
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A multi-step solvent-free mechanochemical route to indium(III) complexes†
Jingyi
Wang
a,
Rakesh
Ganguly
a,
Li
Yongxin
a,
Jesus
Díaz
b,
Han Sen
Soo‡
*acd and
Felipe
García‡
*a
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, 21 Nanyang Link 637371, Singapore. E-mail: hansen@ntu.edu.sg; fgarcia@ntu.edu.sg
bDepartamento de Química Orgánica e Inorgánica, Facultad de Veterinaria, Universidad de Extremadura, Cáceres 10071, Spain
cSingapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE), 1 Create Way, Singapore 138602
dSolar Fuels Laboratory, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798
First published on 15th April 2016
Abstract
Mechanochemistry is well-established in the solid-phase synthesis of inorganic materials but has rarely been employed for molecular syntheses. In recent years, there has been nascent interest in ‘greener’ synthetic methods with less solvent, higher yields, and shorter reaction times being especially appealing to the fine chemicals and inorganic catalyst industries. Herein, we demonstrate that main-group indium(III) complexes featuring bis(imino)acenaphthene (BIAN) ligands are readily accessible through a mechanochemical milling approach. The synthetic methodology reported herein not only bypasses the use of large solvent quantities and transition metal reagents for ligand synthesis, but also reduces reaction times dramatically. These new main-group complexes exhibit the potential to be reduced to indium(I) compounds, which may be employed as photosensitizers in organic catalyses and functional materials.
Mechanochemistry refers to a branch of predominant solid-state processes, in which reactions can be induced by mechanical energy, through grinding, shearing, and ball milling for instance.1 Mechanochemical techniques have historically been mostly applied in the solid-phase syntheses of poorly soluble metal oxides, ceramics,2 and alloys,3 but have increasingly been adopted in modern fields such as nanomaterials.4 Despite some seminal work on cocrystallization of organic compounds in the 1980s,5 there has not been widespread adoption of mechanochemistry in synthetic organometallic chemistry and catalytic reactions until some recent reports on heterogeneous catalysis of cyclopropanation of alkenes using silver foil were reported.6 The use of solid-state synthesis of molecular inorganic compounds has also been reported for transition metal complexes,7 but rarely for main-group inorganic and organometallic systems, despite one previous example of the mechanochemical synthesis of bis(n-propyltetramethylcyclopentadienyl)strontium8 and an aluminium trialkyl complex.9 There has been recent interest in high-yielding, minimal-waste, and low-energy synthetic routes among the communities of green chemistry and the pharmaceutical and fine chemicals industries. More widespread use of mechanochemistry is increasingly attractive and sustainable since the reactions can be solvent-free, rapid, essentially quantitative, and cost-effective.
Our groups have separately been interested in synthetic main-group chemistry,10 and the application of transition metal complexes and metal oxide materials in artificial photosynthesis.11 In view of the dearth of studies on main-group complexes in artificial photosynthesis and photoredox chemistry, we sought to develop an atom economical synthetic route for indium complexes that can potentially function as visible light photosensitizers.12 Currently, the most popular photosensitizers typically comprise heavy-metals, such as in the case of ruthenium oligopyridine complexes, which undergo metal-to-ligand charge transfer (MLCT) transitions and exhibit long-lived excited states.12 Although indium shares the same period, little has been done to explore its heavy atom effect, which could enhance the rate of intersystem crossing from the photoexcited singlet to the triplet state, and hence significantly prolong the lifetime of the photosensitizer. We were particularly attracted to indium complexes primarily due to their ability to mediate organic reactions,13 in addition to other attractive attributes such as their low-toxicity,14 water tolerance,15 and relatively low reduction potential.16
Bis(imino)acenaphthene (BIAN) ligands are highly versatile diimine alternatives to the ubiquitous bipyridine and terpyridine derivatives that have frequently been employed as molecular photosensitizers. Functionalized BIAN compounds have been recognized as robust ligands for transition metal centres, but are synthetically more accessible than polypyridine compounds, via straightforward condensation reactions between various anilines and acenaphthenequinone.17 The combination of the extensive π-system in the acenaphthene ring and the sterically tunable aniline provides a multitude of customizable π-acceptor scaffolds, which give us precise synthetic control over electronic and steric properties. A number of transition metal BIAN complexes have been investigated as versatile catalysts for olefin polymerization18 and organic reactions like cycloaddition of azides and alkynes.19 However, there is only one previous crystallographically characterized example of a monometallic BIAN20 and two of tetrakis(imino)pyracene (TIP)21 indium(III) complexes.
