
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing bistability in FeII and CoII complexes with an unsymmetrically substituted quinonoid ligand†‡
Margarethe
van der Meer§
a,
Yvonne
Rechkemmer§
b,
Frauke D.
Breitgoff
b,
Sebastian
Dechert
c,
Raphael
Marx
b,
María
Dörfel
b,
Petr
Neugebauer
b,
Joris
van Slageren
*b and
Biprajit
Sarkar
*a
aInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34-36, D-14195 Berlin, Germany. E-mail: biprajit.sarkar@fu-berlin.de
bInstitut für Physikalische Chemie, Universität Stuttgart, Pfaffenwaldring 55, D-70569 Stuttgart, Germany. E-mail: ipcjosl@ipc.uni-stuttgart.de
cInstitut für Anorganische Chemie, Georg-August-Universität Göttingen, Tammannstraße 4, D-37077 Göttingen, Germany
First published on 6th April 2016
Abstract
The generation of molecular platforms, the properties of which can be influenced by a variety of external perturbations, is an important goal in the field of functional molecular materials. We present here the synthesis of a new quinonoid ligand platform containing an [O,O,O,N] donor set. The ligand is derived from a chloranilic acid core by using the [NR] (nitrogen atom with a substituent R) for [O] isoelectronic substitution. Mononuclear FeII and CoII complexes have been synthesized with this new ligand. Results obtained from single crystal X-ray crystallography, NMR spectroscopy, (spectro)electrochemistry, SQUID magnetometry, multi-frequency EPR spectroscopy and FIR spectroscopy are used to elucidate the electronic and geometric structures of the complexes. Furthermore, we show here that the spin state of the FeII complex can be influenced by temperature, pressure and light and the CoII complex displays redox-induced spin-state switching. Bistability is observed in the solid-state as well as in solution for the FeII complex. The new ligand presented here, owing to the [NR] group present in it, will likely have more adaptability while investigating switching phenomena compared to its [O,O,O,O] analogues. Thus, such classes of ligands as well as the results obtained on the reversible changes in physical properties of the metal complexes are likely to contribute to the generation of multifunctional molecular materials.
Introduction
The ligand 2,5-dihydroxy-1,4-benzoquinone (dhbq, Fig. 1) containing a symmetrically substituted [O,O,O,O] donor set has been extensively used in coordination chemistry.1 In recent years, the combination of dhbq with CoII showed valence tautomerism (VT).2 A related ligand, chloranilic acid (CA, Fig. 1) and its derivatives, were also used with FeII,3 to generate spin crossover (SCO) properties4 and with CoII,5 to generate VT. In the CoII complexes with dhbq, switching of the spin state was achieved both as a function of temperature and pressure.2 The redox-active nature of the metal centers and the ligands make such systems ideal for investigating redox-induced spin state switching. However, such a process has hardly been investigated for these metal complexes.1–3,5 In both dhbq and CA the ligands contain an all [O] donor set, thus limiting chemical modification of such ligands only to direct substitution on the central ring. We6,7 and others8 have used the [NR] (nitrogen atom with a substituent R) for [O] isoelectronic analogy to modify dhbq and related ligands to generate ligands with symmetrical [O,N,O,N] donor sets (Fig. 1). The “R” handle in the [NR] group provides ample opportunities for steric, electronic and solubility tuning of the metal complexes, a fact that is not possible with dhbq or CA. Thus, functional metal complexes with such [O,N,O,N] donor sets have been useful for investigating electron transfer processes, spin state switching, spin–spin coupling and other properties.6 In these ligands with an [O,N,O,N] donor set, the central six-membered ring is always symmetrically substituted with the heteroatom donors. Examples of potentially bridging quinonoid ligands with unsymmetrically substituted donor groups such as [O,O,O,N] and their metal complexes are rare.6a | ||
| Fig. 1 Potentially bridging quinone ligands with “O” and/or “N” donors. | ||
In our pursuit of generating new quinonoid ligand platforms with one or more [NR] donors, we have now turned our attention to modifying the CA ligand. To the best of our knowledge, CA has never been modified through the [NR] for [O] substitution to generate unsymmetrically substituted ligands with an [O,O,O,N] donor set, even though there has been a recent example of modified CA with a symmetrically substituted [O,N,O,N] donor set3d (Fig. 1). Access to ligands with such unsymmetrical substitution is likely to make studies of preferential coordination of metal centers at one or the other binding site ([O,O] vs. [O,N]) possible. The aforementioned complexes could then be used for generating heterobimetallic complexes, for addressing the targeted switching of a particular metal center in homobimetallic complexes, or for implementing two-qubit gates. In the last of these, the two metal centers that act as the two qubits have to be distinct.9 These goals are important in the field of functional molecular materials but are difficult to achieve with symmetrically substituted quinonoid ligands containing a [O,O,O,O] or [O,N,O,N] donor set.
