
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New sterically-hindered o-quinones annelated with metal-dithiolates: regiospecificity in oxidative addition reactions of a bifacial ligand to the Pd and Pt complexes†
K. A.
Martyanov
*a,
V. K.
Cherkasov
a,
G. A.
Abakumov
a,
M. A.
Samsonov
a,
V. V.
Khrizanforova
b,
Y. H.
Budnikova
b and
V. A.
Kuropatov
 *a
*a
aG.A. Razuvaev Institute of Organometallic Chemistry of Russian Academy of Sciences, 603950, GSP-445, Tropinina str. 49, Nizhny Novgorod, Russia. E-mail: konmart@iomc.ras.ru; viach@iomc.ras.ru
bArbuzov Institute of Organic and Physical Chemistry of Russian Academy of Sciences, 420088, Arbuzov str. 8, Kazan, Russia
First published on 14th March 2016
Abstract
An unusual reactivity of sterically hindered o-quinones with an annelated dithiete ring towards coordination at a dithiolene site has been discovered. New Pd and Pt dithiolate complexes have been synthesized. The reaction proceeds regioselectively, and the quinone site of the parent ligand is not affected even while using an excess of the metal complex. Both Pt and Pd complexes display a square planar surrounding for the metal ion and have very similar NMR, IR and UV/Vis spectra. Surprisingly, being coordinated at the dithiolene site to the metal, the ligand exhibits activity like an o-quinone, it could be reduced with different metals resulting in the corresponding o-semiquinonates which were confirmed by EPR spectroscopy. It was shown that an unpaired electron exhibits HFC with the phosphorus nuclei of phosphine ligands coordinated to the metal ions at the dithiolene site of the molecule.
Introduction
o-Quinones have been a subject of research interest for several decades. Their stable sterically hindered derivatives are utilized in coordination chemistry as redox-active bidentate ligands. Metal o-semiquinonate complexes possess a huge variety of chemical and physical properties, such as valence tautomerism,1 photo and thermomechanical effects,2etc. Studies of metal dithiolenes have grown enormously in the last few years. Their potential applications are in molecular inorganic conductors and superconductors,3 biomimetic catalysts4 and so on. The introduction of heteroatomic functionalities into the o-quinone molecule can drastically change its properties as well as increase the number of coordination sites. In the latter case the quinone becomes a bridging ligand.5 The regioselectivity towards binding of the metal ions at different coordination sites of such ligands is of special interest.Results and discussion
A new sterically-hindered o-quinone 1, annelated with a dithiete ring, has been synthesized recently.6 This species is regarded as a ligand comprising 1,2-dithiolene and 1,2-dioxolene sites ready for metal coordination. Previously we have shown that 1 acts in the reactions with alkali metals, thallium, and copper as a typical o-quinone: it involves a dioxolene site in the reduction leading to the corresponding o-semiquinonates (SQ) or catecholates. As far as the dithiolate face is concerned, it was functionalized using an excess of decacarbonyl dimanganese in toluene (5), but only after quinone was bound via complex 4 (Scheme 2).6 Thus, the regioselective addition to the dithiolene site was not carried out initially. | ||
| Scheme 1 Synthesis of dithiolate complexes 2–3 from ligand 1. | ||
 | ||
| Scheme 2 The reaction of 1 with an excess of decacarbonyl dimanganese. | ||
Herein we present new dithiolate complexes of Pd (2) and Pt (3) with ligand 1. The intriguing feature of these complexes is the vacant o-quinone functionality, therefore the newly synthesized species attract great interest as ligand–precursors of heterobimetallic complexes.
