
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation and decomplexation kinetics of copper(II) complexes with cyclen derivatives having mixed carboxylate and phosphonate pendant arms†
R.
Ševčík
a,
J.
Vaněk
ab,
R.
Michalicová
a,
P.
Lubal
*ab,
P.
Hermann
c,
I. C.
Santos
d,
I.
Santos
d and
M. P. C.
Campello
*d
aDepartment of Chemistry, Faculty of Science, Masaryk University, Kotlářská 2, 611 37 Brno, Czech Republic. E-mail: lubal@chemi.muni.cz; Fax: +420-54949 2494; Tel: +420-54949 5637
bCentral European Institute of Technology (CEITEC), Masaryk University, Kamenice 5, 625 00 Brno, Czech Republic
cDepartment of Inorganic Chemistry, Faculty of Science, Universita Karlova (Charles University), Hlavova 2030, 128 40 Prague 2, Czech Republic
dCentro de Ciências e Tecnologias Nucleares, Instituto Superior Técnico, Universidade de Lisboa, Estrada Nacional 10, 2695-066 Bobadela LRS, Portugal. E-mail: pcampelo@ctn.tecnico.ulisboa.pt; Fax: +351-219946185; Tel: +351-219946233
First published on 8th July 2016
Abstract
The kinetic properties of Cu(II) complexes of H4dota and its analogues with one (H5do3ap), two in the 1,7-position (trans-H6do2a2p), three (H7doa3p) and four (H8dotp) phosphonic acid pendant arms were investigated. The formation of a Cu(II) complex with H4dota, trans-H6do2a2p and H8dotp at a slightly acidic pH is faster for the phosphonic acid derivatives than for H4dota, but with no simple dependence on the number of –CH2PO3H2 substituents (trans-H6do2a2p > H8dotp > H4dota; pH 4–6). Relative differences in the reactivity among the differently protonated species (HnLx−) of the same ligand are successively decreased with the more phosphonic acid groups in the ligand. The faster complexation is probably caused by the higher ability of phosphonates to bind the metal ion and/or to assist in the transfer of protons from the ring amine groups to the bulk water. The acid-assisted decomplexation kinetics of the complexes was followed in highly acidic solutions ([H+] = 0.01–5 M) and at different temperatures (15–70 °C) to determine the activation parameters of the reaction. The kinetic inertness of the Cu(II) complexes follows the order: H4dota > H5do3ap > trans-H6do2a2p > H7doa3p > H8dotp. To obtain information on the influence of additional pendant arms, analogous data were obtained for trans-H2do2a. The ligand is less reactive than H4dota, but the kinetic inertness of its Cu(II) complex is similar to that of the H4dota complex. As it was considered that the published thermodynamics data on the Cu(II)–H8dotp system are probably incorrect, the system was re-investigated. It showed a very high stability for the [Cu(dotp)]6− species and the easy formation of several Cu2L species in the presence of an excess of the metal ion. Also, the structure of the (H6doa3p)− anion in the solid state was determined. These experimental data demonstrate that the substitution of acetic acid pendant arms by methylphosphonic acid ones in H4dota-like ligands increases the rate of complexation but significantly decreases the kinetic inertness of the Cu(II) complexes.
Introduction
Tetraazamacrocyclic ligands with coordinating pendant arms have been studied intensively for a number of years due to their utilization in medicine and molecular biology, e.g. as contrast agents in magnetic resonance imaging (MRI)1 or as metal radioisotope carriers for diagnostic and/or therapeutic purposes.2–4 Generally, metal complexes intended for applications in medicine should exhibit some specific properties, namely high thermodynamic stability and kinetic inertness, desired hydrophilicity/hydrophobicity, etc. To achieve the desired biodistribution/pharmacokinetics of the metal complexes, conjugation to a targeting molecule, such as (oligo)peptides, monoclonal antibodies or their fragments, sugars, etc., is necessary, with ligands bearing a reactive group (bifunctional chelators, BFC)5 usually employed for such conjugations. For metal radioisotopes, the rate of complexation in very dilute solutions is also an important parameter.6 In general, metal complexes of macrocyclic ligands are much more kinetically inert than the complexes of open-chain ligands but their formation is rather slow. The properties of complexes have to be carefully balanced through ligand design and these investigations comprise a vital multidisciplinary field where coordination chemistry plays an important role.In nuclear medicine, a number of metal radioisotopes have been suggested: 44/47Sc, 61/64/67Cu, 68/67Ga, 90Y, 99mTc, 111In, 153Sm, 166Ho, 177Lu, 186/188Re, 213Bi, etc. Among these, copper radioisotopes bring a number of advantages as they are useful both for diagnosis (61/64Cu, β+ emitters for PET imaging) and therapy (67Cu, β− emitter) and their half-life match the pharmacokinetics of different targeting molecules, e.g. oligopeptides or antibody fragments. Research on ligands for copper radioisotopes has continued over the last two decades2,3,7 but an optimal ligand family has not been found, until now. However, due to their commercial availability, bifunctional H4dota-like ligands are the most common chelators used in 64Cu radiopharmaceuticals research, despite them not having optimal complexation properties towards divalent copper.
The properties of macrocyclic ligands can be tuned through the size of the macrocyclic ring, the ring donor atoms and/or the nature of the coordinating pendant arms. The most commonly studied ligands are those based on polyazamacrocycles, namely 1,4,7-triazacyclononane (tacn), 1,4,7,10-tetraazacyclododecane (cyclen) and 1,4,8,11-tetrazacyclotetradecane (cyclam). The most common pendant arm in these ligands is the acetic acid moiety (where the prototype ligand is e.g. H4dota, Fig. 1), though acetamide, propionic acid, alcoholic, methylphosphonic/phosphinic acid and various heterocycle pendant arms have also been suggested. Among these, the acid–base and coordination properties of phosphorus acid groups are the most similar to the properties of the carboxylic group and, therefore, methylphosphonic or methylphosphinic acid pendants are a common choice.8,9 The groups are tetrahedral and more acidic, harder and larger, as well as more hydrophilic than the carboxylic group. For phosphinic acid derivatives, their properties could be further tuned through changing the phosphorus atom substituents and, in addition, by preparing bifunctional chelators with a reactive side group on the phosphorus atom.10–13 With regard the other pendant arms, the heterocycle-containing ones (e.g. methylpicolinate) highly improve Cu(II) binding into the macrocyclic cavity and the complex properties.14,15 Some kinetic data on Cu(II) complexes of the cyclen/cyclam derivatives with pendant arms have been published and the investigations on the complexes of ligands with acetic acid,16–18 methylphosphonic/phosphinic acid13,18–23 or heterocyclic pendant arms14,15 have shown that the kinetic properties are highly influenced by the nature of the pendant arms. Generally, Cu(II) complex formation is accelerated when phosphorus acid or picolinate pendant arms are present, and this is also true with non-carrier-added 64Cu for cyclam13,23,24 as well as for cross-bridged cyclam derivatives;25 in addition, these groups are considered the best pendant arms for radiolabelling.
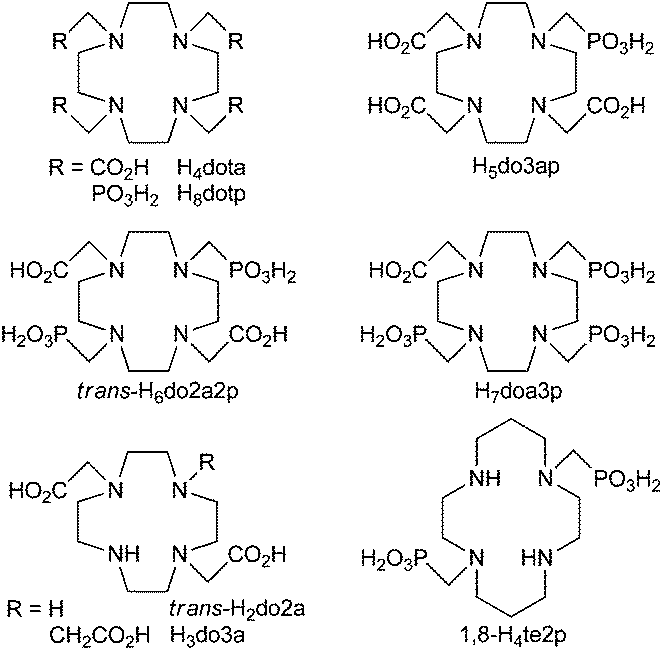 | ||
| Fig. 1 Structures of the discussed ligands. | ||
With the lanthanide(III) complexes of H4dota-like ligands, it has been shown that the presence of one phosphorus acid pendant accelerates complexation, but it is decelerated with more phosphonic acid pendants.11,26,27b In these complexes, the influence of the number of phosphorus acid pendants on decomplexation is not clear.27–31 To obtain analogous data for Cu(II) complexes of cyclen-based ligands, we decided to compare the kinetic behaviour of the complexes with H4dota and its analogues with an increasing number of methylphosphonic acid pendant arms: one (H5do3ap), two in the 1,7-position (trans-H6do2a2p), three (H7do1a3p) and four (H8dotp); see Fig. 1. The coordination chemistry of the symmetric ligands, H4dota and H8dotp, has previously been investigated in a number of studies over a long time; however, for Cu(II), only the dissociation kinetics of its H4dota and H5do3ap complexes18 and partially also the formation kinetics with H4dota16 have been studied. The copper(II) complexes of some methylphosphonic acid cyclen derivatives have also been studied but no kinetic data has been published32,33 except for our recent work on tris(methylphosphonic acid) cyclen derivatives.19
Experimental
The ligands were synthesized according to the published procedures for H4dota,34 H5do3ap,35trans-H6do2a2p,28 H7doa3p,31 H8dotp33 and trans-H2do2a.36 The ligands were used in a zwitterionic form, and any inorganic acids (originating from the ligand syntheses), if present, were removed by ion exchangers. CuCl2·2H2O was purchased from Fluka and its stock solution was standardized chelatometrically.37 The other analytical grade chemicals were purchased from Lachema, Fluka or Merck, and were used as received. The Cu(II) complexes for the spectroscopic and dissociation kinetic studies were prepared by mixing CuCl2 and the appropriate ligand stock solutions in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.2 molar ratio followed by stepwise neutralisation of the solutions to neutral pH.
