
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
When two are better than one: bright phosphorescence from non-stereogenic dinuclear iridium(III) complexes†
Ruth E.
Daniels
a,
Stacey
Culham
a,
Michael
Hunter
a,
Marcus C.
Durrant
a,
Michael R.
Probert
b,
William
Clegg
b,
J. A. Gareth
Williams
 *c and
Valery N.
Kozhevnikov
*a
*c and
Valery N.
Kozhevnikov
*a
aDepartment of Applied Sciences, Northumbria University, Newcastle upon Tyne, NE1 8ST, UK. E-mail: valery.kozhevnikov@northumbria.ac.uk
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
cDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: j.a.g.williams@durham.ac.uk
First published on 17th March 2016
Abstract
A new family of eight dinuclear iridium(III) complexes has been prepared, featuring 4,6-diarylpyrimidines Ly as bis-N^C-coordinating bridging ligands. The metal ions are also coordinated by a terminal N^C^N-cyclometallating ligand LX based on 1,3-di(2-pyridyl)benzene, and by a monodentate chloride or cyanide. The general formula of the compounds is {IrLXZ}2Ly (Z = Cl or CN). The family comprises examples with three different LX ligands and five different diarylpyrimidines Ly, of which four are diphenylpyrimidines and one is a dithienylpyrimidine. The requisite proligands have been synthesised via standard cross-coupling methodology. The synthesis of the complexes involves a two-step procedure, in which LXH is reacted with IrCl3·3H2O to form dinuclear complexes of the form [IrLXCl(μ-Cl)]2, followed by treatment with the diarylpyrimidine LyH2. Crucially, each complex is formed as a single compound only: the strong trans influence of the metallated rings dictates the relative disposition of the ligands, whilst the use of symmetrically substituted tridentate ligands eliminates the possibility of Λ and Δ enantiomers that are obtained when bis-bidentate units are linked through bridging ligands. The crystal structure of one member of the family has been obtained using a synchrotron X-ray source. All of the complexes are very brightly luminescent, with emission maxima in solution varying over the range 517–572 nm, according to the identity of the ligands. The highest-energy emitter is the cyanide derivative whilst the lowest is the complex with the dithienylpyrimidine. The trends in both the absorption and emission energies as a function of ligand substituent have been rationalised accurately with the aid of TD-DFT calculations. The lowest-excited singlet and triplet levels correlate with the trend in the HOMO–LUMO gap. All the complexes have quantum yields that are close to unity and phosphorescence lifetimes – of the order of 500 ns – that are unusually short for complexes of such brightness. These impressive properties stem from an unusually high rate of radiative decay, possibly due to spin–orbit coupling pathways being facilitated by the second metal ion, and to low non-radiative decay rates that may be related to the rigidity of the dinuclear scaffold.
Introduction
The metal centre is an important component in luminescent cyclometallated iridium complexes, normally playing a key role in determining the nature of the luminescence. The high degree of covalency of the Ir–C bond typically leads to efficient spin–orbit coupling (SOC), facilitated by the heavy metal, such that the formally forbidden transitions of intersystem crossing and phosphorescence are enhanced.1 This is an important feature if the complex is to be used as a phosphor in organic light-emitting diodes for example, because it allows both triplet and singlet electro-excitation pathways to contribute to the emission, making the devices more efficient.2 Meanwhile, in bio-imaging, bright phosphorescence opens up time-resolved detection techniques.3,4 The past 15 years have seen increasingly rapid advances in both fields and a large number of such complexes have been thoroughly investigated.5The vast majority of examples to date contain one metal centre. Incorporating two or more metal centres in one luminophore can be an additional tool for influencing photophysical properties, particularly if they are in close proximity.6 Of particular interest is the possibility that the presence of a second metal might increase the radiative rate constant of phosphorescence, kr, compared to a mononuclear complex, by introducing a larger SOC effect than that associated with a single metal centre. Such a notion clearly assumes some synergy between the metal units. However, SOC pathways are complex and poorly understood even in mononuclear complexes,1a and so a largely empirical approach based on experimental data is currently required in order to probe whether such effects are at play in multinuclear systems. The possibility of promoting kr is of particular significance to the design of efficient red emitters, owing to the natural tendency of kr to be lower for low-energy emitters (according to the Einstein coefficient for spontaneous emission which depends on ν3) and to the normally faster rate of competitive non-radiative decay that low-energy excited states are subject to, in line with the energy-gap law.2e
The synthesis of multinuclear complexes as single pure isomers is generally not easy. The D3 or C2 local symmetry of Ir(III) complexes containing bidentate ligands {e.g. those based on Ir(N^C)3 or [Ir(N^C)2(L^X)]+ structures} means that such complexes are chiral and are formed as a racemic mixture of Λ and Δ enantiomers. Since enantiomers have identical [linear] optical properties, this is generally of little consequence for mononuclear complexes,7 but when two or more such metal centres are incorporated into one molecule, a mixture of diastereoisomers necessarily forms (e.g. ΛΛ/ΔΔ + ΛΔ in the case of two metal centres). Diastereoisomers have different properties and need to be separated, a process that can often be exceptionally laborious and time-consuming. For example, Tsuboyama and co-workers described the synthesis of a red-emitting dinuclear iridium complex, related to the archetypal complex Ir(ppy)3, but featuring a bridging 1,4-di(2-pyridyl)benzene ligand (dpb).8 The desired di-iridium complex {Ir(ppy)2}(dpb) could be isolated in only a 3% yield after column chromatography. In many other instances of multinuclear complexes, it has proved impossible to isolate all the isomers in pure form.9
One way to potentially alleviate this problem is to make use of tridentate ligands. For example, the achirality of bis-terpyridyl metal complexes (C2v symmetry) compared to the chirality of D3 tris-bipyridyl analogues has driven extensive research into the supramolecular chemistry of terpyridyl complexes since the 1980s.10 In the field of cyclometallated iridium(III) chemistry, the use of tridentate ligands has lagged far behind that of bidentate ligands, although examples of both charge-neutral and cationic complexes have been reported.11–13 In particular, achiral mononuclear Ir(III) complexes of the form Ir(N^C^N)(N^C)X – comprising derivatives of the tridentate N^C^N-cyclometallating ligand 1,3-bis(2-pyridyl) benzene in conjunction with bidentate N^C-cyclometallating 2-phenylpyridines and monodentate halide or acetylide ligands X – have been found to display excellent luminescent properties, with luminescence quantum yields approaching unity in some case. Some have been successfully incorporated into OLEDs, showing good performance.12d,e
We reasoned that such use of the N^C^N motif, in conjunction with a 4,6-diarylpyrimidine as a bis-N^C-coordinating bridging unit, could provide an attractive route to achiral dinuclear complexes, with none of the problems associated with the formation of additional isomers. Our prototypical dinuclear complex of this new class is {Ir(LACl)2}L1 (Fig. 1). We have also prepared a series of seven other such complexes, which highlight the structural diversity offered by the approach in terms of variation of the N^C^N ligand (labelled alphabetically LA to LC), the bis-N^C coordinating ligand (labelled numerically L1 to L5), and metathesis of the monodentate ligand. Note that the approach requires that the N^C^N ligand be symmetrically substituted, i.e., featuring a C2 axis or mirror plane through the central ring; the use of asymmetrically-substituted ligands would re-introduce chirality.14 The structures of the tri- and bidentate ligands used are shown in Fig. 1a, with the corresponding complexes in Fig. 1b. The synthesis and characterisation of these new luminophores are reported here, together with a study of their impressive luminescence properties interpreted with the aid of TD-DFT calculations.
