
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective formation of silver(I) bis-phospholane macrocycles and further evidence that gold(I) is smaller than silver(I)†
Markus
Streitberger
,
Andy
Schmied
,
Reinhard
Hoy
and
Evamarie
Hey-Hawkins
*
Institute of Inorganic Chemistry, Faculty of Chemistry and Mineralogy, Universität Leipzig, Johannisallee 29, 04103 Leipzig, Germany. E-mail: hey@uni-leipzig.de
First published on 18th May 2016
Abstract
A new synthetic approach to highly flexible bis-phospholanes from 1-benzylphospholane (1) as starting material is described. Silver(I) macrocycles containing 16 ([Ag2(μ-3a)2](BF4)2, 4), 20 ([Ag2(μ-3b)2](BF4)2, 5), 24 ([Ag2(μ-3c)2](BF4)2, 6), and 28 ([Ag2(μ-3d)2](BF4)2, 7) atoms in the ring were obtained in one step from AgBF4 and the respective bis-phospholane (C4H8P)(CH2)n(PC4H8) (n = 5, 7, 9, 11; 3a–d) in excellent yields. Comparison of 6 with the previously reported isomorphous complex [Au2(μ-3c)2](BF4)2 gave further evidence that gold(I) is significantly smaller than silver(I). All complexes were fully characterized by NMR and IR spectroscopy, mass spectrometry, and X-ray diffraction.
Introduction
Bis-phosphines of the general type R2PCH2PR2 (R = Ph, Me) are well known to form macrocyclic dinuclear complexes with silver(I).1 Due to the higher stability of five- or six-membered chelate rings, bis-phosphines R2P(CH2)nPR2 with n = 2 or 3 form mononuclear complexes as well.2 The coordination chemistry of bis-phosphines with larger flexible methylene spacers has been highly neglected so far. Effendy et al. reported a series of silver(I) complexes of Ph2P(CH2)nPPh2 (n = 3–6).3 In this case, ring formation was supported by additional silver(I)–anion coordination. Dinuclear silver(I) macrocycles with bridging Ph2P(CH2)nPPh2 ligands are known, for example, with AgNO3 for n = 3 and 4,4 AgCl for n = 5,5 and AgBF4 with n = 3.6 Crabtree et al. reported the unexpected formation of [Ag2{μ-tBu2P(CH2)3PtBu2}2](BF4)2 as a byproduct when employing AgBF4 for eliminating chlorido ligands from rhodium complexes.7 While gold(I) complexes of highly flexible bis-phosphines with an even number of carbon atoms in the methylene spacer seem to favor polymerization,8 the affinity of silver(I) for coordination numbers three and four enables the formation of macrocycles with 1,6-bis(diphenylphosphino)hexane and bridging perchlorate anions as well.9 The opposite trend of greater influence of the methylene spacer was reported by Puddephatt et al. They combined Ph2P(CH2)nPPh2 (n = 1–6) with trans-1,2-bis(4-pyridyl)ethylene and silver(I) trifluoroacetate and obtained macrocycles for n = 1 and 5 and a one-dimensional polymeric structure for n = 6.10 Recently, we reported the coordination behavior of highly flexible bis-phospholanes with solely aliphatic backbone towards gold(I) ions.11Because the coordination chemistry of bis-phospholanes towards silver(I) has been highly neglected so far, we extended our studies on the coordination chemistry of bis-phospholanes to silver(I) tetrafluoridoborate. The weakly coordinating tetrafluoridoborate anion should allow the formation of argentophilic interactions in the solid state and thus enable comparison with the aurophilic interactions observed in gold(I) complexes.12 Furthermore, we hoped to obtain single crystals that are isomorphous with the previously reported gold(I) complexes, to add further examples to the reported ones13–16 to additionally support the prediction based on theoretical calculations that gold(I) is significantly smaller than silver(I).17
Results and discussion
Improved synthesis of bis-phospholanes
We previously reported a three-step syntheses of the bis-phospholane ligands 3a–3d using a modification of the procedure reported by Haddow et al. starting from 1-phenylphospholane.11,18Treating two equivalents of 1-benzylphospholane (1) with 1,x-dibromoalkanes (x = 5, 7 or 9) in acetonitrile gave the bis-benzylphospholanium salts 2a–c in quantitative yield. In contrast to their phenyl analogues, 2a–c can be treated directly with lithium aluminum hydride to afford the corresponding bis-phospholanes 3a–c (Scheme 1), thus avoiding the harsh conditions required for the basic hydrolysis of the phenyl analogues at high temperature, which can lead to degradation of functional groups.
 | ||
| Scheme 1 New synthetic approach to bis-phospholanes 3a–c. | ||
Although the reductive cleavage of benzyl-substituted phosphonium salts had been described as early as 1975 by Horner et al., only little attention was paid to the benzylphospholane moiety up to now.19
Silver(I) bis-phospholane macrocyles
Addition of AgBF4 to a solution of 3a, 3b, 3c, or 3d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11 in dichloromethane led to macrocycles [Ag2(μ-3a–d)2](BF4)2 (4–7) in excellent yield by self-assembly under thermodynamic control (Scheme 2).