The dithiolate complexes of transition metals have been thoroughly studied due to their unique properties and application in the field of magnetic materials,7 non-linear optics,8 dyes9 and others. One of the most widely used synthetic methods for the preparation of dithiolate complexes is the treatment of dithiols or their alkali salts with metal halides. 1,2-Dithietes also have been described as a potential precursor for the synthesis of dithiolate complexes as alternative species. But taking into account their non-availability, the chemistry of oxidative addition of the metals to the benzodithietes has not been studied yet. There is a review dedicated to metal complexes derived from 3,4-bis(trifluoromethyl)1,2-dithiete,10 and single examples of Pd and Pt interaction with 3,4-di(adamantan-1-yl)-1,2-dithiete11 and 4,4-di-tert-butyl-1,2-dithietan-3-one.12 From this point of view, oxidative addition of metals with simultaneous cleavage of the S–S bond in the dithiete ring of ligand 1 is a possible path for the preparation of dithiolate metal complexes. This synthetic route requires low-valent metal compounds that are easy to be derived for both palladium and platinum.
The reactions of 1 with M(PPh3)4 (M = Pd, Pt) were carried out in the stoichiometric ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in THF at room temperature under vacuum. The colour of the solution turned from violet to green within 30 minutes. Recently, Rawson et al. reported the oxidative addition chemistry of tetrathiocins (equivalent to a dithiete dimer) to group 10 metals leading to the corresponding dithiolates.13 But the reaction with tetrathiocins requires microwave heating for activation.
:1 in THF at room temperature under vacuum. The colour of the solution turned from violet to green within 30 minutes. Recently, Rawson et al. reported the oxidative addition chemistry of tetrathiocins (equivalent to a dithiete dimer) to group 10 metals leading to the corresponding dithiolates.13 But the reaction with tetrathiocins requires microwave heating for activation.
Products 2 and 3 were separated in the crystalline form, and their structures were established by a single-crystal X-ray diffraction study.
Structural studies of 2 show that the palladium center is surrounded by two phosphorus and two sulfur atoms in a slightly tetrahedrally distorted square planar geometry (Fig. 1). The Pd(1) atom deviates from the P(1)P(2)S(1)S(2) plane by 0.065(3) Å. The P–Pd–P, S–Pd–S and P–Pd–S angles vary in the interval 85.03(2)–98.71(2)°. The Pd–S and Pd–P bond distances are 2.2707(6) Å and 2.3230(5)–2.3411(5) Å, respectively. These values correlate with those for the complex Pd(tdt)(PPh3)2 (6) (tdt = 1,2-toluenedithiolate), described by Cao and coworkers (Scheme 3).14 It is noteworthy that a series of Pd complexes with the 4,5-dimethoxybenzenedithiolate ligand and chelate phosphines (7–9) also have similar structural parameters of the metal center surrounding.15
 | ||
| Fig. 1 An ORTEP plot of 2a (top) and its alternative view, illustrating a slightly distored square planar surrounding of the metal center and strong distortion in the quinone ring (bottom). Thermal ellipsoids are drawn at 30% probability. Hydrogen atoms, and tBu groups are omitted and phenyl rings are marked as “Ph” for clarity. | ||
 | ||
| Scheme 3 Square planar dithiolate complexes of Pd(II) with phosphines. | ||
As far as a 6-membered carbon ring is concerned, the steric repulsion between bulky tert-butyl substituents on one hand and sulfur and oxygen atoms on the other hand causes its strong distortion. However, an obvious bond alternation as well as the values of bond lengths O(1)–C(5) and O(2)–C(4) (see Table S1 in the the ESI†) confirm that the species 2 exhibits an o-quinone nature. One more crystal polymorph of 2 was obtained by slow cooling of THF solution (2b). The structural parameters of the metal center are close to 2a, but the crystal lattice contains an additional molecule of THF (Fig. S1†).
The crystals of 3 were produced by slow mixing of THF solution with hexane. An X-ray data study reveals a square planar coordination of 3 (Fig. 2 and S2†). The parameters of 2 are close to a square planar geometry and given in Table S1.† In the quinone ring the distortion is strong: the torsion angle OCCO is 37.36°. An analysis of the structural data allows us to conclude that 3 might be regarded as an o-quinone.
 | ||
| Fig. 2 An ORTEP plot of 3. Thermal ellipsoids are drawn at 30% probability. Hydrogen atoms are omitted for clarity. | ||
The IR spectra of 2 and 3, exhibiting strong absorption, characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups, are very similar. Quite similar signals appear in both the 1H and 13C NMR spectra for 2 and 3. In 31P-NMR of 3 heteronuclear spin–spin coupling between 31P and 195Pt takes place, and a satellite doublet can be observed apart from the main singlet.