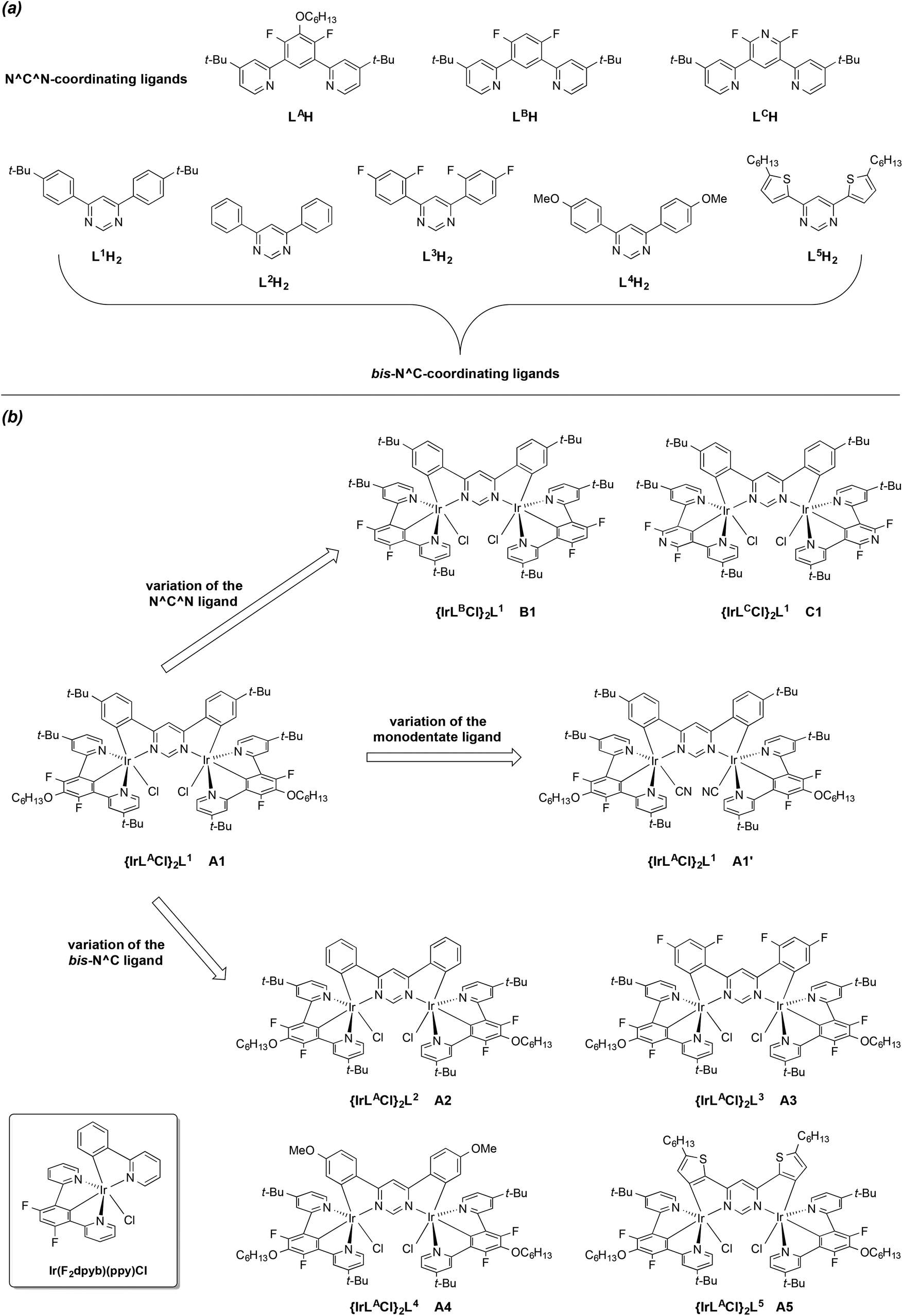 | ||
| Fig. 1 Structural formulae of the ligands (a) and complexes (b) investigated in this work. The structure of Ir(F2dpyb)(ppy)Cl, used as a model mononuclear complex for comparison,12e is shown in the boxed inset at the bottom left. | ||
Results and discussion
Synthesis
Our objectives were to explore the luminescence properties of this new family of complexes, examining the influence of the substitution pattern in the bridging bis-N^C ligand and, independently, in the N^C^N ligand. Thus, we prepared a series of five 4,6-diaryl-substituted pyrimidines L1H2–L5H2, with varying degrees of electron-withdrawing or donating power, ranging from difluorophenyl through to 2-thienyl (Fig. 1a). These proligands were readily accessible using Suzuki–Miyaura cross-coupling reactions between 4,6-dichloropyrimidine and the corresponding arylboronic acids (Scheme 1a). We have previously used similar pyrimidine-based ligands, together with their pyrazine analogues, to access a range of dinuclear Pt(II) complexes with widely-tuneable emission.15 Details of all new syntheses and the characterisation of new compounds are provided in the ESI.†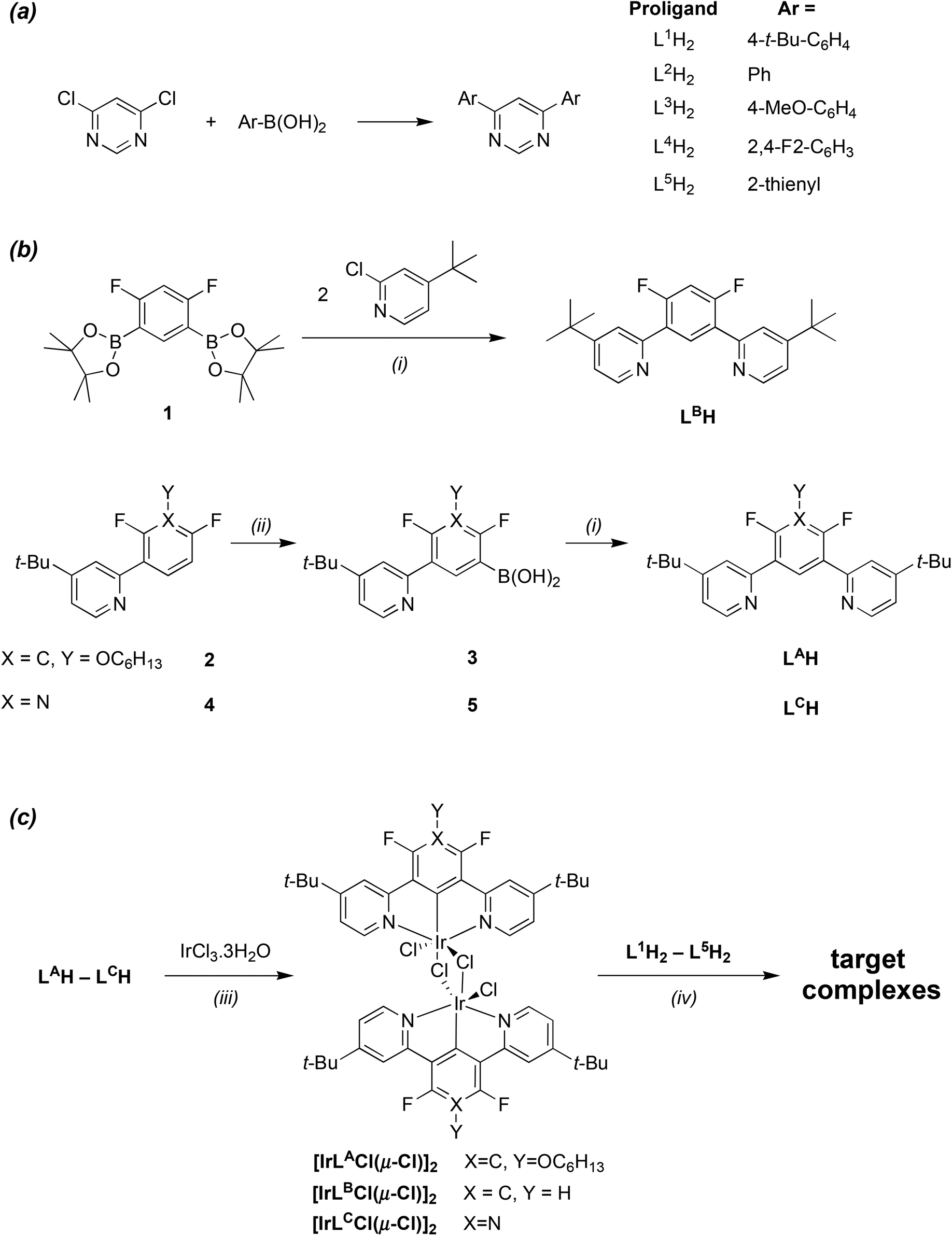 | ||
| Scheme 1 Synthetic routes to (a) the bis-N^C proligands; (b) the N^C^N proligands, and (c) the target complexes (i) Pd cat. (ii) BuLi then B(OiPr)3, work-up with H3O+. (iii) Ethoxyethanol/water. (iv) AgOTf, toluene. | ||
A series of three N^C^N proligands HAL–HLC were also synthesised, each comprising lateral 4-tert-butylpyridine groups, but in which the central ring was varied (Fig. 1a). We chose to include fluorine atoms in the central ring, ortho to the pyridyl rings, since competitive metallation of iridium at these positions has been shown to occur in the absence of substituents, giving N^C-coordinated products as opposed to the desired N^C^N systems.16,17 The fluorine atoms also facilitate the synthesis of these ligands, by ortho-directing the lithiation reactions involved in the synthesis (Scheme 1b). Thus, LAH was prepared from intermediate 2 (whose synthesis we described previously in the preparation of bis-N^C^N-coordinating ligands6h) by conversion to the boronic acid 3 through lithiation and reaction with tri-iso-propylborate, followed by Suzuki–Miyaura cross-coupling of 3 with 4-tert-butyl-2-chloropyridine. LCH was prepared similarly from intermediate 4, in turn obtained from 2,5-difluoropyridine. Meanwhile, LBH was obtained by cross-coupling the diboronate derivative 1 with two equivalents of 4-tert-butyl-2-chloropyridine, using a strategy established earlier in the development of N^C^N-coordinated platinum complexes.18
The preparation of the target complexes was carried out using a two-step procedure similar to that developed previously for related mononuclear complexes (Scheme 1c).12b,e In the first step, reaction of the tridentate proligands with hydrated iridium(III) chloride in a mixture of ethoxyethanol and water generated the dichloro-bridged dimers [Ir(LA–C)Cl(μ-Cl)]2. The presence of the tert-butyl substituents increases the solubility of these products compared to the related complexes of 1,3-dipyridyl-4,6-difluorobenzene, which are insoluble in all common solvents.12e Indeed, the solubility of all three dimers was sufficiently high to allow them to be characterised fully by 1H, 19F and 13C-NMR spectroscopy in CDCl3 solution (see ESI†).
In the second step, the dichloro-bridged dimers were reacted with the bis-N^C proligands L1H2–L5H2 under reflux in toluene, in the presence of silver triflate as a chloride scavenger. After the reaction, the mixture was treated with excess HCl (2 M, aq.) which ensures that the monodentate ligands in the desired products are exclusively Cl.
The identities of the resulting dinuclear complexes were confirmed by 1H, 19F and 13C NMR spectroscopy and by mass spectrometry. It is important to stress that, due to the very strong trans-influence of the formally anionic, metallated carbon atoms (see also Section 2 below), the product which forms in each case is exclusively that in which the pyrimidine ring is disposed trans to the central aryl ring of the N^C^N ligand. There is no evidence for the formation of isomers in which the metallated rings of the N^C and N^C^N units are trans to one another. In other words, the product in each case is uniquely that in which the pyrimidine nitrogen atom lies in the same plane as the N^C^N ligand. Isomeric products in which one or both of the metallated (i.e., C-coordinating) rings of the bis-N^C ligand occupy such a position are not formed. This observation is in line with earlier studies of related mononuclear systems.12b–e,17 Thus, in contrast to most other dinuclear complexes studied to date,6,8,9only one stereoisomer is formed for these complexes. The most characteristic features in the 1H NMR spectra are (i) a singlet with a very high chemical shift δ in the region 11–12 ppm, assigned to H2 of the pyrimidine ring, and (ii) a low-frequency multiplet in the aromatic region, δ 5.6–6.8 ppm, assigned to the protons ortho to the metallated carbon in the aryl ring of the bridging ligand. For ease of discussion in subsequent sections and representation in the figures that follow, the dinuclear complexes {Ir(LXCl)2}Ly (X = A, B or C; y = 1 to 5) will be referred to as Xy; e.g. {Ir(LACl)2}L1 is A1, {Ir(LBCl)2}L2 is B2etc.
A monodentate chloro ligand completes the coordination sphere of each Ir(III) centre in the dimers. Metathesis of this ligand with other monodentate ligands could provide access to a range of other derivatives, potentially allowing further tuning of the properties.17 We have demonstrated the ease with which such metathesis can be accomplished by converting {Ir(LACl)2}L1 (A1) to its cyano derivative {Ir(LACN)2}L1 (which we shall refer to as A1′), simply by treatment with excess KCN.