:1.2 molar ratio followed by stepwise neutralisation of the solutions to neutral pH.
All the kinetic experiments were done at 25.0 ± 0.1 °C or at the other temperatures mentioned in the text. The ionic strength of I = 0.1 M KCl or 5.0 M (Na,H)ClO4 was used in the formation or dissociation kinetic studies, respectively. The kinetics was followed on a diode-array spectrophotometer HP 8453A (Hewlett Packard) and only the fast formation kinetics was measured by a stopped-flow technique (Applied Photophysics Instruments mixer joined with a diode-array detector, J&M and Applied Photophysics SX20). The formation kinetics of the Cu(II) complexes of trans-H6do2a2p, H7doa3p and H8dotp were followed under pseudo-first-order conditions (cCu = (1–3.5) × 10−3 M, cL = 1 × 10−4–5 × 10−5 M) in the pH range 1–1.9. The initial-rate method was applied in the pH range 2.5–6.0 (cCu = 2.5 × 10−4 M, cL = 5 × 10−4 M) in order to eliminate the possible formation of binuclear species in the course of the formation reaction. The dissociation kinetics of all the Cu(II) complexes was studied in 0.1–5 M HClO4 with cCuL = 1 × 10−4 M and in the temperature range 25–65 °C. The wavelength of the maximum of the absorption band in the range 315–325 nm was chosen to monitor the course of the formation/dissociation reactions. The experimental data were processed by PRO-K II (Applied Photophysics) and HP/Excel38 software, giving identical results. The measured values of absorbance were corrected for the background analytical signal. Throughout the text, pH means −log[H+].
UV-Vis spectra were measured (200–1100 nm, 25 °C) in aqueous solution at different pH. Electronic spectra of the individual species in the Cu–L systems were calculated from these data by OPIUM (a software package for the determination of stability constants, involving a module for spectrophotometric data treatment)39 with knowledge of all the complex stability constants, i.e. knowing the abundance of individual species at each pH.
The ligand H7doa3p was prepared as a hydrochloride.31 For the kinetic experiments, HCl was removed by a strong cation exchanger (water elution), where the ligand is retarded more than HCl. Slow evaporation (i.e. weeks) of the first chromatographic fraction containing the ligand gave single crystals suitable for X-ray diffraction analysis.
Results and discussion
In-cage Cu(II) complexes in solution
To compare and interpret the kinetic data, the solution structures and thermodynamic stabilities of the complexes should be known. Speciation diagrams for the systems were calculated from literature data (Fig. S1, S3 and S5†). To obtain directly comparable data through the complex series, solution UV-Vis spectra of mononuclear Cu(II) complexes of H4dota, trans-H6do2a2p and H8dotp were measured at various pH and the electronic spectra of the differently protonated species were calculated from the data (Fig. S2, S4 and S6; Table S1†).The d–d spectra of Cu(II)–H4dota/H8dotp systems agree with the published data.16,32,40 The absorption band maximum of [Cu(dota)]2− (λmax ≈ 740 nm) was not changed with the addition of 1–2 protons to the [Cu(Hndota)](2−n) species. Their structures should be the same as that of [Cu(H2dota)] found in the solid state. There, the Cu(II) ion is bound in the ligand cavity (in-cage complex) with an N2O2 equatorial arrangement and with the two other amines in axial positions; in addition, the two acetate pendants are protonated and non-coordinated.41 With an excess of Cu(II), a polymeric material with Cu2L stoichiometry is formed in the solid state.42 The d–d spectrum of [Cu(dotp)]6− (λmax ≈ 650 nm) is not changed upon the addition of 1–3 protons. The spectrum of the [Cu(H4dotp)]2− species is blue shifted (λmax 628 nm). This could be caused by de-coordination of the axially bound phosphonate group upon the fourth protonation. In the solid state, the [Cu(H2O)(H4dotp)]2− species has a Cu(II) ion in the N4-equatorial plane of a square-pyramid with a water molecule in the axial position and all phosphonate groups are monoprotonated and non-coordinated.19 This structure was expected in acidic solutions from the UV-Vis (above) and EPR data32 of the Cu(II)–H8dotp system as they are similar to those of the [Cu(H2O)(cyclen)]2+ complex.43 The UV band is changed with the complex protonation only in the Cu(II)–H4dota system. Here, the N → Cu LMCT band (λmax ≈ 350 nm) intensity is decreased and the O → Cu LMCT band (λmax ≈ 290 nm) increased due to the lower electron density of the amine groups upon protonation of the non-coordinated carboxylates.
An intermediate behaviour was observed for the Cu(II)–trans-H6do2a2p system (Fig. S2†), whereby the UV/Vis spectrum of the fully deprotonated [Cu(do2a2p)]4− anion resembles that of the [Cu(dotp)]6− species, suggesting the same solution structure. For the [Cu(H2do2a2p)]4− anion (monoprotonated on each phosphonate group; see also below), the spectrum is very similar to that of the [Cu(dota)]2− where two acetates are bound to Cu(II). Therefore, the acetates should be coordinated to the Cu(II) ion and –PO3H− groups, but having a rather low metal-ion binding affinity,8 become free. The other protonated species may have different structures and/or they can be present in solution as a mixture of isomers.
Equilibrium studies of the Cu(II)–ligand systems have been published. The data seem to be reliable except those for the Cu(II)–H8dotp system (see below). The ligand/complex protonation/stability constants for the Cu(II)–ligand systems are given in ESI (Tables S2 and S3†). Generally, complex stability is governed by ring amine basicity (defined as logβ2 = logK1 + logK2), which is increased with a higher number of phosphonic acid pendant arms (Fig. S9†).8 However, the Cu(II)–H8dotp constants33,44,45 do not fit well with this dependence and, therefore, they seem to be incorrect. The free ligand/complex phosphonate groups become monoprotonated in the pH range 5–8, while the carboxylates could be protonated in more acidic solutions. Copper(II) complexes of all the ligands are highly thermodynamically stable, as shown by logKCuL > 20. With Cu(II) excess, weak dinuclear complexes might be formed, whereby uncoordinated pendant arms probably bind the second metal ion (an out-of-cage complexation) and this ability is increasingly pronounced for the ligands with more phosphonate groups (Fig. S10†). The complex species protonated on uncoordinated pendant arms are supposed to be fully thermodynamically stable in decomplexation kinetic experiments (see below) and the kinetically active species should only be those protonated on coordinated pendant arms. However in the Cu(II) complexes of such macrocyclic ligands, even diprotonated phosphonate or protonated carboxylate groups were found to be still coordinated to a central metal ion in the solid state.20,46
Cu(II)–H8dotp system
The stability constants for the Cu(II)–H8dotp system were re-determined as the values in the literature seem to be incorrect (see above). Experimental details on the determination and the found overall protonation/stability constants (Table S4†) as well as the complex species distribution diagrams for the L:Cu 1:1 and 1:2 molar ratios (Fig. S11) are given in ESI.†
First, the H8dotp protonation constants were determined under our experimental conditions (logK2 − logK7: 12.75, 9.20, 8.01, 6.03, 5.18 and 1.74, respectively), and they were in excellent agreement with the published values.47b For treatment of the titration and kinetic data, the first protonation constant (logK1 14.65) was taken from the literature47b as the protonation takes place outside of the potentiometric range. As complexes are almost quantitatively formed even in very acidic solutions (Fig. S11†), the titrations had to start at pH ≈ 1.4. The value of the stability constant obtained for the [Cu(dotp)]6− complex (logK1(CuL) 29.93) is several orders of magnitude higher than the published value44 but fits well into the correlation mentioned above (Fig. S9†). The difference can be attributed to incorrect determination of the first ligand protonation constant in the original paper44 as there are known problems with the determination of the H8dotp protonation constants.47 The first dissociation constant of the complex (pKa4(CuH4L) 4.67; Table S5†) is much lower than that of the other ones (pKa3–1(CuH3–1L) 6.43, 7.20 and 7.96, respectively) and its value is also lower than the corresponding constant in the free ligand. This indirectly points to metal-induced deprotonation of one –PO3H− group in the free ligand connected with its coordination.
