
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enantiopure and racemic radical-cation salts of B(malate)2− anions with BEDT-TTF†
Jordan R.
Lopez
a,
Lee
Martin
 *a,
John D.
Wallis
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b,
Jun-ichi
Yamada
c,
Tomofumi
Kadoya
c,
Simon J.
Coles
d and
Claire
Wilson
d
*a,
John D.
Wallis
a,
Hiroki
Akutsu
b,
Yasuhiro
Nakazawa
b,
Jun-ichi
Yamada
c,
Tomofumi
Kadoya
c,
Simon J.
Coles
d and
Claire
Wilson
d
aSchool of Science and Technology, Nottingham Trent University, Clifton Lane, Clifton, Nottingham, NG11 8NS, UK. E-mail: lee.martin@ntu.ac.uk; Tel: +44 (0)1158483128
bDepartment of Chemistry, Graduate School of Science, Osaka University, 1-1 Machikaneyama-cho, Toyonaka, Osaka 560-0043, Japan
cGraduate School of Material Science, University of Hyogo, Kamigori-cho, Ako-gun, Hyogo 678-1297, Japan
dSchool of Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 10th May 2016
Abstract
We have synthesized the first examples of radical-cation salts of BEDT-TTF with chiral borate anions, [B(malate)2]−, prepared from either enantiopure or racemic bidentate malate ligands. In the former case only one of two diastereoisomers of the borate anion is incorporated, while for the hydrated racemic salt one racemic pair of borate anions containing a R and a S malate ligand is incorporated. Their conducting and magnetic properties are reported. The tight-binding band calculation indicates that the chiral salt has an effective half-filled flat band, which is likely to be caused by the chiral structural feature.
Introduction
Bis(ethylenedithio)tetrathiafulvane (BEDT-TTF) radical-cation salts have been studied extensively following the discovery of a large number of organic metals and superconductors. These salts have the ability to combine more than one physical property via crystal engineering of the conducting BEDT-TTF layers and the insulating anion layers.1 The synthesis of radical-cation salts of this type offers a promising approach to producing crystalline materials which combine the properties of chirality and conductivity.2 Electrical magneto-chiral anisotropy was first observed in bismuth helices,3 and it has also been observed in carbon nanotubes,4 where the resistivity along nanotubes of opposing chirality differs in a coaxial magnetic field. Pop et al. have recently synthesised enantiopure radical-cation salts of (S,S)- and (R,R)-(DM-EDT-TTF)2ClO4.5 These salts, in space groups P6222 and P6422, show metallic behavior down to 40 K and the observation of electrical magneto-chiral anisotropy confirms the chiral nature of charge transport. The DM-EDT-TTF donor has also produced PF6 racemic salts with metal-like conductivity, while the enantiopure salts have a completely different packing and show room temperature charge ordering and semiconducting behaviour.5There are three routes through which chirality can be introduced into radical-cation salts: a chiral donor molecule, chiral anion, or a chiral electrolyte. There have been a large number of enantiopure TTF-based donor molecules synthesised since the first example, tetramethyl-(S,S,S,S)-BEDT-TTF, in 1986.6 Recent promise of the use of chiral donor molecules to produce bulk chiral conductors is evidenced by the aforementioned work of Pop et al.5 Recently a semiconducting TTF-type donor has shown lower activation energy for its racemic salt with BF4 compared to the isostructural analogues for both individual enantiomers.7
Other examples of salts from chiral donor molecules include hydroxyalkyl-BEDT-TTFs,8 bis(oxazoline)-TTFs,9 iodo-TTFs,10 pyrrolo-TTFs,11 and TTF-sulphoxides which have chiral sulphur atoms.12
Racemic and chiral anions have been used to produce radical-cation salts with BEDT-TTF including Fe(croconate)3,13 Cr(2,2′-bipy)(oxalate)2,14 Sb2(L-tartrate)2,15 TRISPHAT16 and Fe(C6O4Cl2)3.17a Fe(C6O4Cl2)3 has also been combined in salts with TM-BEDT-TTF17b in a rare case where a chiral donor and chiral anion have been combined, although the anion crystallised as a racemic mixture. An enantiopure bis-hydroxy-substituted TTF donor has also produced a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt with the meso stereoisomer of the dinuclear [Fe2(oxalate)5]4− anion.8
:1 salt with the meso stereoisomer of the dinuclear [Fe2(oxalate)5]4− anion.8
The most extensive family of BEDT-TTF salts with chiral anions contain tris(oxalato)metallate and these have provided a large number of materials which combine magnetism with conductivity in the same lattice.18 These salts contain a 50:50 mixture of the Δ and Λ enantiomers of [MIII(oxalate)3]3− to give an overall racemic lattice.19 However, the spatial distribution of these two enantiomers within the anion layers determines the donor packing arrangement and thus the conducting properties.
The inclusion of a guest solvent molecule within the lattice of these tris(oxalato)metallate salts introduces the possibility of using a chiral solvent for crystal growth. Crystals containing either enantiopure or racemic sec-phenethyl alcohol molecules show a difference in their conducting properties owing to the disorder of the enantiomers of the guest solvent molecule in the anion layer of the racemic salt.20 Recently, the first examples containing a single enantiomer of tris(oxalato)metallate in the lattice have been produced by using chiral induction through electrocrystallisation from the chiral electrolyte (R)-carvone containing racemic Cr(C2O4)3 or Al(C2O4)3.21
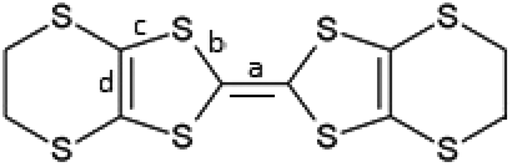
We report here two new BEDT-TTF radical-cation salts incorporating the anion B(malate)2− prepared from either racemic malic acid or enantiopure D-(+)-malic acid. The synthesis of such bis-chelated borate anions offers the prospect of creating complexes with more than one stereogenic centre. In the case of the B(malate)2− reported here the chirality of the bidentate chelated malate ligand is retained but diastereomers are produced through two possible stereochemical configurations at the boron centre which is labile in solution. Therefore, when using enantiopure D-(+)-malic acid ((R)-hydroxybutanedioic acid), a mixture of diastereoisomeric anions will be produced in solution: BSRR with an S boron centre and two R malate ligands, and BRRR which differs only in having an R boron centre (Scheme 1). 1H NMR in deuterio-acetonitrile shows the anions exist in a 2:1 ratio with very similar NMR spectra. When using racemic DL-malic acid, four further diastereomers will be produced: BSSS and BRSS (the mirror images of the anions shown in Scheme 1) and BSRS and BRRS (Scheme 2). 1H NMR in deuterio-acetonitrile is complex but consistent with the presence of these diastereomers. BSRR, BRRR and BSSS, BRSS are racemic pairs and therefore only four signals are expected.
 | ||
| Scheme 1 The two borate anions which can be formed using D-(+)-malic acid. | ||
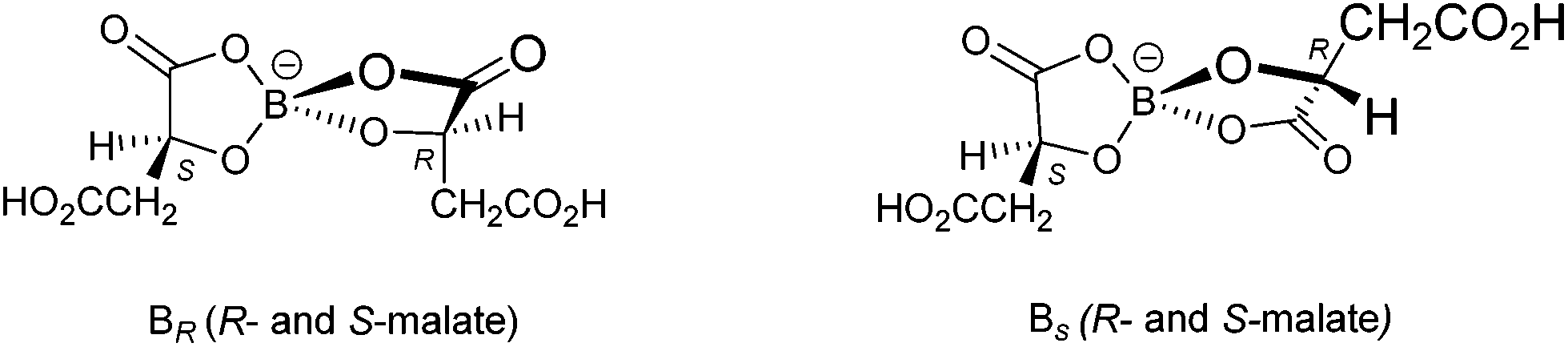 | ||
| Scheme 2 The two borate anions containing both malate enantiomers. | ||
We report here a chiral radical-cation salt of BEDT-TTF, synthesised electrochemically in the presence of chiral BSRR and BRRR anions, in which just the BSRR anion is incorporated into the structure. When repeating this synthesis using a mixture of the six possible diastereomeric borate anions prepared from racemic malic acid a racemic radical-cation salt is obtained but only two of the diastereomeric anions are present in the crystal (BSRS and BRRS).