Crystallography
Crystals of {Ir(LACl)2}L3 (A3) suitable for X-ray diffraction analysis were obtained by slow evaporation of a solution of the complex in CH2Cl2/MeOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). The molecular structure is shown in Fig. 2, with key bond lengths and angles involving the metal listed in Table 1. The molecule has a crystallographic mirror plane, passing through atoms C38 and C39. Two-fold disorder was modelled for atoms C28–C30 (and their H atoms) of the hexyl chain (minor disorder component not shown). Displacement parameters, short contacts, and residual electron density peaks suggest further disorder of this chain and of the t-butyl groups, but this was not modelled.
:1 v/v). The molecular structure is shown in Fig. 2, with key bond lengths and angles involving the metal listed in Table 1. The molecule has a crystallographic mirror plane, passing through atoms C38 and C39. Two-fold disorder was modelled for atoms C28–C30 (and their H atoms) of the hexyl chain (minor disorder component not shown). Displacement parameters, short contacts, and residual electron density peaks suggest further disorder of this chain and of the t-butyl groups, but this was not modelled.
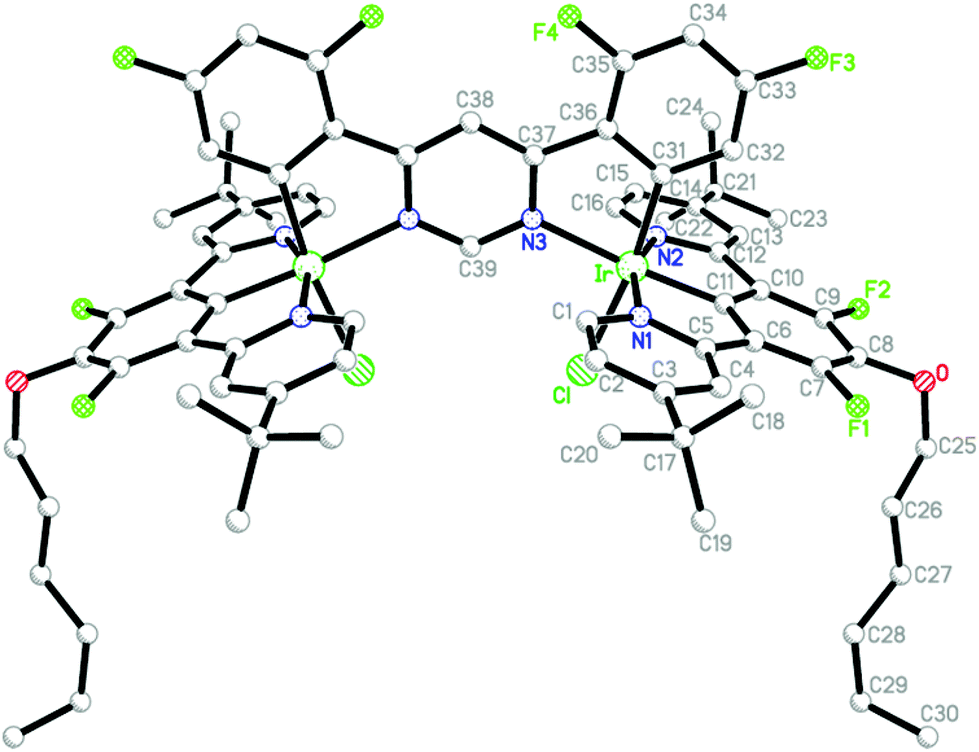 | ||
| Fig. 2 Molecular structure of the dinuclear complex {Ir(LACl)2}L3 (A3) in the crystal. Hydrogen atoms and minor disorder components have been omitted for clarity. | ||
| Bond length (Å) or angle (°) | X-ray data | DFT dataa |
|---|---|---|
| a DFT-derived values in square parentheses are for the geometry with imposed mirror symmetry about the central ligand (see text). | ||
| Ir–N1 | 2.049(5) | 2.090 [2.085] |
| Ir–N2 | 2.048(5) | 2.081 [2.085] |
| Ir–N3 | 2.157(5) | 2.208 [2.215] |
| Ir–C11 | 1.926(7) | 1.936 [1.936] |
| Ir–C31 | 1.996(5) | 2.024 [2.024] |
| Ir–Cl | 2.4406(15) | 2.483 [2.479] |
| N1–Ir–N2 | 160.4(2) | 159.9 [159.9] |
| N1–Ir–C11 | 80.3(2) | 79.9 [80.0] |
| N2–Ir–C11 | 80.1(2) | 80.1 [80.0] |
| N3–Ir–C11 | 173.4(2) | 176.1 [176.5] |
| N3–Ir–C31 | 78.9(2) | 78.3 [78.5] |
| N3–Ir–Cl | 93.66(12) | 92.2 [92.7] |
| C31–Ir–Cl | 172.6(2) | 170.1 [171.1] |
| C11–Ir–C31 | 95.3(3) | 97.9 [98.0] |
It should be noted that the structure contains highly disordered and unidentified solvent in large void volumes (shown in yellow in the a-axis projection in Fig. 3) between the molecules of the dinuclear complex. This was dealt with by the SQUEEZE procedure of the program PLATON.19 Calculations based on unit cell contents do not include this solvent. Full details of the structure are provided in the ESI,† and the structure has been deposited at the CCDC, reference no: 1415318.
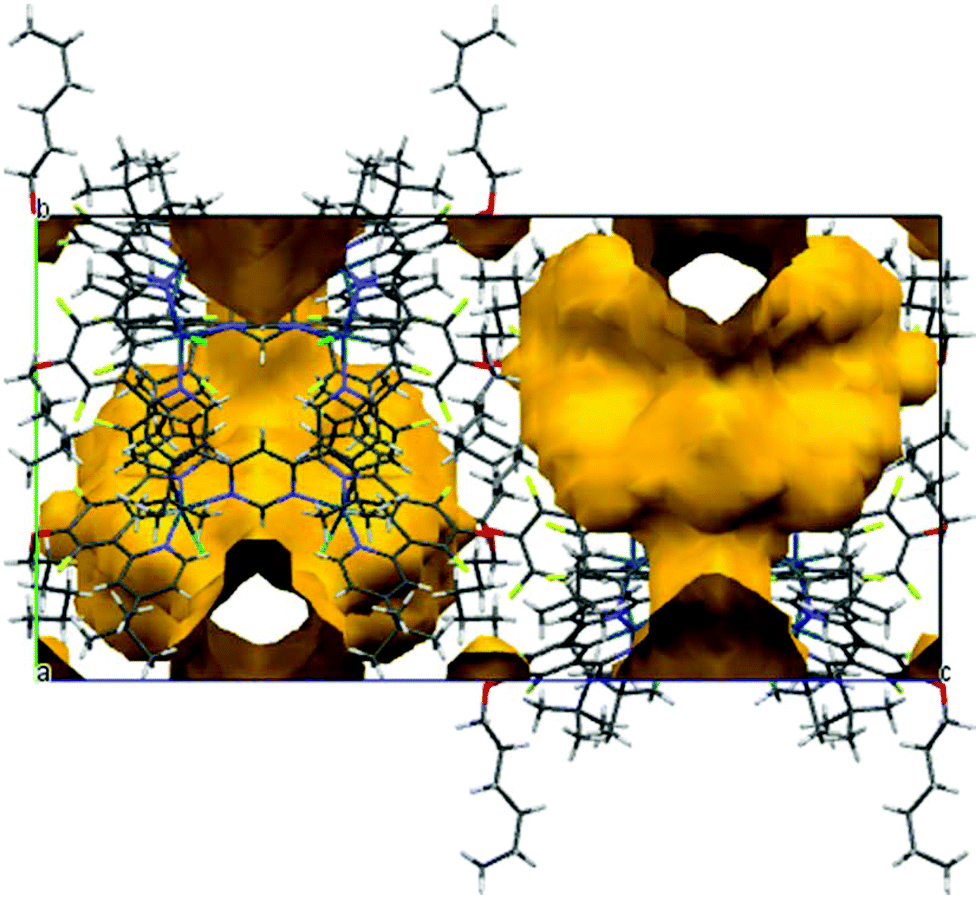 | ||
| Fig. 3 The large voids (shown in yellow) in the crystal structure of the dinuclear complex A3. | ||
By comparing with structurally related mononuclear complexes based on terpyridine rather than an N^C^N-coordinating ligand, the structure of A3 provides evidence for the strong trans influence associated with the central metallated ring of the N^C^N ligand alluded to in section 2. For example, the Ir–N3 bond length of 2.157(5) Å is elongated compared to the Ir–N bond (of N^C ligand) of [Ir(tpy)(MeOmppy)Cl]+, for which the value is 2.068(2) Å,13g and likewise the equatorial Ir–N(bpy) bond of [Ir(tpy)(bpy)Cl]2+, for which the bond length is 2.081(1) Å (Fig. 4).20 In these compounds, the Ir–N bonds are trans to Ir–N as opposed to Ir–C bonds. The strong trans influence of the metallated carbon atoms of the diarylpyrimidine ligand is similarly evident from the longer Ir–Cl bond of 2.4406(15) Å in A3 compared to a value of 2.334(5) Å in [Ir(tpy)(bpy)Cl]2+, where the chloride is trans to a pyridine rather than to a metallated aryl ring.