With an excess of metal ions, dinuclear complexes are very easily formed (Fig. S7 and S10†). In the pH region 3–5, their formation is slow (waiting time 2–2.5 min from each titration point) but still measurable by standard potentiometry. Protonated complexes as well as hydroxido-species with a Cu:L molar ratio of 2:1 had to be involved in the chemical model. The structure of the [Cu2(Hndotp)](8−n)− (n = 0–2) species is probably not different from that of the [Cu(Hndotp)]6−n (n = 0–4) complexes as the d–d transition λmax of the dinuclear complexes are only slightly red shifted (to λmax ≈ 660 nm). Their formation is accompanied by a solution colour change from deep blue to light blue due to the decrease in the absorption coefficients (Table S1†). The UV spectra of the dinuclear species (Fig. S8†) differ from those of the monomeric species. Bands observed at λmax ≈ 225/≈330 nm for the dimeric species can be assigned to O → Cu/N → Cu LMCT bands analogously, as also suggested for the Cu(II) complexes of open-chain imino-bis(methylenephosphonic acid) ligands.48 Similar observations have also been noticed for different Cu:L ratios in the Cu(II)–H4dota system41 (see above). Thus, the dinuclear complexes of H8dotp should contain one in-cage bound Cu(II) ion, while the other one should interact only with oxygen atoms of non-coordinated phosphonate pendant arms. This is also supported by the values of the stepwise equilibrium constants for binding the second Cu(II) ion to the mononuclear complexes (logK2(Cu2L) 5.7–9.3, Table S5†). These values are similar to the values of the formation constants of monoprotonated Cu(II) complexes of simple imino-bis(methylenephosphonates), logK 6.7–10.7, where only oxygen atoms are coordinated.49 The stability constants of all the Cu2L complexes are reasonably correlated with the protonation constants of the uncoordinated pendant arms of the CuL complexes (Fig. S10†). This also indirectly supports the hypothesis that the second Cu(II) ion is bound by pendant arms only.
Formation kinetics
To follow the influence of the number and nature of pendant arms on the Cu(II) complex formation rate, a formation kinetics study was performed with trans-H2do2a, H4dota, trans-H6do2a2p, H7doa3p and H8dotp, where the number of pendant arms was changed (trans-H2do2a vs. H4dota) and the number of phosphonic acids arms was increased (from H4dota to H8dotp). The formation of Cu(II) complexes takes place in a millisecond-to-hour timescale depending on the pH, leading the second-order formation constant to change by over five orders of magnitude (Fig. 2, S12 and S13†). Thus, both conventional (pH 1–3; Fig. S14–S19†) and stopped-flow (pH > 3; Fig. 2, S12 and S13†) techniques had to be employed.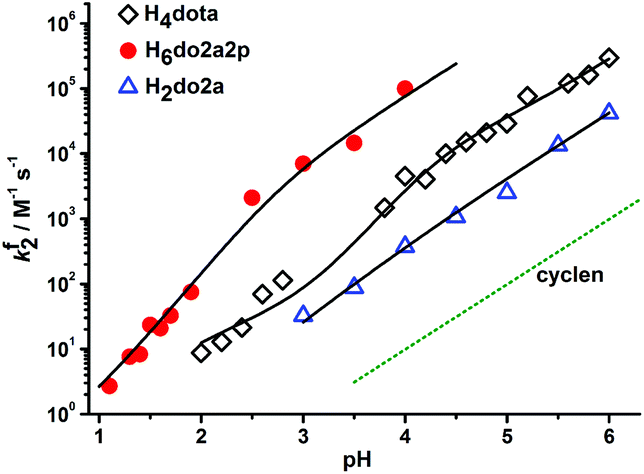 | ||
Fig. 2 Dependence of the second-order formation rate constants, fk2, for the Cu(II) complexes of H4dota (◊), trans-H6do2a2p ( ) and trans-H2do2a ( ) and trans-H2do2a ( ) on pH. The full-line curves were fitted through the experimental data according to eqn (1) and with the parameters given in Table 1. The curve for cyclen (dotted) was simulated on the basis of literature data.50 ) on pH. The full-line curves were fitted through the experimental data according to eqn (1) and with the parameters given in Table 1. The curve for cyclen (dotted) was simulated on the basis of literature data.50 | ||
Differently protonated ligand species, (HnL)n−m (m = 4 and n = 2–5 for H4dota, m = 6 and n = 2–6 for trans-H6do2a2p, m = 7 and n = 4–5 for H7doa3p, and m = 8 and n = 5–7 for H8dotp) are present in the pH range 1–6 used for the investigations (Fig. S1, S3 and S5†) and they can participate in complexation reaction with Cu(II) ion to form the final in-cage complexes, [Cu(HnL)]x− (Scheme 1). These, as well as less protonated ligand species, were involved in the fitting of the experimental data. No reverse reaction could be detected under the used experimental conditions (Fig. S15, S17 and S19†). The particular complex-forming reaction involving the protonated ligand species, (HnL)x−, was characterized by the partial second-order rate constant, fkHnL, where these are related to the observed second-order rate constant fk2 through eqn (1):
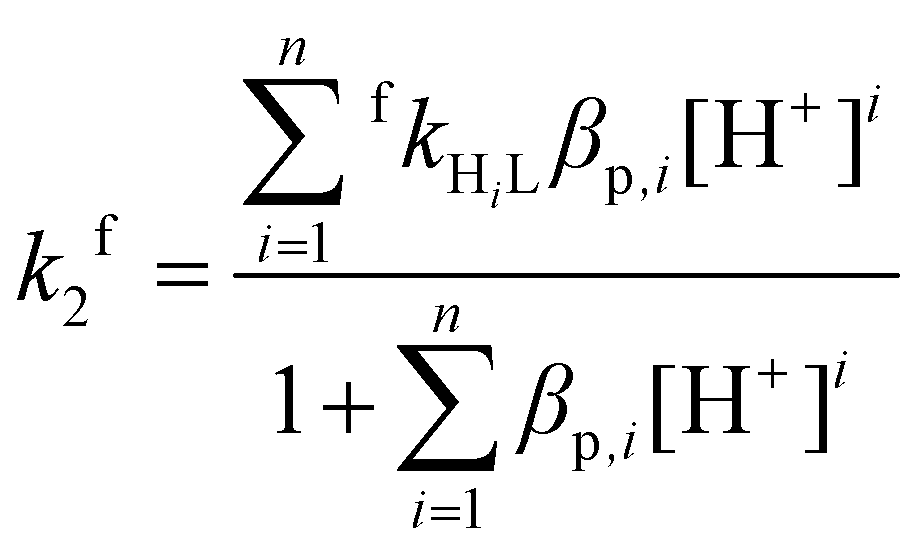 | (1) |
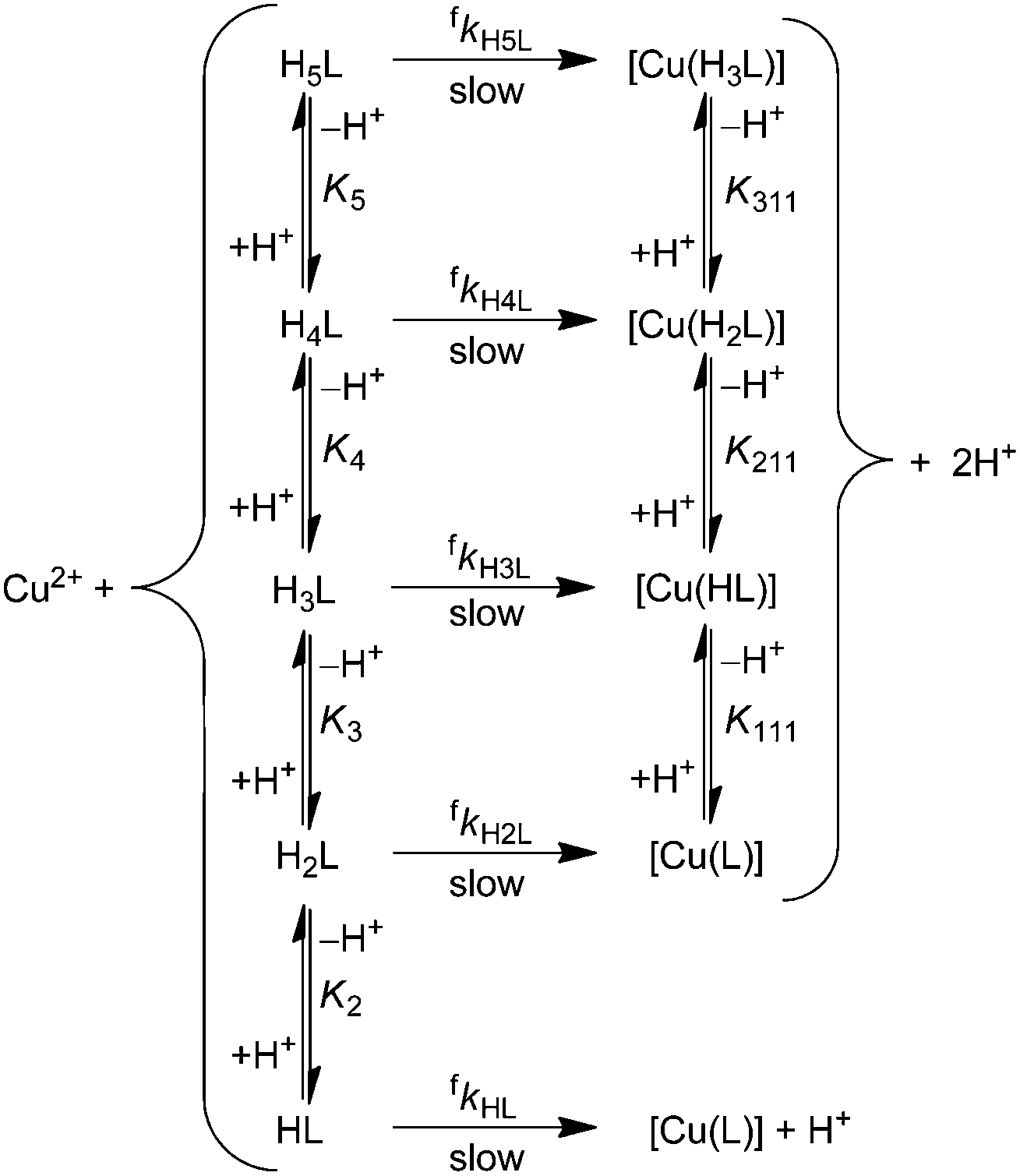 | ||
| Scheme 1 Reaction scheme proposed for the formation of Cu(II) complexes with H4dota, trans-H6do2a2p, H7doa3p or H8dotp; the equilibrium constants K are stepwise ligand/complex protonation constants. Charges of the ligand/complex species are omitted for the sake of clarity. | ||
| Rate constant, M−1 s−1 | Cyclena | trans-H2do2a | H4dota | trans-H6do2a2p | H7doa3p | H8dotp |
|---|---|---|---|---|---|---|
| a Ref. 50 and 52. | ||||||
| f k H5L | — | — | — | 50(3) | 1.60(1) × 103 | 6.50(14) × 104 |
| f k H4L | — | — | 36.9(3) | 9.0(7) × 103 | 3.45(9) × 106 | 3.00(2) × 106 |
| f k H3L | — | — | 7.64(20) × 102 | 1.70(6) × 107 | — | — |
| f k H2L | 0.18–0.6 | 3.2(7) × 102 | 2.32(19) × 104 | — | — | — |
| f k HL | (1.84–4.3) × 106 | 1.47(2) × 108 | 1.43(28) × 109 | — | — | — |
| f k H4L/fkH5L | — | — | — | 180 | 2150 | 46 |
| f k H3L/fkH4L | — | — | 20.7 | 1880 | — | — |
| f k H2L/fkH3L | — | — | 30.3 | — | — | — |
| f k HL/fkH2L | — | 4.55 × 105 | 6.16 × 104 | — | — | — |