Results and discusssion
(BEDT-TTF)2BR/S[(R/S)-malate]2·(H2O)2.85 (I)
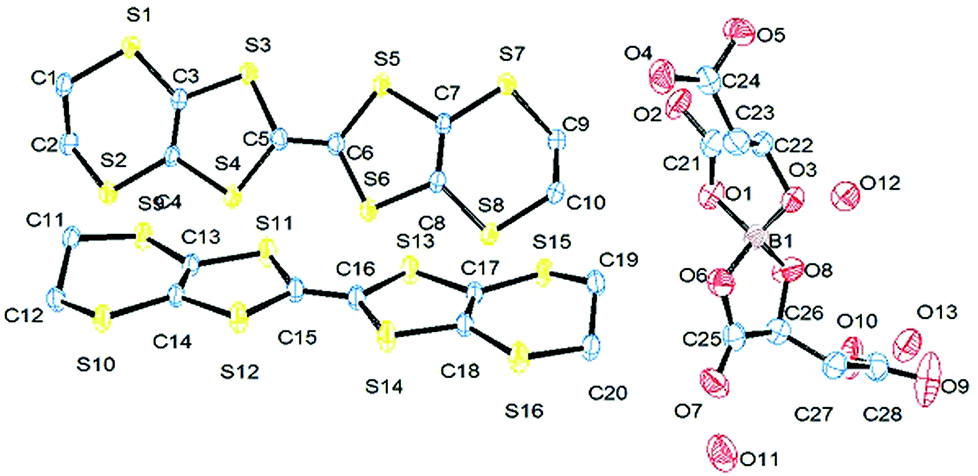
Radical-cation salt I was synthesised by electrocrystallisation of BEDT-TTF in 1,1,2-trichloroethane containing 18-crown-6 and the mixture of potassium borate salts prepared from racemic malic acid. Salt I has the formula (BEDT-TTF)2BR/S[(R/S)-malate]2·(H2O)2.85 with only two of the possible diastereomers present in the radical-cation salt. Salt I crystallises in the triclinic crystal system in the centrosymmetric space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit consists of two crystallographically independent BEDT-TTF molecules, one borate anion and 2.85 water molecules (Fig. 1). The borate anion contains one R- and one S-malate anion. The unit cell thus contains a racemic pair of two such anions, differing only in the configuration at boron (R or S) (Fig. 2). This is the first example of diastereomeric induction within the electrocrystallisation environment without the need for external auxiliaries. Previously chiral radical-cation salts have been synthesised from enantiopure anions,15,16 or from racemates which have been resolved during crystal engineering in chiral solvents.21 The two independent donors are separately stacked along the a axis. The stacks lie side by side in the b direction to form a layer and with an angle of 70.64° between the best planes of the independent donors. This corresponds to the α-type packing arrangement of BEDT-TTF donors. Donor layers are separated by layers containing the borate anions and hydrogen bonded water molecules (Fig. 3). The anion layer contains 2.85 waters per borate anion. The two carboxylic hydrogen atoms are hydrogen bonded to water molecules (1.78–1.85 Å), one of the waters is also hydrogen bonded to two borate ring O atoms (2.05 and 2.15 Å), while the other water is also hydrogen bonded to a ring carbonyl oxygen (2.20 Å) and to a third water molecule (1.99 Å). This third water molecule makes a second hydrogen bond to a ring carbonyl O atom (1.93 Å) (Fig. 4).
. The asymmetric unit consists of two crystallographically independent BEDT-TTF molecules, one borate anion and 2.85 water molecules (Fig. 1). The borate anion contains one R- and one S-malate anion. The unit cell thus contains a racemic pair of two such anions, differing only in the configuration at boron (R or S) (Fig. 2). This is the first example of diastereomeric induction within the electrocrystallisation environment without the need for external auxiliaries. Previously chiral radical-cation salts have been synthesised from enantiopure anions,15,16 or from racemates which have been resolved during crystal engineering in chiral solvents.21 The two independent donors are separately stacked along the a axis. The stacks lie side by side in the b direction to form a layer and with an angle of 70.64° between the best planes of the independent donors. This corresponds to the α-type packing arrangement of BEDT-TTF donors. Donor layers are separated by layers containing the borate anions and hydrogen bonded water molecules (Fig. 3). The anion layer contains 2.85 waters per borate anion. The two carboxylic hydrogen atoms are hydrogen bonded to water molecules (1.78–1.85 Å), one of the waters is also hydrogen bonded to two borate ring O atoms (2.05 and 2.15 Å), while the other water is also hydrogen bonded to a ring carbonyl oxygen (2.20 Å) and to a third water molecule (1.99 Å). This third water molecule makes a second hydrogen bond to a ring carbonyl O atom (1.93 Å) (Fig. 4).
 | ||
| Fig. 1 ORTEP diagram of the asymmetric unit of I showing atomic labelling, hydrogens are omitted for clarity. | ||
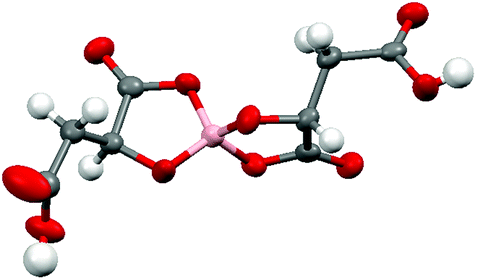 | ||
| Fig. 2 Borate anion in I. | ||
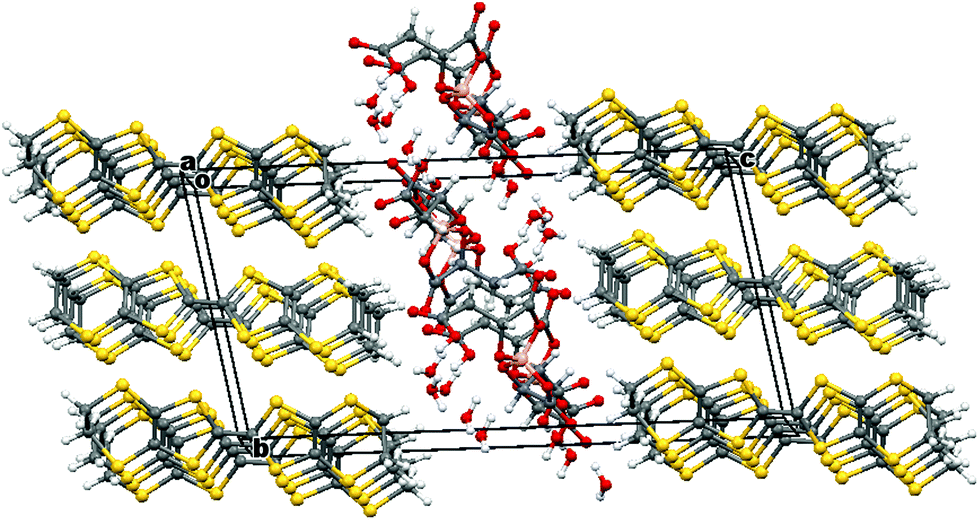 | ||
| Fig. 3 Layered structure of I viewed down the a axis. | ||
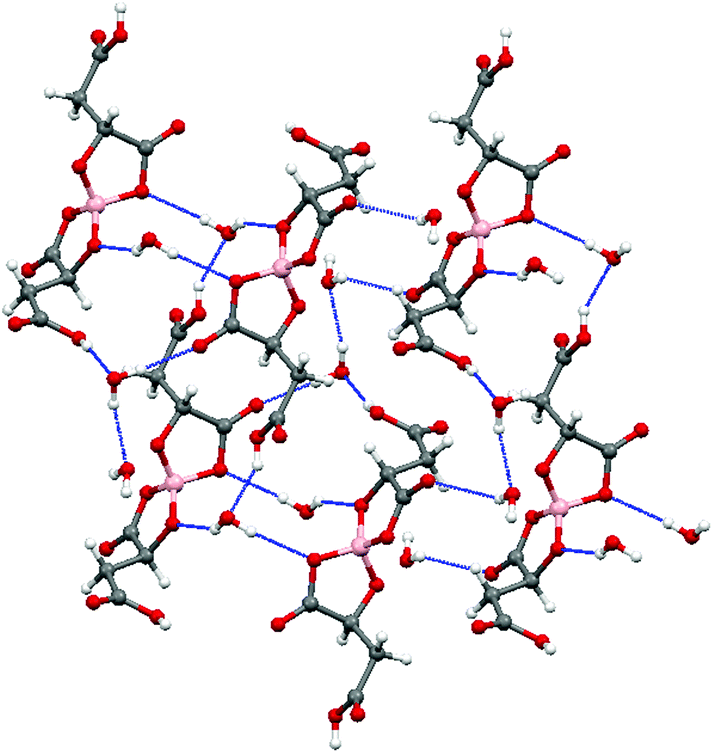 | ||
| Fig. 4 Anion layer of I. | ||
The terminal ethylene carbons of each donor show half chair conformations with each sp3 carbon equidistant from the plane of the TTF unit. The two donors each have thirteen side-to-side S⋯S contacts below the sum of van der Waals radii (Table 1). These contacts are all side-to-side between neighbouring stacks of crystallographically independent donors. Donor A has two short contacts with two of the H2O molecules (S7–O13, 3.285 Å and S2–O11, 3.142 Å) and donor B has one short contact with a borate anion (S10–O4, 3.145 Å) (Fig. 4).
