
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating the anticancer properties of organometallic dendrimers using pyridylferrocene entities: synthesis, cytotoxicity and DNA binding studies†
Preshendren
Govender
a,
Tina
Riedel
b,
Paul J.
Dyson
b and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, Rondebosch, 7701, Cape Town, South Africa. Fax: +27-21-6505195; E-mail: gregory.smith@uct.ac.za
bInstitut des Sciences et Ingénierie Chimique, Ecole Polytechnique Fédérale de Lausanne (EPFL), CH-1015, Lausanne, Switzerland
First published on 19th May 2016
Abstract
A new series of eight first- and second-generation heterometallic ferrocenyl-derived metal–arene metallodendrimers, containing ruthenium(II)–p-cymene, ruthenium(II)–hexamethylbenzene, rhodium(III)–cyclopentadienyl or iridium(III)–cyclopentadienyl moieties have been prepared. The metallodendrimers were synthesized by first reacting DAB-(NH2)n (where n = 4 or 8, DAB = diaminobutane) with salicylaldehyde, and then the Schiff-base dendritic ligands were reacted in a one-pot reaction with the appropriate [(η6-p-iPrC6H4Me)RuCl2]2, [(η6-C6Me6)RuCl2]2, [(η5-C5Me5)IrCl2]2 or [(η5-C5Me5)RhCl2]2 dimers, in the presence of 4-pyridylferrocene. Heterometallic binuclear analogues were prepared as models of the larger metallodendrimers. All complexes have been characterized using analytical and spectroscopic methods. The cytotoxicity of the heterometallic metallodendrimers and their binuclear analogues were evaluated against A2780 cisplatin-sensitive and A2780cisR cisplatin-resistant human ovarian cancer cell lines and against a non-tumorigenic HEK-293 human embryonic kidney cell line. The second generation Ru(II)-η6-C6Me6 metallodendrimer is the most cytotoxic and selective compound. DNA binding experiments reveal that a possible mode-of-action of these compounds involves non-covalent interactions with DNA.
Introduction
Research on the design of heterometallic complexes as possible anticancer agents has flourished.1–6 Due to its favourable electronic properties and ease of functionalization,7–12 ferrocene has been incorporated in various biologically active systems,9,13,14 in an effort to achieve a synergistic effect between the metal centers.15–20 Furthermore, simple derivatives of ferrocene display good activity in vitro, with inhibition of tumors observed in vivo.21,22 In the search for new tamoxifen-like drugs, Jaouen and co-workers23,24 prepared ferrocifens from 1-[4-(2-dimethylaminoethoxy)]-1-(phenyl-2-ferrocenylbut-1-ene), which are highly active ferrocenyl-derivatives of the purely organic breast cancer drug tamoxifen. The increase in activity is attributed to the dual action of the organic drug and the Fenton chemistry (i.e. formation of singlet oxygen) of the Fe centre.25,26 Moreover, the stability of ferrocene in aqueous and aerobic media, the facility with which a large variety of derivatives may be prepared, and its favorable electrochemical properties, has resulted in ferrocene becoming a promising molecule for incorporating in biological applications.12 Furthermore, certain ferrocene-containing compounds exhibit cytotoxicities comparable to the benchmark anticancer drug, cisplatin.Due to the undesirable side-effects displayed by platinum-based anticancer drugs,27,28 researchers have turned their attention to other metals.29,30 One such metal is ruthenium, as ruthenium complexes tend to be less cytotoxic in comparison to platinum compounds,31 and two promising Ru(III) anticancer agents are undergoing clinical trials.32–35 The activity of the Ru(III) anticancer agents is thought to be brought about by reduction of the compounds to a Ru(II) species in vivo,33 and consequently there is a growing interest in the preparation and biological evaluation of ruthenium(II)–arene complexes.36–42 Two anticancer agents are at the forefront of this promising class of organometallic compounds, RAED-C [(η6-p-cym)Ru(ethylene-diamine)Cl]PF6,43,44 and RAPTA-C [(η6-p-cym)Ru(1,3,5-triaza-7-phosphaadamantane)Cl2].45,46 The former is a cytotoxic compound that binds preferentially to DNA,47 whereas the latter is a non-cytotoxic antitumor compound,46,48–51 that displays an antiangiogenic effect52 and binds preferentially to histone proteins.47,49 In addition, a host of Rh(III)–Cp* and Ir(III)–Cp* (where Cp* = cyclopentadienyl) derivatives have previously demonstrated pharmacological properties,53–58 which promote further study of these compounds.
The concept of combining organometallic complexes with compounds of known therapeutic value is a growing area of research, and coupled with the notion of multinuclearity that can result in enhanced therapeutic activity,19,59–71 the combination is an attractive drug-design strategy. Moreover, one way of introducing the notion of multinuclearity is to conjugate therapeutic agents onto dendritic scaffolds,59,72–77 that could potentially exploit the enhanced permeability and retention (EPR) effect.78–80
Recently, we reported tetranuclear and octanuclear N,O- and N,N-ruthenium–arene metallodendrimers, with ferrocene moieties functionalized on the periphery of the dendritic scaffold. The majority of the compounds efficiently inhibit the growth of both A2780 cisplatin-sensitive and A2780cisR cisplatin-resistant human ovarian cancer cells (i.e. IC50 < 5 μM).81 In addition, these complexes displayed no cross-resistance to cisplatin, with the metallodendrimers having a better activity compared to their mononuclear complexes.
On the basis of the above observed results we hypothesized that preparing ferrocenyl-derived metal–arene metallodendrimers, where the ferrocenyl moiety is coordinated directly to the metal center rather than on the periphery, may further enhance the antiproliferative activity of the compounds. In this study, we describe the synthesis of a series of bidentate heterometallic ferrocenyl-derived dendrimers containing ruthenium(II)–p-cymene, ruthenium(II)–HMB (where HMB = hexamethylbenzene), rhodium(III)–Cp* or iridium(III)–Cp* (where Cp* = cyclopentadienyl) moieties [1][PF6]4–[8]PF6]8 (see Scheme 1). The cytotoxicity of the complexes was evaluated against the A2780 and the A2780cisR human ovarian cancer cell lines, as well as against the non-cancerous human embryonic kidney (HEK-293) cell line. In order to investigate whether the antiproliferative activity is size-dependent, binuclear model analogues [9][PF6]–[12][PF6] were also prepared and evaluated.
 | ||
| Scheme 1 Synthesis of ferrocenyl-derived heterometallic metallodendrimers [1][PF6]4–[8][PF6]8. | ||
Results and discussion
Synthesis of ferrocenyl-derived heterometallic metallodendrimers
Ferrocenyl-derived heterometallic metallodendrimers [1][PF6]4–[8][PF6]8 were prepared from the dinuclear precursors [(η6-p-iPrC6H4Me)RuCl2]2, [(η6-C6Me6)RuCl2]2, [(η5-C5Me5)IrCl2]2 or [(η5-C5Me5)RhCl2]2 and 4-ferrocenylpyridine, in the presence of Et3N, functionalized onto known first- and second-generation salicylaldiminepoly(propyleneimine) dendritic ligands (L1, L2)82 (Scheme 1). The metallodendrimers were isolated in moderate yields as hexafluorophosphate salts via a metathesis reaction. Compounds [1][PF6]4–[8][PF6]8 are non-hygroscopic, air- and moisture-stable orange solids, and are soluble in dimethylsulfoxide, acetonitrile or acetone.Compared to non-ferrocenyl neutral derivatives previously reported,74,83 the 1H NMR spectra of [1][PF6]4–[8][PF6]8 show a general downfield shift in signals due the cationic nature of the complexes. More specifically, the imine proton appears slightly more downfield from ca. 7.9 ppm (in the neutral complex)74,83 to ca. 8.2 ppm (in the cationic complex). The shift in the imine proton, the general downfield shift of the aromatic protons and the disappearance of the phenolic proton for [1][PF6]4–[8][PF6]8 compared to the free ligands L1 and L2, suggests coordination to the metal ion via the imine nitrogen and phenolic oxygen. The protons on the substituted and un-substituted Cp rings (of the ferrocenylpyridine moiety) are assigned to the broad singlet and two broad multiplets appearing in the region between 4.0 and 5.0 ppm for [1][PF6]4–[8][PF6]8. The aliphatic protons of the dendritic core and dendritic arms occur as broadened peaks, between 1.2 ppm and 3.0 ppm and at similar chemical shifts to those of the dendritic ligands L1 and L2. All metallodendrimers [1][PF6]4–[8][PF6]8 show a loss of two-fold symmetry around the metal center upon coordination of the N,O-dendritic ligand. Evidence for this is provided by the presence of two sets of signals (one of the signals is masked by the singlet observed for the un-substituted Cp ring) for the –CH2– group adjacent to the imine nitrogen, due to the diastereotopic nature of these protons induced by the chiral metal center. As a result, the methyl protons of the isopropyl group, on the arene ring, of the Ru–p-cymene metallodendrimers [1][PF6]4 and [2][PF6]8 exhibit two broad doublets in the range of 1.2–1.3 ppm. A broad singlet is observed at 2.0 ppm (for [3][PF6]4 and [4][PF6]8) and at 1.6 ppm (for [5][PF6]4 and [8][PF6]8) for the HMB and Cp* protons, respectively.
