
Phenyl substituted tin(IV) phthalocyanine has been obtained in the cationic and neutral form by the reduction of SnIVCl2Pc with sodium tetraphenylborate in the presence of fullerenes C60 and C70. Fullerenes affect the product obtained but are not involved in the crystals. Both compounds [SnPhPc]+(BPh4)−·C6H14 (1) and [SnPhPc]·C6H4Cl2 (2) are well soluble in o-dichlorobenzene and can be obtained as crystals. Salt 1 contains a dianionic Pc2− macrocycle and extra positive charge of SnIV is compensated by the BPh4− anions. Formally neutral compound 2 contains the Pc˙3− macrocycles in the radical trianion state. Unpaired spins are antiferromagnetically coupled within the [SnPhPc]20 dimers. This is a rare example of a formally neutral metal phthalocyanine compound with a radical trianionic phthalocyanine macrocycle. Such compounds are interesting for developing single component conductors or magnets.Experimental
Materials
Tin(IV) phthalocyanine dichloride (SnIVCl2Pc) was purchased from TCI. Sodium tetraphenylborate (NaBPh4, 99.5%) was purchased from Acros. Fullerenes C60 (99.9%) and C70 (99%) were received from MTR Ltd. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure and hexane was distilled over Na/benzophenone. Salts 1 and 2 were synthesized and stored in an MBraun 150B-G glove box with controlled atmosphere containing less than 1 ppm of water and oxygen. Solvents were degassed and stored in the glove box, and KBr pellets used for the IR and UV-visible-NIR analyses were prepared in the glove box. EPR measurements were performed on the polycrystalline samples of 1 and 2 sealed in 2 mm quartz tubes under 10−5 Torr pressure.Synthesis
Crystals of 1 and 2 were obtained by the diffusion technique. A reaction mixture was filtered into a 1.8 cm-diameter, 50 mL glass tube with a ground glass plug, and then 30 mL of n-hexane was layered over the solution. Slow mixing of the solutions resulted in precipitation of crystals over 1–2 months. The solvent was then decanted from the crystals, and they were washed with n-hexane. The compositions of the obtained compounds were determined from the X-ray diffraction analysis on a single crystal. Several crystals from one synthesis were found to consist of a single crystalline phase. For compound 1 the composition was confirmed by elemental analysis: C68H55BN8Sn, calc. C = 73.33, H = 4.94, N = 10.06; found C = 72.89, H = 4.68, N. 10.12. Compound 2 is air sensitive since it contains the Pc˙3− radical anions and adds oxygen during elemental analysis. In this case elemental analysis cannot be used to determine the composition. Nevertheless, the sample contains only selected crystals having appropriate shape. According to X-ray diffraction analysis, these crystals belong to one phase. Therefore, we are sure of the purity of this compound.[SnPhPc](BPh4)·C6H14 (1) was obtained via the reduction of SnIVCl2Pc (29.2 mg, 0.042 mmol) and fullerene C60 (30 mg, 0.042 mmol) with an excess of sodium tetraphenylborate (100 mg, 0.292 mmol) in 16 ml of o-dichlorobenzene by stirring at 100 °C for 24 hours. This results in complete dissolution of SnIVCl2Pc and the formation of a deep blue solution which was cooled down to room temperature and filtered into a tube for diffusion. Black blocks were obtained in 20% yield.
SnPhPc·C6H4Cl2 (2) was obtained via the reduction of SnIVCl2Pc (29.2 mg, 0.042 mmol) and fullerene C70 (35 mg, 0.042 mmol) with an excess of sodium tetraphenylborate (100 mg, 0.292 mmol) in 16 ml of o-dichlorobenzene by stirring at 100 °C for 24 hours. This results in complete dissolution of SnIVCl2Pc and the formation of a deep blue-red solution which was cooled down to room temperature and filtered into a tube for diffusion. Black prisms with copper luster were obtained in 24% yield.
It is interesting that the reduction of SnIVCl2Pc (29.2 mg, 0.042 mmol) with an excess of sodium tetraphenylborate (100 mg, 0.292 mmol) in 16 ml of fullerene free o-dichlorobenzene upon stirring at 100 °C for 24 hours provides only partial dissolution of SnIVCl2Pc, and no crystals are formed under slow mixing with hexane.
General
UV-visible-NIR spectra were recorded in KBr pellets on a Perkin Elmer Lambda 1050 spectrometer in the 250–2500 nm range. FT-IR spectra were obtained in KBr pellets with a Perkin-Elmer Spectrum 400 spectrometer (400–7800 cm−1). EPR spectra were recorded for the polycrystalline samples of 1 and 2 with a JEOL JES-TE 200 X-band ESR spectrometer equipped with a JEOL ES-CT470 cryostat in the temperature range from 293 down to 4 K.Crystal structure determination
Crystal data of 1 at 150(2) K: C68H55BN8Sn, Mr = 1113.70 g mol−1, black block, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.6897(4), b = 14.4413(4), c = 14.6760(4) Å, α = 109.063(2), β = 92.219(2), γ = 96.715(2)°, V = 2714.32(14) Å3, Z = 2, dcalc = 1.363 g cm−3, μ = 0.519 mm−1, F(000) = 1148, 2θmax = 58.022°, reflections measured 22575, unique reflections 12404, reflections with I > 2σ(I) = 11104, parameters refined 703, R1 = 0.0292, wR2 = 0.0743, G.O.F. = 1.028, CCDC 1465069.