| S⋯S | Donor A⋯B |
|---|---|
| Contact/Å | |
| S1⋯S10 | 3.53(1) |
| S1⋯S12 | 3.58(1) |
| S5⋯S16 | 3.47(1) |
| S7⋯S16 | 3.58(1) |
| S2⋯S9 | 3.48(1) |
| S4⋯S9 | 3.56(1) |
| S8⋯S13 | 3.55(1) |
| S8⋯S15 | 3.49(1) |
| S8⋯S10 | 3.48(1) |
| S8⋯S12 | 3.57(1) |
| S1⋯S13 | 3.47(1) |
| S1⋯S15 | 3.46(1) |
| S7⋯S9 | 3.56(1) |
The molecular formula suggests that the two independent donors have a cumulative charge of +1 to balance the charge of the BR/S[(R/S)-malate]2− anion. Using the method of Guionneau et al.22 for estimating oxidation state from molecular geometry the two donors are calculated each to carry a charge of 0.5+ (Table 2). The central C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond lengths are 1.366 Å in both A and B donors.
C bond lengths are 1.366 Å in both A and B donors.
|
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Salt | Donor | a/Å | b/Å | c/Å | d/Å | δ | Q | Charge |
| I at 150 K | A | 1.366 | 1.743 | 1.750 | 1.358 | 0.769 | 0.60+ | |
| B | 1.366 | 1.742 | 1.752 | 1.354 | 0.771 | 0.59+ | 1.19 ± 0.2 = 1 | |
| I at 250 K | A | 1.366 | 1.740 | 1.7525 | 1.3495 | 0.777 | 0.55+ | |
| B | 1.366 | 1.742 | 1.7505 | 1.3535 | 0.773 | 0.58+ | 1.13 ± 0.2 = 1 | |
| II | A | 1.357 | 1.762 | 1.756 | 1.357 | 0.804 | 0.37+ | |
| B | 1.379 | 1.731 | 1.746 | 1.373 | 0.725 | 0.94+ | 1.31 ± 0.3 = 1 | |
The resistivity of I shows exponential temperature dependence only below ∼250 K with an activation energy of 0.050 eV (ρRT = 0.00163 Ω cm) (Fig. 5).
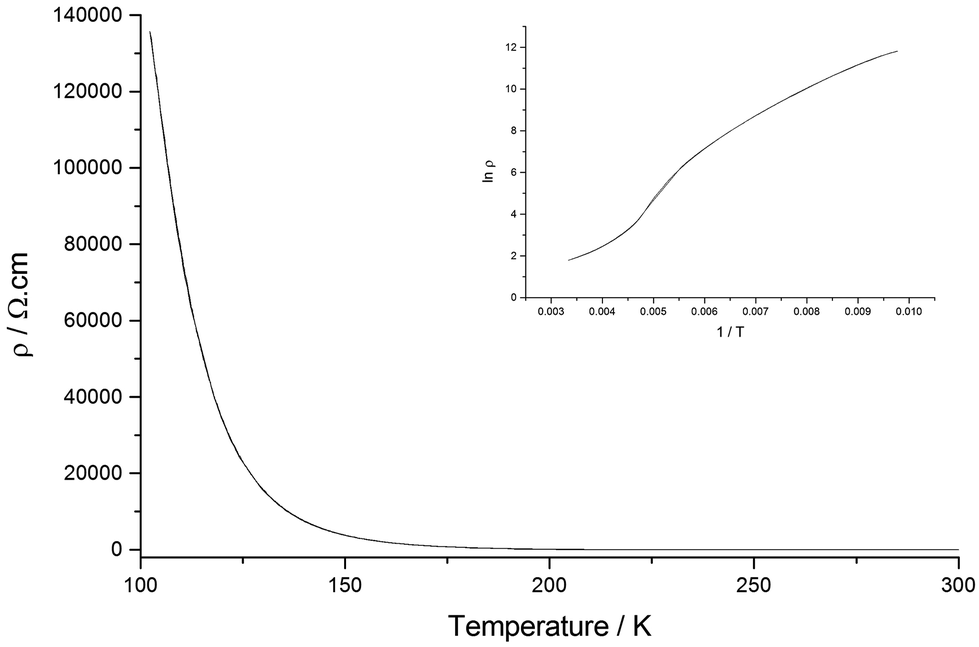 | ||
| Fig. 5 Resistivity data for I. | ||
(BEDT-TTF)2[BS(R-malate)2] (II)
The electrochemical synthesis using the [BR/S(R-malate)2] anion mixture prepared from enantiopure D-(+)-malic acid does not give an isostructural radical-cation salt. Instead, salt II with formula (BEDT-TTF)2[BS(R-malate)2] is synthesized which contains only the borate anion with stereochemistry BSRR and no water (Fig. 6). Salt II crystallises in the orthorhombic crystal system in the space group P212121. The crystal packing is shown in Fig. 7. Two crystallographically independent donors and one borate anion are present in the asymmetric unit (Fig. 8). The two independent donors are organised in an ABAB arrangement in stacks along the a axis, with the adjacent donors lying at an angle of 10°. The stacks lie side by side in an α packing arrangement to form layers perpendicular to the c axis. Adjacent layers are separated by layers composed of the chiral borate anions. The anions form layers held together by hydrogen bonding between the carboxylic acid groups and borate ring O atoms (Fig. 9). The CH2CO2H side chains adopt different conformations, one extended away from the borate and the other folded back so the acid's carbonyl oxygen makes a short contact with the adjacent ring carbonyl carbon (O⋯C: 2.738 Å). There is also a short intermolecular contact from a methylene hydrogen belonging to the extended side chain to a ring carbonyl oxygen (2.37 Å) (Fig. 9). Terminal BEDT-TTF ethylene groups have half chair conformations with sp3 carbons equidistant from the planar TTF core. There is a network of side-to-side S⋯S contacts between donors in neighbouring stacks (Table 3). A short contact is observed between donor B and a borate anion (S2A⋯O17B, 3.043 Å).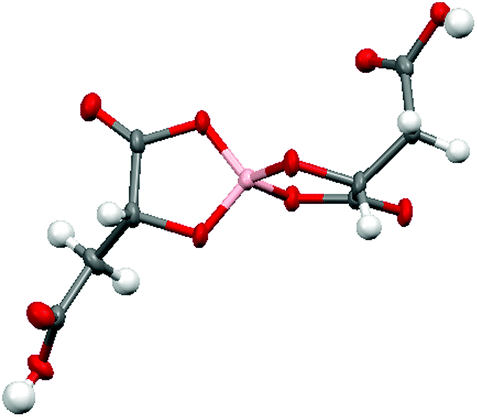 | ||