11 in dichloromethane led to macrocycles [Ag2(μ-3a–d)2](BF4)2 (4–7) in excellent yield by self-assembly under thermodynamic control (Scheme 2).
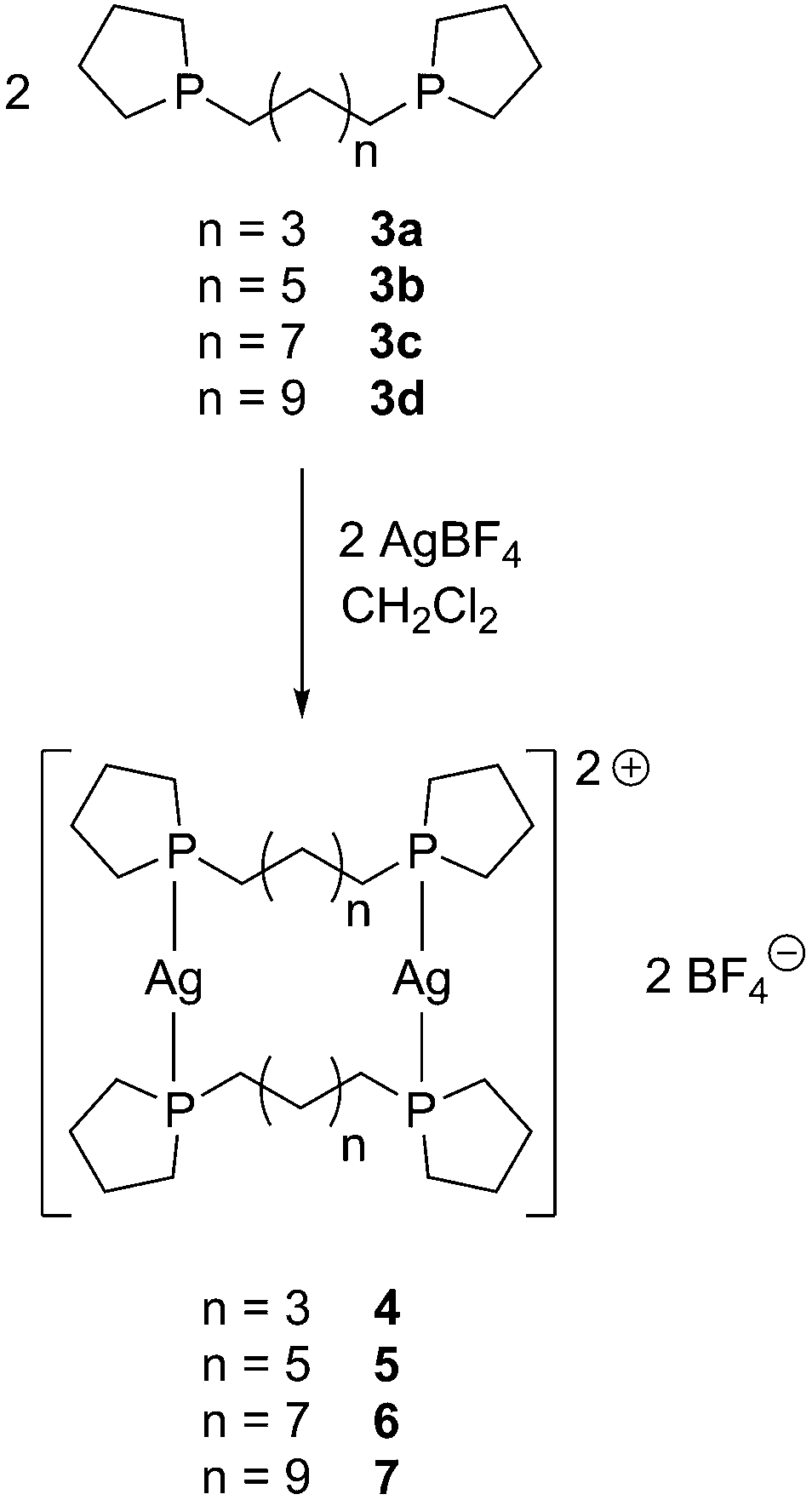 | ||
| Scheme 2 Selective formation of silver(I) macrocycles 4–7. | ||
Complexes 4–7 were obtained as white, light-stable solids. In the 31P{1H} NMR spectra, a downfield shift on coordination from about −27 ppm (3a–d) to about −1 ppm (4–7) occurred. In the mass spectra (ESI(+) mode) [M − BF4]+ and [Ag(3a–d)]+ peaks were observed, indicating the presence of the macrocycles also in solution. Even though the formation of oligomers is conceivable, only macrocycles are observed, which seem to be the thermodynamic products. Coordination-driven self-assembly strategies have been previously employed to obtain well-defined macrocycles.20 One recent example is the selective formation of a dimetallic silver(I) bis-carbene macrocycle (formed selectively in 89% yield after 20 h at 55 °C).21 Solvent effects can also play a major role, as was shown in the solvent-dependent formation of helical polymers and discrete bi-, tri-, and tetranuclear metallacycles from AgX (X = OTf, ClO4, PF6, and BF4) and 9,10-bis(diphenylphosphino)anthracene.22
The complexes 4–7 are soluble in dichloromethane or chloroform, and crystals suitable for X-ray analysis could be obtained over several days from saturated dichloromethane/toluene solutions at room temperature. The crystal structures are shown in Fig. 1, and selected bond lengths and angles are given in Table 1.