, a = 13.6897(4), b = 14.4413(4), c = 14.6760(4) Å, α = 109.063(2), β = 92.219(2), γ = 96.715(2)°, V = 2714.32(14) Å3, Z = 2, dcalc = 1.363 g cm−3, μ = 0.519 mm−1, F(000) = 1148, 2θmax = 58.022°, reflections measured 22575, unique reflections 12404, reflections with I > 2σ(I) = 11104, parameters refined 703, R1 = 0.0292, wR2 = 0.0743, G.O.F. = 1.028, CCDC 1465069.
Crystal data of 2 at 120(2) K: C44H25Cl2N8Sn, Mr = 855.31 g mol−1, black prism, triclinic, P, a = 9.8645(3), b = 12.9714(4), c = 14.1591(6) Å, α = 86.320(3), β = 71.315(3), γ = 85.595(2)°, V = 1709.69(11) Å3, Z = 2, dcalc = 1.661 g cm−3, μ = 0.954 mm−1, F(000) = 858, max. 2θmax = 58.413°, reflections measured 14859, unique reflections 7940, reflections with I > 2σ(I) = 7170, parameters refined 496, R1 = 0.0246, wR2 = 0.0594, G.O.F. = 1.055, CCDC 1465068.
X-ray diffraction data for 1 and 2 were collected on an Oxford diffraction “Gemini-R” CCD diffractometer with graphite monochromated MoKα radiation using an Oxford Instrument Cryojet system. Raw data reduction to F2 was carried out using CrysAlisPro, Oxford Diffraction Ltd. The structures were solved by direct methods and refined by the full-matrix least-squares method against F2 using SHELX-2013.32 Non-hydrogen atoms were refined anisotropically. Positions of hydrogen were calculated geometrically. See the crystallographic data in CIF.
Computational details
DFT and TD-DFT calculations based on the CAM-B3LYP-D3 with the Grimme's D3 dispersion33 and M1134 functionals were performed using the cc-pVDZ (C, H, N, and Cl)35 and cc-pVTZ-PP (Sn)36 basis sets. For the geometries of the [SnPhPc]0 monomer and the [SnPhPc]20 dimer, only the coordinates of the hydrogen atoms were optimized from the X-ray structures with “Opt = Tight”. For the D4h-symmetric [SnCl2Pc]0 and D2h-symmetric [SnCl2Pc]˙−, full geometry optimization was performed at the same level of theory. In the present DFT calculations, “Int = SuperFineGrid” was specified, and the stabilities of the wavefunctions were confirmed by specifying the “Stable = Opt” keyword. Although the M11 functional found that the D4h-symmetric [SnCl2Pc]0 has the restricted-to-unrestricted instability, we considered only the closed-shell D4h-symmetric [SnCl2Pc]0 in this work. As for TD-DFT calculations, sixty and twenty-five excited states were calculated for the [SnPhPc]0 monomer and the [SnPhPc]20 dimer, respectively. The subsequent natural bond orbital (NBO) analysis was performed using the NBO program.37 The computations were performed with the Gaussian 09 program package.38Acknowledgements
The work was supported by RFBR grant no. 13-03-00769 and JSPS KAKENHI Grant Numbers 15K17901, 23225005, and 26288035. Y. N. received a research grant from the JGC-S Scholarship Foundation. Theoretical calculations were performed at the SuperComputer System, Institute for Chemical Research, Kyoto University, and the Research Center for Computational Science, Okazaki, Japan.Notes and references
- T. Nyokong, Coord. Chem. Rev., 2007, 251, 1707 CrossRef CAS.
- T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed.
- J. S. Miller, Inorg. Chem., 2000, 39, 4392 CrossRef CAS.
- C. G. Claessens, W. J. Blau, M. Cook, M. Hanack, R. J. M. Nolte, T. Torres and D. Wöhrle, Monatsh. Chem., 2001, 132, 3 CrossRef CAS.
- I. Beletskaya, V. S. Tyurin, A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659 CrossRef CAS PubMed.
- Y. Diskin-Posner, G. K. Patra and I. Goldberg, Dalton Trans., 2001, 2775 RSC.
- D. V. Konarev, S. S. Khasanov and R. N. Lyubovskaya, Coord. Chem. Rev., 2014, 262, 16 CrossRef CAS.
- C. Cloutour, D. Lafargue, J. A. Richards and J.-C. Pommier, J. Organomet. Chem., 1978, 161, 327 CrossRef CAS.
- C. Cloutour, D. Lafargue and J.-C. Pommier, J. Organomet. Chem., 1980, 190, 35 CrossRef CAS.
- K. M. Kadish, D. Dubois, S. Koeller, J.-M. Barbe and R. Guilard, Inorg. Chem., 1992, 31, 3292 CrossRef CAS.