| Fig. 6 Borate anion in II. | ||
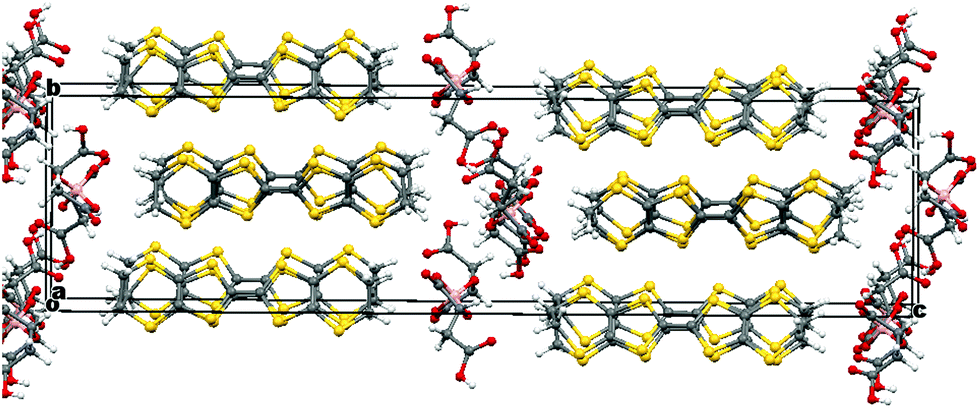 | ||
| Fig. 7 Layered structure of II viewed down the b axis, hydrogens are omitted for clarity. | ||
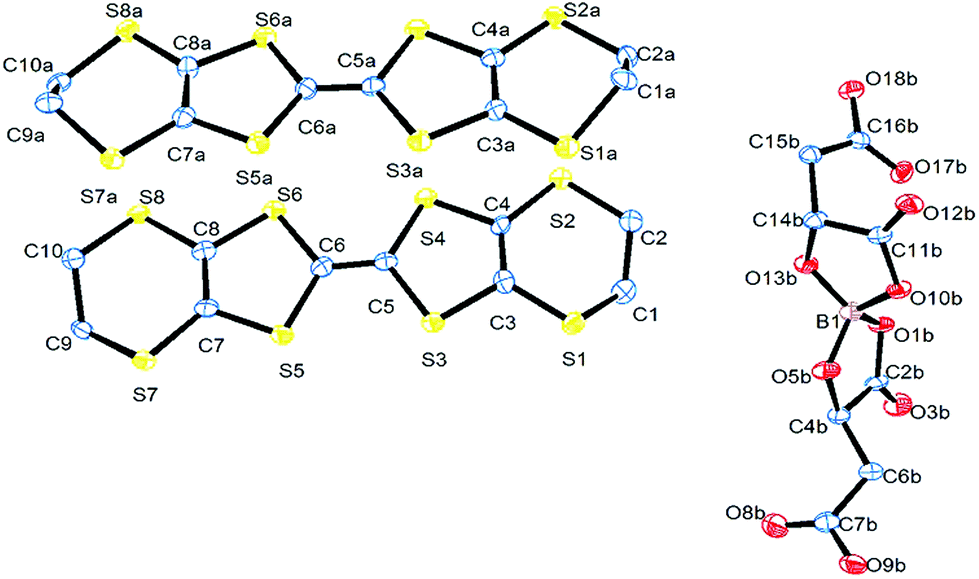 | ||
| Fig. 8 ORTEP diagram of the asymmetric unit of II showing atomic labelling, hydrogens are omitted for clarity. | ||
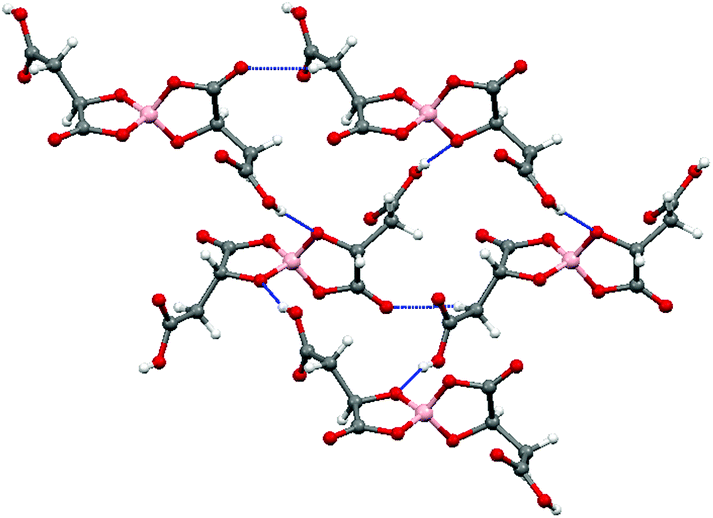 | ||
| Fig. 9 Anion layer of II. | ||
| Donor A⋯B | |
|---|---|
| Contact/Å | |
| S4⋯S7A | 3.48(1) |
| S8⋯S1A | 3.34(1) |
| S8⋯S3A | 3.44(1) |
| S7⋯S2A | 3.45(1) |
| S7⋯S4A | 3.49(1) |
| Donor A⋯A | |
|---|---|
| Contact/Å | |
| S2⋯S7 | 3.45(1) |
| S4⋯S7 | 3.48(1) |
| S3⋯S8 | 3.57(1) |
| Donor B⋯B | |
|---|---|
| Contact/Å | |
| S8A⋯S3A | 3.54(8) |
The formula (BEDT-TTF)2[BS(R-malate)2] suggests that the donors have a cumulative charge of +1 to balance the charge of the borate anion. Using the method of Guionneau et al.22 the two crystallographically independent donors carry charges of 0.94+ and 0.37+, and central CC bond lengths are 1.357 and 1.379 Å. This indicates charge localisation with a 1+ charge on one donor and the other donor being neutral.
Salt II shows low resistivity at room temperature (ρRT = 15.015 Ω cm) and semiconducting behaviour is observed upon cooling from 300 K to 165 K. Hysteresis is also present on heating back up to 300 K giving a less efficient conductor as we observe an increase in Ea (Ea cooling = 0.082 eV, Ea heating 0.099 eV) (Fig. 10).
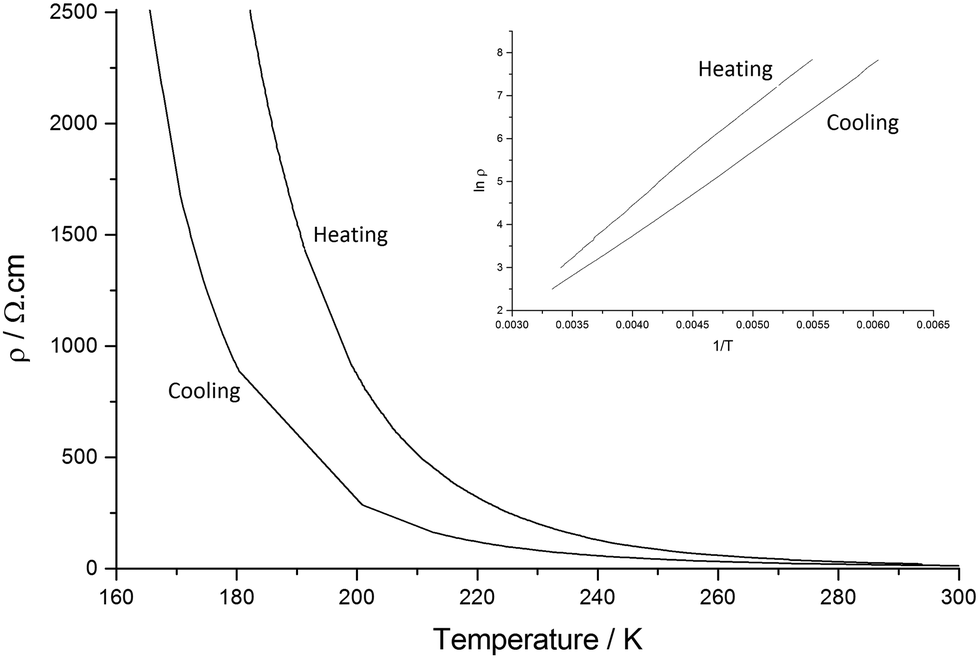 | ||
| Fig. 10 Resistivity data for II. | ||
Comparison of salts I and II
As mentioned above, both salts have similar α-type packing motif and the resistivity behaviours are also similar apart from the anomaly at approximately 190 K on the resistivity curve of I. However, the charge of BEDT-TTF molecules shown in Table 2 indicates that both electronic structures are different, with salt I being metallic at room temperature and salt II in the charge-ordered state at least at 100 K. Therefore, the two salts should have some structural difference. Fig. 11 shows an end-on projection of the donor layers of both salts (I and II), where green and pink coloured molecules are the molecules A and B, respectively. In salt I (Fig. 11a) each crystallographically independent molecule forms a stack along the a axis. On the other hand, salt II (Fig. 11b) has alternating A and B molecules within each stack. We performed a tight binding band calculation of each of the salts.23 The calculation neglected the electron correlation effects so that the band calculations provided Fermi surfaces for many quarter-filled salts although the salts are actually semiconductive. In other words, a semiconducting salt possessing Fermi surfaces on the band calculation indicates that the band gap is caused by the electron correlations. The band dispersions and Fermi surfaces of I at 150 and 250 K are shown in Fig. 12a and b, respectively. There is no significant difference between the structures of I at 150 and 250 K. The band calculation results also have no significant difference between those at 150 and 250 K. Salt I has an electron and a hole pocket, which is similar to β′′-type salts,24 and suggests that salt I is