 | ||
| Fig. 1 Molecular structures of 4, 5, 6 and 7 (from top to bottom). Ellipsoids are drawn at 50% (4, 5, 6) and 30% (7) probability. H atoms, counterions, and solvent molecules are omitted for clarity. See ESI† for the second independent molecule of 7. | ||
| 4 | 5 | 6 |
7a |
|
|---|---|---|---|---|
| a Values for the second independent molecule are given in brackets. | ||||
| Ag1–P1 | 237.21(8) | 239.34(7) | 236.3(1) | 236.4(2) [238.7(2)] |
| Ag1–P2 | 237.02(8) | 238.84(6) | 236.4(1) | 236.6(2) [238.3(2)] |
| Ag2–P3 | 237.8(1) | 238.8(2) [237.5(2)] | ||
| Ag2–P4 | 238.7(1) | 238.2(2) [237.7(2)] | ||
| P1–Ag1–P2 | 176.89(1) | 173.49(1) | 173.81(4) | 170.91(7) [171.20(8)] |
| P3–Ag2–P4 | 172.44(4) | 171.72(7) [174.90(7)] | ||
All complexes show nearly linear coordination at the silver(I) ions with P–Ag–P bond angles between 170.91(7) and 176.89(1)°. Although the shortest Ag–F distances (267.9–300.5 pm) are much longer than the sum of the covalent radii, these weak electrostatic interactions cause a noticeable distortion from linearity. The Ag–P bond lengths range from 236.4(2) to 239.34(7) pm. The Ag–P bond lengths in [Ag2(μ-dppp)2](BF4)2 (dppp = 1,3-bis(diphenylphosphino)propane; Ag–P: 240.16(7), 239.35(7) pm) are slightly increased compared to 4–7, while the P–Ag–P bond angles are significantly smaller (P–Ag–P: 159.66(3)°).6 A P–Ag–P bond angle of 167.3(1)° was observed in [Ag(PPh3)2]BF4 with small Ag–P bond lengths of 232.1(3) pm and 232.2(3) pm.23
In 4–7 the large P–Ag–P bond angles result from electron-rich bis-phospholane ligands stabilizing the oxidation state +I and the weakly coordinating tetrafluoridoborate anions. The electron-rich complex [Ag2{μ-tBu2P(CH2)3PtBu2}2](BF4)2 has large P–Ag–P bond angles as well (169.49(4)°), but due to the bulky tert-butyl groups long Ag–P bonds of 240.6(1) pm.7 The use of electron-rich phosphines (PMe3) and weakly coordinating anions (PF6−) resulted in an Ag–P bond length of 237.5(1) pm and a nearly linear P–Ag–P group with a bond angle of 178.70(4)° in [Ag(PMe3)2]PF6.24
Schmidbaur et al. reported the isomorphous complexes bis(trimesitylphosphine)gold(I) and bis(trimesitylphosphine)silver(I) tetrafluoridoborate with gold(I) being about 6% smaller than silver(I).13 A similar trend is observed for the isomorphous complexes [Au(PPh3)2]BF416a and [Ag(PPh3)2]BF416b for which the Au–P bond is about 4% shorter than the Ag–P bond. A gold(I) radius nearly 7% smaller than in the isomorphous silver(I) complex was observed by Omary et al., on the basis of the M–N bond lengths (M = Ag, Au) in trinuclear pyrazolato complexes.14 Dias et al. reported the same trend in M–C bond lengths for non-isomorphous tris(ethylene)- and tris(cyclooctyne)gold(I) and -silver(I) complexes, and supported this observation by theoretical calculations.15
[Au2(μ-3c)2](BF4)2 and 6 give further evidence that gold(I) is smaller than silver(I) (M–P 229.3(1) and 229.9(1) pm in [Au2(μ-3c)2](BF4)2versus 236.3(1) and 236.4(1) pm in 6) and support theoretical calculations, even though weak Ag⋯F Coloumb interactions are present in both structures.11 These complexes fulfill the criteria (same ligands and counterions, same coordination number and geometry, isomorphous crystal lattice, and same experimental conditions) announced by Schmidbaur et al. for a scientific comparison of bond lengths.13Table 2 presents selected crystal data for 6 and [Au2(μ-3c)2](BF4)2, which underline the close crystallographic resemblance of the two complexes.