The infrared spectra of metallodendrimers [1][PF6]4–[8][PF6]8 display a shift for the (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N)imine stretching vibration from ca. 1630 cm−1 in the ligand (L1 or L2), to lower wavenumbers ca. 1611 cm−1, supporting coordination of the ligand to the metal center via the imine nitrogen. This stretching vibration also overlaps with the vibration observed for the (CN)pyridyl on the ferrocenylpyridine moiety. Inclusion of solvent molecules resulted in elemental analysis data for [1][PF6]4–[8][PF6]8 to within acceptable limits, and is similarly observed with other previously reported metallodendrimers.72,81 High-resolution mass spectrometry data (run in the positive-ion mode) is consistent with the proposed structures of [1][PF6]4–[8][PF6]8, with a base peak observed corresponding to a charged adduct.
N)imine stretching vibration from ca. 1630 cm−1 in the ligand (L1 or L2), to lower wavenumbers ca. 1611 cm−1, supporting coordination of the ligand to the metal center via the imine nitrogen. This stretching vibration also overlaps with the vibration observed for the (CN)pyridyl on the ferrocenylpyridine moiety. Inclusion of solvent molecules resulted in elemental analysis data for [1][PF6]4–[8][PF6]8 to within acceptable limits, and is similarly observed with other previously reported metallodendrimers.72,81 High-resolution mass spectrometry data (run in the positive-ion mode) is consistent with the proposed structures of [1][PF6]4–[8][PF6]8, with a base peak observed corresponding to a charged adduct.
Synthesis of ferrocenyl-derived heterometallic binuclear complexes
Binuclear model complexes [9][PF6]–[12][PF6] were prepared to evaluate whether there is a size dependency correlation between the complexes prepared and their biological activity (see below). Two equivalents of the monomeric ligand L3,84 were reacted with one equivalent of the particular metal-dimer, in the presence of triethylamine, in a one-pot reaction (Scheme 2). Subsequently, 4-ferrocenylpyridine was added to the reaction mixture, which generated a cationic complex, and the products were isolated as red or red-orange hexafluorophosphate salts. The binuclear model complexes have similar solubilities to their dendritic derivatives [1][PF6]4–[8][PF6]8. Furthermore, the synthesis and purity of the heterometallic model (analogues [9][PF6]–[12][PF6] were confirmed by 1H, 13C{1H} NMR and infrared spectroscopy, elemental analysis and mass spectrometry.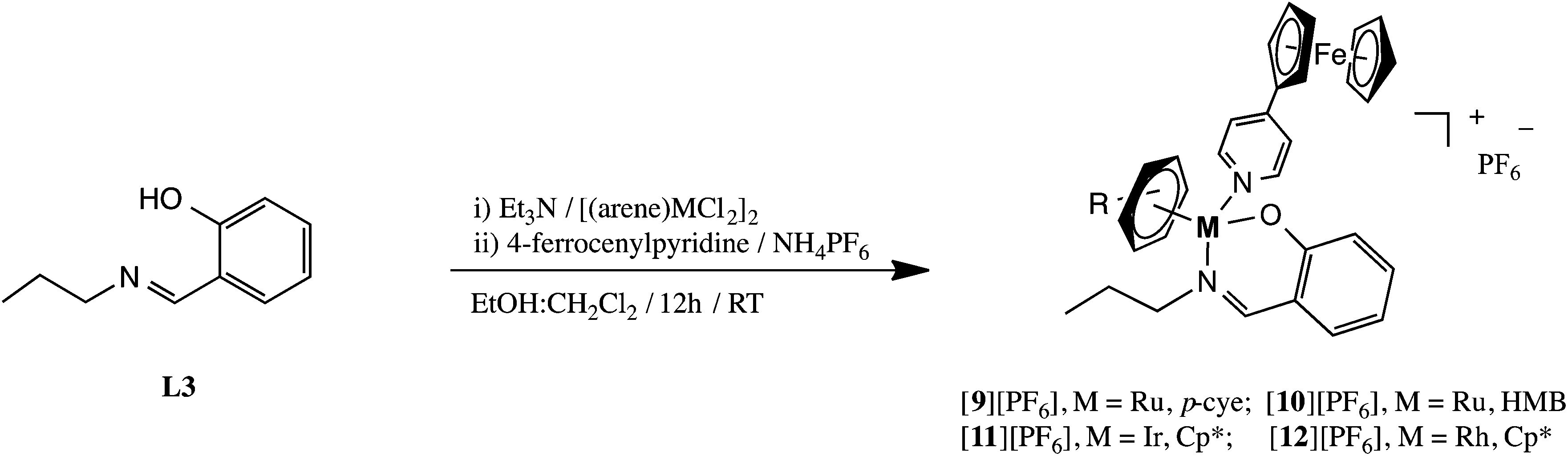 | ||
| Scheme 2 Synthesis of ferrocenyl-derived heterometallic mononuclear complexes [9][PF6]–[12][PF6]. | ||
Biological activity
The antiproliferative activity of the cationic metallodendrimers [1][PF6]4–[8][PF6]8 and the model compounds [9][PF6]–[12][PF6] was established in vitro against the human ovarian cisplatin-sensitive (A2780) and cisplatin-resistant (A2780cisR) cancer cell lines and against the non-tumorigenic human embryonic kidney (HEK-293) cell line (Table 1). The model complexes [9][PF6]–[12][PF6] display modest activity in both cancer cell lines and, notably, they are not cancer cell selective. However, there is a considerable increase in cytotoxicity on moving to the higher generation compounds, in particular, for the ferrocenyl-derived Ru–HMB metallodendrimers ([3][PF6]4 and [4][PF6]8) and Rh–Cp* ([7][PF6]4 and [8][PF6]8). The first-generation metallodendrimers [3][PF6]4 (9.1 μM in A2780; 5.9 μM in A2780cisR) and [7][PF6]4 (6.1 μM in A2780; 12.7 μM in A2780cisR) and second-generation metallodendrimers [4][PF6]8 (4.7 μM in A2780; 3.5 μM in A2780cisR), [6][PF6]8 (11.7 μM in A2780; 7.6 μM in A2780cisR) and [8][PF6]8 (10.7 μM in A2780; 7.9 μM in A2780cisR) are the most active of the series. With the exception of the p-cymene metallodendrimers [1][PF6]4 and [2][PF6]8, and the first-generation Ir–Cp* metallodendrimer [5][PF6]4, the metallodendrimers have similar cytotoxicites in both the sensitive and resistant cancer cell lines, indicating that these compounds operate via a different mechanism of action to that of cisplatin. Similar, IC50 values were previously observed for the p-cymene derivatives (where ferrocene is functionalized onto the periphery of the metallodendrimer),81 were also observed for the p-cymene derivatives [1][PF6]4 and [2][PF6]8, where the ferrocene moiety is directly coordinated to the ruthenium center. The introduction of the ferrocenyl moiety results in comparable cytotoxicities for the Rh and Ir metallodendrimers reported here, to their neutral RhCp*–Cl and IrCp*–Cl analogues reported previously.74 However, incorporation of pyridylferrocene group on [7][PF6]4 improves the activity of the compound compared to the first generation neutral Rh–Cp*–Cl analogue (55 μM in A2780 and 126 μM in A2780cisR).74 Similarly, an improvement in activity for the cationic ferrocenyl-derived Ru–HMB metallodendrimers [3][PF6]4 and [4][PF6]8 was observed compared to their neutral Ru–HMB-Cl analogs (G1-Ru–HMB-Cl = 27 μM (A2780); 25 μM (A2780cisR); G2-Ru–HMB-Cl = 10 μM (A2780); 10 μM (A2780cisR)) reported previously,83 with comparable activities to their cationic Ru–HMB-PTA analogs (G1-Ru–HMB-PTA = 9 μM (A2780); 25 μM (A2780cisR); G2-Ru–HMB-PTA = 6 μM (A2780); 12 μM (A2780cisR)).73 There is a correlation between the size-dependency of the metallodendrimers and the cytotoxicity, with metallodendrimers [3][PF6]4 and [4][PF6]8 being the only compounds to exhibit a significant degree of cancer cell selectivity. A direct comparison between these systems (ferrocene at the metal center) and the previously prepared systems (ferrocene at the periphery)81 cannot be made as different in vitro biological studies were performed. However, coordination of the pyridylferrocene moiety to the Ru center affords more cytotoxic compounds.| Compound | Ma | n | A2780 (IC50, μM) | A2780cisR (IC50, μM) | HEK-293 (IC50, μM) |
|---|---|---|---|---|---|
| a Type of metal(s) present in the complex. b Number of metals within the complex. | |||||
| [1][PF6]4 | Ru–Fe | 8 | 107.2 ± 2.0 | 53.3 ± 0.1 | 138.3 ± 7.3 |
| [2][PF6]8 | Ru–Fe | 16 | 26.6 ± 1.0 | 44.1 ± 1.7 | 20.3 ± 1.5 |
| [3][PF6]4 | Ru–Fe | 8 | 9.1 ± 0.3 | 5.9 ± 0.01 | 11.6 ± 1.1 |
| [4][PF6]8 | Ru–Fe | 16 | 4.7 ± 0.2 | 3.5 ± 0.2 | 5.7 ± 0.2 |
| [5][PF6]4 | Ir–Fe | 8 | 46.4 ± 2.5 | 59.0 ± 0.6 | 35.9 ± 3.4 |
| [6][PF6]8 | Ir–Fe | 16 | 11.7 ± 0.3 | 7.6 ± 1.3 | 13.6 ± 2.0 |
| [7][PF6]4 | Rh–Fe | 8 | 6.1 ± 0.1 | 12.7 ± 1.0 | 19.0 ± 0.6 |
| [8][PF6]8 | Rh–Fe | 16 | 10.7 ± 1.3 | 7.9 ± 0.4 | 9.4 ± 1.4 |
| [9][PF6] | Ru–Fe | 2 | 29.2 ± 1.7 | 33.0 ± 2.4 | 28.7 ± 0.6 |
| [10][PF6] | Ru–Fe | 2 | 19.7 ± 1.3 | 17.4 ± 1.0 | 16.1 ± 1.5 |
| [11][PF6] | Ir–Fe | 2 | 39.6 ± 3.0 | 36.7 ± 0.2 | 31.1 ± 0.3 |
| [12][PF6] | Rh–Fe | 2 | 59.0 ± 0.3 | 72.9 ± 0.3 | 65.5 ± 3.3 |
| cisplatin | Pt | 1 | 1.5 ± 0.5 | 25 ± 5.0 | 10 ± 2.0 |
NMR stability study
Compound stability is important for biological applications and, in the case of metal-based drugs, many compounds are actually prodrugs and hence the identity of the compound that reaches the cell can be important. Following uptake into the cell, related ruthenium–arene-PTA complexes are activated via aquation, generating the aqua species, a process that can be monitored by NMR spectroscopy.85,86 Furthermore, to investigate the influence of the bidentate chelating N,O-dendritic ligands on the stability of the metallodendrimers in solution and to mimic the preparation of solutions prior to biological assays, aqueous stability studies were performed on selected complexes, in a mixture of DMSO-d6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :D2O (50:50% v/v) at 37 °C, using 1H NMR spectroscopy. The HMB-derivatives display the best in vitro activity, thus, the second-generation metallodendrimer [4][PF6]8 and its closest binuclear model analogue [10][PF6] were selected, and studies were undertaken in DMSO-d6:D2O (50:50% v/v) over 24 hours at 37 °C (Fig. 1).