O groups, are very similar. Quite similar signals appear in both the 1H and 13C NMR spectra for 2 and 3. In 31P-NMR of 3 heteronuclear spin–spin coupling between 31P and 195Pt takes place, and a satellite doublet can be observed apart from the main singlet.
The redox characteristics of 2–3 were assessed by using cyclic voltammetry in DMF (Fig. S12–S13†). The measurements were carried out under the same conditions for both Pd and Pt dithiolate as well as for parent o-quinone 1, which reversibly reduced at −0.42 V (Fig. S11†). Reduction of 2 displays two irreversible waves at −0.90 V and −1.20 V, respectively (Ered, vs. Ag/AgCl). Similarly, reduction of 3 displays two irreversible waves at −0.80 V and −0.94 V (Ered, vs. Ag/AgCl). Compared with 1, such results indicate that coordination of the metal fragment at the dithiolene site significantly lowers the electron-acceptor properties of the dioxolene structure: the first reduction potential shifts by 0.48 V and 0.38 V for 2 and 3, respectively. Electrochemical oxidation of 2 and 3 shows two-electron quasi-reversible waves at 0.95 V and 1.13 V, respectively.
The UV/Vis spectra of the complexes in THF are depicted in Fig. S3 and S4.† Broad and relatively low intense absorptions in the 500–700 nm region (ε = 520 M−1 cm−1 (λ = 588 nm) and ε = 580 M−1 cm−1 (λ = 611 nm) in THF for 2 and 3 respectively) are characteristic of o-quinones and correspond to the n–π* transition. Two more characteristics of o-quinones are narrow and intensive lines having the maxima at 422 and 423 nm, respectively, which can be tentatively attributed to π–π* electron transfer (ε = 19800 M−1 cm−1 (2), ε = 19900 M−1 cm−1 (3)).
Summarizing mentioned before, complexes 2 and 3 having an annelated metal dithiolate ring remain o-quinones: their dioxolene sites are ready for metal coordination resulting in heterobimetallic complexes. We have carried out typical o-quinones reactions with alkali metals, thallium amalgam and decacarbonyl dimanganese. The formation of the corresponding o-semiquinonates 10–15 (Scheme 4) was established by EPR spectroscopy (Fig. 3–5 and S16–S18†). The parameters of the spectra are shown in Table 1. It should be noted that all of the reduced derivatives 10–15 were studied only in solution. Their isolation in the crystal phase failed due to their relatively high lability.