semi-metallic. Actually the salt is semi-metallic at room temperature. However, at lower temperature, especially below the transition temperature of 190 K, the resistivity increases with decreasing temperature. Usually a semimetal has no temperature dependence of resistivity. This behaviour indicates that the salt becomes a semiconductor in the low temperature region. The molecular charges shown in Table 2 indicate that the salt is not in the charge disproportionation state, which is paramagnetic, at least down to 150 K. The salt cannot become a paramagnetic Mott insulator because the band dispersion (Fig. 12a) has no Mott gap (see next paragraph). Therefore, it is not likely that the salt has a paramagnetic ground state. Preliminary magnetic susceptibility measurement using only 0.38 mg of powder sample suggests that the data can be fitted by a Curie–Weiss model, we believe that this shows a Curie tail (Fig. S1†), and no magnetic transition was observed. This result strengthens the hypothesis that the ground state is non-magnetic. We speculate at present that the salt becomes a band insulator in the low temperature region by band nesting. Similarly, β′′-(BEDT-TTF)4[(H3O)Cr(C2O4)3]·CH2Cl2 shows semiconducting behaviour although the salt has semimetallic Fermi surfaces.25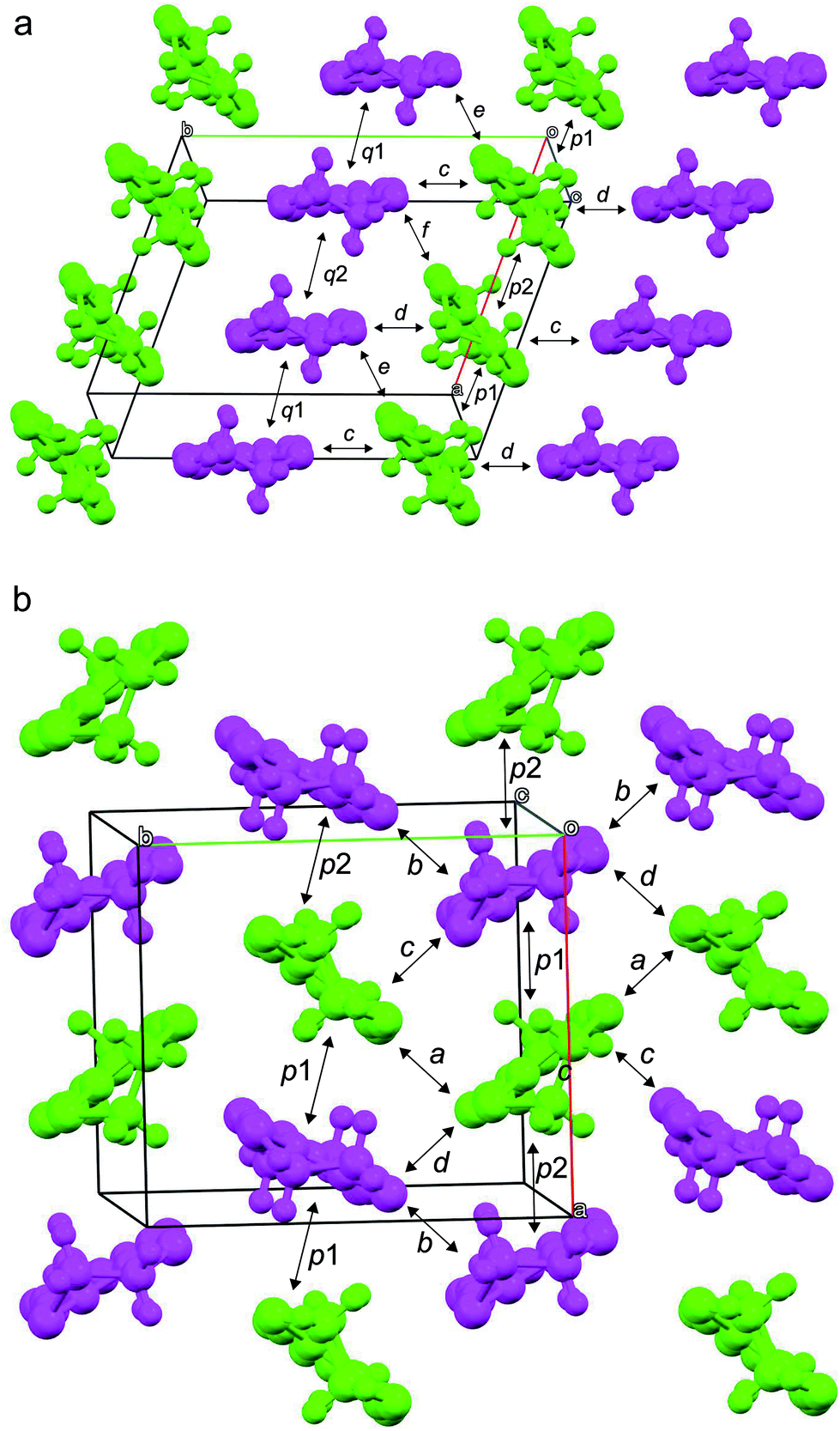 | ||
| Fig. 11 End of the donor layers of I (a) and II (b). Green and pink coloured molecules are molecules A and B, respectively. | ||
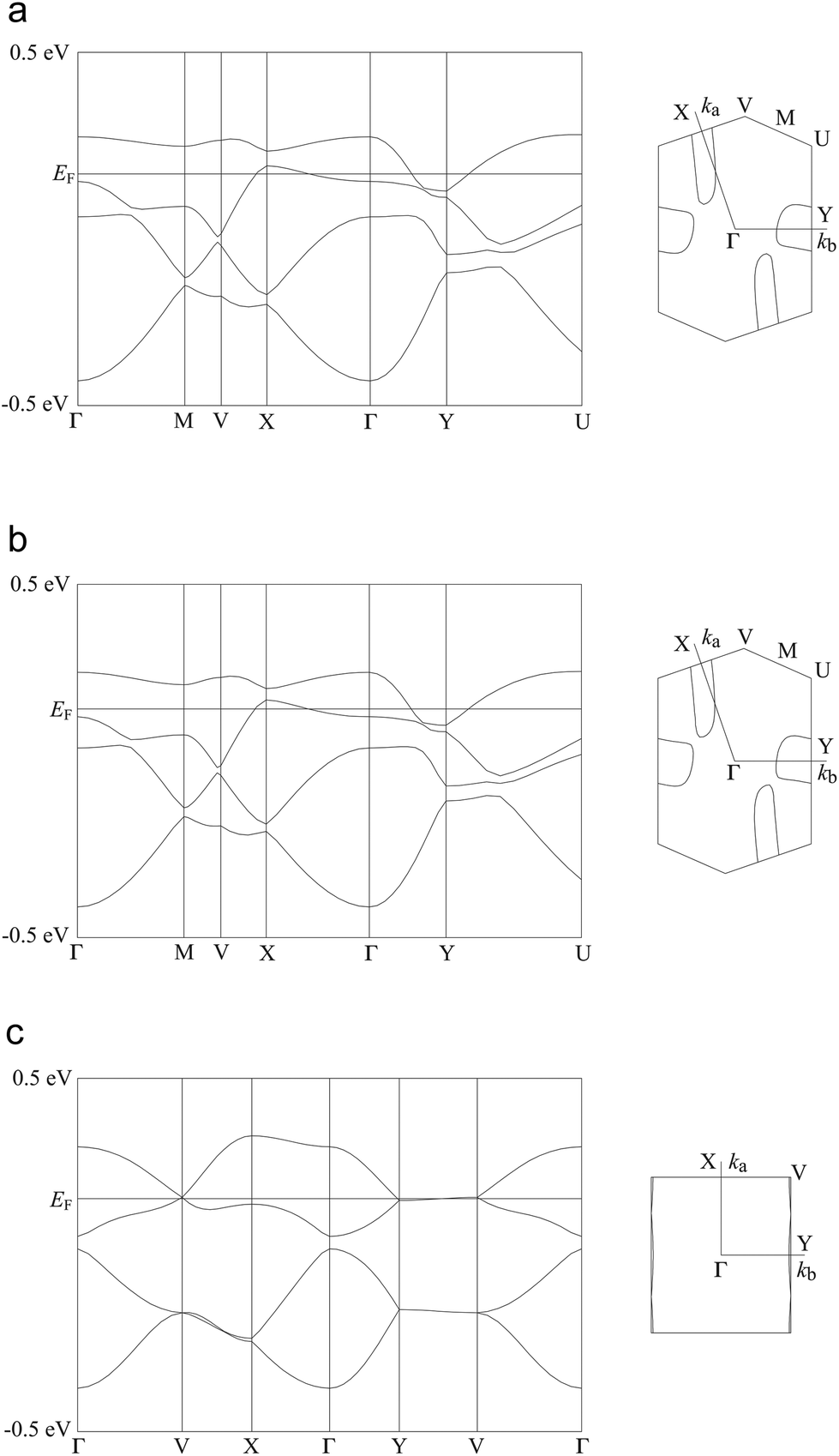 | ||
| Fig. 12 Band dispersions and Fermi surfaces of salts I (a: 150 K; b: 250 K) and II (c). Transfer integrals (×10−3 eV, see also Fig. 11) are p1 = −3.07, p2 = −7.13, q1 = −5.04, q2 = −1.67, c = −7.41, d = −12.21, e = +8.32 and f = +6.58 for salt I at 150 K, p1 = −3.21, p2 = −6.42, q1 = −4.62, q2 = −1.97, c = −7.25, d = −11.42, e = +7.93 and f = +6.59 for salt I at 250 K and p1 = +5.07, p2 = −1.38, a = +5.57, b = +10.64, c = +16.11 and d = +1.10 for salt II. | ||
As mentioned above, although salt II has a similar α-type packing motif to salt I, the arrangement of the green and pink molecules is different (as shown in Fig. 11b). The band dispersions and Fermi surfaces are shown in Fig. 12c, which is very different to that of salt I. The band dispersion has a mid-gap at the centre of the band dispersion, a so-called Mott gap, indicating that the band is effective half filled. So the salt can become a Mott insulator. However, the salt selects a charge-ordered state, in which the charge of the green molecule (A) is almost zero and the charge of the pink molecule (B) is almost one as shown in Table 2. This charge-ordered pattern is the so-called “horizontal stripe”, which is commonly observed in the α-type BEDT-TTF salts. An α-type packing motif can theoretically have many types of charge disproportionation states.26 However, the horizontal stripe type is the most common, which was observed in salt II, and a vertical type, which salt I may have if the salt becomes a charge-ordered state, is not common. Although, salt II has the common horizontal stripe type charge order for α-type BEDT-TTF salts, the band structure is very different from usual α-type BEDT-TTF salts. As shown in Fig. 12c, an effectively half-filled flat band is observed