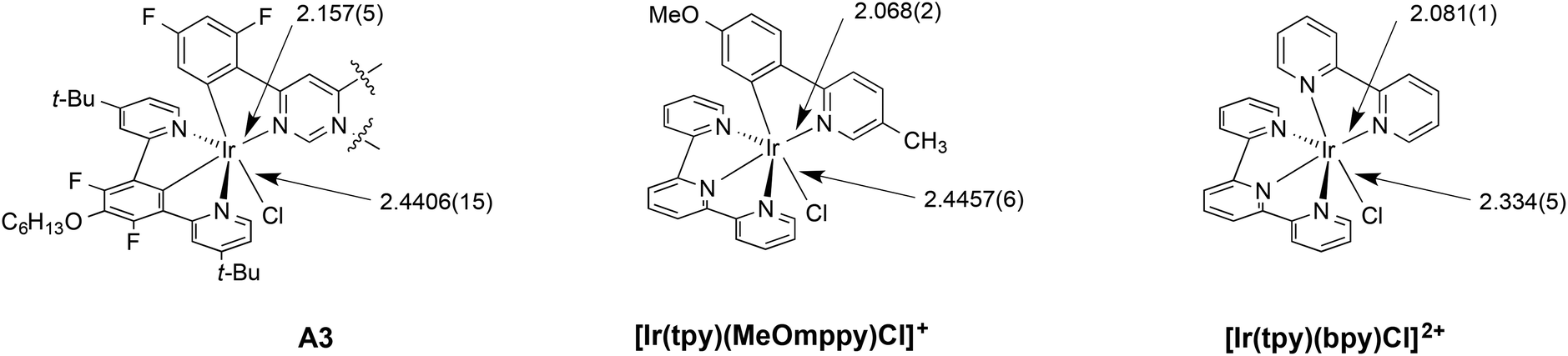 | ||
| Fig. 4 Ir–N and Ir–Cl bond lengths (in Å) in A3 and related complexes highlighting the trans influence of the metallated carbon atoms in elongating trans-disposed bonds. | ||
Density functional theory calculations
Density functional theory (DFT) calculations have been carried out on each of the new complexes, as described in the Experimental section. In order to expedite the calculations, the t-butyl and OC6H13 substituents of the tridentate ligands and the thienyl C6H13 substituents of A5 were truncated to Me, OMe and Me respectively. The t-butyl substituents of L1 were retained in full. After an initial wavefunction stability check, molecules were subjected to full geometry optimisation. Due to the large size of these dinuclear iridium complexes, frequency calculations proved to be intractable and were therefore omitted.All of the optimised geometries of these complexes showed a twist about the central pyrimidine ligand, such that the dihedral angles between the two phenyl rings varied from 7.9° for A2 to 22.8° for A1′. The equivalent dihedral angle between the smaller thiophene rings in A5 was much smaller, at 1.0°. In all cases, this distortion results in rotational symmetry about the central pyrimidine ring. In contrast, the X-ray crystal structure of A3 shows mirror symmetry about the pyrimidine (Fig. 2), such that the central ligand remains planar rather than twisted. This results in different distances between the equivalent pyridine N atoms in the two outer ligands (6.293 and 7.433 Å) in the experimental structure, whereas these distances are equal (at 7.104 Å) in the calculated structure. A geometry optimisation starting from the X-ray crystal structure produced the same calculated structure; hence these slightly different conformations appear to be similar in energy. This conclusion was confirmed by re-optimising the geometry of A3 with imposed mirror symmetry, by the use of z-matrix techniques; this approach gave an optimised structure that was closer to the experimental structure, but just 0.3 kJ mol−1 higher in energy than the rotationally symmetric conformer. Hence, deformation about the central ligand is clearly a very low-energy process that is within both the margin of error of the calculations and the likely effects of molecular packing within the crystal structure.
In all other respects, the calculated structures are as expected (see Table 1 for a comparison of calculated and experimental bond lengths and angles). In each case, the metal is in a distorted octahedral geometry. The Ir–N(pyridine) bond lengths (range 2.079–2.096 Å) are shorter than the Ir–N(pyrimidine) bond lengths (2.208–2.263 Å), as are the Ir–C bond lengths for the tridentate compared to the bidentate cyclometallating ligands (1.917–1.936 Å and 2.017–2.085 Å respectively). These differences mirror those found for related mononuclear complexes and are consistent with the expected trans influences of the various donor atoms involved.12e,17 The Ir–Cl bond lengths cover a narrow range of 2.478–2.489 Å, whilst the Ir–CN bond length in A1′ is 2.062 Å.
UV-visible absorption spectroscopy
In discussing the optical properties of the complexes, it is helpful to divide them into three series, with A1 common to all. In the series A1–A5, the tridentate ligand (LA) is common to each member, and so changes in properties within this series can be ascribed to the influence of the bis-N^C ligand. Meanwhile, in the series A1–C1, the evolution of properties can be related to changing the tridentate ligand. Complex A1 and its cyano derivative A1′ form the third, small series.The UV-visible absorption spectra of the complexes in dichloromethane solution at room temperature are shown in Fig. 5, and absorption maxima and extinction coefficients are tabulated in Table 2. All eight complexes show a series of very intense bands in the UV region (ε in the range 30–80 × 103 M−1 cm−1) together with intense bands in the visible region up to around 500 or 530 nm (ε in the range 5–30 × 103 M−1 cm−1). There is a general trend of decreasing molar absorptivities with increasing wavelength. The spectra are quite typical of cyclometallated iridium(III) complexes,1,2,5 although with higher extinction coefficients than those of related mononuclear complexes of the Ir(N^C^N)(N^C)Cl class, such as Ir(F2dpyb)(ppy)Cl (the structure of which is shown in the inset to Fig. 1 and for which UV-visible absorption data are listed in the footnote to Table 2;12e F2dpybH = 1,3-di(2-pyridyl)-4,6-difluorobenzene).
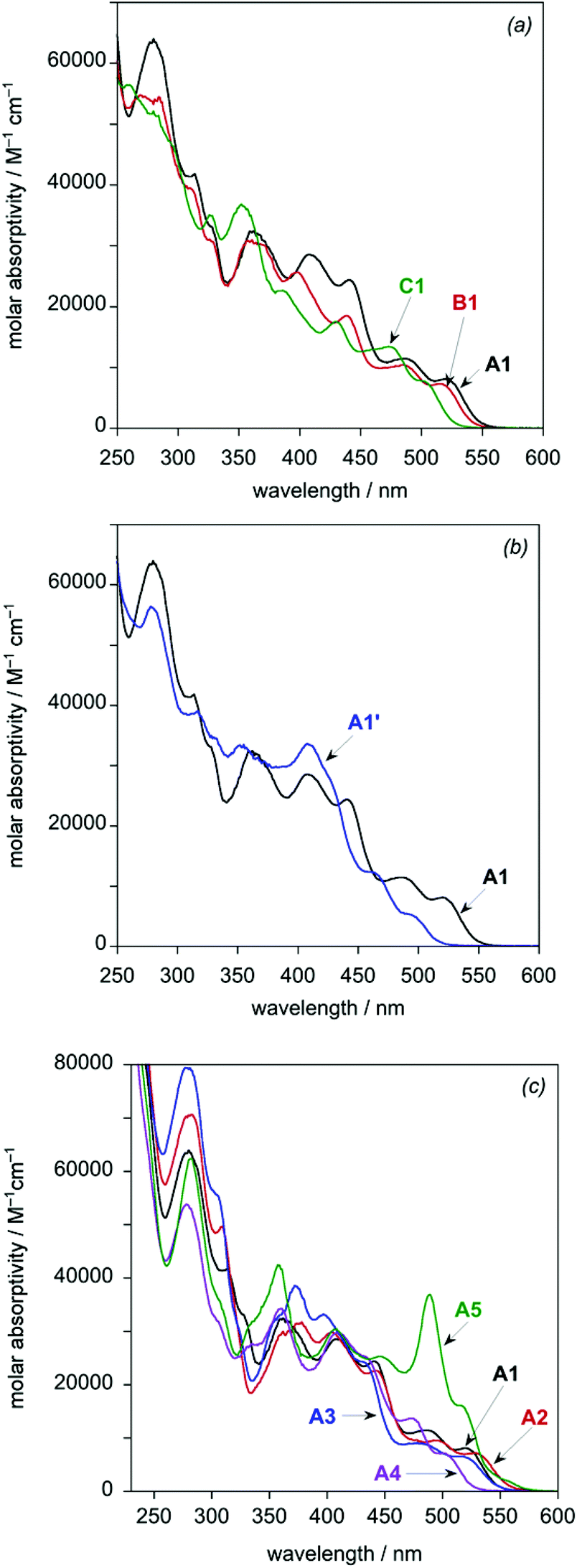 | ||
| Fig. 5 UV-visible absorption spectra of the dinuclear Ir(III) complexes in CH2Cl2 at 298 ± 3 K: (a) the A1–C1 series; (b) A1 and A1′; (c) the A1–A5 series. | ||
| Complexa | λ max/nm (ε/M–1 cm–1) |
|---|---|
|
a For comparison, data for the related mononuclear complex Ir(F2dpyb)(ppy)Cl (Fig. 1 inset) are as follows: λmax/nm (ε/M–1 cm–1) = 260 (44800), 280 (40300), 358 (9100), 381 (11300), 402 (14000), 438 (3640), 473 (1540) [ref. 12e].
|
|
| A1 | 279 (63400), 312 (41500), 360 (32300), 408 (28600), 441 (24400), 484 (11400), 520 (8080) |
| B1 | 270 (54700), 310sh (39500), 326sh (31100), 363 (30400), 398 (25600), 438 (18500), 484 (10300), 514 (7300) |
| C1 | 260 (56500), 281 (51500), 327 (34900), 354 (36500), 385 (22500), 430 (17500), 474 (13400), 500 (7800) |
| A1′ | 279 (56300), 315 (38800), 353 (33000), 409 (33500), 460 (12300), 491 (5390) |
| A2 | 281 (70400), 309 (49600), 376 (31500), 404 (29800), 442 (22600), 494 (9510), 526 (7120) |
| A3 | 279 (79300), 303sh (55800), 358sh (32700), 372 (38600), 397 (33200), 430 (24300), 477 (8970), 514 (6560) |
| A4 | 277 (53600), 335 (27600), 360 (34300), 409 (29700), 433sh (25200), 473 (13700), 498 (7070) |
| A5 | 281 (62300), 305sh (36200), 337sh (31800), 358 (42200), 407 (30300), 446 (25300), 489 (36800), 514 (16100), 554 (2100) |
Within the A1–C1 series, it can be seen that the absorption spectra have very similar profiles, but the bands at λ > 370 nm are increasingly blue-shifted on going from A1 to B1 to C1 (Fig. 5a and Table 2). The change from a difluorophenyl to difluoropyridyl ring (B1 to C1) shifts the bands by around 500 cm−1 to higher energy. On the other hand, the presence of the alkoxy substituent in A1 compared to B1 lowers the energy of the bands by a smaller amount of around 200 cm−1. Meanwhile, changing the monodentate ligand from chloride in A1 to cyanide in A1′ is seen to result in a larger blue-shift of the lowest-energy absorption bands of around 1000 cm−1 (Fig. 5b and Table 2). The overall spectral profile is, however, otherwise similar, and the higher-energy region <350 nm is little affected. For the series A1–A5, in which the bis-N^C ligand is modified, the spectra of A1–A4 are all similar to one another in profile, with the lowest-energy absorption bands increasing in energy in the order A2 < A1 < A3 < A4 (Fig. 5c and Table 2). The bis-thienylpyrimidine complex A5, on the other hand, is rather different from the others at wavelengths >450 nm, in that the two bands which appear in this region have much higher extinction coefficients than those of the other complexes. In addition, this complex displays a weak band at low energy (λmax = 554 nm, ε = 2100 M−1 cm−1), which has no apparent counterpart in the other complexes.