In the following, we present the synthesis of the unsymmetrically substituted ligand 2-[(2′,6′-diisopropyl)anilino)]-3,6-dichloro-5-hydroxy-1,4-benzoquinone (H2L, Fig. 1), its Fe(II) complex [(tmpa)Fe(HL)](BF4), [1]BF4 and its Co(II) complex [(tmpa)Co(HL)](BF4), [2]BF4 (tmpa = tris(2-methylpyridyl)amine). Results from X-ray crystallography, 1H NMR spectroscopy, electrochemistry, multi-frequency EPR spectroscopy, FIR spectroscopy, UV-vis-NIR spectroelectrochemistry, and SQUID magnetometry are discussed below to show the changes in the spin state in these systems as a function of multiple external perturbations and to describe their electronic structures.
Results and discussion
Synthesis and crystal structures
The reaction of CA with 2,6-diisopropylaniline in acetic acid under reflux, followed by recrystallization led to the formation of H2L (Scheme 1 and Experimental section). | ||
| Scheme 1 Synthesis of the ligand (top) and of the metal complexes (bottom). | ||
We had reported earlier on a similar method to prepare ligands with an [O,N,O,N] donor set (Fig. 1) starting from dhbq.6a In those ligands, usually two of the [O] in dhbq are substituted with two [NR] groups leading to the formation of a symmetrically substituted quinonoid ligand as the major product. In the present case the only product that we could isolate was the one where only one of the [O] in CA was substituted with a [NR] group leading to the unsymmetrically substituted ligand H2L that has an [O,O,O,N] donor set. The electron-poor nature of the CA ring because of the presence of the chloro substituents is likely responsible for the sluggishness of CA to undergo such a substitution reaction. The net result however is the formation of an unsymmetrically substituted H2L ligand, examples of which are rare in the literature.6a,h The purity and structure of H2L was established using a combination of 1H and 13C NMR data and elemental analyses (see Experimental section).
The complexes were synthesized in a one-pot reaction using Fe(BF4)2·6H2O or Co(BF4)2·6H2O, tmpa, H2L and NEt3 (Scheme 1 and Experimental section). Recrystallization from CH2Cl2/n-hexane led to the formation of pure, crystalline materials. The initial purity of the complexes was checked through elemental analyses that showed an excellent match between the calculated and experimental values of C, H and N. The final confirmation of the structure came from single crystal X-ray diffraction studies. All further studies on these complexes were performed with the pure crystalline material.
Single crystals of [1]BF4 and [2]BF4 (Fig. 2) were obtained as CH2Cl2 solvates. For both complexes bond length analysis within the (HL−) ligand showed bond localization within the quinone ligand.6,8k This is apparent for example from the bond lengths of 1.367(6) and 1.416(6) Å for the C20–C21 and C21–C22 bonds respectively in [1]BF4 (Table 1). Accordingly, the C20–O1 bond is slightly longer than C22–O3. A similar situation is also observed in the “upper half” of the ligand (HL−). The C22–C23 and C19–C20 bond lengths are around 1.5 Å and in the region of a C–C single bond. The donor atoms are thus best described as being a phenoxide type “O−” and a neutral ketotype “O” donor. One proton is accordingly localized on the N-atom, with the ligand retaining its substituted p-quinone type form. The metal coordination takes place preferentially through the [O,O] donor site which is easier to deprotonate in the H2L ligand. This difference in basicity and less steric crowding at the [O,O] donor site is also the reason for the exclusive formation of mononuclear complexes in the present case. A similar bonding situation is observed within the (HL−) ligand in the Co(II) complex [2]BF4 (Table 1).