As can be observed in Fig. 2 (cyclen vs. trans-H2do2a/H4dota/trans-H6do2a2p), the presence of pendant arms significantly accelerates formation of the Cu(II) complexes and this effect is observable throughout whole pH range. According to Fig. S12 and S13,† the most reactive ligands are trans-H6do2a2p and H7do3ap, which react about ten times faster than H8dotp and 10–100 times faster than H4dota in the pH range 3–5. However, H4dota and H8dotp react similarly fast at pH 1–2. It is commonly accepted that macrocyclic ligands with coordinating pendant arms bind any metal ions in a two-step mechanism: formation of an out-of-cage complex, [Cu(HmL)]*, where mostly pendant arms are coordinated, followed by a rate-determining step, where the proton from the ring amine group(s) is transferred to the bulk solvent and the in-cage complex is simultaneously formed. This mechanism has also been suggested for the formation of Cu(II) complexes with H3do3a51 (Fig. 1) and H4dota.16 As the rate-determining step is ring amine deprotonation, it is useful to compare the reactivity with species with double protonated ring nitrogen atoms and with the same number of protonated pendant arms. This could be done as the –PO3H− group can be stably coordinated to the central atom, similarly to the deprotonated carboxylic group. The reactivity of e.g. (H2dota)2− and (trans-H4do2a2p)2− species, which differ only in the protonation state of the phosphonates, is similar, unlike the reactivity of species with one additional protonated carboxylic group, e.g. (H3dota)− and (trans-H5do2a2p)−. The reactivity is distinctly enhanced (≈3 orders of magnitude; Table 2) once a basic fully deprotonated phosphonate group is present, e.g. see the comparison of (trans-H4do2a2p)2− with (trans-H3do2a2p)3− or (H4doa3p)3−. Considering the most distant members of the ligand series, the (H4dotp)4− species with two monoprotonated phosphonates is as reactive as the (H2dota)2− species with fully deprotonated pendant arms, despite the highest basicity of the ring amine groups in H8dotp. This confirms again the importance of the strong interaction of fully deprotonated phosphonates with the metal ion in the supposed out-of-cage intermediate and/or the better ability of the phosphonates to transfer a proton from the ring amines to the bulk solution. Once the ring amines are only monoprotonated, the reactivity of such species is greatly increased, e.g. the (Htrans-do2a)− or (Hdota)3− anions react much faster than the (H2trans-do2a) or (H2dota)2− species (fkHL/fkH2L = 455000 and 61600, respectively).
| Acidity | trans-H2do2a | H4dota | trans-H6do2a2p | H7doa3p | H8dotp |
|---|---|---|---|---|---|
| pH 6.0 (in ms) | 110 | 15.5 | 0.98 | 1.86 | 3.80 |
| pH 4.0 (in s) | 12.3 | 1.02 | 0.046 | 0.0341 | 0.84 |
| pH 2.0 (in min) | 77.3 | 8.82 | 0.53 | 0.21 | 1.33 |
At most pH values in the studied pH range, complex formation is accelerated with the increasing number of phosphonic acid pendant arms (Table 2). This could be explained by the acid–base and coordination behaviour of the cyclic amines and phosphonic acid group. The phosphonic acid group is more acidic than the carboxylic group and, thus, phosphonates are able to bind metal ions in more acidic solutions. At lower pH, the complexation rate is less decreased for phosphonate-containing ligands. On the other hand, the fully deprotonated phosphonate group is more basic than the carboxylate group and interacts with metal ions more efficiently. Full deprotonation of the phosphonate group(s) also increases the ligand's overall negative charge, thus facilitating the ligand-metal ion electrostatic interactions. With an increasing number of phosphonic acid pendants, the stability of an intermediate out-of-cage complex is increased and this leads to a faster overall complex formation (Table 2). However, this trend could be offset by the rather high basicity of the ring amine groups as well as a too high stability of the intermediate out-of-cage complex, e.g. due to the presence too many well-complexing pendant arms, such as phosphonates. This probably becomes important during Cu(II) complexation with e.g. H8dotp, as the complex is formed more slowly than the complexes with its analogues. In the diprotonated macrocyclic ligand species, the protons are shared among ring amines and the access of a metal ion into the ligand cavity is blocked. Once the first ring amine proton is removed, in-cage complex formation is greatly accelerated. As phosphonates are known to form rather strong hydrogen bonds, they efficiently assist proton removal from the ligand cavity. Here, the difference in reactivity between H2L and HL species (with two or one proton(s) on the ring amine, respectively) of the phosphonate-containing ligands could not be investigated; however, the high reactivity of their even more protonated ligand species indirectly points to their ability to transfer the proton(s). A similar complexation rate enhancement has already been observed for the reaction of Cu(II) with methylphosphonic acid cyclam derivatives.20 There are clear dependences of the partial rate constants on a number of phosphonic acid groups in the ligands or on the values of their first protonation constants (Fig. S20 and S21†).