Luminescence properties
All eight of the new complexes are intensely luminescent in deoxygenated solutions at room temperature. Key emission data are tabulated in Table 3, whilst the emission spectra are illustrated in Fig. 6 (subdivided into the same groups as those of Fig. 5). Most of the spectra show a well-defined maximum, accompanied by a shoulder to the low-energy side; such profiles are typical of rigid chromophores with a small Huang–Rhys factor, in which the emission is concentrated in the highest-energy 0,0 vibrational component band.21 The vibrational structure becomes much more pronounced at 77 K (Fig. 7), under which conditions all of the spectra show at least three components of a vibrational progression of ∼1400 cm−1, typical of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C vibrational modes. The thienyl complex additionally shows bands due to a second, lower-frequency progression. The following trends emerge from the spectra.
C vibrational modes. The thienyl complex additionally shows bands due to a second, lower-frequency progression. The following trends emerge from the spectra.
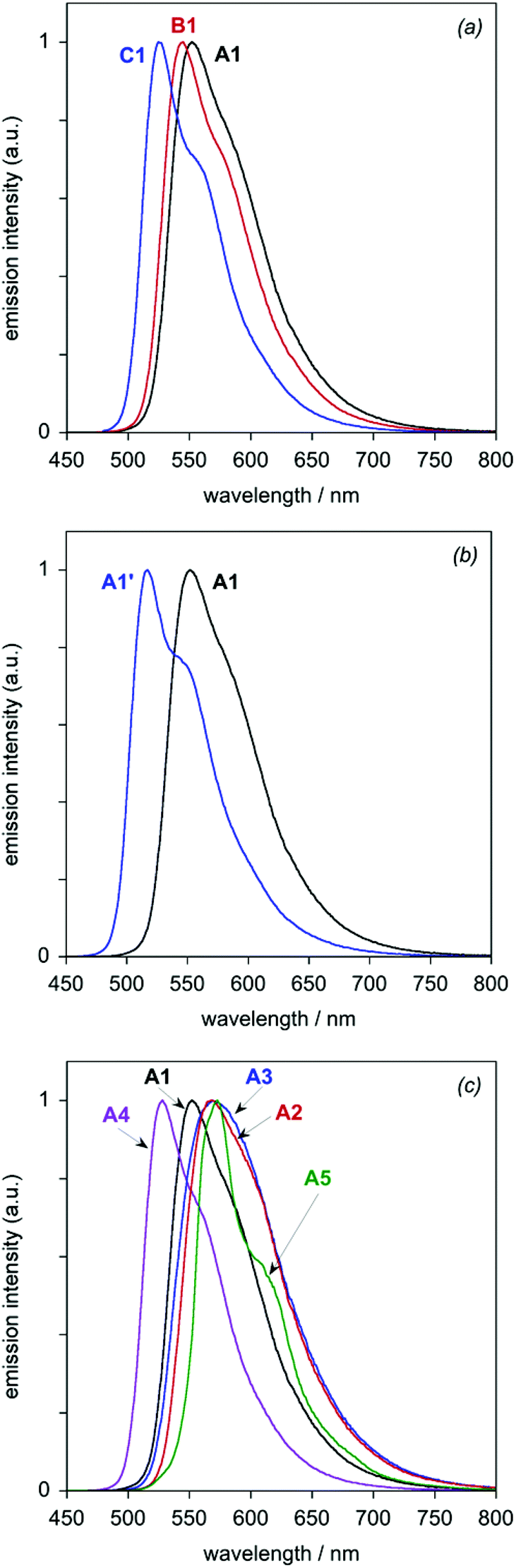 | ||
| Fig. 6 Luminescence spectra of the dinuclear Ir(III) complexes in CH2Cl2 at 298 ± 3 K: (a) the A1–C1 series; (b) A1 and A1′; (c) the A1–A5 series. | ||
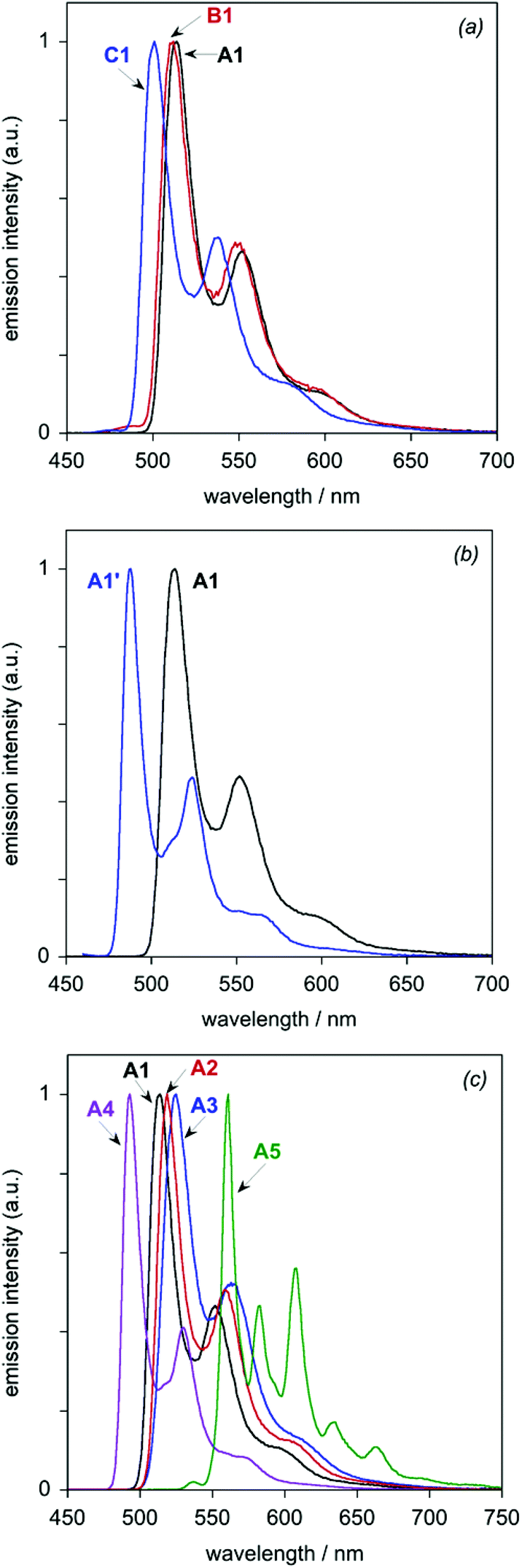 | ||
| Fig. 7 Luminescence spectra of the Ir(III) complexes in diethyl ether/isopentane/ethanol (2:2:1) at 77 K: (a) the A1–C1 series; (b) A1 and A1′; (c) the A1–A5 series. | ||
| Complexa | λ max/nm |
Φ
lumc |
τ/nsd |
k
re/106 s–1 |
∑knr e/106 s–1 |
k
O2Qf/109 M–1 s–1 |
Emission at 77 Kg | |
|---|---|---|---|---|---|---|---|---|
| λ max/nm | τ/ns | |||||||
|
a For comparison, data for the related mononuclear complex Ir(F2dpyb)(ppy)Cl (Fig. 1 inset) are as follows: at 298 K, λmax = 487, 516 nm; Φlum = 0.20; τ = 390 ns; kr = 0.5 × 106 s–1; ∑knr = 2.1 × 106 s–1. At 77 K, λmax = 471, 508, 537, 548, 593 nm; τ = 2.5 μs [ref. 12e].
b EPA = diethyl ether/isopentane/ethanol (2:2:1 v/v).
c Measured using Ir(ppy)3 in 2-MeTHF as the standard, for which Φ = 0.97.
d Values in air-equilibrated solution in parentheses.
e
k
r and ∑knr are the radiative and non-radiative decay rate constants, estimated from the quantum yield and lifetime assuming that the emissive state is formed with unitary efficiency.
f Biomolecular rate constant for quenching by O2, estimated from the luminescence lifetimes in degassed and air-equilibrated solutions, and taking [O2] = 2.2 mM in CH2Cl2 at p = 1 atm air and T = 298 K.
g In EPA: see footnote a.
h Since the measured quantum yield is unity within the error of the measurement, it is not possible to make a meaningful estimate of knr.
|
||||||||
| A1 | 552, 582sh | 0.94 | 440 [210] | 2.14 | 0.14 | 1.13 | 513, 552, 594 | 6650 |
| B1 | 544, 576sh | 1.0 | 550 [160] | 1.82 | —h | 2.01 | 512, 549, 593sh | 7500 |
| C1 | 526, 557sh | 1.0 | 590 [230] | 1.69 | —h | 1.21 | 501, 538, 578 | 7800 |
| A1′ | 517, 547sh | 0.88 | 590 [240] | 1.49 | 0.20 | 1.12 | 488, 523, 560 | 5100 |
| A2 | 568, 599sh | 0.88 | 570 [230] | 1.54 | 0.21 | 1.18 | 519, 559, 602 | 4200 |
| A3 | 570 | 0.91 | 590 [220] | 1.54 | 0.15 | 1.30 | 525, 563, 610sh | 1300 |
| A4 | 528, 557sh | 1.0 | 360 [160] | 2.78 | —h | 1.58 | 493, 530, 569 | 2400 |
| A5 | 572, 611sh | 0.94 | 4000 [380] | 0.24 | 0.015 | 1.08 | 560, 582, 608, 633, 663, 695, 728 | 23000 |
(i) For the A1–C1 series, the emission energy increases in the order A1 < B1 < C1, mirroring the trend observed in absorption. Moreover, based on the 0,0 maxima, the changes in emission energy on going from A1 to B1 (∼200 cm−1) and from B1 to C1 (∼500 cm−1) are very similar to those observed for the lowest-energy absorption band.
(ii) The replacement of Cl by CN in going from A1 to A1′ blue-shifts the emission by ∼1000 cm−1, similar in magnitude to the blue-shift observed in absorption.