 | ||
| Fig. 3 Experimental (top) and simulated (bottom) X-band EPR spectra of 11 in THF, 298 K. | ||
 | ||
| Fig. 4 Experimental (top) and simulated (bottom) X-band EPR spectra of 12 in toluene, 298 K. | ||
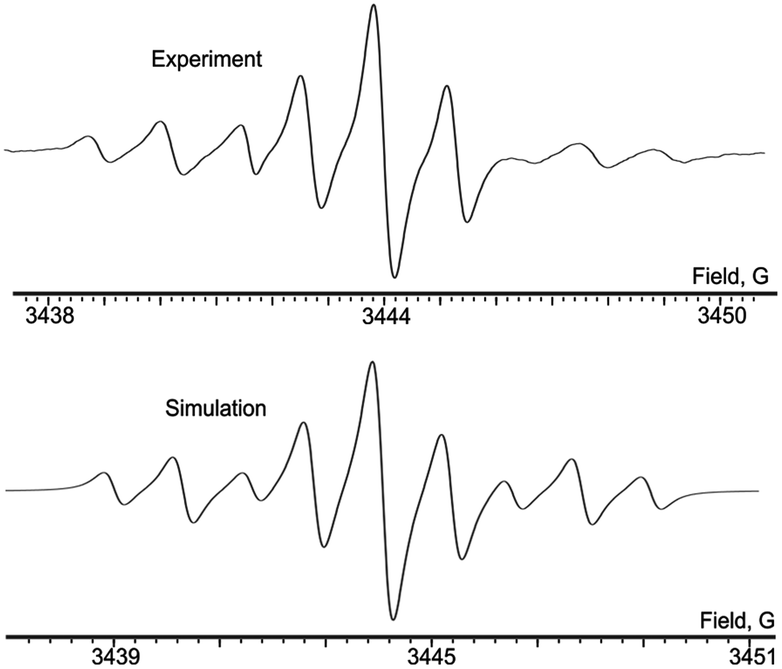 | ||
| Fig. 5 Experimental (top) and simulated (bottom) X-band EPR spectra of 14 in THF, 298 K. | ||
 | ||
| Scheme 4 Heterobimetallic o-semiquinonates. | ||
| No. | M | M′ | Solvent | g iso | a M′ [G] | a L [G] |
|---|---|---|---|---|---|---|
| a Satellite splitting of 195Pt nucleus at the dithiolene site of the compound. | ||||||
| 10 | Pd | Tl | THF | 1.9964 | 30.3 | 1.4 (2 × P) |
| Toluene | 1.9963 | 18.4 | 1.4 (2 × P) | |||
| 11 | Pd | K | THF | 2.0045 | — | 0.7 (2 × P) |
| 12 | Pd | Mn(CO)3PPh3 | Toluene | 2.0026 | 5.8 | 26.8 (P) |
| 1.4 (2 × P) | ||||||
| 13 | Pt | Tl | THF | 1.9933 | 31.7;6.4a | 1.8 (2 × P) |
| Toluene | 1.9928 | 19.2;6.1a | 1.8 (2 × P) | |||
| 14 | Pt | K | THF | 2.0024 | 7.5a | 1.3 (2 × P) |
| 15 | Pt | Mn(CO)3PPh3 | Toluene | 1.9990 | 5.8;6.2a | 26.7 (P) |
| 1.7 (2 × P) | ||||||
EPR studies show that an unpaired electron undergoes hyperfine coupling (HFC) with two equivalent phosphorus nuclei of the phosphine ligands coordinated to the metal ion at the dithiolene site of the molecule. At the same time, the coupling constants of the metal nucleus at the dioxolene coordination site are considerably lower than those for mononuclear o-semiquinonates. Thus, the HFC constant of 203,205Tl nucleus in THF decreases from 52.5 G (for thallium semiquinonate (SQ) with a non-coordinated dithiolene site)6 to 30.3 G (10) and 31.7 G (13). It is a known fact that the value of the splitting constant of the metal nuclei in semiquinonates depends on the redox potential of the quinone ligand.16 Taking this into account, its small values confirm that the oxidative ability of the dioxolene structure becomes lower after metal addition to the dithiolene site. This is consistent with the data of electrochemical measurements, which showed a significant shift of the first reduction potential for 2 and 3 compared to parent quinone 1. Moreover, treatment of 2 with additional equivalents of a mild reductant such as Pd(PPh3)4 does not yield binuclear species (Scheme 1), although the formation of catecholates easily proceeds in the case of a better acceptor o-chloranil17 or 3,6-di-tert-butyl-o-benzoquinone.
Further reduction of semiquinonates 10 and 13 with an excess of thallium amalgam doesn't lead to formation of the corresponding catecholates: the process proceeds with destruction of the dithiolate metal center. Thus, the preparative path for the synthesis of SQTl species by preliminary exhaustive reduction of the quinone with an excess of thallium amalgam to catecholate followed by addition of an equimolar amount of o-quinone18 is not applicable for 2 and 3 (Scheme S1 in ESI†).