11
| 6 | [Au2(μ-3c)2](BF4)2 | |
|---|---|---|
| Formula | C34H68Ag2B2F8P4·0.5C7H8 | C34H68Au2B2F8P4·0.5C7H8 |
| Crystal system | Triclinic | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
| Z | 2 | 2 |
| a [pm] | 1034.8(5) | 1037.3(5) |
| b [pm] | 1143.1(5) | 1105.6(5) |
| c [pm] | 2051.2(5) | 2090.2(5) |
| V [nm3] | 2.364(2) | 2.388(2) |
Steric interactions between the phospholane moieties are not observed. In bis(trimesitylphosphine)silver(I) tetrafluoridoborate, for example, the Ag–P bond lengths (244.09(9) pm) are increased by more than 10 pm compared to [Ag(PPh3)2]BF4 (Ag–P: 232.1(3) pm, 232.2(3) pm), and Alyea et al. observed significant intramolecular methyl–methyl interactions in bis(trimesityl)silver(I) cations.25 Furthermore, the sterically demanding mesityl groups in [M(PMes3)2]BF4 (M = Ag, Au) prohibit any deviation from linearity.
Summary
We have presented a new and simplified route to bis-phospholane ligands using 1-benzylphospholane (1) as a starting material. Furthermore, the fascinating coordination behavior of 3a–d towards silver(I) tetrafluoridoborate was investigated. A comparison of the crystal structures of the isomorphous complexes 6 and [Au2(μ-3c)2](BF4)2 give further evidence that gold(I) is significantly smaller than silver(I). Argentophilic interactions were not observed in the solid state.Experimental section
Materials and methods
All reactions were carried out in a nitrogen atmosphere by using standard Schlenk techniques and anhydrous solvents, which were purified with an MB SPS-800 solvent purification system from MBRAUN or as mentioned in the literature. Benzylphosphine,26 1,3,2-dioxathiepane-2,2-dioxide,27 and 3d11 were prepared according to the literature. All other chemicals were used as purchased. NMR spectra were recorded at 298 K with a Bruker AVANCE DRX 400 spectrometer. The chemical shifts δ of 1H, 13C, 31P are reported in parts per million (ppm) at 400.12, 100.63 and 162.02 MHz, respectively, with tetramethylsilane as an internal standard and referencing to the unified scale. Coupling constants J are given in Hz. FTIR spectra were recorded with a PerkinElmer Spectrum 2000 FTIR spectrometer, scanning between 400 and 4000 cm−1, by using KBr pellets. Wavenumbers ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) are reported in cm−1. Mass spectra were recorded with ESQUIRE 3000 plus (ESI) and Finnigan MAT 8230 (EI) spectrometers. Elemental analyses were carried out with a Heraeus VARIO EL oven. Melting points were measured in sealed capillaries by using a variable heater from Gallenkamp.
are reported in cm−1. Mass spectra were recorded with ESQUIRE 3000 plus (ESI) and Finnigan MAT 8230 (EI) spectrometers. Elemental analyses were carried out with a Heraeus VARIO EL oven. Melting points were measured in sealed capillaries by using a variable heater from Gallenkamp.
Crystallographic data for compounds 4, 5, 6 and 7 were collected with an Oxford Diffraction CCD Xcalibur-S diffractometer (data reduction with CrysAlis Pro,28 including the program SCALE 3 ABSPACK29 for empirical absorption correction) by using MoKα irradiation (λ = 71.073 pm) and ω-scan rotation. Structures were solved with the SIR tool.30 Refinement was performed with SHELXL97.31 Non-hydrogen atoms were refined anisotropically. Hydrogen atoms were refined by constrained methods using the riding model. The refinement was carried out with the least-squares method on F2. Final R indices were calculated as follows: R1 = ∑||Fo|−|Fc||/∑|Fo| and wR2 = {∑[w(Fo2 − Fc2)2]/∑w(Fo2)2}1/2. Figures were drawn with ORTEP.32 CCDC 1439660 (4), 1439661 (5), 1439662 (6) and 1439663 (7) contain the supplementary crystallographic data for this paper.