Decomplexation kinetics
The decomplexation kinetics of the Cu(II) complexes was studied under acid catalysis in order to follow this process in a reasonable time. NaClO4 was used as a background electrolyte as we have recently shown that nitrate anions, other commonly considered ‘inert’ anions, can interact with macrocyclic ligands and/or complexes.23 Examples of data for the dissociation of Cu(II)–trans-H6do2a2p/trans-H2do2a complexes are shown in Fig. 3 (for more data, see Fig. S23–S27†). | ||
| Fig. 3 Pseudo-first-order rate constants as a function of proton concentration for acid-assisted decomplexation of the [Cu(trans-do2a2p)]4− (A) and [Cu(trans-do2a)] (B) complexes (I = 5.0 M (Na,H)ClO4). | ||
As given above, some complex species protonated on the pendant arm(s) are thermodynamically stable and, thus, kinetically inactive. Therefore, additional proton(s) have to be bound to the complex species to promote their dissociation. On the basis of the kinetics data for the complexes of a number of macrocyclic ligands, the dissociation mechanism in Scheme 2 can be postulated, which leads to eqn (2):
 | (2) |
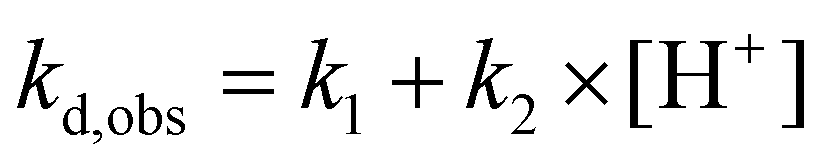 | (3) |
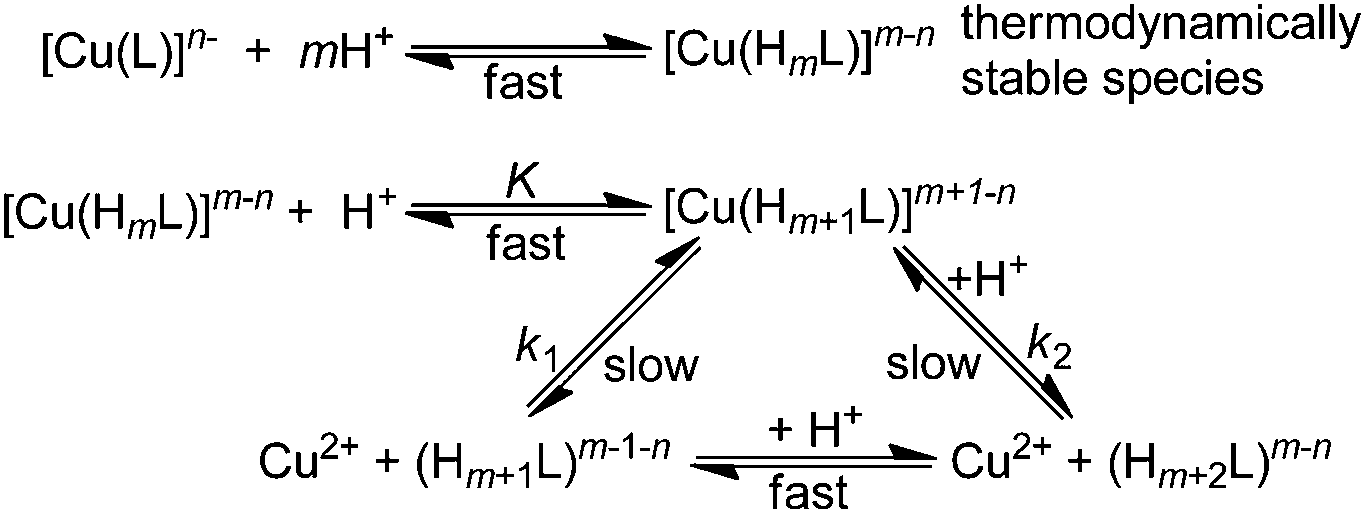 | ||
| Scheme 2 Proposed reaction scheme for acid-assisted decomplexation of the Cu(II) complexes. The charges of the ligand/complex species are omitted for clarity and a number of protons in the scheme (m) vary for different ligands. | ||
Linear dependence (eqn (3)) was found for complexes of the octadentate ligands (H4dota–H8dotp), while eqn (2) had to be used for the trans-H2do2a complex (Fig. 3). The protonation constant for formation of the reaction intermediate could be calculated only for the trans-H2do2a complex. The complete set of results is shown in ESI (Table S5†). In addition, the dependences of the rate constants k1 and k2 on temperature were used to estimate the activation parameters for each reaction pathway, and the results are given in Table 3.
| Parameter | Rate constant | ΔH≠ kJ mol−1 | ΔS≠ J mol−1 K−1 |
|---|---|---|---|
| a Taken from ref. 18 for ligand structures, see Fig. 1. b Extrapolated from the temperature dependence using the activation/thermodynamic parameters. | |||
| H4dotaa | |||
| k 1/10−5, s−1 | 0.584 | 65 | −128 |
| k 2/10−5, M−1 s−1 | 0.0206 | 107 | −15 |
| k 2/k1, M−1 | 0.035 | ||
| τ 1/2 (pH 2), h | 33.0 | ||
| H5do3apa | |||
| k 1/10−5, s−1 | 7.84 | 66(1) | −101(4) |
| k 2/10−5, M−1 s−1 | 0.43 | 105(2) | −3(5) |
| k 2/k1, M−1 | 0.055 | ||
| τ 1/2 (pH 2), h | 2.45 | ||
| trans-H6do2a2p | |||
| k 1/10−3 s−1 | 0.411(8) | 73(1) | −64(4) |
| k 2/10−3 M−1 s−1 | 0.173(3) | 62(3) | −110(10) |
| k 2/k1, M−1 | 0.42(1) | ||
| τ 1/2 (pH 2), h | 0.47 | ||
| H7doa3p | |||
| k 1/10−3, s−1 | 1.51(4) | 80(2) | −32(8) |
| k 2/10−3, M−1 s−1 | 0.87(1) | 74(5) | −55(16) |
| k 2/k1, M−1 | 0.58(2) | ||
| τ 1/2 (pH 2), min | 7.60 | ||
| H8dotp | |||
| k 1/10−3, s−1 | 4.33(7) | 72(3) | −50(9) |
| k 2/10−3, M−1 s−1 | 1.12(3) | 75(5) | −50(16) |
| k 2/k1, M−1 | 0.26(1) | ||
| τ 1/2 (pH 2), min | 2.70 | ||
| trans-H2do2a | |||
| k 1/10−5, s−1 | 0.605 | 68(5) | −117(16) |
| k 2/10−5, M−1 s−1 | 0.0563 | 78(2) | −102(6) |
| k 2/k1, M−1 | 0.093 | ||
| logK |
1.35 | −8 | 2 |
| τ 1/2 (pH 2), h | 31.5 | ||
| H3do3aa | |||
| k 1/10−5, s−1 | 0.907 | 49 | −178 |
| k 2/10−5, M−1 s−1 | 0.168 | 73 | −110 |
| k 2/k1, M−1 | 0.093 | ||
| τ 1/2 (pH 2), h | 21.2 | ||
To compare the effect of an increasing number of phosphonate pendant arms in the ligand series on the kinetic inertness of their Cu(II) complexes, dkobsvs. [H+] plots were compared (Fig. 4 and S28†). The most kinetically inert is the Cu(II)–H4dota complex, while the inertness of the Cu(II) complexes is monotonously decreased with the higher number of phosphonic acid groups at any solution acidity. The constants k1 and k2, representing both reaction pathways, were 104- and 103 times higher for H8dotp than for H4dota, respectively (Fig. 5). Acceleration of the acid-assisted decomplexation of the Cu(II) complexes is more pronounced in the H4dota–H5do3ap–trans-H6do2a2p series than for H7doa3p/H8dotp with more phosphonic acid pendants (Fig. 4 and 5). In addition, direct proton attack (k2 pathway) is slower than the simple proton transfer pathway (k1 pathway), and this difference is more significant for H4dota than for H8dotp (Table 3 and Fig. 5).