from Y to V, on which the Fermi level is located. The resultant Fermi surfaces are straight line like a 1D band located on the edge of the Brillouin zone. The band structure of the usual α-type BEDT-TTF salts consists of quasi-1D and 2D Fermi surfaces, similar to the κ- or λ-type Fermi surface. The comparison of the transfer integrals indicates that the b and c (see Fig. 11b) values are almost twice as large as those of normal α-type BEDT-TTF salts. This indicates that salt II has strong 1D nature along the side-by-side, b-axis direction. If we change the value of the transfer integrals of b and c into the half values, the calculation provides the common α-type Fermi surfaces. Magnetic susceptibility measurement using 2.70 mg of powder sample of II (Fig. 13) suggests that the magnetic curve obeys a 2D Heisenberg model with J = −71.1 K but with a spin concentration of only 25%. A residual temperature independent magnetic susceptibility of 5.7 × 10−4 emu mol−1 was observed, suggesting that some fraction of the electrons are still itinerant. The strong side-by-side interaction may make some percentage (75%?) of spin freedom. The observation of the separation of the charge and spin freedom may be due to the polar nature of the crystal (see below). In addition, materials having a half-filled flat band can become ferromagnetic, which is so called Flat-Band Ferromagnetism.28 Salt II may be a candidate for a Flat-Band Ferromagnet. Now the sample has a band gap in the middle of the effective upper band caused by charge ordering. Applying static or uniaxial pressure to the sample can make the band gap become null, and the sample may become a ferromagnet. Moreover, a flat band material may have such a large density of state that it may have a large Seebeck coefficient.29 It is therefore a candidate for a thermoelectric transducer. Further research is now in progress.
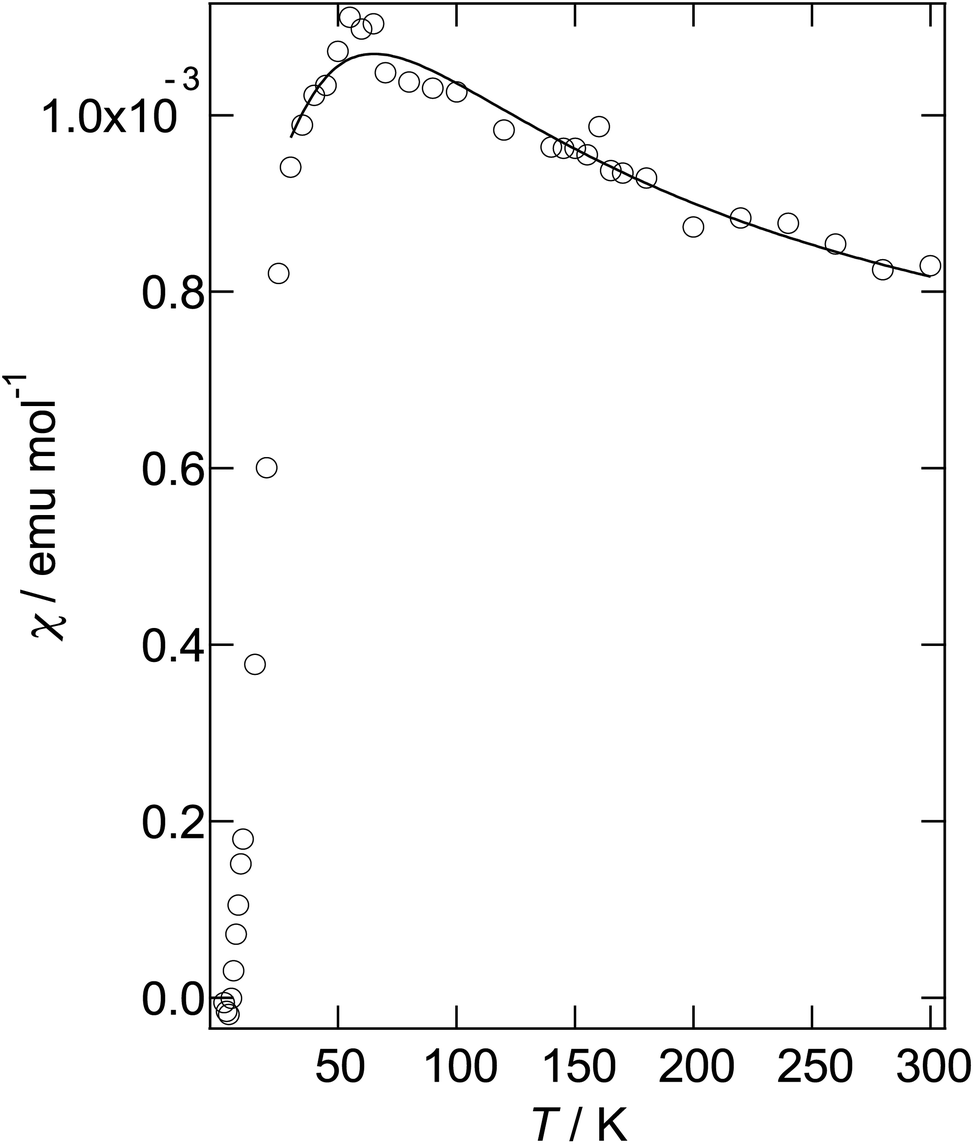 | ||
| Fig. 13 Magnetic susceptibility of II after subtracting a Curie tail of 0.80% of S = 1/2 spin. The solid line is calculated on the basis of a 2D Heisenberg model.27 | ||
We believe that the unique electronic structure of salt II is owing to its chiral and polar anion. We calculated the dipole moment of the chiral anion using Winmostar Ver.4.10430 (MOPAC AM1) using atomic coordinates of the crystal structure. A dipole moment of 5.104 Debye is calculated, which directs to approximately the crystallographic c axis (Fig. S2a†). However, there are two anions in the anion layer, both of which appear to be arranged to cancel both dipole moments. Therefore the calculation of the dipole moment of both anions was performed. A dipole moment of 3.103 Debye was calculated, which was still large. The dipole vector directs to approximately the -a direction (Fig. S2b†). For the cancellation of the dipole moment along the a axis, the donor layer appears to go into the 0101 charge-ordered state along the a axis. Finally we calculated the dipole moment of four anions that surround a donor layer. A residual dipole moment of 1.431 Debye was calculated, which directs to the –b direction. The dipole moment can be converted into a voltage using the equation, ΔΦ = μNcosθ/εε0 (ref. 31) = 0.14 eV, where ε ≈ 2 and θ = 0.0° were assumed.