Synthesis and characterisation
Yield: 6.6 g (64%). Colorless oil. Found C, 73.96; H, 8.24. Calc. for C11H15P: C, 74.14; H, 8.48. IR: = 2941 (s), 1698 (s), 1601 (s), 1494 (m), 1452 (m), 1265 (s), 869 (m), 769 (m), 700 (s), 656 (m) cm−1. 1H NMR (CDCl3): δ = 7.25 (m, 2H), 7.15 (m, 3H), 2.65 (s, 2H), 1.65 ppm (m, 8H). 13C{1H} NMR (CDCl3): δ = 138.9 (d, JCP = 4.9), 128.9 (d, JCP = 5.5), 128.3 (s), 125.5 (d, JCP = 2.1), 35.4 (d, JCP = 20.5), 27.6 (d, JCP = 4.0), 25.2 ppm (d, JCP = 13.6). 31P{1H} NMR (CDCl3): δ = −16.7 ppm (s). MS (EI): m/z (%) = 178 [M]+ (5), 150 [M − C2H4]+ (15), 91 [M − C4H8P]+ (100), 77 [M − C5H10P]+ (5), 65 [M − C6H10P]+ (15).
Preparation of phosphonium salts
Dibromoalkane (4.0 mmol) was added to a stirred solution of 1-benzylphospholane (8.0 mmol) in acetonitrile (10 mL). After stirring for two days at 80 °C, the clear solution was cooled to rt and the product precipitated. After filtration, the residue was washed with n-pentane (5 mL). The resulting white solids (2a–c) were used without further purification. = 3060 (m), 3029 (m), 2906 (s), 2792 (w), 1601 (m), 1494 (s), 1456 (s), 1399 (s), 1266 (m), 1111 (m), 1028 (m), 889 (m), 831 (m), 774 (m), 701 (s), 573 (m), 518 (m), 490 (s) cm−1. 1H NMR (CD3CN): δ = 7.43 (m, 10H), 3.99 (d, 4H, JHP = 16.0), 2.49 (m, 4H), 2.31 (m, 8H), 1.91 (m, 4H,), 1.67 (m, 4H), 1.58 ppm (m, 6H). 13C{1H} NMR (CD3CN): δ = 131.1 (d, JCP = 5.1), 130.5 (d, JCP = 3.2), 130.2 (d, JCP = 9.1), 129.3 (d, JCP = 3.7), 31.0 (t, JCP = 16.1), 28.7 (d, JCP = 41.1), 26.8 (d, JCP = 5.1), 21.4 (d, JCP = 40.3), 21.2 (d, JCP = 4.4), 21.0 ppm (d, JCP = 50.2). 31P{1H} NMR (CD3CN): δ = 54.2 ppm (s). MS (ESI(+), CH3OH): m/z = 505 [M − Br]+, 213 [M − 2Br]2+.
= 3028 (m), 2999 (m), 2922 (s), 2877 (s), 1601 (m), 1495 (s), 1455 (s), 1404 (s), 1266 (m), 1117 (m), 1088 (m), 1074 (m), 1023 (w), 888 (m), 708 (s), 571 (w), 487 (m) cm−1. 1H NMR (CD2Cl2): δ = 7.48 (m, 4H), 7.39 (m, 6H), 4.24 (d, 4H, JHP = 15.6), 2.86 (m, 4H), 2.42 (m, 8H), 1.93 (m, 4H), 1.70 (m, 4H), 1.58 ppm (m, 10H). 13C{1H} NMR (CD3OD): δ = 131.2 (d, JCP = 5.1), 130.6 (d, JCP = 2.1), 130.2 (d, JCP = 9.1), 129.5 (d, JCP = 3.5), 31.1 (d, JCP = 15.9), 28.8 (s), 28.7 (d, JCP = 40.9), 27.0 (d, JCP = 4.9), 22.7 (d, JCP = 4.4), 21.6 (d, JCP = 43.0), 21.0 (d, JCP = 50.3). 31P{1H} NMR (CD2Cl2): δ = 54.0 ppm (s). MS (ESI(+), CH3OH): m/z = 533 [M − Br]+, 227 [M − 2Br]2+.