Here, we report the successful application of mechanochemical milling for both the preparation of BIAN ligands – bearing both electron-donating and electron-withdrawing groups – and the subsequent formation of the corresponding indium(III) complexes. These compounds were further characterized for future applications as main-group photosensitizers.22 Our novel mechanochemical approach not only allowed us to bypass the use of transition metal reagents during the synthesis of the BIAN ligands, but also provided a more atom efficient, essentially solvent-free, and time-efficient access to such compounds.
Generally, substituted BIAN ligands are synthesized by condensation of acenaphthoquinone with the corresponding aniline under acidic conditions. In many cases, complexation with either ZnCl2 or NiBr2 is required before the removal of the metal ion to furnish the free ligand.23 For example, p-MeOAr-BIAN (1) and p-BrAr-BIAN (2) are synthesized by refluxing acenaphthoquinone with the corresponding aniline in acetic acid in the presence of ZnCl2, followed by de-metallation with K2CO3 or Na2C2O4 (Scheme 1).24
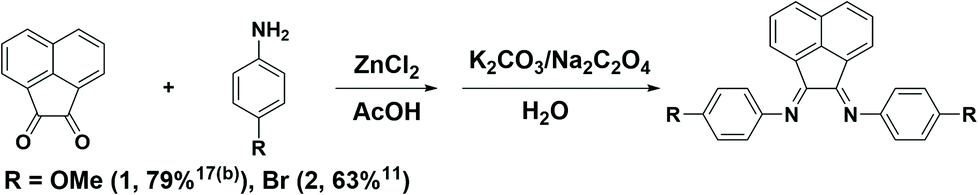 | ||
| Scheme 1 Zinc-templated synthesis of the Ar-BIAN ligands. | ||
Milling acenaphthoquinone with the corresponding aniline in the presence of a catalytic amount of additive (p-anisidine with acetic acid, and p-bromoaniline with trifluoroacetic anhydride, for 1 and 2, respectively) produced Ar-BIAN ligands, 1 (84%) and 2 (64%). This method avoids the use of ZnCl2 as the templating agent and therefore circumvents the need for de-metallation (Scheme 2). As a comparison, reactions were performed thermally at 180 °C in the same reaction vessel without milling. This led to decomposition of both 1 and 2 during the course of 4 h. Based on the 1H NMR spectra the reaction is almost quantitative (ESI, Fig. S3 and S6†) and analytically pure products can be obtained after recrystallization from suitable solvents. The acid-catalyzed mechanochemical synthesis of BIAN ligands described herein presents advantages over established solution-based methods due to short reaction times, virtually solvent-free conditions, and versatility in application to both electron-rich and electron-poor anilines (in 1 and 2, respectively). Indium(III) BIAN complexes p-MeOAr-BIAN-InCl3 (3, 72%) and p-BrAr-BIAN-InCl3 (4, 84%) were obtained by milling equimolar quantities of the corresponding BIAN ligand and InCl3 in a grinder jar equipped with a 10 mm ball for 40 min and 2 h, respectively. Similarly, the solid state thermal reactions of neat 1 and 2 with InCl3 at 180 °C for 2.5 h without milling led to decomposition products and no reaction was observed in the attempted syntheses of 3 and 4, respectively. These reaction times represent noticeable improvements from the related previously reported synthesis of Mes-BIAN-InCl3 (Mes = mesityl) in THF where the reaction was stirred for 12 hours prior to isolation.20