:D2O (50:50% v/v) at 37 °C, using 1H NMR spectroscopy. The HMB-derivatives display the best in vitro activity, thus, the second-generation metallodendrimer [4][PF6]8 and its closest binuclear model analogue [10][PF6] were selected, and studies were undertaken in DMSO-d6:D2O (50:50% v/v) over 24 hours at 37 °C (Fig. 1).
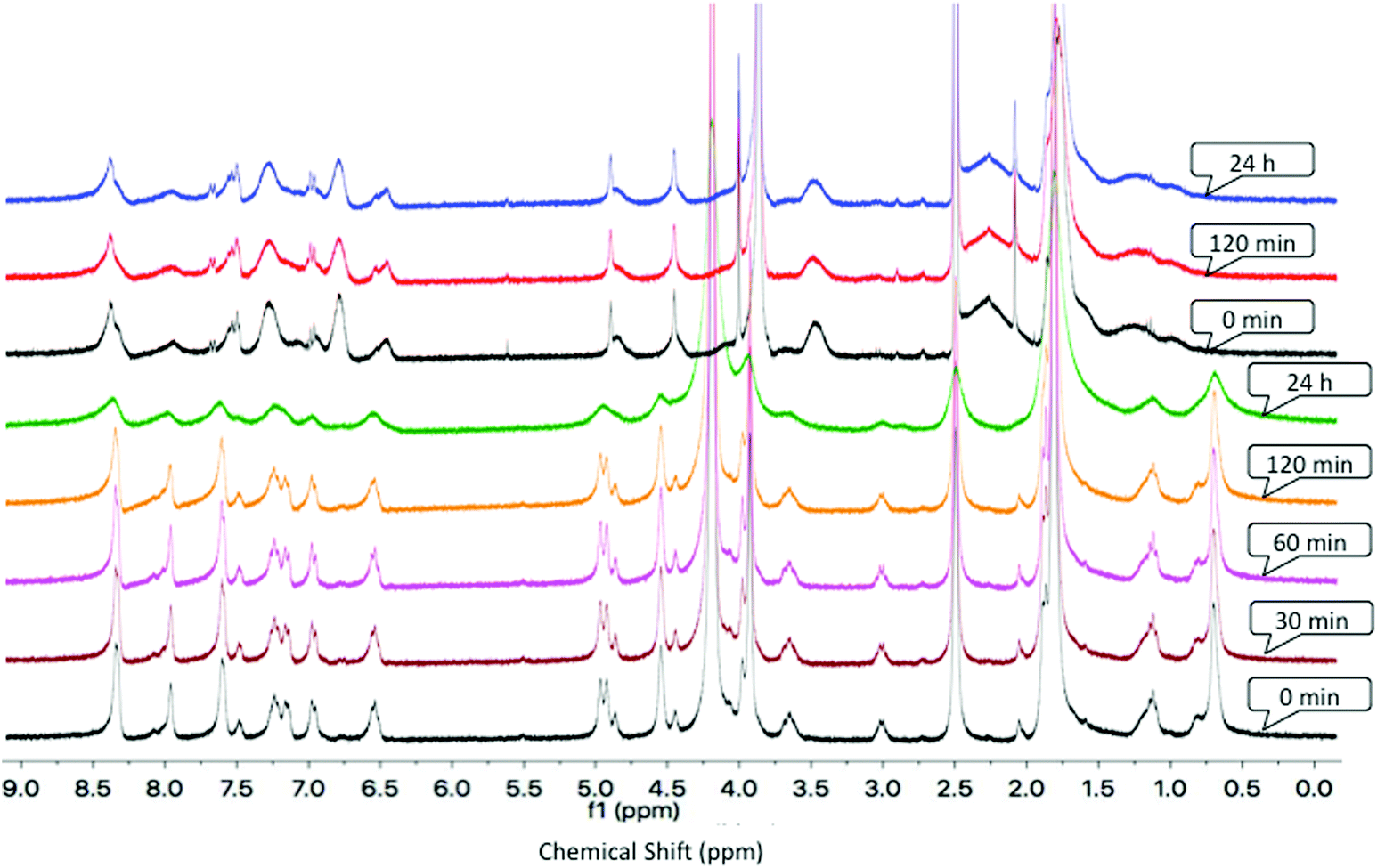 | ||
| Fig. 1
1H NMR spectra of [4][PF6]8 (top three spectra) and [10][PF6] (bottom 5 spectra) in DMSO-d6:D2O (50:50% v/v) over 24 hours at 37 °C. | ||
The 1H NMR spectra of [4][PF6]8 and [10][PF6] (Fig. 1) show that the compounds are stable in solution, with no side-products or the formation of the aqua-species observed over a period of 24 hours. In addition, these data show that the complexes are stable in deuterated dimethylsulfoxide, relevant during the time period between preparation of the compound stock solutions and dosing of the cancer cells in the assay.
UV-Vis study: interactions with a DNA model
DNA is a potential drug target for ruthenium–arene compounds and is an important target in cancer therapy.87 Furthermore, some of the most cytotoxic ruthenium drugs act as DNA intercalators upon coordination to the suitable ancillary ligand.40 In order to correlate the antiproliferative activity of the metallodendrimers to possible interactions with DNA, a UV-vis study was performed. The interactions of [4][PF6]8 (the most active compound) and its model analogue [10][PF6] were selected for the study and their interaction with Red Salmon testes DNA monitored by UV-vis spectroscopy (Fig. 2). | ||
| Fig. 2 UV-vis absorption spectra of 0.15 mM [10][PF6] (left) and 0.23 mM [4][PF6]8 (right) in 0.2 mM HEPES buffer, pH 6.3 at 37 °C, in H2O in the absence and then in the presence of increasing concentration of Red Salmon testes DNA. | ||
The UV-vis spectra for both [4][PF6]8 and [10][PF6] display hypochromic shifts (upon DNA addition) as the absorbance at λmax = 320 nm (for [4][PF6]8) and λmax = 310 nm (for [10][PF6]) decrease, with no substantial red- or blue-shift observed for λmax for both systems (Fig. 2). Similar hypochromic shifts were reported with Cu(II), Co(II) and Ni(II) complexes upon addition of increasing concentration of DNA,88 whereas norepinephrine (DNA binding agent) displays a hyperchromic shift upon DNA addition.89 A hyperchromic shift is usually associated with the partial uncoiling of the DNA double helix, which exposes more of the DNA base-pairs, which in turn results in the increase in absorbance at λmax. However, hypochromic effects are also associated with non-covalent interactions between the complex and DNA, such as intercalative binding,90 indicating that DNA could be a relevant target for these compounds.