= 3029 (m), 2924 (s), 2909 (s), 2858 (m), 1631 (w), 1602 (w), 1495 (m), 1456 (m), 1406 (m), 1265 (m), 1114 (s), 1077 (m), 831 (m), 707 (s), 489 (m) cm−1. 1H NMR (CD3CN): δ = 7.51 (m, 4H), 7.39 (m, 6H), 4.32 (d, 4H, JHP = 15.6), 2.77 (m, 4H), 2.41 (m, 8H), 1.87 (m, 4H), 1.65 (m, 8H), 1.43 ppm (m, 10H). 13C{1H} NMR (CD3OD): δ = 131.2 (d, JCP = 5.1), 130.6 (d, JCP = 3.1), 130.2 (d, JCP = 9.0), 129.5 (d, JCP = 3.6), 31.6 (d, JCP = 15.7), 29.9 (s), 29.7 (s), 28.7 (d, JCP = 41.0), 27.0 (d, JCP = 4.9), 22.9 (d, JCP = 4.7), 22.1 (d, JCP = 42.9), 21.0 (d, JCP = 50.3). 31P{1H} NMR (CD3CN): δ = 53.7 (s). MS (ESI(+), CH3OH): m/z = 561 [M − Br]+, 241 [M − 2Br]2+.
Preparation of bis-phospholanes 3a–c
2.0 mmol of the respective bis-phosphonium salt (2a–c) were added to a suspension of 0.12 g (3.0 mmol) LiAlH4 in THF (40 mL). The reaction mixture was stirred at rt overnight and then quenched carefully with 10 mL of degassed methanol. The solvent was removed under reduced pressure and the residue extracted with hexanes (3 × 10 mL). The solvent was removed under reduced pressure and pure bis-phospholanes were obtained by bulb-to-bulb distillation at 10−3 mbar as colorless oils. The spectroscopic data of 3a–c are in agreement with those reported previously.11Preparation of compounds 4, 5, 6, 7
AgBF4 (97 mg; 0.50 mmol) was added to a stirred solution of the bis-phospholane (3a–d) (0.50 mmol) in CH2Cl2 (10 mL). After stirring for 3 h at rt, the solution was filtered and volatile compounds were removed in vacuo. The white solid obtained was washed with n-pentane (3 × 10 mL). Suitable crystals for X-ray diffraction studies could be obtained from saturated dichloromethane/toluene solutions at rt as colorless prisms (4–6) or needles (7). All compounds were dried in vacuo prior to elemental analysis. = 2936 (s), 2858 (m), 1637 (w), 1447 (w), 1411 (w), 1055 (s), 854 (w), 718 (w), 519 (w), 487 (w) cm−1. 1H NMR (CD2Cl2): δ = 2.12 (m, 8H), 1.86 (m, 24H), 1.66 (m, 8H), 1.55 ppm (m, 12H). 13C{1H} NMR (CD2Cl2): δ = 32.7 (br s), 27.6 (br s), 27.1 (br d, JCP = 16.1), 26.9 (br s), 25.1 ppm (br d, JCP = 19.1). 11B{1H} NMR (CD2Cl2): δ = −1.2 ppm (s). 31P{1H} NMR (CD2Cl2): δ = −0.3 ppm (s). MS (ESI(+), CH2Cl2, CH3CN): m/z = 791 [M − BF4]+, 351 [Ag(3a)]+.
= 2925 (s), 2851 (s), 2100 (w), 1810 (w), 1466 (s), 1448 (m), 1410 (s), 1306 (m), 1282 (m), 1183 (m), 1067 (s), 950 (m), 870 (m), 852 (m), 719 (m), 688 (m), 518 (s), 493 (m), 454 (m) cm−1. 1H NMR (CD2Cl2): δ = 2.12 (m, 8H), 1.86 (m, 28H), 1.63 (m, 8H), 1.47 (m, 12H), 1.34 ppm (m, 4H). 13C NMR (CD2Cl2): δ = 30.6 (br d, JCP = 12.0), 29.2 (br s), 27.4 (br s), 27.1 (br d, JCP = 16.2 Hz), 26.9 (br s), 25.1 ppm (br d, JCP = 19.6). 11B NMR (CD2Cl2): δ = −1.2 ppm (s). 31P{1H} NMR (CD2Cl2): δ = −0.8 ppm (s). MS (ESI(+), CH2Cl2, CH3CN): m/z = 847 [M − BF4]+, 379 [Ag(3b)]+.
= 2924 (s), 2848 (s), 1466 (m), 1449 (m), 1413 (m), 1304 (w), 1054 (s), 918 (w), 855 (m), 741 (w), 721 (w), 698 (m), 519 (m), 471 (m) cm−1. 1H NMR (CD2Cl2): δ = 2.11 (m, 8H), 1.88 (m, 24H), 1.64 (m, 8H), 1.51 (m, 16H), 1.32 ppm (m, 12H). 13C{1H} NMR (CD2Cl2): δ = 32.8 (s), 30.3 (br s), 29.4 (br s), 28.7 (s), 27.0 (s), 26.9 (s), 25.2 ppm (br s). 11B{1H} NMR (CD2Cl2): δ = −1.2 ppm (s). 31P{1H} NMR (CD2Cl2): δ = −2.4 ppm (s). MS (ESI(+), CH2Cl2, CH3CN): m/z = 903 [M − BF4]+, 407 [Ag(3c)]+.