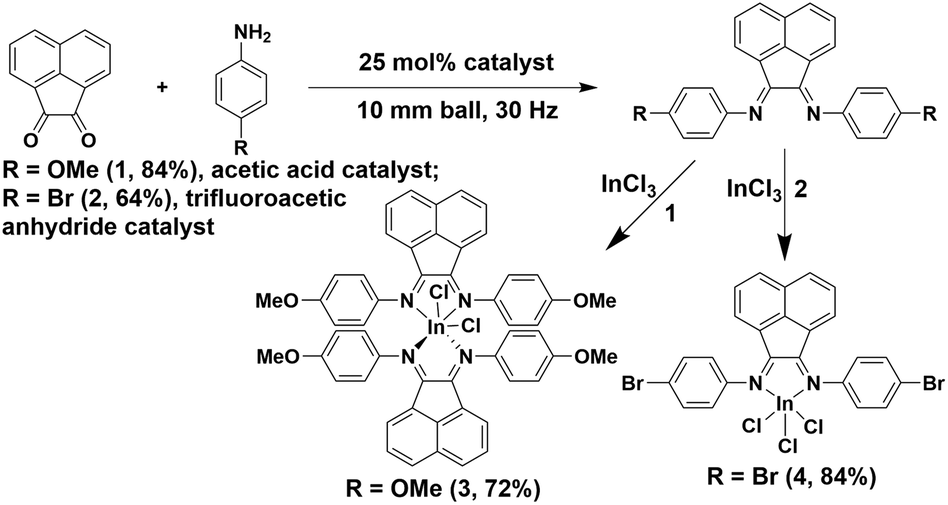 | ||
| Scheme 2 Mechanochemical synthesis of the Ar-BIAN ligands and indium(III) complexes. | ||
Complexes 3 and 4 were successfully recrystallized to provide crystals suitable for X-ray structural analyses. Furthermore, all products have been comprehensively characterized by NMR spectroscopy and found to be indistinguishable from those produced by the usual solution methods (ESI, Schemes S7 and S8†). Direct formation of the indium(III) complexes by a one-pot process from each crude product of the mechanochemical ligand synthesis was also attempted. In the case of 3, complex formation can be identified from the 1H NMR spectrum, which was isolated in 57% yield after recrystallization. However, in the case of 4, the single crystal X-ray data show that 4 was formed, but the 1H NMR spectrum of the crude mixture revealed a mixture of products. The one-pot reaction described herein – without ligand isolation – is a rare example of a multistep mechanochemical synthesis. The previously reported examples typically start from “preformed” ligands and metal complexes25 rather than precursors to ligands and metal salts. Our current report thus further raises the orthogonality of multi-step mechanochemical synthesis and widens its applicability.
Both indium(III) complexes adopt distorted octahedral geometries (Fig. 1). The C–N and C–C bond distances on the diimine framework are consistent with the typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and C–C bond lengths previously reported.20 Therefore each complex consists of neutral BIAN ligands coordinating via dative bonds to the indium(III) metal center. Complex 4 contains only one bound BIAN with In–N bond distances of 2.390(3) and 2.301(3) Å. In contrast, 3 crystallized out as a bis(BIAN) complex, with [InCl4]− as the counter anion. The average In–N bond distance in 3 (2.313(3) Å) is shorter than that measured in 4 (2.346(4) Å), reflecting the stronger binding and hence more electron rich nature of the imine N atoms in 3. The In–Cl bond distances of the bis(BIAN) cation in complex 3 are 2.4126(5) and 2.3963(5) Å whereas those in complex 4 range from 2.4185(9) to 2.4336(9) Å. It is interesting to note that the average In–Cl bond distance in 3 is slightly shorter than that in the related bromo derivative, despite the fact that both are significantly longer than the In–Cl distance in InCl3(THF)3 (2.331(3) Å).26 A lengthening of the In–Cl bond distances in our indium(III) complexes is expected due to the increased electron donation from the Ar-BIAN ligands compared to InCl3(THF)3. However, the slight increase of the In–Cl bond distance in 4 over that in 3 is surprising, and may be due to extra electron density donated by the N,N-dimethylformamide (DMF) molecule to the metal center.