(iii) Within the series A1–A5, the trend in emission energy at room temperature is A5 ∼A3 ∼A2 < A1 < A4. At this temperature, the lower-energy vibrational shoulder is only just detectable for A3 and essentially not at all for A2. At 77 K, the 0,0 components are clearly resolved, however, and the order of emission energy is A5 < A3 < A2 < A1 < A4.
The excitation spectra closely match the absorption spectra in each case, indicating that the same emissive state is formed with high efficiency, irrespective of the excitation wavelength. Such behaviour is quite typical of cyclometallated Ir(III) and Pt(II) complexes, where the processes of internal conversion and intersystem crossing to the lowest-lying triplet state are all several orders of magnitude faster than the rate of fluorescence from the singlet manifold.22 The luminescence quantum yields in solution at room temperature are very high for all of the complexes, approaching unity, within the uncertainty on the measurement. The quantum yields are higher than those of previously explored members of the mononuclear Ir(N^C^N)(N^C)Cl series (e.g., see footnote to Table 3).12e The values are comparable to those of fac-Ir(ppy)3, despite the emission being significantly further towards the red than this archetypal complex (apart from A1′).23 With the exception of the thienyl complex A5, the luminescence lifetimes are in the range 360–590 ns at room temperature, which are significantly shorter than that of fac-Ir(ppy)3, for which a value of 1.6 μs has been reported under comparable conditions.23 Given that the quantum yields are comparable to that of fac-Ir(ppy)3, this observation is suggestive of a significantly higher rate of radiative decay in the dinuclear complexes. Assuming that the emissive state is formed with unitary efficiency upon photo-excitation, the radiative and non-radiative rate constants, kr and ∑knr respectively, can be estimated through the relationships.
| kr = Φ/τ and ∑knr = (1 − Φ)/τ |
The values of the rate constants estimated in this way are included in Table 3. It is evident that, apart from A5, the kr values are increased by a factor of 2–5 compared to that of Ir(ppy)3, for which kr has been estimated to be 6 × 105 s−1.23
The luminescence of all the complexes is quenched by oxygen, with a bimolecular rate constant of the order of 109 M−1 s−1, quite typical of cyclometallated Ir(III) and Pt(II) complexes. However, since kr values are unusually high and the lifetimes correspondingly short (again with the exception of A5), the effect of oxygen at atmospheric pressure of air is unusually small, reducing the lifetimes and emission intensities by a factor of only about 2. Complex A5 is anomalous in that it has a much longer lifetime than the other complexes, both at room temperature and at 77 K (τ = 4 and 23 μs respectively). Its emission in solution is correspondingly more sensitive to O2, being quenched by a factor of ∼10 under air-equilibrated conditions.
Interpretation of the photophysical data with reference to TD-DFT results
Time-dependent DFT (TD-DFT) calculations were carried out at the optimised geometries obtained as described in section 3. The main excitation energies, their oscillator strengths and the respective predominant molecular orbital contributions are compiled in Tables S1–S8 of the ESI.† Visible spectra were calculated in vacuum, and in dichloromethane solution using the polarised continuum model for the solvent24 (Fig. S1 and S2†). In all cases, the general effect of solvent is to blue-shift the frequency of the highest energy singlet band by 15 to 38 nm and to increase its intensity. The experimental spectra show more structure in the absorption bands than those simulated using the TD-DFT data. Such structure is likely to arise as a result of absorption to vibrationally excited levels of the electronically excited states, which is not modelled by the theory. For example, the lowest-energy bands of A1 are separated by around 1400 cm−1, which is typical of aromatic CC vibrations. This mirrors the vibrational progression observed in the emission spectra (albeit arising in that case from transitions from the triplet state to the vibrationally excited levels of the ground state).
The calculations provide very useful insight into the trends in absorption and emission energies discussed earlier. For all of the complexes, the trend in the energy of the lowest-energy absorption band determined experimentally correlates well with the HOMO–LUMO gap, as does the trend in the experimental emission energy, as illustrated in Fig. 8c. Plots of the calculated S1 and T1 energies in vacuum and in solvent are shown in the ESI,† and likewise show a very good correlation with the experimentally determined data (Fig. S3†). Thus, it is appropriate to seek to understand the influence of the substituents in terms of their effects on the HOMO and LUMO energies. Molecular orbital plots of the HOMO and LUMO of all of the complexes (as well as HOMO–1 and LUMO+1) are shown in the ESI (Fig. S4–S11†). Typically for mononuclear cyclometallated Ir(III) and Pt(II) complexes with aryl-heterocycle derivatives (e.g. ppy), the LUMO is based largely on the heterocyclic ring (pyridine in the case of ppy), whilst the HOMO spans the metallated ring and the metal atom, reflecting the high degree of covalency of the M–C bond.2e,25 In the case of the series of dinuclear complexes reported here, one might intuitively expect the more electron-deficient pyrimidine heterocycle of the bis-N^C ligand to offer lower-energy π* orbitals than the pyridine rings of the tridentate ligand, and indeed the calculations confirm that the LUMO is predominantly localised on the pyrimidine ring in all eight complexes (Fig. S4–S11†). On the other hand, the HOMO spans the metal and the metallated ring of either the N^C^N or the N^C ligand, according to the identity of the two ligands.
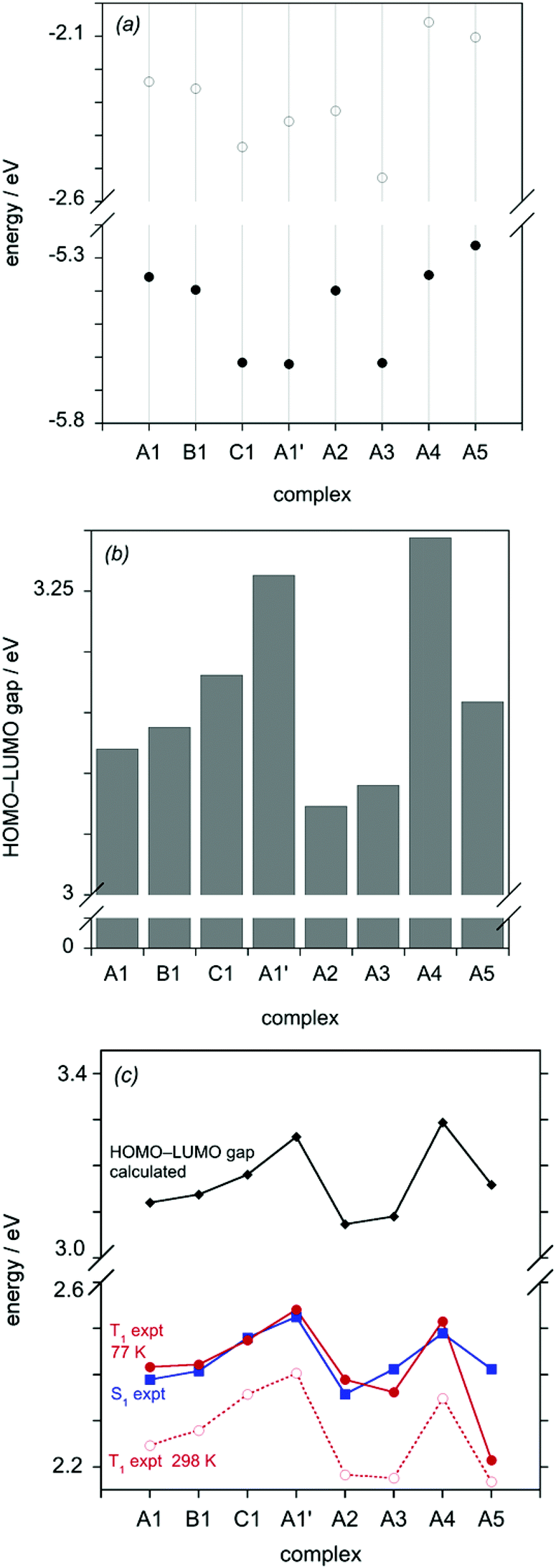 | ||
| Fig. 8 (a) Calculated HOMO and LUMO energies for the complexes in vacuo, in eV (filled and empty circles respectively). (b) The HOMO–LUMO gap (eV) calculated from the respective energies in (a). (c) Plot of the experimental S1 energy (blue squares), estimated based on λmax of the lowest-energy absorption band (excluding the weak band at 554 nm for A5, which is concluded to be a triplet transition, S0 → T1 – see text); the T1 energy estimated from experimental λmax (0,0) values in the emission spectra at 298 K (open red circles) and at 77 K (filled red circles); and the calculated HOMO–LUMO gap (black diamonds). The lines between points are provided solely as a guide to aid the eye. | ||
Fig. 8a shows the calculated HOMO and LUMO energies of the complexes and Fig. 8b illustrates the magnitude of the resulting gap. The blue-shift in absorption and emission that is observed on going from A1 through B1 to C1 can be seen to arise from a larger stabilisation of the HOMO than the LUMO. Examining the HOMO orbital plots, it can be seen that the HOMO of A1 involves the aryl ring of the N^C^N ligand with only a minor contribution from that of the N^C unit (Fig. S4†). Removal of the electron-donating alkoxy group on going to B1 switches round this distribution of electron density in the HOMO: it now involves almost primarily the N^C aryl ring with only a minor contribution from the N^C^N (Fig. S5†). In C1, in which the central aryl ring of the N^C^N ligand is replaced by a more electron-deficient pyridyl ring, the contribution of the tridentate ligand to the HOMO becomes negligible (Fig. S6†).
The replacement of the chloride ligand by cyanide in (A1 to A1′) can similarly be seen to have a much larger stabilising effect on the HOMO than the LUMO (Fig. 8b), accounting for the large blue-shift observed experimentally both in absorption and emission. This is consistent with the observation that the metal typically makes a much more significant contribution to the HOMO than the LUMO.
The small red-shift in absorption and emission on going from A1 to A2 can be seen from Fig. 8a to arise primarily from a stabilisation of the LUMO, larger than the effect on the HOMO, upon removal of the t-butyl substituents in the N^C ligand. Meanwhile, the data in Fig. 8b would suggest that A3 should be very similar to A2, which is indeed the case for the emission, see Fig. 6c (in absorption, a small blue shift is observed experimentally, Fig. 5c). At first sight, this is a somewhat surprising result, given that fac-Ir(dfppy)3 is well-known to be strongly blue-shifted relative to fac-Ir(ppy)3 for example, attributed to the effect of the fluorine atoms in stabilising the HOMO.26 However, from Fig. 8a, it is apparent that the electron-withdrawing fluorine atoms in A3 stabilise the LUMO and HOMO to similar extents, leading to the lack of significant effect on the optical properties. In contrast, the significant blue-shift observed in the absorption and emission of A4 compared to A1 is seen from Fig. 8a to arise almost exclusively from the destabilisation of the N^C-based LUMO. In this regard, it is pertinent to note that the electron-donating methoxy substituents of A4 are located para to the electron-deficient pyrimidine ring. Interestingly, inspection of the frontier orbitals of A4 (Fig. S10†) shows that the methoxy substituent is sufficiently electron-rich to shift the HOMO to the N^C as opposed to the N^C^N ligand.