 | ||
| Fig. 2 Perspective view of [1]BF4·2CH2Cl2 (left) and [2]BF4·2CH2Cl2 (right). The ellipsoids are drawn at 50% probability. Hydrogen atoms (except N–H), solvent molecules and counter-ions are omitted for clarity. | ||
| [1]BF4·2CH2Cl2 | [2]BF4·2CH2Cl2 | [1]BF4·2CH2Cl2 | [2]BF4·2CH2Cl2 | ||
|---|---|---|---|---|---|
| a M = Fe for [1]BF4·2CH2Cl2 and M = Co for [2]BF4·2CH2Cl2. | |||||
| M–N1a | 1.984(3) | 2.193(4) | C21–C22 | 1.418(5) | 1.413(6) |
| M–N2 | 1.973(4) | 2.107(4) | C22–C23 | 1.521(6) | 1.546(6) |
| M–N3 | 1.927(3) | 2.065(4) | C23–C24 | 1.392(5) | 1.380(6) |
| M–N4 | 1.957(4) | 2.090(4) | C24–C19 | 1.411(5) | 1.415(5) |
| M–O1 | 1.936(3) | 2.034(3) | C20–O1 | 1.285(5) | 1.275(5) |
| M–O2 | 1.978(3) | 2.158(3) | C19–O2 | 1.263(5) | 1.240(5) |
| C19–C20 | 1.490(5) | 1.524(6) | C22–O3 | 1.241(5) | 1.233(5) |
| C20–C21 | 1.368(6) | 1.369(6) | C23–N5 | 1.342(5) | 1.327(5) |
| N1–M–O1a | 176.5(1) | 172.6(1) | N2–M–N3 | 89.2(2) | 94.4(2) |
| N2–M–N4 | 167.0(2) | 153.7(2) | N2–M–O1 | 93.3(1) | 96.7(1) |
| N3–M–O2 | 175.4(1) | 171.7(1) | N2–M–O2 | 91.1(1) | 107.4(1) |
| N1–M–N2 | 83.5(1) | 78.2(1) | N3–M–N4 | 91.6(2) | 94.6(2) |
| N1–M–N3 | 85.3(1) | 80.8(1) | N3–M–O1 | 93.5(1) | 94.4(1) |
| N1–M–N4 | 83.7(1) | 78.9(1) | N4–M–O1 | 99.7(1) | 107.2(1) |
| O1–M–O2 | 81.8(1) | 77.5(1) | N4–M–O2 | 89.1(1) | 85.9(1) |
The Fe–O and Fe–N bond distances in [1]BF4 are all shorter than 2 Å (Table 1). These values indicate the existence of a low spin (LS) state (LS-t62g configuration in a simplified model for an octahedral complex) for the Fe(II) center at the measured temperature of 133 K.4 Accordingly, the octahedron around the Fe(II) center is a compact one as can be seen from only a small deviation of the angles from the ideal 90° (Table 1). In contrast, the Co–O and Co–N distances in [2]BF4 are all longer than 2 Å, and the octahedron around the Co(II) centers displays large distortions as can be seen from the large deviations of bond angles around it from the ideal 90° (Table 1). These data are an indication of a distorted, six-fold coordinated high spin (HS) Co(II) center ( configuration in a simplified model for an octahedral complex).6d
configuration in a simplified model for an octahedral complex).6d
Electrochemistry and UV-vis-NIR spectroelectrochemistry
The electron transfer properties of the two complexes were investigated by a combination of electrochemistry and UV-vis-NIR spectroelectrochemistry. Both complexes display a quasi-reversible one electron reduction, the potential of which is close to −1 V vs. Fc/Fc+ (Fig. 3). UV-vis-NIR spectroelectrochemical measurements did not lead to the regeneration of the starting spectrum on scanning the potential back, precluding the spectroscopic characterization of the one-electron reduced form. However, the appearance of the reduction step at similar potentials for both complexes, together with earlier reports on similar compounds,6d makes an assignment of the reduction step on the (HL−) ligand reasonable. | ||
| Fig. 3 Cyclic voltammogram of [1]+ (in CH2Cl2/0.1 M Bu4NPF6) and [2]+ (in CH3CN/0.1 M/Bu4NPF6) measured at 295 K. | ||