 | ||
| Fig. 4 Comparison of the decomplexation rate constants, dkobs, of the studied Cu(II) complexes (25 °C, I = 5.0 M (Na,H)ClO4). Lines for H3do3a, H4dota and H5do3ap are based on data from ref. 18. | ||
 | ||
| Fig. 5 Dependence of the rate constants, k1 and k2, for the acid-assisted dissociation of Cu(II) complexes on a number of phosphonic acid groups (nP) in H4dota and its analogues as obtained at 25 °C. The experimental data for H4dota and H5do3ap complexes were taken from ref. 18. | ||
The k1 and k2 rate constant values could be correlated (Fig. S29 and S30†) with the ring amine basicity of the ligands (logK2) as well as with the overall protonation constants of the CuL complexes (logβp(CuL)) as these values represent the capability of the Cu(II) complexes to be protonated on the free pendant arms. Both dependences indirectly prove that proton transfer from protonated phosphonate group(s) to the ring amines (k1) is more important with an increased number of phosphonates, as well as that direct proton attack (k2) can be mediated by the phosphonate pendants through the formation of hydrogen bonds between an arm oxygen atom and a ring nitrogen atom. Direct proton attack (k2 pathway) seems to be more sensitive to the ligand and/or Cu(II) complex basicity than proton transfer (k1 pathway). In addition, phosphonate groups are also able to interact with metal ions even in acidic solutions.20a,46,54,55 Therefore, an increasing number of phosphonate groups assists better in removal of the metal ion from the ligand cavity. Isokinetic plots (Fig. S31†) demonstrate the analogous reaction mechanism for the k1 pathway, while the trend for the k2 pathway is different for the decomplexation of the Cu(II)–H4dota and Cu(II)–H5do3ap complexes. Overall, the kinetic inertness expressed as decomplexation half-times (Table 3) significantly decreases with the increasing number of phosphonate groups.
The H4dota-like ligands have four pendant arms, but only one or two of them can be coordinated in the in-cage Cu(II) complexes. To investigate the influence of the presence of an “excess” of pendant arms, the dissociation kinetics of the Cu(II)–trans-H2do2a complex (Fig. 3) was also studied. The high kinetic inertness (τ1/2 ≈ 20–30 h at pH 2) is retained in the trans-H2do2a–H3do3a–H4dota series, since both k1 and k2 parameters are almost the same. This highlights that the presence of non-coordinated carboxylates does not have an important effect on the acid-assisted decomplexation. On the other hand, the kinetic inertness decreases by one order or two orders of magnitude for the H3do3a/H5do3ap and trans-H2do2a/H6do2a2p pairs, respectively, due to the presence of phosphonate groups.
There are some published dissociation kinetic data for Cu(II) complexes of other cyclen derivatives, e.g. the complex of N,N′,N′′,N′′′-tetrakis(2 hydroxoethyl)cyclen with weakly coordinating alcohol pendant arms is very kinetically inert,56 while complexes of tetrakis(phosphinate) cyclen derivatives are similarly as labile as the Cu(II)–H8dotp complex as the phosphinate groups are able to bind the metal ion in acidic solutions.21,22
Solid-state structure of KH6doa3p·3H2O
A single crystal of KH6doa3p·3H2O was obtained by slow evaporation of the solvent from an acidic aqueous solution of H7doa3p. The experimental and fitting details are given in Table S6.† The molecular structure of the ligand anion is shown in Fig. 6 and the crystal packing in Fig. S32,† while the most relevant bond lengths and angles of the ligand anion are presented in Table S7.† | ||
| Fig. 6 Molecular structure of (H6doa3p)− anion as found in the solid-state structure of KH6doa3p·3H2O. Intramolecular hydrogen bonds are shown as dashed lines. | ||
In the structure of the (H6doa3p)− anion, two ring nitrogen atoms are protonated, and each pendant arm is monoprotonated on its oxygen atom. The protonated oxygen atoms are clearly distinguished through the bond lengths (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O 1.208 Å vs. C–OH 1.313 Å, and P–O 1.48–1.53 Å vs. P–OH 1.55–1.59 Å; Table S7†). The ring nitrogen atoms are almost coplanar and all the pendant arms are oriented to the same side of the plane. Two phosphonate pendant arms attached to the non-protonated nitrogen atoms are positioned just above the N4-plane. They are connected by a strong hydrogen bond (O1–H⋯O7: d(D–A) 2.51 Å, ∠(D–H-A) 162°) and are involved in the intramolecular hydrogen bond network (Fig. 6 and Table S7†). One of the phosphonate groups forms a medium-strong hydrogen bond to a protonated ring amine adjacent over the ethylene chain (N10–H⋯O7: d(D–A) 2.93 Å, ∠(D–H-A) 145°) and this causes asymmetry in the network of hydrogen bonds between the ring amine groups (d(D–A) 2.84–3.9 Å). Such an arrangement has been found in zwitterionic H8dotp,57 where the ring nitrogen and pendant arm protonation scheme as well as connectivity are the same; thus, the intramolecular hydrogen bond parameters are analogous. Different arrangements were found in the zwitterionic forms of H5do3ap26 and trans-H6do2a2p,28 where the protonated nitrogen atom bears a monoprotonated phosphonate group, which is involved in an intramolecular hydrogen bond with this >NH+ group, while the other pendants are directed outside. The other pendant arms of the (H6doa3p)− anion are bound to the protonated nitrogen atoms and are oriented out of the macrocycle ring. All the pendant arms form a complex system of strong intermolecular hydrogen bonds (d(D–A) 2.47–2.68 Å, ∠(D–H-A) 166–174°; Fig. S32 and Table S7†). This leads to double-chains of ligand molecules linked by the strong hydrogen bonds interconnected through K+ cation layers.
O 1.208 Å vs. C–OH 1.313 Å, and P–O 1.48–1.53 Å vs. P–OH 1.55–1.59 Å; Table S7†). The ring nitrogen atoms are almost coplanar and all the pendant arms are oriented to the same side of the plane. Two phosphonate pendant arms attached to the non-protonated nitrogen atoms are positioned just above the N4-plane. They are connected by a strong hydrogen bond (O1–H⋯O7: d(D–A) 2.51 Å, ∠(D–H-A) 162°) and are involved in the intramolecular hydrogen bond network (Fig. 6 and Table S7†). One of the phosphonate groups forms a medium-strong hydrogen bond to a protonated ring amine adjacent over the ethylene chain (N10–H⋯O7: d(D–A) 2.93 Å, ∠(D–H-A) 145°) and this causes asymmetry in the network of hydrogen bonds between the ring amine groups (d(D–A) 2.84–3.9 Å). Such an arrangement has been found in zwitterionic H8dotp,57 where the ring nitrogen and pendant arm protonation scheme as well as connectivity are the same; thus, the intramolecular hydrogen bond parameters are analogous. Different arrangements were found in the zwitterionic forms of H5do3ap26 and trans-H6do2a2p,28 where the protonated nitrogen atom bears a monoprotonated phosphonate group, which is involved in an intramolecular hydrogen bond with this >NH+ group, while the other pendants are directed outside. The other pendant arms of the (H6doa3p)− anion are bound to the protonated nitrogen atoms and are oriented out of the macrocycle ring. All the pendant arms form a complex system of strong intermolecular hydrogen bonds (d(D–A) 2.47–2.68 Å, ∠(D–H-A) 166–174°; Fig. S32 and Table S7†). This leads to double-chains of ligand molecules linked by the strong hydrogen bonds interconnected through K+ cation layers.
Conclusions
We showed that the reactivity of H4dota-like ligands towards the Cu(II) ion is significantly altered after substitution of the acetate pendant arms(s) by methylphosphonic acid one(s). This can be explained by the easier formation of an out-of-cage intermediate24 due to the higher charge and high complexing ability of the deprotonated phosphonate groups and/or easier transfer of proton(s) from the ring amine group(s) to the bulk solvent assisted by the phosphonates. The rate of decomplexation of the Cu(II) complexes is also highly influenced by the presence of phosphonates since they probably affect the transfer of proton(s) from both the solvent and the already protonated pendant arms into the ligand cavity. In addition, the basicity of the amine groups is increased with the number of phosphonic acid pendant arms and, therefore, the amino groups are more prone to bind proton(s). Stability constants of the Cu(II)–H8dotp system reported in the literature were re-evaluated. The stability constant of the [Cu(dotp)]5− complex is one the highest determined till now and is due to the very high ligand basicity and stable dinuclear complexes, where the additional Cu(II) ion is bound to non-coordinated phosphonate groups that are formed even in slightly acidic solutions.The results in the present study can be utilized in the design of ligands for the complexation of copper radioisotopes. These and other published results indicate that the presence of phosphonic acid group(s) increases the Cu(II) complexation rates of both cyclen and cyclam derivatives. However, their influence on decomplexation rates is less clear. Here, the presence of phosphonic acid groups highly facilitates decomplexation. Unlike complexes of the title cyclen derivatives, Cu(II) complexes of the cyclam methylphosphonic acid derivatives (e.g. 1,8-H4te2p; Fig. 1) with not fully substituted ring nitrogen atoms are kinetically inert, often even more so than the complexes of the analogous ligands with acetic acid pendants. However as shown here, substitution of the cyclen skeleton with methylphosphonic acids is not a suitable way to obtain good ligands for copper radioisotopes.