- D. Y. Dawson, J. C. Sanyalang and J. Arnold, J. Am. Chem. Soc., 1996, 118, 6082 CrossRef CAS.
- G. Du, A. Ellern and L. K. Woo, Inorg. Chem., 2004, 43, 2379 CrossRef CAS PubMed.
- D. Y. Dawson, J. C. Sanyalang, J. Arnold, A. Auger, P. M. Barnham, I. Chambrier, M. J. Cook and D. L. Hughes, J. Mater. Chem., 2005, 15, 168 RSC.
- C. Lecomte, J. Protas, P. Cocolios and R. Guilard, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1980, 36, 2769 CrossRef.
- A. L. Balch, L. Latos-Grazynski, B. C. Noll and S. L. Phillips, Inorg. Chem., 1993, 32, 1124 CrossRef CAS.
- A. G. DiPasquale and J. M. Mayer, J. Am. Chem. Soc., 2008, 130, 1812 CrossRef CAS PubMed.
- R. D. Arasasingham, A. L. Balch, M. M. Olmstead and S. L. Phillips, Inorg. Chim. Acta, 1997, 263, 161 CrossRef CAS.
- M. Hanack, G. Renz, J. Strahle and S. Schmid, J. Org. Chem., 1991, 56, 3501 CrossRef CAS.
- M. J. Chen, J. W. Rathke and J. C. Huffman, Organometallics, 1993, 12, 4673 CrossRef CAS.
- W. Galezowski and M. Kubicki, Inorg. Chem., 2005, 44, 9902 CrossRef CAS PubMed.
- M. Tahiri, P. Doppelt, J. Fischer and R. Weiss, Inorg. Chem., 1988, 27, 2897 CrossRef CAS.
- D. V. Konarev, S. V. Simonov, S. S. Khasanov and R. N. Lyubovskaya, Dalton Trans., 2011, 40, 9176 RSC.
- D. V. Konarev, S. I. Troyanov, M. Ishikawa, M. Faraonov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, J. Porphyrins Phthalocyanines, 2014, 18, 1157 CrossRef CAS.
- D. V. Konarev, L. V. Zorina, S. S. Khasanov, A. L. Litvinov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2013, 42, 6810 RSC.
- D. V. Konarev, A. V. Kuzmin, M. A. Faraonov, M. Ishikawa, Y. Nakano, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Chem. – Eur. J., 2015, 21, 1014 CrossRef CAS PubMed.
- D. V. Konarev, A. V. Kuzmin, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Dalton Trans., 2014, 43, 13061 RSC.
- D. V. Konarev, A. V. Kuzmin, Y. Nakano, M. A. Faraonov, S. S. Khasanov, A. Otsuka, H. Yamochi, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2016, 55, 1390 CrossRef CAS PubMed.
- J. Janczak and R. Kubiak, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m237 Search PubMed.
- (a) M.-H. Whangbo and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 6093 CrossRef CAS; (b) J. Ren, W. Liang and M.-H. Whangbo, Crystal and Electronic Structure Analysis Using CAESAR, Prime Color Software, Inc., 1998. (This book can be downloaded free of charge from the website: http://www.PrimeC.com/). Default parameters were used Search PubMed.
- J. S. Smart, in Magnetism III, ed. G. T. Rado and H. Suhl, Academic Press, NY, 1963, p. 63 Search PubMed.
- (a) J. Mack, N. Kobayashi and M. J. Stillman, J. Porphyrins Phthalocyanines, 2006, 10, 1219 CrossRef CAS; (b) A. Rosa and G. Ricciardi, Can. J. Chem., 2009, 87, 994 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- (a) T. Yanai, D. Tew and N. Handy, Chem. Phys. Lett., 2004, 393, 5 CrossRef; (b) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- R. Peverati and D. G. Truhlar, J. Phys. Chem. Lett., 2011, 2, 2810 CrossRef CAS.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- (a) K. A. Peterson, D. Figgen, M. Dolg and H. J. Stoll, Chem. Phys., 2007, 126, 124101 Search PubMed; (b) K. A. Peterson, J. Chem. Phys., 2003, 119, 11099 CrossRef CAS; (c) D. J. Feller, Comput. Chem., 1996, 17, 1571 CrossRef CAS; (d) K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045 CrossRef CAS PubMed.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, Version 3.1 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- G. Saito and Y. Yoshida, Top. Curr. Chem., 2012, 312, 67 CrossRef CAS PubMed.
- J. A. Cissell, T. P. Vaid and A. L. Rheingold, Inorg. Chem., 2006, 45, 2367 CrossRef CAS PubMed.
- J. A. Cissell, T. P. Vaid, A. G. DiPasquale and A. L. Rheingold, Inorg. Chem., 2007, 46, 7713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The IR spectra of the starting and reference compounds and salts 1 and 2, the EPR spectrum of salt 1, and details of the DFT calculations for compound 2. CCDC 1465069 and 1465068. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01132b |
| This journal is © The Royal Society of Chemistry 2016 |