In contrast to the reduction step, the oxidation of both compounds appears at different potentials, and has very different features. For [1]+, an electrochemically and chemically reversible oxidation step with a half-wave potential of 0.32 V is observed. All features of this peak, together with results from UV-vis-NIR spectroelectrochemistry experiments (see below) show a chemically as well as an electrochemically reversible electron transfer step, leading likely to the generation of a HS FeIII center at the measured temperatures in fluid solution.3d For [2]+, the forward wave is observed at 0.66 V (Fig. 3) and the re-reduction wave appears at −0.30 V. In UV-vis-NIR spectroelectrochemical experiments, the starting spectrum of [2]+ is re-generated on scanning the potential back to −0.30 V after the oxidation step (see below). These results are an indication of an electron transfer-chemical (EC) reaction or more complicated mechanism accompanied by spin state changes. Starting from a HS CoII center in [2]+, that is a distorted octahedral d7 center, oxidation is expected to lead to a LS CoIII species (see NMR discussion below). Such a low-spin d6 system (LS-t62g configuration in a simplified model) will certainly contain shorter Co–ligand bond lengths and display smaller distortions from a perfect octahedral geometry as compared to the HS-CoII form. The aforementioned process is usually associated with large reorganization energy. Additionally, there is a possibility of the formation of different isomers during the oxidation process (see discussion below). All these phenomena contribute to the EC mechanism. The HS CoII form is however regenerated on re-reducing the complex, making the process chemically reversible.
In the UV-vis-NIR spectrum, [1]+ displays a weak long wavelength band at 845 nm and a weak shoulder at 620 nm (Fig. 4). For [2]+ the weak long wavelength band appears at 561 nm. Quinonoid ligands such as H2L usually display weak π to π* transitions in the visible region.6a For the present complexes we tentatively assign these bands to a mixture of such quinone ligand centered transitions and metal to ligand charge transfer (MLCT) transitions from filled d orbitals based on the metal to low lying empty orbitals on the quinone ligand. The bands at 351 nm for [1]+ and 342 nm for [2]+ are likely MLCT bands where the target orbitals are higher-lying empty orbitals based on the ligands. Further bands at higher energies are all ligand based transitions (Table 2).
 | ||
| Fig. 4 Changes in the UV-vis-NIR spectrum of [1]+ in CH2Cl2/0.1 M Bu4NPF6 (top) and of [2]+ in CH3CN/0.1 M Bu4NPF6 (bottom) during oxidation. | ||
| λ/nm (ε/103 M−1 cm−1) | |
|---|---|
| a From OTTLE spectroelectrochemistry in CH2Cl2 for [1]+ and CH3CN for [2]+, 0.1 M Bu4NPF6. | |
| [1]+ | 255 (27.1); 351 (28.1); 620 sh, 845 (1.37) |
| [1]2+ | 232 (31.4); 320 (17.1); 398 (11.0); 554 (3.68) |
| [2]+ | 231 (13.6); 257 (12.6); 342 (11.7); 561 (1.25) |
| [2]2+ | 235 (14.4); 255 (13.3); 305 (6.13); 388 (9.87); 613 (1.50) |
On one electron oxidation, the long wavelength bands for both complexes lose intensity and new bands appear at 554 nm for [1]2+ and at 613 nm for [2]2+ (Fig. 4). Furthermore, both the bands at 351 nm ([1]+) and 342 nm ([2]+) for the starting complexes lose intensity on one electron oxidation. This fact is in accordance with the assignment of these bands as MLCT bands (metal centered oxidation, see below). New bands appear at 398 nm for [1]2+ and at 388 nm for [2]2+, which are assigned to ligand to metal charge transfer (LMCT) transitions from filled orbitals on the quinone ligand to empty d orbitals on the oxidized metal centers. As expected, the oxidation steps do not have much influence on the bands that appear at further higher energies (Fig. 4). For both complexes, scanning the potential back, led to the quantitative regeneration of the spectra corresponding to the starting species, thus indicating the reversible nature of the oxidation steps.