N and C–C bond lengths previously reported.20 Therefore each complex consists of neutral BIAN ligands coordinating via dative bonds to the indium(III) metal center. Complex 4 contains only one bound BIAN with In–N bond distances of 2.390(3) and 2.301(3) Å. In contrast, 3 crystallized out as a bis(BIAN) complex, with [InCl4]− as the counter anion. The average In–N bond distance in 3 (2.313(3) Å) is shorter than that measured in 4 (2.346(4) Å), reflecting the stronger binding and hence more electron rich nature of the imine N atoms in 3. The In–Cl bond distances of the bis(BIAN) cation in complex 3 are 2.4126(5) and 2.3963(5) Å whereas those in complex 4 range from 2.4185(9) to 2.4336(9) Å. It is interesting to note that the average In–Cl bond distance in 3 is slightly shorter than that in the related bromo derivative, despite the fact that both are significantly longer than the In–Cl distance in InCl3(THF)3 (2.331(3) Å).26 A lengthening of the In–Cl bond distances in our indium(III) complexes is expected due to the increased electron donation from the Ar-BIAN ligands compared to InCl3(THF)3. However, the slight increase of the In–Cl bond distance in 4 over that in 3 is surprising, and may be due to extra electron density donated by the N,N-dimethylformamide (DMF) molecule to the metal center.
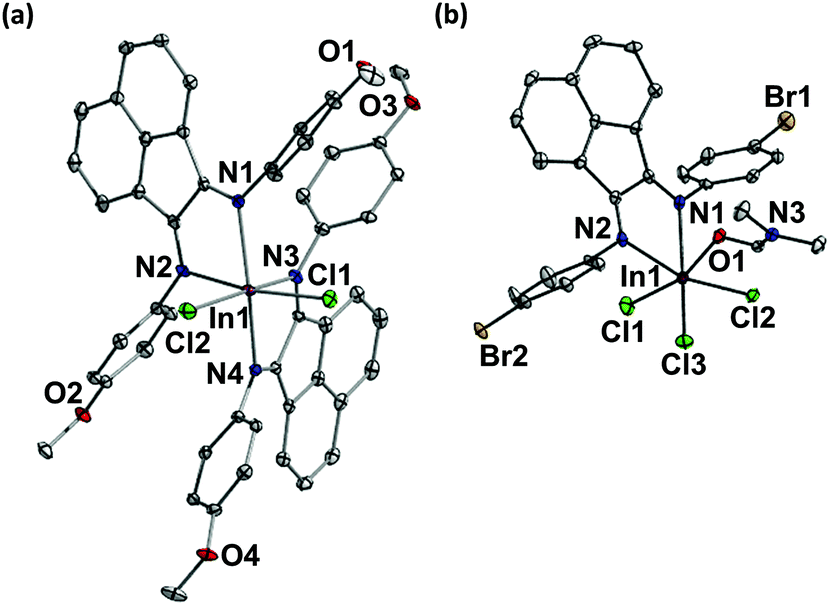 | ||
| Fig. 1 Thermal ellipsoid plots (50%) of (a) [(p-MeOAr-BIAN)2InCl2]+ (3) and (b) (p-BrAr-BIAN)InCl3 (4) (DMF solvate molecule excluded). Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): (a) In1–N1 2.296(2), In1–N2 2.324(2), In1–N3 2.311(2), In1–N4 2.323(2), In1–Cl1 2.413(5), In1–Cl2 2.396(5). (b) In1–N1 2.390(3), In1–N2 2.301(3), In1–Cl1 2.434(9), In1–Cl2 2.419(9), In1–Cl3 2.429(9), In1–O1 2.258(2). | ||
At 25 °C, the 1H NMR spectrum of 3 displays broadened signals in the aromatic region, which may be the result of cis–trans isomerization taking place in solution on the NMR timescale. To understand the behaviour of 3 in solution, variable temperature NMR spectroscopy was carried out in acetone-d6 (ESI, Fig. S7†). As the temperature was reduced, the isomerization process slowed down and the signals from the two isomers started to be resolved. Three singlets were observed with an intensity ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 at around 4 ppm at −70 °C. They correspond to the three separate proton environments of the –OCH3 groups in the trans- (one singlet) and cis-isomers (two singlets). In the aromatic region, two sets of signals could be observed below −60 °C, with one set being assigned to the trans-isomer and the other set to the cis-isomer. According to the integration of the signals, the ratio between the trans and cis isomers varies with temperature. An enthalpy change of +19.7 kJ mol−1 and an entropy change of +75.8 J mol−1 K−1 for the cis to trans isomerization were calculated from the Van't Hoff plot (ESI, Fig. S8†).