Conclusions
A series of bidentate heterometallic ferrocenyl-derived dendrimers containing ruthenium(II)–p-cymene, ruthenium(II)–HMB, rhodium(III)–cyclopentadienyl or iridium(III)–cyclopentadienyl moieties have been successfully synthesized and characterized. Heterometallic model complexes were also prepared and characterized (for comparison with the larger metallodendrimers). Antiproliferative studies performed on the metallodendrimers and the model binuclear complexes show that the model complexes display limited activity, whereas the metallodendrimers display higher cytotoxicities, particularly the second-generation ferrocenyl-derived ruthenium(II)–hexamethylbenzene metallodendrimer, which displayed the best activity of the series. The presence of ferrocene leads to more cytotoxic compounds compared to previously reported non-ferrocenyl derivatives. In addition, these metallodendrimers displayed no cross-resistance to cisplatin and some are cancer cell selective, being less cytotoxic to the non-tumorigenic cells included in the study. Spectroscopic studies illustrate that these systems are stable in solution and additional DNA binding studies suggest that a possible mode-of-action of these metallodendrimers (at least for the most active derivative) involves possible non-covalent interactions (illustrated by UV-vis experiments) with the DNA.Experimental
General details
All solvents and reagents were purchased from Sigma-Aldrich; DAB-G2-PPI-(NH2)8 was purchased from SyMO-Chem and used without further purification. Ruthenium trichloride trihydrate, iridium trichloride trihydrate and rhodium trichloride trihydrate were obtained from Heraeus Limited. L1,82L2,82L3,84 [(η6-p-iPrC6H4Me)RuCl2]2,91 [(η6-C6Me6)RuCl2]2,92 [(η5-C5Me5)IrCl2]2,93 [(η5-C5Me5)RhCl2]2,93 and 4-ferrocenylpyridine (4-FcPyr)16 were prepared according to literature procedures. Infrared (IR) spectra determined in the solid state on a Perkin-Elmer Spectrum 100 FT-IR spectrometer equipped with a SMART iTR ATR unit. The intensity of stretching vibrations are marked as strong (s), medium (m) and weak (w). Nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury XR300 spectrometer (1H: 300.08 MHz; 13C{1H}: 75.46 MHz) or Bruker Biospin GmbH spectrometer (1H: 400.22 MHz; 13C{1H}: 100.65 MHz) at ambient temperature. Chemical shifts δ in ppm indicate a downfield shift relative to tetramethylsilane (TMS) and were referenced relative to the signal of the solvent.94 Individual peaks are marked as singlet (s), doublet (d), doublet-of-doublet (dd), triplet (t), or multiplet (m). High-resolution electrospray ionization-mass spectrometry (HR-ESI-MS) was carried out on a Waters Synapt mass spectrometer. Data were recorded in positive ion mode. Elemental analysis (C, H, N) was carried out using a Thermo Flash 1112 Series CHNS-O Analyzer. For certain metallodendrimers, the analyses are outside acceptable limits, which is ascribed to the encapsulation of solvent molecules and/or other inorganic salts by the dendritic compounds. UV-vis absorption studies were carried out on a Cary UV-vis spectrophotometer using a 1 cm path length quartz cuvette to carry out the measurements.Compound synthesis
:DCM (50:50, 30 mL) mixture. The resulting yellow-suspension was stirred at room temperature for 0.5 hour. Next, [(η6-p-iPrC6H4Me)RuCl2]2 (0.0815 g, 0.133 mmol for [1][PF6]4; 0.0794 g, 0.130 mmol for [2][PF6]8) or [(η6-C6Me6)RuCl2]2 (0.0870 g, 0.129 mmol for [3][PF6]4; 0.0901 g, 0.133 mmol for [4][PF6]8) was added to the reaction mixture. The reaction mixture was stirred overnight at room temperature, then, the reaction mixture was filtered and the filtrate reduced to ca. 5 mL. This was followed by the addition 4-ferrocenylpyridine (0.0692 g, 0.263 mmol for [1][PF6]4; 0.0678 g, 0.258 mmol for [2][PF6]8; 0.0669 g, 0.254 mmol for [3][PF6]4; 0.0696 g, 0.265 mmol for [4][PF6]8) and the solution was stirred at RT for 2 hours and filtered. NH4PF6 (0.0429 g, 0.263 mmol for [1][PF6]4; 0.0420 g, 0.258 mmol for [2][PF6]8; 0.0414 g, 0.254 mmol for [3][PF6]4; 0.0431 g, 0.264 mmol for [4][PF6]8) was added to the filtrate at 0 °C and stirred for 4 hours. An orange-red precipitate was observed. The solid was isolated by filtration, washed with isopropanol and finally with Et2O. The solid was dried in vacuo.
[DAB-G1-PPI-{(η6-p-cymene)Ru((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}4][PF6]4 ([1][PF6]4). Orange solid. Yield: 0.120 g, 57%. IR (ATR): ν (cm−1) = 1611 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 0.96–1.21 & 1.24–1.32 (br m, 24H, CH(CH3)2 p-cymene), 1.71–2.26 (overlapping m, 24H, NCH2CH2 core, NCH2CH2 core, NCH2CH2CH2Nbranch, NCH2CH2CH2Nbranch), 2.51–3.11 (br m, 16H, CH3 p-cymene, CH(CH3)2 p-cymene), 3.96 (br s, 20H, Cp–CHunsubst. ring), 4.32–4.64 (overlapping br m, 16H, NCH2CH2CH2Nbranch, Cp–CH), 4.70–4.98 (m, 8H, Cp–CH), 5.54–5.76 (m, 8H, Arp-cymene), 5.81–6.03 (m, 8H, Arp-cymene), 6.27–6.41 (m, 4H, Ar), 6.87–7.02 (br m, 8H, 2 × Ar), 7.09–7.24 (m, 4H, Ar), 7.39–7.63 (m, 8H, Pyr), 8.11 (br s, 4H, CHimine), 8.39–8.55 (m, 8H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 17.0, 21.5, 22.1 (CH3 p-cymene); 50.7, 66.4, 69.7, 70.2, 71.7 (CH2); 67.5, 71.6 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.0 (CCp); 30.8, 83.1, 84.1, 84.5, 88.8 (CHp-cymene); 99.8, 119.9 (Cp-cymene); 115.0, 121.3, 135.0, 135.1 (CHAr); 120.0 (Cpyr); 121.7, 152.5 (CHpyr); 152.9, 164.9 (CAr); 165.9 (CHimine). Elemental analysis for C140H160F24Fe4N10O4P4Ru4·2DCM (3424.2436): Found C, 49.86; H, 4.47; N, 3.94%; calcd C, 49.81; H, 4.83; N, 4.09%. MS (HR-ESI-TOF, m/z): 417.8664 [M − 4(4-FcPyr)]4+ (where M = [1][PF6]4 − 4PF6).
[DAB-G2-PPI-{(η6-p-cymene)Ru((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}8][PF6]8 ([2][PF6]8). Orange solid. Yield: 0.1207 g, 56%. IR (ATR): ν (cm−1) = 1612 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.02–1.20 & 1.23–1.35 (br m, 48H, CH(CH3)2 p-cymene), 1.56–3.17 (overlapping m, 64H, NCH2CH2 core, NCH2CH2 core, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N2nd branch, NCH2CH2CH2N2nd branch), 2.87 (br m, 32H, CH3 p-cymene, CH(CH3)2 p-cymene), 3.96 (br s, 40H, Cp–CHunsubst. ring), 4.44–4.70 (br m, 32H, NCH2CH2CH2N2nd branch, Cp–CH), 4.78–4.98 (m, 16H, Cp–CH), 5.57–5.81 (m, 16H, Arp-cymene), 5.86–6.04 (m, 8H, Arp-cymene), 6.32–6.42 (m, 8H, Ar), 6.97–7.11 (br m, 16H, 2 × Ar), 7.16–7.27 (m, 8H, Ar), 7.43–7.61 (m, 16H, Pyr), 8.16 (br s, 8H, CHimine), 8.40–8.58 (m, 16H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 17.0, 21.5, 22.1 (CH3 p-cymene); 49.0, 52.4, 66.6, 69.8 (CH2); 67.6, 71.7 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.0 (CCp); 30.8, 83.2, 84.2, 84.6, 89.2 (CHp-cymene); 99.9, 120.0 (Cp-cymene); 115.0, 121.7, 135.0, 134.8, 135.1 (CHAr); 120.8 (Cpyr); 121.3, 152.2 (CHpyr); 153.5, 164.7 (CAr); 165.5 (CHimine). Elemental analysis for C296H336F48Fe8N22O8P8Ru8·9DCM (7509.4676): Found C, 48.51; H, 4.67; N, 4.39%; calcd C, 48.78; H, 4.75; N, 4.10%. MS (HR-ESI-TOF, m/z): 533.0239 [M − 3(4-FcPyr) + H]9+ (where M = [2][PF6]8 − 8PF6).
[DAB-G1-PPI-{(η6-HMB)Ru((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}4][PF6]4 ([3][PF6]4). Orange solid. Yield: 0.0749 g, 35%. IR (ATR): ν (cm−1) = 1610 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.27–1.55 (br m, 4H, NCH2CH2 core), 1.77–1.88 (m, 8H, NCH2CH2CH2Nbranch), 2.03 (br s, 72H, ArHMB), 2.38–2.71 (overlapping m, 12H, NCH2CH2 core, NCH2CH2CH2Nbranch), 3.59–3.75 (br m, 8H, NCH2CH2CH2Nbranch), 4.05 (br s, 20H, Cp–CHunsubst. ring), 4.53–4.62 (m, 8H, Cp–CH), 4.90–5.00 (m, 8H, Cp–CH), 6.51–6.62 (m, 4H, Ar), 6.84–6.96 (m, 4H, Ar), 7.06–7.23 (m, 4H, Ar), 7.28–7.42 (m, 4H, Ar), 7.66–7.80 (m, 8H, Pyr), 8.14 (br s, 4H, CHimine), 8.45–8.63 (m, 8H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 14.7 (CH3 HMB); 25.4, 51.0, 66.9, 69.8, 70.2 (CH2); 67.5, 71.7 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.0 (CCp); 94.2 (CHMB); 115.4, 123.3, 134.4, 134.6 (CHAr); 121.8 (Cpyr); 122.1, 151.4 (CHpyr); 153.2, 165.1 (CAr); 165.7 (CHimine). Elemental analysis for C156H196N18F24Fe4O4P8Ru4·2IPOH (3486.7828): Found C, 53.54; H, 5.23; N, 4.49%; calcd C, 53.05; H, 5.55; N, 4.02%. MS (HR-ESI-TOF, m/z): 428.1369 [M − (4-FcPyr) + 2H]6+ (where M = [3][PF6]4 − 4PF6).