Investigation of spin state changes through multiple external perturbations
Temperature- and field-dependent magnetic susceptibility measurements of [1]BF4 (Fig. 5 and S1‡) show a sudden decrease in the χT product from a value of 1.96 cm3 K mol−1 at room temperature to a value of 0.52 cm3 K mol−1 at 140 K. This behaviour is indicative of spin crossover from a high spin state (HS, S = 2) at high temperatures to a low spin state (LS, S = 0) at low temperature, with a concurrent decrease of the magnetic moment. On further lowering of the temperature, χT remains constant until it rapidly drops below 8 K which we attributed to zero-field splitting. χT does not reach the expected value of an S = 2 ion in the HS state (3.0 cm3 K mol−1 for g = 2), nor is the χT product zero at low temperatures. These observations indicate an incomplete SCO in the solid state. Such incomplete SCO processes have been observed previously.3d,4d,e To simulate the susceptibility data, an Ising-like model was applied,10 with the best fit shown in Fig. 5. The simulation revealed a transition temperature of T1/2 = 251 ± 1 K. The cooperativity parameter, which is a measure of the influence of cooperative intermolecular interactions on the SCO process was determined to be J = 167 ± 2 cm−1. Furthermore, the energy gap between HS and LS states was found to be Δ = 226 ± 15 cm−1. The HS molar fraction is 60% at 300 K and 16% at low temperature (Fig. 5). | ||
| Fig. 5 Temperature dependence of χT for [1]BF4 measured at 1000 Oe (black symbols), photo-induced change in magnetization (red symbols) by irradiation with green laser light (λ = 532 nm). The solid line is the fit (see text), and the dashed line is the high-spin fraction (right hand scale). The inset shows the temperature dependence of χT obtained from solution 1H NMR data. | ||
Photomagnetic experiments were carried out to study whether the conversion from LS to HS in [1]BF4 can also be induced by irradiation, known as light induced excited spin state trapping (LIESST).11 Upon excitation with green laser light (532 nm) the magnetic susceptibility increases (Fig. 5). The higher χT value corresponds to a higher fraction of the sample being present in the HS state, hence the conversion from the LS to HS state was induced by irradiation. The relative change in magnetic moment on irradiation is rather weak. This is typically due to the high-spin form also absorbing light at the irradiation wavelength. This has two consequences: (i) the light does not penetrate very far into the sample, (ii) reverse SCO can occur, leading to a steady state distribution of high- and low-spin species. After switching off the laser and measuring the magnetic susceptibility while slowly warming the sample to 300 K the χT product first remains stable and then decreases until it coincides with the data without irradiation. This indicates that [1]BF4 undergoes thermal relaxation at 42 K. This characteristic temperature is called TLIESST or critical temperature in the literature. The occurrence of LIESST was further confirmed by an isothermal relaxation measurement at 15 K, where the influence of sample heating immediately after commencing the irradiation can be clearly seen (Fig. S2‡). Upon discontinuation of the irradiation, the magnetic moment rapidly increases due to sample cooling, followed by a slow decay due to thermal relaxation.
The complex [1]+ also displays SCO behaviour in a CD2Cl2 solution (Fig. 5, inset). At 303 K, a 1H NMR spectrum is obtained which spans the region between 0 and 75 ppm, confirming the paramagnetic nature of the FeII center (Fig. S3‡). On cooling to 183 K all signals are observed within the 0 to 10 ppm range confirming the conversion of [1]+ to the diamagnetic, LS FeII form. A T1/2 of 240 K was determined for the SCO process in solution.
To study the electronic structure of [1]BF4, EPR spectra were recorded at X-band frequency (Fig. S4‡), where a clear resonance line attributed to the HS species is observed. In the high-frequency EPR (HFEPR), no corresponding signal was observed. This could be due to HS to LS conversion as a result of the 1.5 tons of pressure used in preparing the HFEPR pellet. An enhancement of the LS species under pressure has been previously found.12 We have used the standard Hamiltonian for Zeeman splitting and second rank axial (D) and rhombic (E) zero-field splitting to simulate the data: ![[script letter H]](https://www.rsc.org/images/entities/char_e142.gif) = gμBB + D(Ŝz2 − S(S + 1)/3) + E(Ŝx2 − Ŝy2). Combining the results of magnetometric and spectroscopic measurements the best simulations were obtained with g = 2.16 ± 0.05, D = 2 ± 0.5 cm−1 and E = 0.047 ± 0.0002 cm−1 (Fig. S4‡). These values are not untypical for HS iron(II).