Relatively high natural abundance of 195Pt allows for observation of satellite splitting. The EPR spectrum demonstrating these signals is registered for potassium o-semiquinonate 14 (Fig. 5). Apart from the main triplet attributed to HFC with phosphorus, a less intensive satellite doublet of triplets appears due to splitting of nucleus 195Pt (33.8%, I = 1/2, aPt = 7.4 G). The lines in the right component of doublet are wider because of anisotropy, which is typical of platinum complexes.19 Comparing the HFS constant value at the nucleus of platinum in the binuclear complex 14 with those in the corresponding semiquinonate complexes, where the platinum nucleus is situated in close proximity of the unpaired electron containing dioxolene coordination site, one can easily see that the constant differs by less than two times (12.7–13.2 G in Pt semiquinonates).19,20 This confirms a substantial delocalization of the spin density to the periphery of the binuclear complex.
It's known that the interaction of quinones with Mn2(CO)10 in toluene leads to the formation of SQMn(CO)4, and the subsequent addition of triphenylphosphine to the starting mixture gives the species SQMn(CO)3PPh3.21 The reaction of Mn2(CO)10 with 2 and 3 results in the formation of SQMn(CO)3PPh3 regardless of the fact whether PPh3 is present in the starting mixture or not. We suppose that the presence of some amount of phosphine molecules in the solution could be explained by partial dissociation of parent complexes (Scheme 5). Then this phosphine interacts with manganese carbonyl resulting in the corresponding adducts 12 and 15.
 | ||
| Scheme 5 Dissociation of 2 and 3 with subsequent formation of SQMn(CO)3PPh3 (16–17). | ||
To summarize the coordination properties of the bifacial ligand 1, the regioselectivity of oxidative addition depends on the metallic nature of the reducing agent. Strong oxidants, in which the ions possess high Lewis acidity and are singly-charged, attack presumably at the dioxolene coordination site, whereas divalent cations with mild Lewis acidity form complexes at the dithiolene site of the ligand. The dioxolene coordination site in the dithiolate Pt and Pd complexes exhibits o-quinone properties and remains ready for the metal coordination. Thus, both the coordination sites of the ligand could be involved in the binding of the metal ions. An unpaired electron localized mainly on the semiquinone site of the molecule acts as a spin label reporting information about the arrangement of the coordination sphere of the metal ion at the periphery. This may be useful in the study of dynamic processes in the coordination sphere of dithiolene complexes. The potential application of such a bifacial ligand might be construction of an electric junction between the metal surface and coordination molecules in the solution: the dithiolene coordination site could be used for anchoring to the metal cluster or even surface or whereas the dioxolene node is suitable for building the functional coordination polymeric chain. Other areas of potential application for such combined dithiolene and dioxolene systems are molecular devices, non-linear optics, biological sensors and so on.