[DAB-G2-PPI-{(η6-HMB)Ru((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}8][PF6]8 ([4][PF6]8). Orange solid. Yield: 0.0764 g, 33%. IR (ATR): ν (cm−1) = 1610 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.56–3.33 (overlapping m, 64H, NCH2CH2 core, NCH2CH2 core, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N2nd branch, NCH2CH2CH2N2nd branch), 2.00 (br s, 144H, ArHMB), 3.57–3.75 (br m, 16H, NCH2CH2CH2N2nd branch), 4.05 (br s, 40H, Cp–CHunsubst. ring), 4.52–4.61 (m, 16H, Cp–CH), 4.91–5.01 (m, 16H, Cp–CH), 6.50–6.61 (m, 8H, Ar), 6.80–6.97 (m, 8H, Ar), 7.13–7.22 (m, 8H, Ar), 7.27–7.40 (m, 8H, Ar), 7.68–7.78 (m, 16H, Pyr), 8.09 (br s, 8H, CHimine), 8.49–8.65 (m, 16H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 14.7 (CH3 HMB); 21.9, 51.0, 51.9, 61.8, 69.8 (CH2); 67.5, 71.7 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.4 (CCp); 94.2 (CCp*); 115.4, 123.4, 134.4, 134.5 (CHAr); 120.5 (Cpyr); 122.1, 151.4 (CHpyr); 152.8, 165.0 (CAr); 165.8 (CHimine). Elemental analysis for C296H336F48Fe8N22O8P8Ru8·3DCM (7224.2996): Found C, 52.20; H, 5.24; N, 4.49%; calcd C, 52.37; H, 5.22; N, 4.27%. MS (HR-ESI-TOF, m/z): 562.0537 [M − 5(4-FcPyr)]8+ (where M = [4][PF6]8 − 8PF6).
[DAB-G1-PPI-{(η5-C5Me5)Ir((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}4][PF6]4 ([5][PF6]4). Orange solid. Yield: 0.151 g, 61%. IR (ATR): ν (cm−1) = 1612 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.26–1.60 (overlapping m, 72H, ArCp*, NCH2CH2 core, NCH2CH2CH2Nbranch), 2.23–2.66 (overlapping m, 12H, NCH2CH2 core, NCH2CH2CH2Nbranch), 4.06 (br s, 20H, Cp–CHunsubst. ring), 4.15–4.39 (br m, 8H, NCH2CH2CH2Nbranch), 4.51–4.67 (m, 8H, Cp–CH), 4.89–5.07 (m, 8H, Cp–CH), 6.52–6.67 (m, 4H, Ar), 6.98–7.09 (m, 4H, Ar), 7.21–7.33 (m, 4H, Ar), 7.38–7.52 (m, 4H, Ar), 7.69–7.88 (m, 8H, Pyr), 8.13 (br s, 4H, CHimine), 8.58–9.02 (m, 8H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 7.76 (CH3 Cp*); 24.0, 49.8, 53.4, 63.9, 69.7, 71.7 (CH2); 67.7, 71.6 (Cp–CH); 70.4 (Cp–CHunsubst. ring); 77.9 (CCp); 94.2 (CCp*); 116.4, 122.7, 134.4, 135.0 (CHAr); 122.3 (Cpyr); 122.1, 150.3 (CHpyr); 153.8, 164.1 (CAr); 163.6 (CHimine). Elemental analysis for C140H164F24Fe4Ir4N10O4P4·3DCM (3877.808): Found C, 44.49; H, 4.45; N, 3.63%; calcd C, 44.29; H, 4.42; N, 3.61%. MS (HR-ESI-TOF, m/z): 626.0876 [M + K]5+ (where M = [5][PF6]4 − 4PF6).
[DAB-G2-PPI-{(η5-C5Me5)Ir((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}8][PF6]8 ([6][PF6]8). Orange solid. Yield: 0.177 g, 70%. IR (ATR): ν (cm−1) = 1613 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.32–3.08 (overlapping m, 64H, NCH2CH2 core, NCH2CH2 core, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N2nd branch, NCH2CH2CH2N2nd branch), 1.51 (br s, 120H, ArCp*), 4.05 (br s, 40H, Cp–CHunsubst. ring), 4.21–4.38 (br m, 16H, NCH2CH2CH2N2nd branch), 4.52–4.66 (m, 16H, Cp–CH), 4.91–5.07 (m, 16H, Cp–CH), 6.48–6.66 (m, 8H, Ar), 6.97–7.10 (m, 8H, Ar), 7.25–7.31 (m, 8H, Ar), 7.33–7.40 (m, 8H, Ar), 7.66–7.88 (m, 16H, Pyr), 8.19 (br s, 8H, CHimine), 8.59–8.83 (m, 16H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 7.8 (CH3 Cp*); 23.8, 49.9, 51.4, 63.7, 69.7 (CH2); 67.7, 71.9 (Cp–CH); 70.4 (Cp–CHunsubst. ring); 77.6 (CCp); 87.1 (CCp*); 116.4, 122.7, 134.5, 135.0 (CHAr); 122.3 (Cpyr); 122.1, 150.3 (CHpyr); 153.5, 163.9 (CAr); 163.5 (CHimine). Elemental analysis for C296H344F48Fe8Ir8N22O8P8·10DCM (8331.6636): Found C, 44.01; H, 4.00; N, 3.89%; calcd C, 44.11; H, 4.40; N, 3.70%. MS (HR-ESI-TOF, m/z): 626.0886 [M − 5(4-FcPyr)]8+ (where M = [6][PF6]8 − 8PF6).
[DAB-G1-PPI-{(η5-C5Me5)Rh((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}4][PF6]4 ([7][PF6]4). Orange solid. Yield: 0.126 g, 56%. IR (ATR): ν (cm−1) = 1611 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.24–1.73 (overlapping m, 72H, ArCp*, NCH2CH2 core, NCH2CH2CH2Nbranch), 2.17–3.04 (overlapping m, 12H, NCH2CH2 core, NCH2CH2CH2Nbranch), 4.06–4.24 (br m, 28H, Cp–CHunsubst. ring, NCH2CH2CH2Nbranch), 4.50–4.61 (m, 8H, Cp–CH), 4.91–5.00 (m, 8H, Cp–CH), 6.52–6.73 (m, 4H, Ar), 7.01–7.14 (m, 4H, Ar), 7.19–7.26 (m, 4H, Ar), 7.31–7.48 (m, 4H, Ar), 7.67–7.86 (m, 8H, Pyr), 8.22 (br s, 4H, CHimine), 8.53–8.76 (m, 8H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 8.02 (CH3 Cp*); 24.5, 50.2, 60.9, 69.7, 71.7 (CH2); 67.6, 71.7 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.3 (CCp); 95.1 (CCp*); 115.5, 123.3, 134.8, 135.0 (CHAr); 122.0 (Cpyr); 122.7, 150.6 (CHpyr); 153.2, 164.7 (CAr); 166.2 (CHimine). Elemental analysis for C140H164F24Fe4N10O4P4Rh4·2DCM (3435.6352): Found C, 49.46; H, 4.66; N, 3.88%; calcd C, 49.64; H, 4.93; N, 4.08%. MS (HR-ESI-TOF, m/z): 617.8252 [M − (4-FcPyr)]4+ (where M = [7][PF6]4 − 4PF6).
[DAB-G2-PPI-{(η5-C5Me5)Rh((C7H5NO)-κ2-N,O)-4-ferrocenylpyridine)}8][PF6]8 ([8][PF6]8). Orange solid. Yield: 0.119 g, 56%. IR (ATR): ν (cm−1) = 1611 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.25–3.41 (overlapping m, 64H, NCH2CH2 core, NCH2CH2 core, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N1st branch, NCH2CH2CH2N2nd branch, NCH2CH2CH2N2nd branch), 1.52 (br s, 120H, ArCp*), 3.97–4.33 (br m, 56H, Cp–CHunsubst. ring, NCH2CH2CH2N2nd branch), 4.49–4.59 (m, 16H, Cp–CH), 4.90–4.98 (m, 16H, Cp–CH), 6.53–6.70 (m, 8H, Ar), 7.03–7.16 (m, 8H, Ar), 7.24–7.51 (br m, 16H, 2 × Ar), 7.69–7.94 (m, 16H, Pyr), 8.25 (br s, 8H, CHimine), 8.53–8.68 (m, 16H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 7.79 (CH3 Cp*); 24.6, 50.1, 51.8, 63.7, 69.5 (CH2); 67.6, 71.7 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.0 (CCp); 95.1 (CCp*); 115.8, 122.9, 134.7, 134.9 (CHAr); 122.4 (Cpyr); 122.7, 152.0 (CHpyr); 153.1, 165.9 (CAr); 166.0 (CHimine). Elemental analysis for C296H344F48Fe8N22O8P8Ru8·14DCM (7956.9148): Found C, 46.13; H, 4.99; N, 3.55%; calcd C, 46.79; H, 4.71; N, 3.87%. MS (HR-ESI-TOF, m/z): 400.1145 [M + 6H]14+ (where M = [8][PF6]8 − 8PF6).