:1:1 at around 4 ppm at −70 °C. They correspond to the three separate proton environments of the –OCH3 groups in the trans- (one singlet) and cis-isomers (two singlets). In the aromatic region, two sets of signals could be observed below −60 °C, with one set being assigned to the trans-isomer and the other set to the cis-isomer. According to the integration of the signals, the ratio between the trans and cis isomers varies with temperature. An enthalpy change of +19.7 kJ mol−1 and an entropy change of +75.8 J mol−1 K−1 for the cis to trans isomerization were calculated from the Van't Hoff plot (ESI, Fig. S8†).
The UV-vis spectra for both BIAN ligands 1 and 2 and their corresponding indium(III) complexes 3 and 4 were recorded at 298 K and are illustrated in Fig. 2. The extinction coefficient decreases when the electron-donating –OCH3 group is replaced by the electron withdrawing –Br group, consistent with the trend reported by Hasan.17 The high energy bands are assigned to the π–π* ligand-centered transitions between the arylimine and the acenaphthene moieties, whereas the longer wavelength bands are due to the n–π* transition from the diimine to the acenaphthene (vide infra). The absorption spectrum of 3 is red-shifted in comparison with the free ligand 1. However the absorption spectra for 4 and the uncoordinated free ligand 2 are very similar. Nevertheless, both the electrochemical studies and the DFT calculations were consistent with the formation of the proposed indium(III) complex (vide infra).
 | ||
| Fig. 2 UV-Vis spectra for 1 in acetonitrile (ACN, black), 3 in ACN (red), 2 in DMF (blue), and 4 in DMF (green). | ||
TD-DFT calculations were used to predict the electronic absorption spectra of the complexes isolated. The calculations were carried out with different basis sets (B3LYP/6-31+G* and B3LYP/6-31++G**) and the spectra have been recorded either under vacuum or with the polarizable continuum model to account for solvent effects. To validate our choice of basis sets, geometry optimizations were performed at the B3LYP/6-31+G* level of theory, with the pseudo potential LANL2DZ for the indium, chlorine, and bromine atoms. The agreement between the experimentally determined and the computed structures supports the method used (Table S11, ESI†). The calculated predominant contributions to the absorption maximum of complex 3 (λmax = 461 nm) corresponds to electronic transitions from the HOMO → LUMO, HOMO−2 → LUMO, HOMO−1 → LUMO, and HOMO−1 → LUMO+1. On the other hand, the visible absorption maximum of complex 4 (λmax = 400 nm) corresponds exclusively to the HOMO → LUMO transition (see the ESI†). As illustrated in Fig. 3, the HOMO of 4 consists largely of contributions from the arylimino fragments, whereas the LUMO is delocalized over the atoms of the acenaphthene and imine moieties. This supports our assignments of the UV-vis spectral bands above.
 | ||
| Fig. 3 Electron density distributions of the molecular orbitals involved in the UV-Vis spectra for 4 calculated at the B3LYP/6-31+G(d) level of theory. | ||
The electrochemical behavior of both indium complexes at 298 K was studied using cyclic voltammetry (CV) (Table 1). The voltammograms of 3 in ACN and 4 in DMF are shown in Fig. 4. During the cathodic scan of 4, a series of reduction waves were observed between −1.0 V to −2.2 V. The first two reduction waves appeared to be quasi-reversible on the timescale of the experiment. We attribute these first two reduction waves to the reduction of the indium(III) center on 4, since this process may involve some structural changes due to the dissociation of Cl− anions. The third reduction wave occurred at −1.6 V and subsequent processes were probably due to reduction of ligand 2. The anodic processes occurring between −1.2 V and −0.1 V likely result from re-oxidation of the ligand-bound indium intermediate products.