= gμBB + D(Ŝz2 − S(S + 1)/3) + E(Ŝx2 − Ŝy2). Combining the results of magnetometric and spectroscopic measurements the best simulations were obtained with g = 2.16 ± 0.05, D = 2 ± 0.5 cm−1 and E = 0.047 ± 0.0002 cm−1 (Fig. S4‡). These values are not untypical for HS iron(II).
In the case of [2]BF4, we have used far-infrared (FIR) spectroscopy to determine the zero-field splitting (ZFS). The spectra (Fig. 6) show a clear feature at 66 cm−1. Due to the strong distortion of the octahedral coordination of CoII we can assume the orbital angular momentum to be quenched, resulting in spin-only magnetism with S = 3/2.6d The excitation energy corresponds to 2|D| and therefore, |D| = 33 ± 2 cm−1. EPR spectra of [2]BF4 were recorded at 9.5 and 300 GHz (Fig. 7). At the X band frequency signals at 150 and 340 mT were observed. The resonance lines in the HFEPR (300 GHz) spectrum are located at 3.8, 4.1, 4.9 and 5.4 T and at 10.3 and 10.7 T. Preliminary fits showed that the EPR spectra can only be simulated by assuming two different species A and B, each showing a different set of anisotropic g values, but the same D value. The spectra were modelled with a positive zero-field splitting parameter D of +33(2) cm−1, g values of 2.78(5), 2.14(2) and 2.08(3) for species A and g-values of 2.58(3), 1.93(4) and 2.00(2) for species B. HFEPR is very sensitive to small differences in the structure that might not be observable in magnetic or crystallographic measurements. Also, the low temperature structure of [2]BF4 is not known, but might feature two slightly different molecules in the unit cell. A hyperfine coupling of A∥ = 180 MHz was included in order to better reproduce the X-band signal at higher fields (Fig. 7). Two crystallographic positions for CoII semiquinonate complexes have been observed by others as well.13
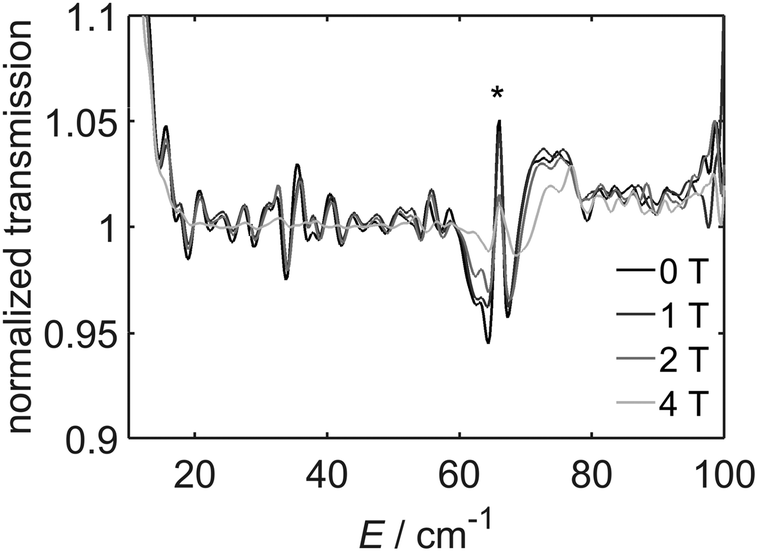 | ||
| Fig. 6 FIR spectra of [2]BF4 recorded at 9 K and 0–4 T. The spectra have been normalized by dividing by the spectrum recorded at 6 T. A measurement artefact is marked with *. The transition at 65 cm−1 which changes intensity with the magnetic field corresponds to 2D in the absence of rhombic zero-field splitting. | ||
 | ||
| Fig. 7 X-band EPR (top) and HFEPR (300 GHz, bottom) spectra recorded on pressed powder samples of [2]BF4. The light grey lines are the fits using parameters given in the text. | ||
Magnetic susceptibility measurements were carried out to study whether the complex [2]BF4 displays valence tautomerism (VT). The χT product decreases gradually with T from 300 to 50 K. Below 50 K the decline is steeper due to zero-field splitting until a value of 1.44 cm3 mol−1 K is reached at 1.8 K (Fig. S5‡). As the decline of χT with T is gradual we conclude that complex [2]BF4 does not show VT to any substantial extent in the temperature range studied here. χT was simulated, assuming high spin cobalt(II), with D = +33 cm−1, g1 = 1.58, g2 = 1.93, and g3 = 2.00, the gradual decline of χT being modelled as temperature independent paramagnetism (χTIP = 900 × 10−6 cm3 mol−1) (Fig. S5‡). The experimental values are in good agreement with the simulated values supporting the assumption of spin-only magnetism and correspond well with the reported values for HS CoII.14 The magnetization curve measured at 1.8 K shows a saturation value of 2.06μB (inset in Fig. S5‡), and was also modelled successfully with the same parameters. The complex [2]+ was chemically oxidized with NOBF4. The resulting [2]2+ form was found to be diamagnetic and delivers a 1H NMR spectrum that fits with a LS CoIII form (Fig. 8). This result thus confirms a metal-centered oxidation as discussed in the spectroelectrochemistry section above. Whereas the freshly dissolved