[CH3CH2CH2-(η6-p-cymene)Ru(C7H5NO)-κ2-N,O)-4-ferrocenylpyridine][PF6] ([9][PF6]). Red solid. Yield: 0.2152 g, 91%. IR (ATR): ν (cm−1) = 1609 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 1.09 (t, 3J = 7.3 Hz, 3H, NCH2CH2CH3), 1.31 & 1.23 (d, 3J = 6.9 Hz, 6H, CH(CH3)2 p-cymene), 1.82–1.91 & 2.03–2.11 (m, 2H, NCH2CH2CH3), 2.80 (s, 3H, CH3 p-cymene), 2.83–2.90 (m, 1H, CH(CH3)2 p-cymene), 3.97 (s, 5H, Cp–CHunsubst. ring), 4.43–4.49 & 4.53–4.59 (m, 2H, NCH2CH2CH3), 4.58 (t, 3J = 1.9 Hz, 2H, Cp–CH), 4.97 (t, 3J = 1.9 Hz, 2H, Cp–CH), 5.70–5.81 (m, 2H, Arp-cymene), 5.98–6.10 (m, 2H, Arp-cymene), 6.40 (ddd, 3J = 7.9, 8.9, 1.1 Hz, 1H, Ar), 6.94 (d, 3J = 8.4 Hz, 1H, Ar), 7.01 (dd, 3J = 7.9, 1.8 Hz, 1H, Ar), 7.22 (ddd, 3J = 8.5, 6.9, 1.8 Hz, 1H, Ar), 7.57 (d, 3J = 6.8 Hz, 2H, Pyr), 8.12 (s, 1H, CHimine), 8.53 (d, 3J = 6.8 Hz, 2H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 10.6 (CH3); 8.1, 21.4, 22.0 (CH3 p-cymene); 25.0, 66.8 (CH2); 67.5, 71.6 (Cp–CH); 70.2 (Cp–CHunsubst. ring); 77.9 (CCp); 30.8, 83.1, 84.2, 84.5, 89.2 (CHp-cymene); 99.9, 119.9 (Cp-cymene); 114.9, 121.3, 134.8, 135.0 (CHAr); 120.3 (Cpyr); 121.6, 152.5 (CHpyr); 149.6, 164.8 (CAr); 165.9 (CHimine). Elemental analysis for C35H39F6FeN2OPRu (805.5831): Found C, 52.01; H, 4.81; N, 3.37%; calcd C, 52.18; H, 4.88; N, 3.48%. MS (HR-ESI-TOF, m/z): 398.1064 [M − (4-FcPyr)]+ (where M = [9][PF6] − PF6).
[CH3CH2CH2-(η6-HMB)Ru(C7H5NO)-κ2-N,O)-4-ferrocenylpyridine][PF6] ([10][PF6]). Red solid. Yield: 0.0330 g, 13%. IR (ATR): ν (cm−1) = 1611 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 0.93 (t, 3J = 7.3 Hz, 3H, NCH2CH2CH3), 1.18–1.25 & 1.51–1.58 (m, 2H, NCH2CH2CH3), 2.04 (s, 18H, CH3 HMB), 3.98–4.05 & 4.32–4.39 (m, 2H, NCH2CH2CH3), 4.07 (s, 5H, Cp–CHunsubst. ring), 4.63 (s, 2H, Cp–CH), 5.03 (d, 3J = 9.2 Hz, 2H, Cp–CH), 6.59 (t, 3J = 7.8 Hz, 1H, Ar), 7.10 (d, 3J = 8.4 Hz, 1H, Ar), 7.22 (d, 3J = 9.4 Hz, 1H, Ar), 7.28–7.34 (m, 1H, Ar), 7.76 (d, 3J = 6.6 Hz, 2H, Pyr), 8.15 (s, 1H, CHimine), 8.61 (d, 3J = 6.5 Hz, 2H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 10.3 (CH3); 14.7 (CH3HMB); 25.3, 65.4 (CH2); 67.5, 71.6 (Cp–CH); 70.2 (Cp–CHunsubst. ring); 78.3 (CCp); 94.3 (CHMB); 115.4, 123.3, 134.4, 134.6 (CHAr); 121.9 (Cpyr); 122.1, 151.4 (CHpyr); 153.0, 164.9 (CAr); 165.8 (CHimine). Elemental analysis for C36H43F6FeN2OPRu (821.6257): Found C, 52.51; H, 5.21; N, 3.37%; calcd C, 52.63; H, 5.27; N, 3.41%. MS (HR-ESI-TOF, m/z): 426.1373 [M − (4-FcPyr)]+ (where M = [10][PF6] − PF6).
[CH3CH2CH2-(η5-C5Me5)Ir(C7H5NO)-κ2-N,O)-4-ferrocenylpyridine][PF6] ([11][PF6]). Red-orange solid. Yield: 0.0682 g, 55%. IR (ATR): ν (cm−1) = 1612 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 0.89 (t, 3J = 7.3 Hz, 3H, NCH2CH2CH3), 1.49–1.60 (m, 2H, NCH2CH2CH3), 1.61 (s, 15H, CH3 Cp*), 4.07 (s, 5H, Cp–CHunsubst. ring), 4.21–4.29 (m, 2H, NCH2CH2CH3), 4.64 (t, 3J = 1.9 Hz, 2H, Cp–CH), 5.04 (d, 3J = 1.9 Hz, 2H, Cp–CH), 6.61 (t, 3J = 7.0 Hz, 1H, Ar), 7.03 (d, 3J = 8.4 Hz, 1H, Ar), 7.31 (dd, 3J = 7.7, 1.7 Hz, 1H, Ar), 7.43 (ddd, 3J = 8.6, 7.1, 1.6 Hz, 1H, Ar), 7.77 (d, 3J = 6.5 Hz, 2H, Pyr), 8.24 (s, 1H, CHimine), 8.72 (d, 3J = 6.8 Hz, 2H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 7.73 (CH3 Cp*); 10.4 (CH3); 24.6, 67.6 (CH2); 67.7, 71.8 (Cp–CH); 70.3 (Cp–CHunsubst. ring); 78.1 (CCp); 87.2 (CCp*); 116.4, 122.5, 134.3, 135.0 (CHAr); 122.5 (Cpyr); 122.1, 150.3 (CHpyr); 150.5, 163.9 (CAr); 163.6 (CHimine). Elemental analysis for C34H40F6FeIrN2OP (885.7300): Found C, 45.98; H, 4.74; N, 3.37%; calcd C, 46.11; H, 4.55; N, 3.16%. MS (HR-ESI-TOF, m/z): 490.1714 [M − (4-FcPyr)]+ (where M = [11][PF6] − PF6).
[CH3CH2CH2-(η5-C5Me5)Rh(C7H5NO)-κ2-N,O)-4-ferrocenylpyridine][PF6] ([12][PF6]). Red solid. Yield: 0.0582 g, 49%. IR (ATR): ν (cm−1) = 1610 (s & br, imine & pyridine, C
N). 1H NMR ((CD3)2CO): δ (ppm) = 0.88 (t, 3J = 7.4 Hz, 3H, NCH2CH2CH3), 1.37–1.41 (m, 2H, NCH2CH2CH3), 1.64 (s, 15H, CH3 Cp*), 4.08 (s, 5H, Cp–CHunsubst. ring), 4.10–4.15 (m, 2H, NCH2CH2CH3), 4.63 (t, 3J = 1.9 Hz, 2H, Cp–CH), 5.04 (d, 3J = 1.9 Hz, 2H, Cp–CH), 6.59–6.63 (m, 1H, Ar), 7.08 (d, 3J = 8.4 Hz, 1H, Ar), 7.29 (dd, 3J = 7.8, 1.8 Hz, 1H, Ar), 7.36 (dd, 3J = 8.5, 6.9, 1.7 Hz, 1H, Ar), 7.80 (d, 3J = 6.6 Hz, 2H, Pyr), 8.23 (s, 1H, CHimine), 8.67 (d, 3J = 6.5 Hz, 2H, Pyr). 13C{1H} NMR ((CD3)2CO): δ (ppm) = 7.68 (CH3 Cp*); 10.4 (CH3); 24.5, 65.0 (CH2); 67.6, 71.6 (Cp–CH); 70.2 (Cp–CHunsubst. ring); 75.3 (CCp); 95.1 (CCp*); 115.3, 123.2, 134.8, 135.0 (CHAr); 122.7 (Cpyr); 122.4, 150.5 (CHpyr); 152.0, 165.5 (CAr); 166.2 (CHimine). Elemental analysis for C34H40F6FeN2OPRh (693.5100): Found C, 59.08; H, 5.74; N, 4.37%; calcd C, 58.89; H, 5.81; N, 4.04%. MS (HR-ESI-TOF, m/z): 400.1151 [M − (4-FcPyr)]+ (where M = [12][PF6] − PF6).