The dipole moment caused by the anion layer should be cancelled, for which the donor layers should also be polarized. The donor layers would be in a unique charge disproportionate state to produce dipole moments also along the b-axis. In the circumstance, we speculate that the strong 1D interaction of salt II along the b axis, which provides the unique flat band, is owing to the residual dipole moment, whose origin is the chirality of the borate anion, because a crystal including a chiral molecule cannot possess any inversion centres or mirror and glide planes and become chiral and polar. Thus we believe that chirality provides a significant effect upon the electronic structure.
Experimental
Synthesis and purification of starting materials
BEDT-TTF was purchased from Sigma Aldrich and recrystallised from chloroform. 1,1,2-trichloroethane, L-(−)-malic acid, D-(+)-malic acid, D/L-malic acid, boric acid, sodium hydroxide and 18-crown-6 were purchased from Sigma Aldrich and used as received.Synthesis of K[B(malate)2]32
Boric acid (2.47 g, 40.0 mmol, 1.00 equiv.) was added to a solution of racemic D/L-malic acid (10.73 g, 80.0 mmol, 2.00 equiv.) in water (10 ml). To this was added a solution of KOH (2.08 g, 40.0 mmol, 1.00 equiv.) in water (30 ml) and the reaction mixture stirred at 95 °C. After evaporation of the water K[B(malate)2] was isolated as a white powder. The same method was used replacing D/L-malic acid with either L-(−)-malic acid or D-(+)-malic acid. Yields >99% yield in all cases.Racemic Salt; δH (400 MHz, CD3CN): four diastereomers, 4.44–4.51 (2H, m) (2 × OCH), 2.65–2.74 (2H, m) (2 × CHαHβCO2H), 2.46–2.55 (2H, m) (2 × CHαHβCO2H); δC (100 MHz, CD3CN): 178.3 & 178.4 (2 × CO2B), 171.8 (2 × CO2H), 72.2 & 72.4 (2 × OCH), 38.87, 38.91, 39.11 & 39.14 (2 × CH2CO2H). CHN: K[B(C4O5H4)2]·H2O; expected: C 28.95%, H 3.01%; found: C 28.94%, H 3.04%.
Chiral Salt; δH (400 MHz, CD3CN): two diastereomers in ratio 2:1, 4.47 (2H, dd, J = 7.9, 4.3 Hz, major) & 4.49 (2H, dd, J = 8.6, 4.2 Hz, minor) (2 × OCH), 2.67 (2H, dd, J = 15.6, 4.2 Hz, minor) & 2.71 (2H, dd, J = 15.8, 4.3 Hz, major) (2 × CHαHβCO2H), 2.49 (2H, dd, J = 15.6, 8.6 Hz, minor) & 2.52 (2H, dd, J = 15.8, 7.9 Hz, major) (2 × CHαHβCO2H); δC (100 MHz, CD3CN): 178.2 & 178.3 (2 × CO2B), 171.8 (2 × CO2H), 72.1 & 72.3 (2 × OCH), 38.8 & 39.0 (2 × CH2CO2H). CHN: K[B(C4O5H4)2]; expected: C 30.6%, H 2.57%; found: C 30.32%, H 2.72%.
Synthesis of radical-cation salts
Crystals of I and II‡ were grown on platinum electrodes by electrocrystallisation in 40 ml H-shaped electrochemical cells.
Platinum electrodes were cleaned by applying a voltage across the electrodes in 1 M H2SO4 in each direction to produce H2 and O2 at the electrodes, then washed with distilled water and thoroughly dried. K[B(malate)2] (100 mg) and 18-crown-6 (200 mg) were dissolved in 1,1,2-trichloroethane (30 ml) with stirring overnight before filtering into an H-cell containing BEDT-TTF (10 mg) in the anode compartment. Salt I was prepared using K[B(malate)2] synthesized from racemic malic acid. Salt II was prepared using K[B(malate)2] synthesized from D-(+)-malic acid. H-cells were placed in a dark box on a vibration-free bench at a constant current of 0.2 μA and after 21 days a large number of black hexagonal crystals were harvested from the anode of the H-cell containing the racemic anion. A large number of black plates were collected from the H-cell containing the enantiopure anion. Attempts were also made to grow crystals using K[B(malate)2] prepared from L-(−)-malic acid, but despite repeated attempts using a variety of conditions it was not possible to obtain single crystals.
Physical measurements
Four-probe DC transport measurements were made on crystals using a HUSO HECS 994C multi-channel conductometer. Gold wires (15 μm diameter) were attached to the crystal, and the attached wires were connected to an integrated circuit plug with carbon conductive cement.SQUID magnetometry
Magnetic susceptibility measurements were performed with a Quantum Design MPSM2 SQUID magnetometer using randomly orientated polycrystalline material encased in aluminium foil. Magnetization was recorded from 2 to 300 K at 0.3 and 0.1 Tesla for I and II.Conclusions
Two new radical-cation salts of BEDT-TTF with B(malate)2− have been synthesised. These are the first examples of radical-cation salts of BEDT-TTF with borate anions. The two salts differ by having either chiral or racemic bidentate malate ligands. Both salts have novel packing arrangements of the B(malate)2− diastereomers and show preference for certain diastereomers over others in the electrochemical synthesis. Both salts show α-type BEDT-TTF packing but differ in the distribution of the crystallographically independent donor molecules. The racemic salt has donor stacks which contain only a single crystallographically independent donor molecule and is metallic at room temperature. The chiral salt has donor stacks which contain both donors in alternating ABAB fashion, and this salt is in the charge-ordered state showing semiconducting behaviour.We have synthesised a variety of borate anions with numerous chiral ligands and are continuing to synthesise further salts with BEDT-TTF to closely examine the effect of chirality upon the physical properties. We are also performing experiments using chiral hydroxylalkyl-BEDT-TTF derivatives with the aim of transmitting the chirality between anion and conducting donor layers through hydrogen-bonding interactions.
Acknowledgements
This work has been supported by the Royal Society [Research Grants (RG100853 and RG081209), International Exchange Scheme (IE130367), and International Joint Project (JP0869972)]. We thank EPSRC for funding the National Crystallography Service. JRL and LM thank NTU for a PhD studentship.Notes and references
- For a series of reviews, see: Chem. Rev., 2004, 104, passim; J. Phys. Soc. Jpn., 2006, 75, passim; J. M. Williams, J. R. Ferraro, R. J. Thorn, K. D. Carlson, U. Geiger, H. H. Wang, A. M. Kini and M. H. Whangbo, Organic Superconductors: Synthesis, Structure, Properties and Theory, Prentice Hall, Englewood Cliffs, NJ, 1992 Search PubMed.
- N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061 RSC.
- G. L. J. A. Rikken and E. Raupach, Nature, 1997, 390, 493 CrossRef CAS; G. L. J. A. Rikken, J. Folling and P. Wyder, Phys. Rev. Lett., 2001, 87, 236602 CrossRef PubMed.
- V. Krstic, S. Roth, M. Burghard, K. Kern and G. L. J. A. Rikken, J. Chem. Phys., 2002, 117, 11315 CrossRef CAS; V. Krstic and G. L. J. A. Rikken, Chem. Phys. Lett., 2002, 364, 51 CrossRef; G. L. J. A. Rikken, Science, 2011, 331, 864 CrossRef PubMed.
- F. Pop, P. Auban-Senzier, E. Canadell, G. L. J. A. Rikken and N. Avarvari, Nat. Commun., 2014, 5, 3757, DOI:10.1038/ncomms4757; F. Pop, P. Auban-Senzier, A. Frackowiak, K. Ptaszyński, I. Olejniczak, J. D. Wallis, E. Canadell and N. Avarvari, J. Am. Chem. Soc., 2013, 135, 17176–17186 CrossRef CAS PubMed.
- J. D. Dunitz, A. Karrer and J. D. Wallis, Helv. Chim. Acta, 1986, 69, 69 CrossRef.
- L. Martin, J. D. Wallis, M. A. Guziak, J. Oxspring, J. R. Lopez, S.-i. Nakatsuji, J.-i. Yamada and H. Akutsu, CrystEngComm, 2014, 16, 5424 RSC.
- I. Awheda, S. Krivickas, S. Yang, L. Martin, M. A. Guziak, A. C. Brooks, F. Pelletier, M. Le Kerneau, P. Day, P. Horton, H. Akutsu and J. D. Wallis, Tetrahedron, 2013, 69, 8738 CrossRef CAS; S. J. Krivickas, C. Hashimoto, J. Yoshida, A. Ueda, K. Takahashi, J. D. Wallis and H. Mori, Beilstein J. Org. Chem, 2015, 11, 1561 CrossRef PubMed; F. Leurquin, T. Ozturk, M. Pilkington and J. D. Wallis, J. Chem. Soc., Perkin Trans. 1, 1997, 3173–3178 RSC.
- F. Riobé and N. Avarvari, Chem. Commun., 2009, 3753 RSC.