 | ||
| Fig. 4 Cyclic voltammograms of (a) 3 in ACN solution and (b) 4 in DMF solution at 298 K with 0.10 M (nBu4N)PF6 as the supporting electrolyte and glassy carbon (3 mm in diameter) as the working electrode. All potentials reported are referenced to Fc+/Fc. Scan rate: 100 mV s−1. | ||
| Compound | E oxp/Eredp (V vs. Fc+/Fc) | |
|---|---|---|
| 3 | 4 | |
| Ligand reduction | −1.02/−1.13 | −1.23/−1.57 |
| −/−1.74 | −2.10/−2.21 | |
| −2.17/−2.29 | — | |
| Ligand oxidation | +1.41/− | — |
| +1.75/− | — | |
| In(III) reduction | −0.74/−0.81 | −0.96/−1.03 |
| −/−1.46 | −1.26/−1.29 | |
In the case of 3 in ACN, two irreversible anodic waves were observed at +1.4 V and +1.7 V, which arise from oxidation of the ligand.17 During the cathodic scan, a total of six reduction waves were observed but we could not assign the origin for each because of the equilibrium processes of 3 in solution (vide supra). The first reduction at −0.8 V is electrochemically reversible with ΔEp = 60 mV, while the next two reduction waves appear to be chemically reversible. The electrochemical reversibility of the first reduction wave suggested an increased stability of [(p-MeOAr-BIAN)2InCl2]+, since no reversible redox cycle was observed in 4.
The voltammograms of both indium(III) complexes show considerably different behavior from their corresponding Ar-BIAN ligand (CV of 2 in DMF in ESI, Fig. S15†; CV of 1 in ACN can be found in ref. 17). In particular, the indium(III) complexes exhibit more reduction waves due to electrochemical processes at the indium(III) center. This indicates that 3 and 4 can be readily converted to their indium(I) congeners by using mild reducing agents with reduction potentials between −1.0 V to −2.0 V.27
Herein we have introduced an essentially solvent-free, atom-efficient route to the synthesis of Ar-BIAN ligands and their indium(III) complexes through a mechanochemical milling approach. This methodology circumvents the ZnCl2 templating step frequently used in the synthesis of Ar-BIAN ligands, and reduces the reaction time for the synthesis of the indium(III) complexes. This protocol can be generally applied to electron-donating and electron-withdrawing variants of the BIAN ligands and complexes. Both complexes absorb visible light, although the complex bearing the –OMe group absorbs further in the red region. The electrochemical studies support the feasibility of reduction of the indium(III) complexes to their indium(I) derivatives, both of which will be investigated as potential photosensitizers in the future.
F. G. would like to thank the NTU start-up grant (M4080552) and the MOE Tier 1 grant (M4011441) for financial support. H. S. S. is supported by a NTU start-up grant (M4081012), a MOE Tier 1 grant (M4011144), and the Nanyang Assistant Professorship (M4081154). H. S. S. also thanks the Solar Fuels Laboratory at NTU and the Singapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE) CREATE Programme.
References
- (a) E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC; (b) N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- C. X. Cui, J. Su and R. H. Baughman, Method for the mechanochemical preparation of high performance ferroelectric ceramics, US pat, US6136229A, 2000 Search PubMed.
- (a) V. Sepelak, A. Duvel, M. Wilkening, K.-D. Becker and P. Heitjans, Chem. Soc. Rev., 2013, 42, 7507–7520 RSC; (b) C. Suryanarayana, Prog. Mater. Sci., 2001, 46, 1–184 CrossRef CAS; (c) S. L. James, et al. , Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- (a) P. Balaz, et al. , Chem. Soc. Rev., 2013, 42, 7571–7637 RSC; (b) T. K. Purkait, Ph.D. thesis, Tulane University, 2013 Search PubMed.
- (a) M. R. Caira, L. R. Nassimbeni and A. F. Wildervanck, J. Chem. Soc., Perkin Trans. 2, 1995, 2213–2216 RSC; (b) M. C. Etter and D. A. Adsmond, J. Chem. Soc., Chem. Commun., 1990, 589–591 RSC; (c) M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS; (d) A. O. Patil, D. Y. Curtin and I. C. Paul, J. Am. Chem. Soc., 1984, 106, 348–353 CrossRef CAS; (e) V. R. Pedireddi, W. Jones, A. P. Chorlton and R. Docherty, Chem. Commun., 1996, 987–988 RSC; (f) F. Toda, K. Tanaka and A. Sekikawa, J. Chem. Soc., Chem. Commun., 1987, 279–280 RSC.