Finally, we turn to complex A5. We noted its anomalously long luminescence lifetime of 4 μs in section 5. At 77 K, the lifetime of this complex increases to 23 μs, which is much longer than that of typical 3MLCT states in Ir(III) complexes, whilst its spectrum at 77 K displays much more vibrational structure. Such changes are consistent with an excited state of more thienyl-pyrimidine localised 3π–π* parentage, mirroring conclusions made on Ir(thpy)2(acac) versus Ir(ppy)2(acac) for example {and similarly for Pt(thpy)(acac) versus Pt(ppy)(acac)}.27 It seems likely that at 298 K, states of predominant 3MLCT and 3π–π* character are close in energy, but the former is displaced to higher energy at 77 K, leading to the distinctly 3π–π*-like spectrum and lifetime observed at low temperature.28 It is notable that the simplistic correlation with the HOMO–LUMO gap works well for the S1 state of A5 (from the absorption spectrum) but less well for the emission data, particularly at 77 K (Fig. 8c). In contrast, the more rigorous calculation of the T1 energy captures the trend well, with A5 predicted to have much the lowest-energy emission amongst all the complexes, as observed at 77 K (Fig. S3†). Indeed, we surmise that the additional weak absorption band observed at 554 nm for A5 may be the direct excitation to the 3π–π* state.29
What can be concluded about the influence of a second metal ion? It is clear from comparison with the most closely related mononuclear complexes available (see data in Table 3 and footnote) that the radiative rate constant in the dinuclear complexes is enhanced whilst the non-radiative decay is suppressed. This observation is all the more remarkable given that the excited state energies of these new complexes are significantly lower than in the model mononuclear complexes: typically, for a given class of complex, non-radiative decay processes increase with decreasing excited state energy, according to the energy-gap law, whilst a decrease in kr may be anticipated, according to the Einstein coefficient which depends on ν3. The consistency with which kr is enhanced in our recently studied multinuclear complexes featuring such bis-cyclometallating ligands supports the notion that spin–orbit coupling pathways may be enhanced by the presence of a second heavy metal centre.6d,h,15 That non-radiative decay is suppressed may, on the other hand, arise from the greater conformational rigidity of the dinuclear systems.
Concluding remarks
In summary, we have shown that 4,6-diarylpyrimidines are attractive bis-cyclometallating ligands that can be used to bind simultaneously to two iridium ions via N^C coordination to each metal centre. The use of a tridentate, N^C^N-coordinating, terminal ligand ensures that the resulting dinuclear complexes are achiral. This contrasts with the mixtures of stereoisomers that are formed when tris-bidentate metal units are employed. Moreover, the strong trans influence of cyclometallated aryl rings leads to one unique product, in which the pyrimidine ring is trans to the central ring of the N^C^N ligand at both metal centres. The resulting products are very intensely luminescent, with quantum yields close to unity and the emission energy being tuneable through substituents in either the bidentate or tridentate ligand or metathesis of the monodentate ligand. Whilst offering strong light absorption and intense emission in the visible region, dinuclear cyclometallated complexes clearly offer huge potential of structural diversity. The versatility of the design strategy, and ease of synthesis of suitable tri- and bidentate ligands, can be expected to lead to further new compounds, for example, designed as water-splitting catalysts or for specific interactions with biomolecules, in both of which fields multinuclear complexes are increasingly recognised to offer unique opportunities.30,31Experimental
Synthesis and characterisation
Full details are provided in the ESI.†1H and 13C NMR spectra were recorded on JEOL Delta-270 or JEOL ECS-400 spectrometers operating at the frequencies indicated in the ESI.† Chemical shifts (δ) are in ppm, referenced to tetramethylsilane Si(CH3)4, and coupling constants are in Hertz. Mass spectra were recorded at the EPSRC National Mass Spectrometry Service Centre on a Thermo Scientific LTQ Orbitrap XL Mass Spectrometer using low resolution electrospray (ESI) or high resolution nano-electrospray (NSI) ionisation techniques. Proligands L1H2–L5H2 were prepared from 4,6-dichloropyrimidine, using the procedure we reported previously.15Photophysical measurements
Absorption spectra were measured on a Biotek Instruments XS spectrometer, using quartz cuvettes of 1 cm path length. Steady-state luminescence spectra were measured using a Jobin Yvon FluoroMax-2 spectrofluorimeter, fitted with a red-sensitive Hamamatsu R928 photomultiplier tube; the spectra shown are corrected for the wavelength dependence of the detector, and the quoted emission maxima refer to the values after correction. Samples for emission measurements were contained within quartz cuvettes of 1 cm path length modified with appropriate glassware to allow connection to a high-vacuum line. Degassing was achieved via a minimum of three freeze–pump–thaw cycles whilst connected to the vacuum manifold; final vapour pressure at 77 K was <5 × 10−2 mbar, as monitored using a Pirani gauge. Luminescence quantum yields were determined using fac-Ir(ppy)3 in degassed 2-methyltetrahydrofuran solution as the reference (Φlum = 0.97 (ref. 23)); estimated uncertainty in relative values of Φlum is ±10% or better. The luminescence lifetimes of the complexes were measured by time-correlated single-photon counting, following excitation at 405 nm with an EPL-405 pulsed-diode laser. The emitted light was detected at 90° using a Peltier-cooled R928 PMT after passage through a monochromator. The lifetime of A5 at 77 K was measured using the same detector operating in multi-channel scaling mode, following excitation with a microsecond pulsed xenon flash-lamp. The estimated uncertainty in the quoted lifetimes is ±10% or better. Bimolecular rate constants for quenching by molecular oxygen, kQ, were determined from the lifetimes in degassed and air-equilibrated solution, taking the concentration of oxygen in CH2Cl2 at 0.21 atm O2 to be 2.2 mmol dm−3.Density functional theory calculations
All DFT calculations were carried out using Gaussian 09W.32 The B3LYP hybrid functional was used,33 together with the LanL2DZ basis set for iridium,34 and 6-31+G(d) for all other atom types. Geometries were fully optimised in vacuo. UV-visible absorption spectra were obtained by single point TD-DFT calculations at the optimised geometries, using the same functional and basis sets, either in vacuo or with a PCM solvent correction for dichloromethane.24 The molecular orbitals and spectra were visualised using GaussView 5.0 with Gaussian line shapes and an arbitrary half-width at half-height of 1000 cm−1 for the latter.Acknowledgements
We thank EPSRC for funding (grants EP/I014942/1 and EP/H051902/01), the EPSRC National Mass Spectrometry Service Centre, Swansea for recording mass spectra, and Diamond Light Source for access to synchrotron facilities (beamline I19).References
- (a) A. F. Rausch, H. H. H. Homeier, P. I. Djurovich, M. E. Thompson and H. Yersin, Proc. SPIE, 2007, 6655, F6550 CrossRef; (b) H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS; (c) P. T. Chou, Y. Chi, M. W. Chung and C. C. Lin, Coord. Chem. Rev., 2011, 255, 2653 CrossRef CAS; (d) B. J. Powell, Coord. Chem. Rev., 2015, 295, 46 CrossRef CAS.
- (a) M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151 CrossRef CAS; (b) Highly Efficient OLEDs with Phosphorescent Materials, ed. H. Yersin, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (c) Y. Chi and P. T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC; (d) L. Murphy and J. A. G. Williams, Top. Organomet. Chem., 2010, 28, 75 CrossRef CAS; (e) L. F. Gildea and J. A. G. Williams, Iridium and platinum complexes for OLEDs, in Organic Light-Emitting Diodes: Materials, Devices and Applications, ed. A. Buckley, Woodhead, Cambridge, 2013 Search PubMed.
- For a review of the concept of time-resolved bioimaging using phosphorescent metal complexes, see: E. Baggaley, J. A. Weinstein and J. A. G. Williams, Struct. Bonding, 2015, 165, 205 CrossRef.
- (a) L. Murphy, A. Congreve, L.-O. Palsson and J. A. G. Williams, Chem. Commun., 2010, 46, 8743 RSC; (b) S. Zhang, M. Hosaka, T. Yoshihara, K. Negishi, Y. Ilida, S. Tobita and T. Takeuchi, Cancer Res., 2010, 70, 4490 CrossRef CAS PubMed; (c) T. Yoshihara, Y. Yamaguchi, M. Hosaka, T. Takeuchi and S. Tobita, Angew. Chem., Int. Ed., 2012, 51, 4148 CrossRef CAS PubMed; (d) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS; (e) T. Yoshihara, S. Murayama and S. Tobita, Sensors, 2015, 15, 13503 CrossRef CAS PubMed; (f) Q. Zhao, Y. H. Liu, Y. F. Cao, W. Lv, Q. Yu, S. J. Liu, X. M. Liu, M. Shi and W. Huang, Adv. Opt. Mater., 2015, 3, 233 CrossRef CAS.