- J. Lieffrig, R. Le Pennec, O. Jeannin, P. Auban-Senzier and M. Fourmigué, CrystEngComm, 2013, 15, 4408 RSC.
- S. Yang, A. C. Brooks, L. Martin, P. Day, H. Li, P. Horton, L. Male and J. D. Wallis, CrystEngComm, 2009, 11, 993 RSC.
- M. Chas, M. Lemarié, M. Gulea and N. Avarvari, Chem. Commun., 2008, 220 RSC; M. Chas, F. Riobé, R. Sancho, C. Minguıllón and N. Avarvari, Chirality, 2009, 21, 818 CrossRef CAS PubMed.
- C. J. Gómez-García, E. Coronado, S. Curreli, C. Giménez-Saiz, P. Deplano, M. L. Mercuri, L. Pilia, A. Serpe, C. Faulmann and E. Canadell, Chem. Commun., 2006, 4931 RSC.
- A. M. Madalan, E. Canadell, P. Auban-Senzier, D. Brânzea, N. Avarvari and M. Andruh, New J. Chem., 2008, 32, 333 RSC.
- E. Coronado, J. R. Galán-Mascarós, C. J. Gómez-García, A. Murcia-Martinez and E. Canadell, Inorg. Chem., 2004, 43, 8072 CrossRef CAS PubMed.
- M. Clemente-León, E. Coronado, C. J. Gómez-García, A. Soriano-Portillo, S. Constant, R. Frantz and J. Lacour, Inorg. Chim. Acta, 2007, 360, 955 CrossRef; F. Riobé, F. Piron, C. Réthoré, A. M. Madalan, C. J. Gómez-García, J. Lacour, J. D. Wallis and N. Avarvari, New J. Chem., 2011, 35, 2279–2286 RSC.
- (a) S. Benmansour, E. Coronado, C. Giménez-Saiz, C. J. Gómez-García and C. Rößer, Eur. J. Inorg. Chem., 2014, 24, 3949 CrossRef; (b) M. Atzori, F. Pop, P. Auban-Senzier, C. J. Gómez-García, E. Canadell, F. Artizzu, A. Serpe, P. Deplano, N. Avarvari and M. L. Mercuri, Inorg. Chem., 2014, 53, 7028 CrossRef CAS PubMed; (c) M. Atzori, F. Pop, P. Auban-Senzier, R. Clérac, E. Canadell, M. L. Mercuri and N. Avarvari, Inorg. Chem., 2015, 54, 3643–3653 CrossRef CAS PubMed.
- M. Kurmoo, A. W. Graham, P. Day, S. J. Coles, M. B. Hursthouse, J. L. Caulfield, J. Singleton, F. L. Pratt, W. Hayes, L. Ducasse and P. Guionneau, J. Am. Chem. Soc., 1995, 117, 12209 CrossRef CAS; L. Martin, S. S. Turner, P. Day, P. Guionneau, J. K. Howard, D. E. Hibbs, M. E. Light, M. B. Hursthouse, M. Uruichi and K. Yakushi, Inorg. Chem., 2001, 40, 1363 CrossRef PubMed; T. G. Prokhorova, S. S. Khasanov, L. V. Zorina, L. I. Buravov, V. A. Tkacheva, A. A. Baskakov, R. B. Morgunov, M. Gener, E. Canadell, R. P. Shibaeva and E. B. Yagubskii, Adv. Funct. Mater., 2003, 13, 403 CrossRef; E. Coronado, S. Curreli, C. Giménez-Saiz and C. J. Gómez-García, Inorg. Chem., 2012, 51, 1111 CrossRef PubMed.
- L. Martin, S. S. Turner, P. Day, F. E. Mabbs and E. J. L. McInnes, J. Chem. Soc., Chem. Commun., 1997, 1367 RSC; L. Martin, S. S. Turner, P. Day, K. M. A. Malik, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Chem. Commun., 1999, 513 RSC; L. Martin, S. S. Turner and P. Day, Synth. Met., 1999, 102, 1638 CrossRef CAS.
- L. Martin, P. Day, H. Akutsu, J.-i. Yamada, S.-i. Nakatsuji, W. Clegg, R. W. Harrington, P. N. Horton, M. B. Hursthouse, P. McMillan and S. Firth, CrystEngComm, 2007, 10, 865 RSC.
- L. Martin, P. Day, S.-i. Nakatsuji, J.-i. Yamada, H. Akutsu and P. Horton, CrystEngComm, 2010, 12, 1369 RSC; L. Martin, S.-i. Nakatsuji, J.-i. Yamada, H. Akutsu and P. Day, J. Mater. Chem., 2010, 20, 2738–2742 RSC; L. Martin, H. Akutsu, P. N. Horton, M. B. Hursthouse, R. W. Harrington and W. Clegg, Eur. J. Inorg. Chem., 2015, 11, 1865 CrossRef CAS; L. Martin, H. Akutsu, P. N. Horton and M. B. Hursthouse, CrystEngComm, 2015, 17, 2783 RSC.
- P. Guionneau, C. J. Kepert, D. Chasseau, M. R. Truter and P. Day, Synth. Met., 1997, 86, 1973 CrossRef CAS.
- T. Mori, A. Kobayashi, Y. Sasaki, H. Kobayashi, G. Saito and H. Inokuchi, Bull. Chem. Soc. Jpn., 1984, 57, 627 CrossRef CAS . (See also http://www.op.titech.ac.jp/lab/mori/lib/program.html).
- T. G. Prokhorova, S. S. Khasanov, L. V. Zorina, L. I. Buravov, V. A. Tkacheva, A. A. Baskakov, R. B. Morgunov, M. Gener, E. Canadell, R. P. Shibaeva and E. B. Yagubskii, Adv. Funct. Mater., 2003, 13, 403 CrossRef CAS.
- S. Rashid, S. S. Turner, D. LePevelen, P. Day, M. E. Light, M. B. Hunthouse, S. Firth and R. J. H. Clark, Inorg. Chem., 2001, 40, 5304 CrossRef CAS PubMed.
- H. Seo, J. Phys. Soc. Jpn., 2000, 69, 805 CrossRef CAS.
- M. E. Lines, J. Chem. Phys. Solids, 1970, 31, 101 CrossRef CAS.
- R. Arita, Y. Sugawa, K. Kuroki and H. Aoki, Phys. Rev. Lett., 2002, 88, 127202 CrossRef PubMed; A. Mielke and H. Tasaki, Commun. Math. Phys., 1993, 158, 341 CrossRef.
- K. Mori, H. Usui, H. Sakakibara and K. Kuroki, AIP adv., 2012, 2, 042108 CrossRef.
- N. Senda, Idemitsu Tech. Rep., 2006, 49, 106 Search PubMed.
- M. Suda, N. Kameyama, A. Ikegami and Y. Einaga, J. Am. Chem. Soc., 2009, 131, 865 CrossRef CAS PubMed.
- R. Gausepohl, P. Buskens, J. Kleinen, A. Bruckmann, C. W. Lehmann, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2006, 45, 3689 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1468076–1468078. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt01038e |
| ‡ Crystal data: I at 150 K: C28H29.44O12.72S16B1, M = 1093.26, black plate, a = 8.6620(4), b = 11.9323(5), c = 21.7062(7) Å, α = 76.855(5), β = 84.526(6), γ = 70.436(5)°, U = 2058.16(16) Å3, T = 150 K, space group P, Z = 2, μ = 0.901 mm−1, reflections collected = 19802, independent reflections = 15717, R1 = 0.0450, wR2 = 0.1125 [F2 > 2σ(F2)], R1 = 0.0623, wR2 = 0.1205 (all data). Crystal data: I at 250 K: C28H29.43O12.72S16B1, M = 1093.25, black plate, a = 8.7239(3), b = 12.0104(3), c = 21.7699(6) Å, α = 76.913(5), β = 84.377(6), γ = 70.239(5)°, U = 2090.34(14) Å3, T = 250 K, space group P Crystal data: II: C28H24O10S16B1, M = 1044.24, black plate, a = 9.1696(8), b = 10.2343(9), c = 41.196(4) Å, α = β = γ = 90°, U = 3866.0(6) Å3, T = 100 K, space group P212121, Z = 4, μ = 0.950 mm−1, reflections collected = 44 Data for I were collected on a Rigaku R-AXIS VII imaging plate system with FR-E SuperBright High-Brilliance Rotating Anode Generator with confocal monochromated MoKα radiation, using a Rapid Auto software for control and processing. Data for II were collected on a Rigaku AFC12 diffractometer with Mo rotating anode, using standard control and processing software. All structures were solved and refined with programs from the SHELX family of computer programs. CCDC 1468076–1468078 contains supplementary X-ray crystallographic data for I and II. |
| This journal is © The Royal Society of Chemistry 2016 |