solid delivered a spectrum corresponding to only one isomer, on leaving the solution for several hours, the formation of a second positional isomer in solution was observed (Fig. 8 and S6‡). These results thus confirm the discussion on reorganization after the oxidation step as stated in the electrochemistry section above. The spin state of the [2]+ species can thus be switched from S = 3/2 for a HS-CoII center to S = 0 for a LS-CoIII center by a simple reversible one-electron step.6d The potentially bridging ligand, HL−, the types of which are known to take part in electron transfer, does not participate in the oxidation process of [2]+. The quintessentially non-innocent quinone ligand thus acts as innocent for the present case, thus displaying the need for specifying a particular metal–ligand combination when discussing ligand non-innocence.15
 | ||
| Fig. 8 1H-NMR of [2]2+ directly measured after dissolving the sample. | ||
Conclusions
Summarizing, we have presented here the synthesis of a rare unsymmetrically substituted, potentially bridging quinone ligand. A mononuclear FeII and a CoII complex with this ligand has been synthesized and structurally characterized. The FeII complex displays SCO in the solid state and in solution with a transition temperature of 251 K in the solid state. The spin state of this complex can be switched either by varying the temperature or pressure. The same complex also shows a LIESST effect with a thermal relaxation at 42 K. The CoII complex displays a redox-induced spin state switching, delivering a LS CoIII complex on performing a reversible one electron transfer step. The potentially non-innocent (HL−) ligand thus remains innocent on the oxidative side. FIR spectroscopy delivers a D value of +33 cm−1 for [2]BF4. We have used a combination of crystallographic, (spectro)electrochemical, spectroscopic and magnetic methods to probe the electronic structures and the changes in magnetic properties of the molecules in various states. In particular, the combination of FIR spectroscopy, magnetism and (HF)EPR spectroscopy has allowed us to determine the spin Hamiltonian parameters of the complexes accurately. The results presented here show the potential of these newly synthesized quinone ligands for generating complexes displaying bistability as a function of multiple external perturbations. Future work will target complexes with higher nuclearity with an aim of incorporating multiple switching units within the same molecule.Experimental section
General considerations
Tris(2-methylpyridyl)amine (tmpa) was prepared according to a literature known procedure.16 All other reagents were commercially available and were used as received. All solvents were dried and distilled using common techniques unless otherwise mentioned. All reactions are carried out under a N2-atmosphere.Instrumentation
Cyclic voltammetry was carried out in 0.1 M Bu4NPF6 solution using a three-electrode configuration (Pt or glassy carbon working electrode, Pt counter electrode, Ag wire as pseudoreference) and PAR Versa STAT 4 potentiostat. The ferrocene/ferrocenium (Fc/Fc+) couple served as an internal reference. UV-vis-NIR absorption spectra were recorded on an Avantes spectrometer system: Ava Light-DH-BAL (light source), AvaSpec-ULS2048 (UV-vis-detector) and AvaSpec-NIR256-2.5TEC (NIR-detector). Spectroelectrochemical measurements were carried out using an optically transparent thin layer electrochemical (OTTLE) cell.17 Elemental analysis was performed on a Perkin Elmer Analyser 240. Mass spectrometry experiments were carried out on a Bruker Daltonics Mictrotof-Q mass spectrometer and mass spectrum simulations were done with mmass.X-ray crystallography