- L. Chen, M. O. Bovee, B. E. Lemma, K. S. M. Keithley, S. L. Pilson, M. G. Coleman and J. Mack, Angew. Chem., Int. Ed., 2015, 54, 11084–11087 CrossRef CAS PubMed.
- (a) C. J. Adams, M. A. Kurawa, M. Lusi and A. G. Orpen, CrystEngComm, 2008, 10, 1790–1795 RSC; (b) V. P. Balema, J. W. Wiench, M. Pruski and V. K. Pecharsky, Chem. Commun., 2002, 1606–1607 RSC; (c) G. A. Bowmaker, N. Chaichit, C. Pakawatchai, B. W. Skelton and A. H. White, Dalton Trans., 2008, 2926–2928 RSC; (d) G. A. Bowmaker, B. W. Skelton and A. H. White, Inorg. Chem., 2009, 48, 3185–3197 CrossRef CAS PubMed; (e) T. Chen, B. Liang and X. Xin, J. Solid State Chem., 1997, 132, 291–293 CrossRef CAS; (f) G. Hihara, M. Satoh, T. Uchida, F. Ohtsuki and H. Miyamae, Solid State Ionics, 2004, 172, 221–223 CrossRef CAS; (g) P. J. Nichols, C. L. Raston and J. W. Steed, Chem. Commun., 2001, 1062–1063 RSC; (h) D. N. T. Ohshita, A. Tsukamoto, N. Tsuchiya, T. Isobe and N. Y. A. H. I. M. Senna, Ann. Chim. Sci. Mater., 2002, 27, 91–101 CrossRef; (i) S. K. Thabet, H. A. Tayim and M. U. Karkanawi, Inorg. Nucl. Chem. Lett., 1972, 8, 211–213 CrossRef CAS; (j) X. Yao, L. Zheng and X. Xin, J. Solid State Chem., 1995, 117, 333–336 CrossRef CAS; (k) A. Kobayashi, T. Hasegawa, M. Yoshida and M. Kato, Inorg. Chem., 2016, 55, 1978–1985 CrossRef CAS PubMed.
- D. W. Peters and R. G. Blair, Faraday Discuss., 2014, 170, 83–91 RSC.
- N. R. Rightmire, T. P. Hanusa and A. L. Rheingold, Organometallics, 2014, 33, 5952–5955 CrossRef CAS.
- (a) Y. X. Shi, R. Z. Liang, K. A. Martin, D. G. Star, J. Diaz, X. Y. Li, R. Ganguly and F. Garcia, Chem. Commun., 2015, 51, 16468–16471 RSC; (b) Y. X. Shi, R. Z. Liang, K. A. Martin, N. Weston, S. Gonzalez-Calera, R. Ganguly, Y. Li, Y. Lu, A. J. M. Ribeiro, M. J. Ramos, P. A. Fernandes and F. García, Inorg. Chem., 2015, 54, 6423–6432 CrossRef CAS PubMed; (c) M. Wu. M, A. M. Gill, L. Yunpeng, L. Falivene, L. Yongxin, R. Ganguly, L. Cavallo and F. Garcia, Dalton Trans., 2015, 44, 15166–15174 RSC.