Experimental
Reagents and solvents were used as purchased or purified by standard procedures.22 All synthetic procedures were carried out under vacuum. 2,5-Di-tert-butyl-7,8-dithiabicyclo[4.2.0]octa-1,5-diene-3,4-dione was prepared according to a literature procedure.6 Syntheses of tetrakis(triphenylphosphine)palladium as well as tetrakis(triphenylphosphine)platinum were carried out according to ref. 23 and 24 respectively.Instrumentation
IR spectra were recorded on a FSM-1201 FTIR spectrometer. UV/Vis spectra were recorded on a Perkin-Elmer Lambda 25 spectrometer in 0.1 cm quartz cells. 1H, 13C and 31P NMR spectra were obtained on a Bruker Avance III 400 MHz instrument. The chemical shifts are expressed in parts per million downfield. X-band EPR spectra were recorded with a Bruker EMX spectrometer. The samples for EPR study were prepared according to the procedures described by Prokof'ev et al.25 Elemental analysis was carried out with a EURO EA machine. CV experiments were carried out using an e2P Epsilon potentiostat equipped with a three-electrode cell, C3. Conditions: 100 mV s−1 scan rate, 0.1 M Bu4NBF4, glassy carbon working electrode.C of phenyl and o-benzoquinone rings), 185.78 ppm (CO); 31P NMR (161.97 MHz, CDCl3, 25 °C): δ = 26.29 ppm (PPh3); IR (Nujol): 1624s (CO), 1435, 1307, 1216, 1189, 1159, 1091, 1026, 997, 914, 840, 813, 754, 692 cm−1; elemental analysis: calculated (%): C, 65.75; H, 5.30; P, 6.78; Pd, 11.65; S, 7.02; found (%):C, 66.23; H, 5.57; P, 7.12; Pd, 12.03; S, 6.74;
C of phenyl and o-benzoquinone rings), 185.96 ppm (CO); 31P NMR (161.97 MHz, CDCl3, 25 °C): δ = 19.17 ppm (PPh3, with 195Pt satellites: δ = 10.27, 28.30 ppm); IR (Nujol): 1624s (CO), 1438, 1313, 1217, 1163, 1096, 1067, 1029, 996, 912, 841, 816, 754, 741, 691 cm−1; elemental analysis: calculated (%): C, 59.93; H, 4.83; P, 6.18; Pt, 19.47; S, 6.40; found (%):C, 59.85; H, 4.61; P, 6.78; Pt, 20.01; S, 6.43;
Potassium semiquinonates 11 and 14 were generated directly before recording of EPR spectra by shaking of the ampoule with 5 ml of degassed 1 × 10−4 M solution of 2 or 3 and a small piece of metal potassium.
Thallium semiquinonates 10 and 13 were prepared in a similar manner using thallium amalgam as a reducing agent. Manganese carbonyl phosphine semiquinonates12 and 15 were generated by irradiation of 1 × 10−4 M toluene solution of 2 or 3 in the presence of an equivalent amount of Mn2(CO)10. The reaction mixture was irradiated with the focused light of the halogen lamp directly into the EPR spectrometer cavity.
Acknowledgements
This work was supported by the Russian Foundation for Basic Research (grant 15-43-02350 r_povolzhie_a) and the Program for support of Leading Scientific Schools NSh-271.2014.3e.Notes and references
- C. G. Pierpont, Coord. Chem. Rev., 2001, 216–217, 99 CrossRef CAS.
- G. A. Abakumov and V. I. Nevodchikov, Dokl. Akad. Nauk SSSR, 1982, 266(6), 1407 CAS.
- L. Valade and H. Tanaka, in Molecular Materials, ed. D. W. Bruce, D. O'Hare and R. I. Walton, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- (a) T. Xu, D. Chen and X. Hu, Coord. Chem. Rev., 2015, 303, 32 CrossRef CAS; (b) R. Hidalgo, P. A. Ash, A. J. Healy and K. A. Vincent, Angew. Chem., Int. Ed., 2015, 54, 7110 CrossRef CAS PubMed; (c) S. Ott, M. Kritikos, B. Åkermark, L. Sun and R. Lomoth, Angew. Chem., Int. Ed., 2004, 43, 1006 CrossRef CAS PubMed.
- (a) V. Kuropatov, S. Klementieva, G. Fukin, A. Mitin, S. Ketkov, Y. Budnikova, V. Cherkasov and G. Abakumov, Tetrahedron, 2010, 66, 7605 CrossRef CAS; (b) N. O. Chalkov, V. K. Cherkasov, G. A. Abakumov, G. V. Romanenko, S. Y. Ketkov, I. V. Smolyaninov, A. G. Starikov and V. A. Kuropatov, Eur. J. Org. Chem., 2014, 4571 CrossRef CAS.
- V. A. Kuropatov, G. A. Abakumov, G. K. Fukin, S. V. Klementieva and V. K. Cherkasov, Chem. – Eur. J., 2012, 18, 13821 CrossRef PubMed.
- P. Cassoux, Coord. Chem. Rev., 1999, 185, 213 CrossRef.