Acknowledgements
We are grateful for support to the Grant Agency of the Czech Republic (13-08336S), the Ministry of Education of the Czech Republic (MSM0021620857, MUNI/A/1500/2015), C2TN (UID/Multi/04349/2013), European Union (ERASMUS and CEITEC 2020, grant no. LQ1601) and GRICES (travelling grant to PL). We also thank to Prof. Manuel Martinéz (Department of Inorganic Chemistry, Universitat de Barcelona, Spain) for the opportunity to carry out some initial experiments on a stopped-flow apparatus.References
- (a) The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Chichester, 2nd edn, 2013 Search PubMed; (b) P. Hermann, J. Kotek, V. Kubíček and I. Lukeš, Dalton Trans., 2008, 3027–3047 RSC; (c) C. F. G. C. Geraldes and S. Laurent, Contrast Media Mol. Imaging, 2009, 4, 1–23 CrossRef CAS PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- M. Shokeen and C. J. Anderson, Acc. Chem. Res., 2009, 42, 832–841 CrossRef CAS PubMed.
- (a) C. S. Cutler, H. M. Hemmkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS PubMed; (b) C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC; (c) E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC.
- (a) S. Liu, Adv. Drug Delivery Rev., 2008, 60, 1347–1370 CrossRef CAS PubMed; (b) L. Lattuada, A. Barge, G. Cravotto, G. B. Giovenzana and L. Tei, Chem. Soc. Rev., 2011, 40, 3019–3049 RSC; (c) L. Frullano and P. Caravan, Curr. Org. Synth., 2011, 8, 535–565 CrossRef CAS PubMed.
- E. Brücher, G. Tircsó, Z. Baranyai, Z. Kovács and A. D. Sherry, in The Chemistry of Contrast Agents in Medical Magnetic Resonance Imaging, ed. A. Merbach, L. Helm and É. Tóth, Wiley, Chichester, U.K., 2nd edn, 2013, pp. 157–208 Search PubMed.
- (a) C. J. Anderson and R. Ferdani, Cancer Biother. Radiopharm., 2009, 24, 379–393 CrossRef CAS PubMed; (b) T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed; (c) B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168–6195 RSC; (d) M. T. Ma and P. S. Donnelly, Curr. Top. Med. Chem., 2011, 11, 500–520 CrossRef CAS PubMed.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216–217, 287–312 CrossRef.
- R. Delgado, V. Félix, L. M. P. Lima and D. W. Price, Dalton Trans., 2007, 2734–2745 RSC.
- (a) J. Rudovský, J. Kotek, P. Hermann, I. Lukeš, V. Mainero and S. Aime, Org. Biomol. Chem., 2005, 3, 112–117 RSC; (b) P. Řezanka, V. Kubíček, P. Hermann and I. Lukeš, Synthesis, 2008, 1431–1435 Search PubMed.
- M. Försterová, I. Svobodová, P. Lubal, P. Táborský, J. Kotek, P. Hermann and I. Lukeš, Dalton Trans., 2007, 535–549 RSC.
- (a) J. Notni, P. Hermann, J. Havlíčková, J. Kotek, V. Kubíček, J. Plutnar, N. Loktionova, P. J. Riss, F. Rösch and I. Lukeš, Chem. – Eur. J., 2010, 16, 7174–7185 CrossRef CAS PubMed; (b) J. Šimeček, J. Notni, O. Zemek, P. Hermann and H.-J. Wester, ChemMedChem, 2012, 7, 1375–1378 CrossRef PubMed.
- M. Paúrová, J. Havlíčková, A. Pospíšilová, M. Vetrík, I. Císařová, H. Stephan, H.-J. Pietzsch, M. Hrubý, P. Hermann and J. Kotek, Chem. – Eur. J., 2015, 21, 4671–4687 CrossRef PubMed.
- (a) L. M. P. Lima, D. Esteban-Gomez, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2012, 51, 6916–6927 CrossRef CAS PubMed; (b) L. M. P. Lima, Z. Halime, R. Marion, N. Camus, R. Delgado, C. Platas-Iglesias and R. Tripier, Inorg. Chem., 2014, 53, 5269–5279 CrossRef CAS PubMed; (c) A. Rodriguez-Rodriguez, Z. Garda, E. Ruscsák, D. Esteban-Gómez, A. de Blas, T. Rodríguez-Blas, L. M. P. Lima, M. Beyler, R. Tripier, G. Tircsó and C. Platas-Iglesias, Dalton Trans., 2015, 44, 5017–5031 RSC.
- A. Rodríguez-Rodríguez, Z. Halime, L. M. P. Lima, M. Beyler, D. Deniaud, N. Le Poul, R. Delgado, C. Platas-Iglesias, V. Patinec and R. Tripier, Inorg. Chem., 2016, 55, 619–632 CrossRef PubMed.
- S. P. Kasprzyk and R. G. Wilkins, Inorg. Chem., 1982, 21, 3349–3352 CrossRef CAS.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. J. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 4829–4833 CrossRef CAS.
- I. Voráčová, J. Vaněk, J. Pasulka, Z. Střelcová, P. Lubal and P. Hermann, Polyhedron, 2013, 61, 99–104 CrossRef.
- (a) L. M. P. Lima, C. V. Esteves, R. Delgado, P. Hermann, J. Kotek, R. Ševčíková and P. Lubal, Eur. J. Inorg. Chem., 2012, 2533–2547 CrossRef CAS.
- (a) J. Kotek, P. Lubal, P. Hermann, I. Císařová, I. Lukeš, T. Godula, I. Svobodová, P. Táborský and J. Havel, Chem. – Eur. J., 2003, 9, 233–248 CrossRef CAS PubMed; (b) I. Svobodová, P. Lubal, J. Plutnar, J. Kotek, J. Havlíčková, P. Hermann and I. Lukeš, Dalton Trans., 2006, 5184–5197 RSC; (c) I. Svobodová, J. Havlíčková, J. Plutnar, P. Lubal, J. Kotek and P. Hermann, Eur. J. Inorg. Chem., 2009, 3577–3592 CrossRef.
- P. Lubal, M. Kývala, P. Hermann, J. Holubová, J. Rohovec, J. Havel and I. Lukeš, Polyhedron, 2001, 20, 47–55 CrossRef CAS.
- R. Ševčík, J. Vaněk, P. Lubal, Z. Kotková, J. Kotek and P. Hermann, Polyhedron, 2014, 67, 449–455 CrossRef.
- T. David, V. Kubíček, O. Gutten, P. Lubal, J. Kotek, H.-J. Pietzsch, L. Rulíšek and P. Hermann, Inorg. Chem., 2015, 54, 11751–11766 CrossRef CAS PubMed.
- M. Frindel, N. Camus, A. Rauscher, M. Bourgeois, C. Alliot, L. Barré, J.-F. Gestin, R. Tripier and A. Faivre-Chauvet, Nucl. Med. Biol., 2014, 41, e49–e57 CrossRef CAS PubMed.
- (a) D. J. Stigers, R. Ferdani, G. R. Weisman, E. H. Wong, C. J. Anderson, J. A. Golen, C. Moore and A. L. Rheingold, Dalton Trans., 2010, 39, 1699–1701 RSC; (b) R. Ferdani, D. J. Stigers, A. L. Fiamengo, L. Wei, B. T. Y. Li, J. A. Golen, A. L. Rheingold, G. R. Weisman, E. H. Wong and C. J. Anderson, Dalton Trans., 2012, 41, 1938–1950 RSC; (c) Y. Guo, R. Ferdani and C. J. Anderson, Bioconjugate Chem., 2012, 23, 1470–1477 CrossRef CAS PubMed; (d) M. Jiang, R. Ferdani, M. Shokeen and C. J. Anderson, Nucl. Med. Biol., 2013, 40, 245–251 CrossRef CAS PubMed; (e) D. Zeng, Q. Ouyang, Z. Cai, X.-Q. Xie and C. J. Anderson, Chem. Commun., 2014, 50, 43–45 RSC; (f) Z. Cai, Q. Ouyang, D. Zeng, K. N. Nguyen, J. Modi, L. Wang, A. G. White, B. E. Rogers, X.-Q. Xie and C. J. Anderson, J. Med. Chem., 2014, 57, 6019–6029 CrossRef CAS PubMed; (g) Z. Cai, B. T. Y. Li, E. H. Wong, G. R. Weisman and C. J. Anderson, Dalton Trans., 2015, 44, 3945–3948 RSC; (h) N. Bhatt, N. Soni, Y. S. Ha, W. Lee, D. N. Pandya, S. Sarkar, J. Y. Kim, H. Lee, S. H. Kim, G. I. An and J. Yoo, ACS Med. Chem. Lett., 2015, 6, 1162–1166 CrossRef CAS PubMed.