Cytotoxicity studies
The human A2780 and A2780cisR ovarian carcinoma cell lines were obtained from the European Collection of Cell Cultures (Salisbury, UK). Non-tumorigenic HEK-293 cells were obtained from ATCC (Sigma, Switzerland). Cells were grown routinely in RPMI-1640 GlutaMax medium with 10% fetal bovine serum (FBS, Pan Biotech, Germany) and 1% antibiotics at 37 °C and 5% CO2. Cytotoxicity was determined using the MTT assay (MTT = 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) as previously described.83 Briefly, cells were seeded in 96-well plates as monolayers with 100 μL of cell solution (approximately 10000 cells) per well and pre-incubated for 24 h in medium supplemented with 10% FBS. Compounds were prepared as DMSO solution, then dissolved in the culture medium and immediately serially diluted to the appropriate concentration, to give a final DMSO concentration of 0.5%. A 100 μL portion of drug solution was added to each well and the plates were incubated for another 72 h. Subsequently, MTT (5 mg mL−1 solution in PBS) was added to the cells and the plates were incubated for a further 4 h. The culture medium was aspirated, and the purple formazan crystals formed by the mitochondrial dehydrogenase activity of vital cells were dissolved in DMSO. The optical density, directly proportional to the number of surviving cells, was quantified at 590 nm using a multiwell plate reader and the fraction of surviving cells was calculated from the absorbance of untreated control cells. The IC50 values were determined by fitting the plot of the log of the ratio between the percentages of surviving cells, divided by the amount of dead cells, against the log of drug concentration using a linear threshold function. Evaluation is based on means from at least two independent experiments, each comprising four microcultures per concentration level.
NMR studies
For the stability/aquation studies, second generation metallodendrimer [4][PF6]8 and its closest binuclear analogue [10][PF6] was dissolved in DMSO-d6 and D2O (50:50% v/v, because of limited solubility), warmed at 37 °C (to mimic physiological temperature) before samples were monitored using 1H and 31P{1H} NMR experiments at 37 °C over 24 hours on a Bruker Biospin GmbH spectrometer (1H: 400.22 MHz, 31P{1H}: 162.00 MHz).
UV-Vis studies
Absorption studies of a 0.23 mM and 0.15 mM solution of [4][PF6]8 and [10][PF6] respectively in H2O with 0.22 mM HEPES buffer, pH 6.3 were performed in the range 225–345 nm. Solutions of Red Salmon testes DNA sodium salt were prepared fresh before each experiment using milli-Q water. 50 μL aliquots of 50 nM DNA stock solution were added to [4][PF6]8 and [10][PF6] to make the DNA concentrations: 2.38, 4.55, 6.52, 8.33, 10.0, 11.5, 13.0 and 14.3 nM (for [4][PF6]8 & [10][PF6]) and a further 15.5 and 16.7 nM (for [10][PF6]).Conflict of interest
The authors declare no competing financial interest.Acknowledgements
Financial support from the University of Cape Town, the National Research Foundation (NRF) of South Africa and EPFL is acknowledged. This work is based on the research supported wholly/in part by the National Research Foundation of South Africa (Grant Number: 90500).References
- N. Lease, V. Vasilevski, M. Carreira, A. de Almeida, M. Sanau, P. Hirva, A. Casini and M. Contel, J. Med. Chem., 2013, 56, 5806–5818 CrossRef CAS PubMed.
- M. P. Donzello, E. Viola, C. Ercolani, Z. Fu, D. Futur and K. M. Kadish, Inorg. Chem., 2012, 51, 12548–12559 CrossRef CAS PubMed.
- H. Charvatova, T. Riedel, I. Cisarova, P. J. Dyson and P. Stepnicka, J. Organomet. Chem., 2016, 802, 21–26 CrossRef CAS.
- L. Massai, J. Fernandez-Gallardo, A. Guerri, A. Arcangeli, S. Pillozzi, M. Contel and L. Messori, Dalton Trans., 2015, 44, 11067–11076 RSC.
- J. Fernandez-Gallardo, B. T. Elie, M. Sanau and M. Contel, Chem. Commun., 2016, 52, 3155–3158 RSC.
- M. Serratice, L. Maiore, A. Zucca, S. Stoccoro, I. Landini, E. Mini, L. Massai, G. Ferraro, A. Merlino, L. Messori and M. A. Cinellu, Dalton Trans., 2016, 45, 579–590 RSC.
- D. R. van Staveren and N. Metzler-Nolte, Chem. Rev., 2004, 104, 5931–5985 CrossRef CAS PubMed.
- M. F. R. Fouda, M. M. Abd-Elzaher, R. A. Abdelsamaia and A. A. Labib, Appl. Organomet. Chem., 2006, 21, 613–625 CrossRef.
- S. S. Braga and A. M. S. Silva, Organometallics, 2013, 32, 5626–5639 CrossRef CAS.
- P. Pigeon, S. Top, A. Vessières, M. Huché, E. A. Hillard, E. Salmon and G. Jaouen, J. Med. Chem., 2005, 48, 2814–2821 CrossRef CAS PubMed.
- A. Vessières, S. Top, W. Beck, E. A. Hillard and G. Jaouen, Dalton Trans., 2006, 4, 529–541 RSC.
- C. Ornelas, New J. Chem., 2011, 35, 1973–1985 RSC.
- K. Heinze and H. Lang, Organometallics, 2013, 32, 5623–5625 CrossRef CAS.
- D. Plazuk, J. Zakrzewski, M. Salmain, A. Blauz, B. Rychlik, P. Strzelczyk, A. Bujacz and G. Bujacz, Organometallics, 2013, 32, 5774–5783 CrossRef CAS.
- H. Goitia, Y. Nieto, D. Villacampa, C. Kasper, A. Laguna and C. Gimeno, Organometallics, 2013, 32, 6069–6078 CrossRef CAS.
- J. Rajput, J. R. Moss, A. T. Hutton, D. T. Hendricks, C. E. Arendse and C. Imrie, J. Organomet. Chem., 2004, 689, 1553–1568 CrossRef CAS.
- J. Schulz, J. Tauchman, I. Císařová, T. Riedel, P. J. Dyson and P. Stěpnička, J. Organomet. Chem., 2014, 751, 604–609 CrossRef CAS.
- J. Tauchman, G. Süss-Fink, P. Štěpnička, O. Zava and P. J. Dyson, J. Organomet. Chem., 2013, 723, 233–238 CrossRef CAS.
- M. Auzias, J. Gueniat, B. Therrien, G. Süss-Fink, A. K. Renfrew and P. J. Dyson, J. Organomet. Chem., 2009, 694, 855–861 CrossRef CAS.
- M. Auzias, B. Therrien, G. Süss-Fink, P. Štěpnička, W. H. Ang and P. J. Dyson, Inorg. Chem., 2008, 47, 578–583 CrossRef CAS PubMed.
- L. V. Popova, V. N. Babin, Y. A. Belousov, Y. S. Nekrasov, A. E. Snegueva, N. P. Borodina, G. M. Shaposhnikova, O. B. Bychenko and P. M. Raevskii, Appl. Organomet. Chem., 1993, 7, 85–94 CrossRef CAS.
- P. Köpf-Maier, H. Köpf and E. W. Neuse, J. Cancer Res. Clin., 1984, 108, 336–340 CrossRef.
- A. Nguyen, A. Vessieres, E. A. Hillard, S. Top, P. Pigeon and G. Jaouen, Chimia, 2007, 61, 716–724 CrossRef CAS.
- S. Top, A. Vessières, C. Cabestaing, I. Laios, G. Leclereq, C. Provot and G. Jaouen, J. Organomet. Chem., 2001, 637–639, 500–506 CrossRef CAS.
- E. A. Hillard, A. Vessières, F. Le Bideau, D. Plazul, D. Spera, M. Huché and G. Jaouen, ChemMedChem, 2006, 1, 551–559 CrossRef CAS PubMed , and references therein.
- P. Wardman and L. P. Candeias, Radiat. Res., 1996, 145, 523–531 CrossRef CAS PubMed.
- M. Gordon and S. Hollander, J. Med. Chem., 1993, 24, 209–265 CAS.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- C. S. Allardyce and P. J. Dyson, Dalton Trans., 2016, 45, 3201–3209 RSC.
- G. Palermo, A. Magistrato, T. Riedel, T. von Erlach, C. A. Davey, P. J. Dyson and U. Rothlisberger, ChemMedChem, 2015 DOI:10.1002/cmdc.201500478.
- M. Pongratz, P. Schluga, M. A. Jakupec, V. B. Arion, C. G. Hartinger, G. Allmaier and B. K. Keppler, J. Anal. At. Spectrom., 2004, 19, 46–51 RSC.
- P. Heffeter, B. Atil, K. Kryeziu, D. Groza, G. Koellensperger, W. Korner, U. Jungwirth, T. Mohr, B. K. Keppler and W. Berger, Eur. J. Cancer, 2013, 49, 3366–3375 CrossRef CAS PubMed.
- M. J. Clarke, F. C. Zhu and D. R. Frasca, Chem. Rev., 1999, 99, 2511–2533 CrossRef CAS PubMed.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- S. Leijen, S. A. Burgers, P. Baas, D. Pluim, M. Tibben, E. Van Werkhoven, E. Alessio, G. Sava, J. H. Beijnen and J. H. M. Schellens, Invest. New Drugs, 2015, 33, 201–214 CrossRef CAS PubMed.
- A. L. Noffke, A. Habtemariam, A. M. Pizarro and P. J. Sadler, Chem. Commun., 2012, 48, 5219–5246 RSC.
- G. Süss-Fink, Dalton Trans., 2010, 39, 1673–1688 RSC.
- A. A. Nazarov, C. G. Hartinger and P. J. Dyson, J. Organomet. Chem., 2014, 751, 251–260 CrossRef CAS.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and B. K. Keppler, in Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, 2011, pp. 151–174 Search PubMed.
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- M. A. Jakupec, M. Galanski, V. B. Arion, C. G. Hartinger and B. K. Keppler, Dalton Trans., 2008, 183–194 RSC.
- W. H. Ang, A. Casini, G. Sava and P. J. Dyson, J. Organomet. Chem., 2011, 696, 989–998 CrossRef CAS.