= 2924 (s), 2850 (s), 1467 (s), 1449 (m), 1412 (s), 1304 (m), 1282 (m), 1262 (m), 1052 (s), 950 (m), 897 (w), 855 (m), 803 (m), 722 (m), 689 (m), 518 (s), 487 (m) cm−1. 1H NMR (CD2Cl2): δ = 2.21–1.76 (m, 32H), 1.75–1.18 ppm (m, 44H). 13C NMR (CD2Cl2): δ = 30.7 (d, JCP = 14.6), 30.7 (s), 29.2 (s), 29.0 (d, JCP = 12.3), 27.1 (d, JCP = 18.6), 26.8 (s), 26.6 (s), 25.3 ppm (d, JCP = 22.2). 11B{1H} NMR (CD2Cl2): δ = −1.2 ppm (s). 31P{1H} NMR (CD2Cl2): δ = −1.4 ppm (s). MS (ESI(+), CH2Cl2, CH3CN): m/z = 959 [M − BF4]+, 435 [Ag(3d)]+.
Acknowledgements
We gratefully acknowledge financial support from the Fonds der Chemischen Industrie (FCI, doctoral grant to A. S.), the European Union and the Free State of Saxony (ESF, project no. 100148808; R. H.), the Graduate School Leipzig School of Natural Sciences – Building with Molecules and Nano-objects (BuildMoNa), and the COST Action CM1302 Smart Inorganic Polymers (SIPs). We thank M. Böhlmann, F. Seifert, S. Märcker, and R. Zäbe for measurements of IR and NMR spectra and Dr P. Lönnecke, Dr K. Zeckert, and Dr R. Frank for help with structure solutions.References
- (a) D. Perreault, M. Drouin, A. Michel, V. M. Miskowski, W. P. Schaefer and P. D. Harvey, Inorg. Chem., 1992, 31, 695–702 CrossRef CAS; (b) Effendy, C. di Nicola, M. Nitiatmodjo, C. Pettinari, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2005, 358, 735–747 CrossRef CAS; (c) Effendy, J. V. Hanna, F. Marchetti, D. Martini, C. Pettinari, R. Pettinari, B. W. Skelton and A. H. White, Inorg. Chim. Acta, 2004, 357, 1523–1537 CrossRef CAS; (d) D. M. Ho and R. Bau, Inorg. Chem., 1983, 22, 4073–4079 CrossRef CAS; (e) S. P. Neo, Z.-Y. Zhou, T. C. W. Mak and T. S. A. Hor, Inorg. Chem., 1995, 34, 520–523 CrossRef CAS.
- P. Di Bernardo, G. Dolcetti, R. Portanova, M. Tolazzi, G. Tomat and P. Zanonato, Inorg. Chem., 1990, 29, 2859–2862 CrossRef CAS.
- Effendy, C. di Nicola, M. Fianchini, C. Pettinari, B. W. Skelton, N. Somers and A. H. White, Inorg. Chim. Acta, 2005, 358, 763–795 CrossRef CAS.
- E. R. T. Tiekink, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 1933–1934 CrossRef.
- A. Cassel, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 2521–2523 CrossRef.
- M. L. Smith, P. M. Almond, T. E. Albrecht-Schmitt and W. E. Hill, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2003, 59, m1013–m1014 CAS.
- S. P. Crabtree, A. S. Batsanov, J. A. Howard and M. Kilner, Polyhedron, 1998, 17, 367–372 CrossRef CAS.
- (a) M.-C. Brandys and R. J. Puddephatt, Chem. Commun., 2001, 1280–1281 RSC; (b) M.-C. Brandys, M. C. Jennings and R. J. Puddephatt, J. Chem. Soc., Dalton Trans., 2000, 4601–4606 RSC.
- S. Kitagawa, M. Kondo, S. Kawata, S. Wada, M. Maekawa and M. Munakata, Inorg. Chem., 1995, 34, 1455–1465 CrossRef CAS.
- M.-C. Brandys and R. J. Puddephatt, Chem. Commun., 2001, 1508–1509 RSC.
- M. Streitberger, A. Schmied and E. Hey-Hawkins, Inorg. Chem., 2014, 53, 6794–6804 CrossRef CAS PubMed.
- (a) C. Y. Chen, J. Y. Zeng and H. M. Lee, Inorg. Chim. Acta, 2007, 360, 21–30 CrossRef CAS; (b) L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2008, 47, 230–240 CrossRef CAS PubMed; (c) X. Liu, G.-C. Guo, M.-L. Fu, X.-H. Liu, M.-S. Wang and J.-S. Huang, Inorg. Chem., 2006, 45, 3679–3685 CrossRef CAS PubMed; (d) L. Dobrzanska, H. G. Raubenheimer and L. J. Barbour, Chem. Commun., 2005, 5050–5052 RSC; (e) H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 127, 756–797 CrossRef; (f) H. Schmidbaur and A. Schier, Angew. Chem., Int. Ed., 2015, 54, 746–784 CrossRef CAS PubMed.