- Reviews and selected recent representative examples include: (a) I. M. Dixon, J. P. Collin, J. P. Sauvage, L. Flamigni, S. Encinas and F. Barigelletti, Chem. Soc. Rev., 2000, 29, 385 RSC; (b) E. Baranoff, J. P. Collin, L. Flamigni and J. P. Sauvage, Chem. Soc. Rev., 2004, 33, 147 RSC; (c) M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970 CrossRef CAS PubMed; (d) L. Flamigni, A. Barbieri, C. Sabatini, B. Ventura and F. Barigelletti, Top. Curr. Chem., 2007, 281, 143 CrossRef CAS; (e) E. Kim and S. B. Park, Chem. – Asian J., 2009, 4, 1646 CrossRef CAS PubMed; (f) G. Zhou, W. Y. Wong and X. Yang, Chem. – Asian J., 2011, 6, 1706 CrossRef CAS PubMed; (g) Q. Zhang, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (h) K. K. W. Lo, S. P. Y. Li and K. Y. Zhang, New J. Chem., 2011, 35, 265 RSC; (i) Z. Liu, W. He and Z. Guo, Chem. Soc. Rev., 2013, 42, 1568 RSC; (j) G. R. Freeman and J. A. G. Williams, Top. Organomet. Chem., 2013, 40, 89 CrossRef CAS; (k) C. E. Welby, L. Gilmartin, R. R. Marriott, A. Zahid, C. R. Rice, E. A. Gibson and P. I. P. Elliott, Dalton Trans., 2013, 42, 13527 RSC; (l) S. Ladouceur and E. Zysman-Colman, Eur. J. Inorg. Chem., 2013, 2985 CrossRef CAS; (m) J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, M. K. Nazeeruddin and E. Baranoff, Dalton Trans., 2014, 43, 5667 RSC; (n) C. L. Ho, H. Li and W. Y. Wong, J. Organomet. Chem., 2014, 751, 261 CrossRef CAS; (o) C. J. Xu, A. Guenet, N. Kyritsakas, J. M. Planeix and M. W. Hosseini, Inorg. Chem., 2015, 54, 10429 CrossRef CAS PubMed; (p) J. Fernandez-Cestau, N. Gimenez, E. Lalinde, P. Montano, M. T. Moreno and S. Sanchez, Organometallics, 2015, 34, 1766 CrossRef CAS; (q) E. Baranoff and B. F. E. Curchod, Dalton Trans., 2015, 44, 8318 RSC; (r) K. Beydoun, M. Zaarour, J. A. G. Williams, T. Roisnel, V. Dorcet, A. Planchat, A. Boucekkine, D. Jacquemin, H. Doucet and V. Guerchais, Inorg. Chem., 2013, 52, 12416 CrossRef CAS PubMed.
- (a) S. Serroni, A. Juris, S. Campagna, M. Venturi, G. Denti and V. Balzani, J. Am. Chem. Soc., 1994, 116, 9086 CrossRef CAS; (b) S. Campagna, S. Serroni, A. Juris, M. Venturi and V. Balzani, New J. Chem., 1996, 20, 773 CAS; (c) G. Di Marco, M. Lanza, A. Mamo, I. Stefio, C. Di Pietro, G. Romeo and S. Campagna, Anal. Chem., 1998, 70, 5019 CrossRef CAS PubMed; (d) V. N. Kozhevnikov, M. C. Durrant and J. A. G. Williams, Inorg. Chem., 2011, 50, 6304 CrossRef CAS PubMed; (e) E. Baranoff, E. Orselli, L. Allouche, D. Di Censo, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Chem. Commun., 2011, 47, 2799 RSC; (f) L. Donato, C. E. McCusker, F. N. Castellano and E. Zysman-Colman, Inorg. Chem., 2013, 52, 8495 CrossRef CAS PubMed; (g) Y. Zheng, A. S. Batsanov, M. A. Fox, H. A. Al-Attar, K. Abdullah, V. Jankus, M. R. Bryce and A. P. Monkman, Angew. Chem., Int. Ed., 2014, 53, 11616 CrossRef CAS PubMed; (h) P. H. Lanoë, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831 RSC.
- Note that chirally-resolved complexes display unique chiroptical properties such as circular dichroism and circularly polarised luminescence; e.g. (a) X. Chen, Y. Okamoto, T. Yano and J. Otsuki, J. Sep. Sci., 2007, 30, 713 CrossRef CAS PubMed; (b) C. Schaffner-Hamann, A. von Zelewsky, A. Barbieri, F. Barigelletti, G. Muller, J. P. Riehl and A. Neels, J. Am. Chem. Soc., 2004, 30, 9339 CrossRef PubMed; (c) F. J. Coughlin, M. S. Westrol, K. D. Oyler, N. Byrne, C. Kraml, E. Zysman-Colman, M. S. Lowry and S. Bernhard, Inorg. Chem., 2008, 47, 2039 CrossRef CAS PubMed; (d) L. R. Yang, A. von Zelewsky, H. P. Nguyen, G. Muller, G. Labat and H. Stoeckli-Evans, Inorg. Chim. Acta, 2009, 362, 3853 CrossRef CAS PubMed.
- A. Tsuboyama, T. Takiguchi, S. Okada, M. Osawa, H. Hoshino and K. Ueno, Dalton Trans., 2004, 1115 RSC.
- For example: A. Auffrant, A. Barbieri, F. Barigelletti, J. Lacour, P. Mobian, J. P. Collin, J. P. Sauvage and B. Ventura, Inorg. Chem., 2007, 46, 6911 CrossRef CAS PubMed.
- (a) J. P. Sauvage, J. P. Collin, J. C. Chambron, S. Guillerez, C. Coudret, V. Balzani, F. Barigelletti, L. De Cola and L. Flamigni, Chem. Rev., 1994, 94, 993 CrossRef CAS; (b) E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133 RSC.
- J. A. G. Williams, A. J. Wilkinson and V. L. Whittle, Dalton Trans., 2008, 2081 RSC.
- (a) A. J. Wilkinson, A. E. Goeta, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2004, 43, 6513 CrossRef CAS PubMed; (b) A. J. Wilkinson, H. Puschmann, J. A. K. Howard, C. E. Foster and J. A. G. Williams, Inorg. Chem., 2006, 45, 8685 CrossRef CAS PubMed; (c) S. Obara, M. Itabashi, F. Okuda, S. Tamaki, Y. Tanabe, Y. Ishii, K. Nozaki and M. Haga, Inorg. Chem., 2006, 45, 8907 CrossRef CAS PubMed; (d) J. Kuwabara, T. Namekawa, M. Haga and T. Kanbara, Dalton Trans., 2012, 41, 44 RSC; (e) P. Brulatti, R. J. Gildea, J. A. K. Howard, V. Fattori, M. Cocchi and J. A. G. Williams, Inorg. Chem., 2012, 51, 3813 CrossRef CAS PubMed.
- (a) A. J. S. Bexon and J. A. G. Williams, C. R. Chim., 2005, 8, 1326 CrossRef CAS; (b) T. Yutaka, S. Obara, S. Ogawa, K. Nozaki, N. Ikeda, T. Ohno, Y. Ishii, K. Sakai and M. Haga, Inorg. Chem., 2005, 44, 4737 CrossRef CAS PubMed; (c) A. Auffrant, A. Barbieri, F. Barigelletti, J. P. Collin, L. Flamigni, C. Sabatini and J. P. Sauvage, Inorg. Chem., 2006, 45, 10990 CrossRef CAS PubMed; (d) V. L. Whittle and J. A. G. Williams, Inorg. Chem., 2008, 47, 6596 CrossRef CAS PubMed; (e) V. L. Whittle and J. A. G. Williams, Dalton Trans., 2009, 3929 RSC; (f) L. L. Tinker and S. Bernhard, Inorg. Chem., 2009, 48, 10507 CrossRef CAS PubMed; (g) D. N. Chirdon, W. J. Transue, H. N. Kagalwala, A. Kaur, A. B. Maurer, T. Pintauer and S. Bernhard, Inorg. Chem., 2014, 53, 1487 CrossRef CAS PubMed.
- One example of a mononuclear complex of the type Ir(N^C^N)(N^C)Cl featuring an asymmetric N^C^N ligand has been resolved into its constituent enantiomers: M. Ashizawa, L. Tang, K. Kobayashi, H. Sato, A. Yamagishi, F. Okuda, T. Harada, R. Kuroda and M. Haga, Dalton Trans., 2009, 1700 RSC.
- S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef CAS PubMed.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783 RSC.
- Bis-quinolylbenzene analogues give predominantly N^C^N coordination, even in the absence of such substituents: L. F. Gildea, A. S. Batsanov and J. A. G. Williams, Dalton Trans., 2013, 42, 10388 RSC.
- (a) L. Murphy, P. Brulatti, V. Fattori, M. Cocchi and J. A. G. Williams, Chem. Commun., 2012, 48, 5817 RSC; (b) E. Rossi, L. Murphy, P. L. Brothwood, A. Colombo, C. Dragonetti, D. Roberto, R. Ugo, M. Cocchi and J. A. G. Williams, J. Mater. Chem., 2011, 21, 15501 RSC; (c) M. Cocchi, D. Virgili, V. Fattori, J. A. G. Williams and J. Kalinowski, Appl. Phys. Lett., 2007, 90, 023506 CrossRef.
- A. L. Spek, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 9 CrossRef CAS PubMed.
- N. Yoshikawa, S. Yamabe, N. Kanehisa, Y. Kai, H. Takashima and K. Tsukahara, Inorg. Chim. Acta, 2009, 362, 361 CrossRef CAS.
- A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2009, 48, 11407 CrossRef CAS PubMed.
- (a) G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2009, 113, 2 CrossRef CAS PubMed; (b) G. J. Hedley, A. Ruseckas and I. D. W. Samuel, J. Phys. Chem. A, 2010, 114, 8961 CrossRef CAS PubMed; (c) F. Messina, E. Pomarico, M. Silatini, E. Baranoff and M. Chergui, J. Phys. Chem. Lett., 2015, 6, 4475 CrossRef CAS PubMed.
- T. Sajato, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard III and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813 CrossRef PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- P. J. Hay, J. Phys. Chem. A, 2002, 106, 1634 CrossRef CAS.
- A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed.
- D. N. Kozhevnikov, V. N. Kozhevnikov, M. Z. Shafikov, A. M. Prokhorov, D. W. Bruce and J. A. G. Williams, Inorg. Chem., 2011, 50, 3804 CrossRef CAS PubMed.
- W. Leslie, A. S. Batsanov, J. A. K. Howard and J. A. G. Williams, Dalton Trans., 2004, 623 RSC.
- Weak but well-defined S0 → T1 absorption bands have similarly been observed for Pt(II) complexes of dipyridylbenzene ligands; J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609 CrossRef CAS PubMed.
- A. Petronilho, J. A. Woods, S. Bernhard and M. Albrecht, Eur. J. Inorg. Chem., 2014, 708 CrossRef CAS.
- A. Wragg, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem. – Eur. J., 2014, 20, 14004 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, 2009 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterisation of new compounds; TD-DFT results and correlation with experimental data; X-ray crystallography of complex A3. CCDC 1415318. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00881j |
| This journal is © The Royal Society of Chemistry 2016 |