- R. E. Morris, R. E. Aird, P. D. S. Murdoch, H. Chen, J. Cummings, N. D. Hughes, S. Parsons, A. Parkin, G. Boyd, D. I. Jodrell and P. J. Sadler, J. Med. Chem., 2001, 44, 3616–3621 CrossRef CAS PubMed.
- R. E. Aird, J. Cummings, A. A. Ritchie, M. Muir, R. E. Morris, H. Chen and P. J. Sadler, Br. J. Cancer, 2002, 86, 1652–1657 CrossRef CAS PubMed.
- C. Scolaro, A. Bergamo, L. Brescacin, R. Delfino, M. Cocchietto, G. Laureneczy, T. J. Geldbach, G. Sava and P. J. Dyson, J. Med. Chem., 2005, 48, 4161–4171 CrossRef CAS PubMed.
- A. Bergamo, A. Masi, P. J. Dyson and G. Sava, Int. J. Oncol., 2008, 33, 1281–1289 CAS.
- Z. Adhireksan, G. E. Davey, P. Campomanes, M. Groessl, C. M. Clavel, H. Yu, A. A. Nazarov, C. H. F. Yeo, W. H. Ang, P. Droge, U. Rothlisberger, P. J. Dyson and C. A. Davey, Nat. Commun., 2014, 5, 3462 Search PubMed.
- S. Chatterjee, S. Kundu, A. Bhattacharyya, C. G. Hartinger and P. J. Dyson, J. Biol. Inorg. Chem., 2008, 13, 1149–1155 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, M. Muller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4728 RSC.
- A. Weiss, X. Ding, J. R. van Beijnum, I. Wong, T. J. Wong, R. H. Berndsen, O. Dormond, M. Dallinga, L. Shen, R. O. Schlingemann, R. Pili, C.-M. Ho, P. J. Dyson, H. van den Bergh, A. W. Griffioen and P. Nowak-Sliwinska, Angiogenesis, 2015, 18, 233–244 CrossRef CAS PubMed.
- A. Weiss, D. Bonvin, R. H. Berndsen, E. Scherrer, T. J. Wong, P. J. Dyson, A. W. Griffioen and P. Nowak-Sliwinska, Sci. Rep., 2015, 5, 8990 CrossRef CAS PubMed.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- I. Romero-Canelon and P. J. Sadler, Inorg. Chem., 2013, 52, 12276–12291 CrossRef CAS PubMed.
- L. Roeglin, E. H. M. Lempens and E. W. Meijer, Angew. Chem., Int. Ed., 2011, 50, 102–112 CrossRef CAS PubMed.
- O. Dömötör, S. Aicher, M. Schmidlehner, M. S. Novak, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and E. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef PubMed.
- J. Ruiz, V. Rodriguez, N. Cutillas, K. G. Samper, M. Capdevilla, O. Palacios and A. Espinosa, Dalton Trans., 2012, 41, 12847–12856 RSC.
- O. Domotor, S. Aicher, M. Schmidlehner, A. Roller, M. A. Jakupec, W. Kandioller, C. G. Hartinger, B. K. Keppler and E. A. Enyedy, J. Inorg. Biochem., 2014, 134, 57–65 CrossRef CAS PubMed.
- G. Gupta, G. Sharma, B. Koch, S. Park, S. S. Lee and J. Kim, New J. Chem., 2013, 37, 2573–2581 RSC.
- P. Govender, B. Therrien and G. S. Smith, Eur. J. Inorg. Chem., 2012, 2853–2862, DOI:10.1002/ejic.201200161.
- G. S. Smith and B. Therrien, Dalton Trans., 2011, 40, 10793–10800 RSC.
- S. K. Singh and D. S. Pandey, RSC Adv., 2014, 4, 1819–1840 RSC.
- K. Wang and E. Gao, Anti-Cancer Agents Med. Chem., 2014, 14, 147–169 CrossRef CAS PubMed.
- C. G. Hartinger, A. D. Phillips and A. A. Nazarov, Curr. Top. Med. Chem., 2011, 11, 2688–2702 CrossRef CAS PubMed.
- M. G. Mendoza-Ferri, C. G. Hartinger, M. A. Mendoza, M. Groessl, A. Egger, R. E. Eichinger, J. B. Mangrum, N. P. Farrell, M. Maruszak, P. J. Bednarski, F. Klein, M. A. Jakupec, A. A. Nazarov, K. Severin and B. K. Keppler, J. Med. Chem., 2009, 52, 916–925 CrossRef CAS PubMed.
- M. G. Mendoza-Ferri, C. G. Hartinger, R. Eichinger, N. Stolyarova, K. Severin, M. A. Jakupec, A. A. Nazarov and B. K. Keppler, Organometallics, 2008, 27, 2405–2407 CrossRef CAS.
- N. Farrell, Met. Ions Biol. Syst., 2004, 42, 251–296 CAS.
- A. Casini, M. A. Cinellu, G. Minghetti, C. Gabbiani, M. Coronnello, E. Mini and L. Messori, J. Med. Chem., 2006, 49, 5524–5531 CrossRef CAS PubMed.
- C. Gabbiani, A. Casini, L. Messori, A. Guerri, M. A. Cinellu, G. Minghetti, M. Corsini, C. Rosani, P. Zanello and M. Arca, Inorg. Chem., 2008, 47, 2368–2379 CrossRef CAS PubMed.
- M. A. Cinellu, L. Maiore, M. Manassero, A. Casini, M. Arca, H. H. Fiebig, G. Kelter, E. Michelucci, G. Pieraccini, C. Gabbiani and L. Messori, Med. Chem. Lett., 2010, 1, 336–339 CrossRef CAS PubMed.
- B. C. E. Makhubela, M. Meyer and G. S. Smith, J. Organomet. Chem., 2014, 772–773, 229–241 CrossRef CAS.
- A. R. Burgoyne, B. C. E. Makhubela, M. Meyer and G. S. Smith, Eur. J. Inorg. Chem., 2015, 1433–1444 CrossRef CAS.
- P. Govender, F. Edafe, B. C. E. Makhubela, P. J. Dyson, B. Therrien and G. S. Smith, Inorg. Chim. Acta, 2014, 409, 112–120 CrossRef CAS.
- P. Govender, L. C. Sudding, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2013, 42, 1267–1277 RSC.
- R. Payne, P. Govender, B. Therrien, C. M. Clavel, P. J. Dyson and G. S. Smith, J. Organomet. Chem., 2013, 729, 20–27 CrossRef CAS.
- L. C. Sudding, R. Payne, P. Govender, F. Edafe, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, J. Organomet. Chem., 2014, 774, 79–85 CrossRef CAS.
- T. Ahamad, S. F. Mapolie and S. M. Alshehri, Med. Chem. Res., 2010, 138, 171–179 CAS.
- S. El Kazzouli, N. El Brahmi, S. Mignani, M. Bousmina, M. Zablocka and J.-P. Majoral, Curr. Med. Chem., 2012, 19, 4995–5010 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS PubMed.
- H. Kobayashi, B. Turkbey, R. Watanabe and P. L. Choyke, Bioconjugate Chem., 2014, 25, 2093–2100 CrossRef CAS PubMed.
- H. Nakamura, F. Jun and H. Maeda, Expert Opin. Drug Delivery, 2015, 12, 53–64 CrossRef CAS PubMed.
- P. Govender, H. Lemmerhirt, A. T. Hutton, B. Therrien, P. J. Bednarski and G. S. Smith, Organometallics, 2014, 33, 5535–5545 CrossRef CAS.
- R. Malgas, S. F. Mapolie, S. O. Ojwach, G. S. Smith and J. Darkwa, Catal. Commun., 2008, 9, 1612–1617 CrossRef CAS.
- P. Govender, A. K. Renfrew, C. M. Clavel, P. J. Dyson, B. Therrien and G. S. Smith, Dalton Trans., 2011, 40, 1158–1167 RSC.
- M. A. Torzilli, S. Colquhoun, D. Doucet and R. H. Beer, Polyhedron, 2002, 21, 697–704 CrossRef CAS.
- C. Scolaro, C. G. Hartinger, C. S. Allardyce, B. K. Keppler and P. J. Dyson, J. Inorg. Biochem., 2008, 102, 1743–1748 CrossRef CAS PubMed.
- C. Gossens, A. Dorcier, P. J. Dyson and U. Rothlisberger, Organometallics, 2007, 26, 3969–3975 CrossRef CAS.
- F. Wang, J. Bella, J. A. Parkinson and P. J. Sadler, J. Biol. Inorg. Chem., 2005, 10, 147–155 CrossRef CAS PubMed.
- R. Gomathi, A. Ramu and A. Murugan, Bioinorg. Chem. Appl., 2014, 2014, 1–12 CrossRef PubMed.
- M. Aslanoglu and N. Oge, Turk. J. Chem., 2005, 29, 477–485 CAS.
- N. Shahabadi, S. Kashanian, M. Khosravi and M. Mahdavi, Transition Met. Chem., 2010, 35, 699–705 CrossRef CAS.
- M. A. Bennett and A. K. Smith, J. Chem. Soc., Dalton Trans., 1974, 2, 233–241 RSC.
- M. A. Bennett, T. W. Matheson, G. B. Robertson, A. K. Smith and P. A. Tucker, Inorg. Chem., 1980, 19, 1014–1021 CrossRef CAS.
- C. White, A. Yates and P. M. Maitlis, Inorg. Synth., 1992, 29, 228–234 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6dt00849f |
| This journal is © The Royal Society of Chemistry 2016 |