- P. Táborský, P. Lubal, J. Havel, J. Kotek, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2005, 70, 1909–1942 CrossRef.
- (a) Z. Piskula, I. Svobodová, P. Lubal, S. Lis, Z. Hnatejko and P. Hermann, Inorg. Chim. Acta, 2007, 360, 3748–3755 CrossRef CAS; (b) P. Táborský, I. Svobodová, P. Lubal, Z. Hnatejko, S. Lis and P. Hermann, Polyhedron, 2007, 26, 4119–4130 CrossRef; (c) I. Svobodová, Z. Piskula, P. Lubal, S. Lis and P. Hermann, J. Alloys Compd., 2008, 451, 42–45 CrossRef.
- M. P. C. Campello, S. Lacerda, I. C. Santos, G. A. Pereira, C. F. G. C. Geraldes, J. Kotek, P. Hermann, J. Vaněk, P. Lubal, V. Kubíček, É. Tóth and I. Santos, Chem. – Eur. J., 2010, 16, 8446–8465 CrossRef CAS PubMed.
- L. M. P. Lima, R. Delgado, P. Hermann, R. Ševčík, P. Lubal, H. F. Carvalho, A. F. Martins, É. Tóth and C. F. G. C. Geraldes, Eur. J. Inorg. Chem., 2012, 2548–2559 CrossRef CAS.
- F. K. Kálmán, Z. Baranyai, I. Tóth, I. Bányai, R. Király, E. Brücher, S. Aime, X. Sun, A. D. Sherry and Z. Kovács, Inorg. Chem., 2008, 47, 3851–3862 CrossRef PubMed.
- M. P. C. Campello, M. Balbina, I. Santos, P. Lubal, R. Ševčíková and R. Ševčík, Helv. Chim. Acta, 2009, 92, 2398–2413 CrossRef CAS.
- C. F. G. C. Geraldes, M. P. M. Marques, B. de Astro and E. Pereira, Eur. J. Inorg. Chem., 2000, 559–565 CrossRef CAS.
- X. Sun, M. Wuest, Z. Kovacs, A. D. Sherry, R. Motekaitis, Z. Wang, A. E. Martell, M. J. Welch and C. J. Anderson, J. Biol. Inorg. Chem., 2003, 8, 217–225 CrossRef CAS PubMed.
- R. Delgado and J. J. R. F. da Silva, Talanta, 1982, 29, 815–822 CrossRef CAS PubMed.
- J. Rudovský, P. Cígler, J. Kotek, P. Hermann, P. Vojtíšek, I. Lukeš, J. A. Peters, L. V. Elst and R. N. Muller, Chem. – Eur. J., 2005, 11, 2375–2384 CrossRef PubMed.
- J. M. Weeks, M. R. Tailor and K. P. Wainwright, Dalton Trans., 1997, 317–322 RSC.
- (a) R. Přibyl, Analytical Applications of EDTA and Related Compounds, Pergamon Press, Oxford, 1972 Search PubMed; (b) G. Schwarzenbach and H. Flaschka, Complexometric Titrations, Methuen, London, 1969 Search PubMed.
- E. J. Billo, Excel for Chemists, Wiley-VCH, New York, 2001 Search PubMed.
- (a) M. Kývala and I. Lukeš, International Conference on Chemometrics ‘95, Pardubice, Czech Republic, 1995, p. 63; full version of “OPIUM” is available (free of charge) on http://www.natur.cuni.cz/~kyvala/opium.html; (b) M. Kývala, P. Lubal and I. Lukeš, IX. Spanish-Italian, and Mediterranean Congress on Thermodynamics of Metal Complexes (SIMEC 98), Girona, Spain, 1998.
- R. Delgado, J. J. R. Frausto da Silva and M. C. T. A. Vaz, Talanta, 1986, 33, 285–287 CrossRef CAS PubMed.
- A. Riesen, M. Zahnder and T. A. Kaden, Helv. Chim. Acta, 1986, 69, 2067–2073 CrossRef CAS.
- A. Riesen, M. Zehnder and T. A. Kaden, Helv. Chim. Acta, 1986, 69, 2074–2080 CrossRef CAS.
- M. C. Styka, R. C. Smierciak, E. L. Blinn, R. E. DeSimone and J. V. Passariello, Inorg. Chem., 1978, 17, 82–86 CrossRef CAS.
- I. M. Kabachnik, T. Ya. Medved, F. I. Belskii and S. A. Pisareva, Izv. Akad. Nauk SSSR, Ser. Khim., 1984, 844–849 CAS.
- T. Ya. Medved, M. I. Kabachnik, F. I. Belskii and S. A. Pisareva, Izv. Akad. Nauk SSSR, Ser. Khim., 1988, 2103–2107 CAS.
- S. Füzerová, J. Kotek, I. Císařová, P. Hermann, K. Binnemans and I. Lukeš, Dalton Trans., 2005, 2908–2915 RSC.
- (a) R. Delgado, L. C. Siegfried and T. A. Kaden, Helv. Chim. Acta, 1990, 73, 140–148 CrossRef CAS; (b) R. Delgado, J. Costa, K. P. Guerra and L. M. P. Lima, Pure Appl. Chem., 2005, 77, 569–579 CAS.
- B. Kurzak, A. Kamecka, K. Kurzak, J. Jezierska and P. Kafarski, Polyhedron, 1998, 17, 4403–4413 CrossRef CAS.
- K. Popov, H. Rönkkömäki and L. H. J. Lajunen, Pure Appl. Chem., 2001, 73, 1641–1677 CrossRef CAS.
- R. Ševčíková, P. Lubal, M. P. C. Campello and I. Santos, Polyhedron, 2013, 62, 268–273 CrossRef.
- H.-Z. Cai and T. A. Kaden, Helv. Chim. Acta, 1994, 77, 383–398 CrossRef CAS.
- (a) M. Kodama and E. Kimura, J. Chem. Soc., Dalton Trans., 1976, 116–120 RSC; (b) A. P. Leuger, L. Hertli and T. A. Kaden, Helv. Chim. Acta, 1978, 61, 2296–2306 CrossRef.
- W.-J. Lan and C.-S. Chung, J. Chem. Soc., Dalton Trans., 1994, 191–194 RSC.
- (a) P. Táborský, I. Svobodová, P. Lubal, Z. Hnatejko, S. Lis and P. Hermann, Polyhedron, 2007, 26, 4119–4130 CrossRef; (b) J. Kotek, P. Hermann, I. Císařová, J. Rohovec and I. Lukeš, Inorg. Chim. Acta, 2001, 317, 324–330 CrossRef CAS; (c) J. Kotek, P. Vojtíšek, I. Císařová, P. Hermann and I. Lukeš, Collect. Czech. Chem. Commun., 2001, 66, 363–381 CrossRef CAS.
- K. L. Nash, R. D. Rogers, J. Ferraro and J. Zhang, Inorg. Chim. Acta, 1998, 269, 211–223 CrossRef CAS.
- M. L. Turonek, P. A. Duckworth, G. S. Laurence, S. F. Lincoln and K. P. Wainwright, Inorg. Chim. Acta, 1995, 230, 51–57 CrossRef CAS.
- I. Lázár, C. H. Duane, W. Kim, C. E. Kiefer and A. D. Sherry, Inorg. Chem., 1992, 31, 4422–4424 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Distribution diagrams and spectral profiles of species present in the Cu(II)–H4dota, Cu(II)–trans-H6do2a2p, Cu(II)–H8dotp systems; primary titration data for the Cu(II)–H8dotp system; additional formation kinetics data and selected figures; additional acid-assisted decomplexation kinetics data and selected figures; experimental and additional data for the solid-state structure of K(H6doa3p)·), 3H2O and a figure of its crystal packing. CCDC 1469446. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01127f |
| This journal is © The Royal Society of Chemistry 2016 |