- (a) A. Agiral, H. S. Soo and H. Frei, Chem. Mater., 2013, 25, 2264–2273 CrossRef CAS; (b) S. Gazi, W. K. H. Ng, R. Ganguly, A. M. P. Moeljadi, H. Hirao and H. S. Soo, Chem. Sci., 2015, 6, 7130–7142 RSC; (c) H. S. Soo, A. Agiral, A. Bachmeier and H. Frei, J. Am. Chem. Soc., 2012, 134, 17104–17116 CrossRef CAS PubMed; (d) S. K. Muduli, S. Wang, S. Chen, C. F. Ng, C. H. A. Huan, T. C. Sum and H. S. Soo, Beilstein J. Nanotechnol., 2014, 5, 517–523 CrossRef CAS PubMed; (e) H. Shao, S. K. Muduli, P. D. Tran and H. S. Soo, Chem. Commun., 2016, 52, 2948–2951 RSC; (f) H. S. Soo, A. C. Komor, A. T. Iavarone and C. J. Chang, Inorg. Chem., 2009, 48, 10024–10035 CrossRef CAS PubMed; (g) H. S. Soo, M. L. Macnaughtan, W. W. Weare, J. Yano and H. M. Frei, J. Phys. Chem. C, 2011, 115, 24893–24905 CrossRef CAS; (h) H. S. Soo, M. T. Sougrati, F. Grandjean, G. J. Long and C. J. Chang, Inorg. Chim. Acta, 2011, 369, 82–91 CrossRef CAS; (i) J. W. Kee, Y. Y. Ng, S. A. Kulkarni, S. K. Muduli, K. Xu, R. Ganguly, Y. Lu, H. Hirao and H. S. Soo, Inorg. Chem. Front., 2016, 3 10.1039/C5QI00221D , Adv. Article.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- P. Cintas, Synlett, 1995, 1087–1096 CrossRef CAS.
- L. A. Paquette, in Green Chemistry. Frontiers in Benign Chemical Synthesis and Processes, ed. P. T. Anastas and T. C. Williamson, Oxford University Press, Oxford, 1998, 250 Search PubMed.
- J. Podlech and T. C. Maier, Synthesis, 2003, 0633–0655 CrossRef CAS.
- C. J. Li and T. H. Chan, Tetrahedron Lett., 1991, 32, 7017–7020 CrossRef CAS.
- K. Hasan and E. Zysman-Colman, J. Phys. Org. Chem., 2013, 26, 274–279 CrossRef CAS.
- M. Shiotsuki, P. S. White, M. Brookhart and J. L. Templeton, J. Am. Chem. Soc., 2007, 129, 4058–4067 CrossRef CAS PubMed.
- L. Li, P. S. Lopes, V. Rosa, C. A. Figueira, M. A. N. D. A. Lemos, M. T. Duarte, T. Aviles and P. T. Gomes, Dalton Trans., 2012, 41, 5144–5154 RSC.
- N. J. Hill, G. Reeske, J. A. Moore and A. H. Cowley, Dalton Trans., 2006, 4838–4844 RSC.
- K. V. Vasudevan and A. H. Cowley, New J. Chem., 2011, 35, 2043–2046 RSC.
- (a) L. A. Cameron, J. W. Ziller and A. F. Heyduk, Chem. Sci., 2016, 7, 1807–1814 RSC; (b) D. A. Evans, L. M. Lee, I. Vargas-Baca and A. H. Cowley, Organometallics, 2015, 34, 2422–2428 CrossRef CAS; (c) W. W. Kramer, L. A. Cameron, R. A. Zarkesh, J. W. Ziller and A. F. Heyduk, Inorg. Chem., 2014, 53, 8825–8837 CrossRef CAS PubMed.
- F. Ragaini, S. Cenini, S. Tollari, G. Tummolillo and R. Beltrami, Organometallics, 1999, 18, 928–942 CrossRef CAS.
- (a) A. L. Gottumukkala, J. F. Teichert, D. Heijnen, N. Eisink, S. van Dijk, C. Ferrer, A. van den Hoogenband and A. J. Minnaard, J. Org. Chem., 2011, 76, 3498–3501 CrossRef CAS PubMed; (b) C. S. K. Mak, H. L. Wong, Q. Y. Leung, W. Y. Tam, W. K. Chan and A. B. Djurišić, J. Organomet. Chem., 2009, 694, 2770–2776 CrossRef CAS.
- (a) E. H. H. Chow, F. C. Strobridge and T. Friščić, Chem. Commun., 2010, 46, 6368–6370 RSC; (b) J. G. Hernández, I. S. Butler and T. Friščić, Chem. Sci., 2014, 5, 3576–3582 RSC.
- B. R. Whittlesey and I. P. Ittycheriah, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1994, 50, 693–695 CrossRef.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1439070 and 1439071. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00978f |
| ‡ These authors jointly supervised this work. |
| This journal is © The Royal Society of Chemistry 2016 |