- (a) C. S. Winter, S. N. Oliver, R. J. Manning, J. D. Rush, C. A. S. Hill and A. E. Underhill, J. Mater. Chem., 1992, 2(4), 443 RSC; (b) J.-L. Zuo, T.-M. Yao, F. You, X.-Z. You, H.-K. Fun and B.-C. Yip, J. Mater. Chem., 1996, 6(10), 1633 RSC.
- (a) E. A. M. Geary, L. J. Yellowlees, L. A. Jack, I. D. H. Oswald, S. Parsons, N. Hirata, J. R. Durrant and N. Robertson, Inorg. Chem., 2005, 44, 242 CrossRef CAS PubMed; (b) A. Islam, H. Sugihara, K. Hara, L. P. Singh, R. Katoh, M. Yanagida, Y. Takahashi, S. Murata and H. Arakawa, Inorg. Chem., 2001, 40, 5371 CrossRef CAS PubMed.
- A. Davison, R. H. Holm, R. E. Benson and W. Mahler, in Inorganic Syntheses, ed. E. L. Muetterties, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1967, vol. 10 Search PubMed.
- Y. Ono, Y. Sugihara, A. Ishii and J. Nakayama, J. Am. Chem. Soc., 2003, 125, 12114 CrossRef CAS PubMed.
- T. Shigetomi, K. Okuma, N. Nagahora and Y. Yokomori, Bull. Chem. Soc. Jpn., 2009, 82, 855 CrossRef CAS.
- J. D. Wrixon, J. J. Hayward, O. Raza and J. M. Rawson, Dalton Trans., 2014, 43, 2134 RSC.
- R. Cao, M. Hong, F. Jiang, B. Kang, X. Xie and H. Liu, Polyhedron, 1996, 15, 2661 CrossRef CAS.
- J. D. Wrixon, J. J. Hayward and J. M. Rawson, Inorg. Chem., 2015, 54(19), 9384 CrossRef CAS PubMed.
- G. A. Razuvaev, V. K. Cherkasov and G. A. Abakumov, J. Organomet. Chem., 1978, 160, 361 CrossRef CAS.
- A. L. Balch and Y. S. Sohn, J. Am. Chem. Soc., 1971, 93, 1290 CrossRef CAS.
- V. A. Muraev, G. A. Abakumov and G. A. Razuvaev, Dokl. Akad. Nauk SSSR, 1974, 217(5), 1083 CAS.
- G. A. Abakumov, I. A. Teplova, V. K. Cherkasov and K. G. Shalnova, Inorg. Chim. Acta Lett., 1979, 32, L57 CrossRef CAS.
- J. J. Loughrey, N. J. Patmore, A. Baldansuren, A. J. Fielding, E. J. L. McInnes, M. J. Hardie, S. Sproules and M. A. Halcrow, Chem. Sci., 2015, 6, 6935 RSC.
- G. A. Abakumov, V. K. Cherkasov, K. G. Shalnova, I. A. Teplova and G. A. Razuvaev, J. Organomet. Chem., 1982, 236, 333 CrossRef CAS.
- A. Weissberger, E. S. Proskauer, J. A. Riddick and E. E. Toops, in Organic Solvents. Physical Properties and Methods of Purification, Interscience, New-York, London, 1955 Search PubMed.
- J. R. Malpass, D. A. Hemmings, A. L. Wallis, S. R. Fletcher and S. Patel, J. Chem. Soc., Perkin Trans. 1, 2001, 1044–1050 RSC.
- R. Ugo, F. Cariati, G. L. Monica and J. J. Mrowca, in Inorganic Syntheses, John Wiley & Sons, Inc., Hoboken, NJ, USA, 1968, vol. 11 Search PubMed.
- A. I. Prokofiev, S. P. Solodovnikov, I. S. Belostotskaya and V. V. Ershov, Russ. Chem. Bull., 1974, 23, 189–191 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1446632–1446634. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00769d |
| This journal is © The Royal Society of Chemistry 2016 |