- A. Bayler, A. Schier, G. A. Bowmaker and H. Schmidbaur, J. Am. Chem. Soc., 1996, 118, 7006–7007 CrossRef CAS.
- M. A. Omary, M. A. Rawashdeh-Omary, M. W. A. Gonser, O. Elbjeirami, T. Grimes, T. R. Cundari, H. V. K. Diyabalanage, C. S. P. Gamage and H. V. R. Dias, Inorg. Chem., 2005, 44, 8200–8210 CrossRef CAS PubMed.
- (a) M. Fianchini, C. F. Campana, B. Chilukuri, T. R. Cundari, V. Petricek and H. V. R. Dias, Organometallics, 2013, 32, 3034–3041 CrossRef CAS; (b) H. V. R. Dias, M. Fianchini, T. R. Cundari and C. F. Campana, Angew. Chem., Int. Ed., 2008, 120, 566–569 CrossRef; (c) H. V. R. Dias, M. Fianchini, T. R. Cundari and C. F. Campana, Angew. Chem., Int. Ed., 2008, 47, 556–559 CrossRef CAS PubMed; (d) A. Das, C. Dash, M. A. Celik, M. Yousufuddin, G. Frenking and H. V. R. Dias, Organometallics, 2013, 32, 3135–3144 CrossRef CAS; (e) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 51, 3940–3943 CrossRef CAS PubMed; (f) A. Das, C. Dash, M. Yousufuddin, M. A. Celik, G. Frenking and H. V. R. Dias, Angew. Chem., Int. Ed., 2012, 124, 4006–4009 CrossRef.
- (a) J.-C. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 611–613 CrossRef; (b) R. E. Bachman and D. F. Andretta, Inorg. Chem., 1998, 37, 5657–5663 CrossRef CAS PubMed.
- (a) P. Schwerdtfeger, P. D. W. Boyd, A. K. Burrell, W. T. Robinson and M. J. Taylor, Inorg. Chem., 1990, 29, 3593–3607 CrossRef CAS; (b) P. Schwerdtfeger, M. Dolg, W. H. E. Schwarz, G. A. Bowmaker and P. D. W. Boyd, J. Chem. Phys., 1989, 91, 1762–1774 CrossRef CAS; (c) P. Pyykko, Chem. Rev., 1988, 88, 563–594 CrossRef CAS; (d) P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123–17135 CrossRef CAS PubMed.
- M. F. Haddow, A. J. Middleton, A. G. Orpen, P. G. Pringle and R. Papp, Dalton Trans., 2009, 202–209 RSC.
- (a) L. Horner, P. Walach and H. Kunz, Phosphorus, 1975, 6, 63 CAS; (b) L. Horner, P. Walach and H. Kunz, Phosphorus Sulfur Relat. Elem., 1978, 5, 171–184 CrossRef CAS.
- E. M. López-Vidal, V. Blanco, M. D. García, C. Peinador and J. M. Quintela, Org. Lett., 2012, 14, 580–583 CrossRef PubMed.
- C.-X. Wang, Y. Gao, Y.-X. Deng, Y.-J. Lin, Y.-F. Han and G.-X. Jin, Organometallics, 2015, 34, 5801–5806 CrossRef CAS.
- R. Lin and J. H. K. Yip, Inorg. Chem., 2006, 45, 4423–4430 CrossRef CAS PubMed.
- J.-C. Wang, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, 52, 611–613 CrossRef.
- E. C. Alyea, S. Kannan and P. R. Meehan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, m365–m367 Search PubMed.
- E. C. Alyea, G. Ferguson and A. Somogyvari, Inorg. Chem., 1982, 21, 1369–1371 CrossRef CAS.
- L. Horner, H. Hoffmann and P. Beck, Chem. Ber., 1958, 91, 1583–1588 CrossRef CAS.
- I. Bonnaventure and A. B. Charette, J. Org. Chem., 2008, 73, 6330–6340 CrossRef CAS PubMed.
- CrysAlis Pro, Oxford Diffraction Ltd, Oxfordshire, UK, 2010 Search PubMed.
- SCALE3 ABSPACK, Oxford Diffraction Ltd, Oxfordshire, UK, 2010 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, SHELX97: Programs for Crystal Structure Analysis (Release 97-2), Universität Göttingen, Göttingen, 1997 Search PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1439660–1439663. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00945j |
| This journal is © The Royal Society of Chemistry 2016 |