Crystal data and details of the data collections are given in Table S1.‡ X-ray data were collected on a STOE IPDS II diffractometer and BRUKER AXS (graphite monochromated Mo-Kα radiation, λ = 0.71073 Å) by the use of ω scans. The structures were solved by direct methods (SHELXS-97) and refined on F2 using all reflections with SHELXL-2014.18a Non-hydrogen atoms were refined anisotropically. Most hydrogen atoms were placed in calculated positions and assigned to an isotropic displacement parameter of 1.2/1.5Ueq(C). The nitrogen-bound hydrogen atoms were refined freely. Both compounds contain disordered CH2Cl2 solvent molecules. In the case of [1]BF4·2CH2Cl2 two CH2Cl2 are disordered about inversion centers. One was refined at 1/2 occupancy, the other one shows an additional positional disorder and each part was refined at 1/4 occupancy. The occupancy factors of the third solvent molecules were refined to 0.880(6)/0.120(6). DFIX restraints (dC–Cl = 1.79 Å, dCl⋯Cl = 2.8 Å) and EADP constraints were applied to model the disorder. In the case of [2]BF4·2CH2Cl2 two CH2Cl2 are disordered about inversion centers. Both were refined at 1/2 occupancy. The occupancy factors of the third solvent molecules were refined to 0.777(6)/0.223(6). DFIX restraints (dC–Cl = 1.79 Å) and EADP constraints were used to model the disorder. Furthermore the BF4− anion of [1]BF4·2CH2Cl2 is disordered (occupancy factors: 0.637(6)/0.363(6)). EADP constraints were used for anisotropic refinement of the counter ion.Face-indexed absorption corrections for [1]BF4·2CH2Cl2 were performed numerically with the program X-RED.18b SADABS,18c CCDC 971157 ([1]BF4) and 1052194 ([2]BF4) contain the cif files for this work.
Magnetic susceptibility measurements
The magnetic measurements on powder samples were performed with a MPMS 3 SQUID magnetometer for [2]BF4 and with a MPMS-XL7 SQUID magnetometer for [1]BF4, both from Quantum Design. Samples were slightly pressed, Teflon wrapped powder pellets. For the temperature-dependent measurements below 50 K an external magnetic field of 1000 Oe was applied; for measurements above 40 K 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe was applied. The molar susceptibility data were corrected for diamagnetic contributions by subtracting χdiam = −0.5 × MM × 10−6 cm3 mol−1. The molar susceptibility data of [1]BF4 have been corrected for diamagnetic impurities as well. Magnetic susceptibility measurements in solution were analyzed using the Evans method.19
000 Oe was applied. The molar susceptibility data were corrected for diamagnetic contributions by subtracting χdiam = −0.5 × MM × 10−6 cm3 mol−1. The molar susceptibility data of [1]BF4 have been corrected for diamagnetic impurities as well. Magnetic susceptibility measurements in solution were analyzed using the Evans method.19
For the photomagnetic experiments an external magnetic field of 1000 Oe was applied, [1]BF4 was cooled down to 1.8 K and then heated to 15 K. Irradiation of the sample was performed by coupling a 532 nm laser beam through an optical fiber to the cavity of the MPMS-XL7 SQUID magnetometer. The sample was irradiated until no further increase in susceptibility was observed. After switching off the laser the magnetic susceptibility was measured while warming the sample to 300 K at a rate of 1 K min−1 (inset in Fig. 5). The photomagnetic experiments were carried out on a thin layer of sample in vacuum grease and scaled to match the data measured for a heavier and thus more accurately weighed sample of [1]BF4.
EPR spectroscopy
X-band EPR spectra at 9.47 GHz were recorded using a Bruker EMX spectrometer equipped with a continuous flow cryostat. HFEPR spectra were measured with a home-built spectrometer at frequencies between 95 and 380 GHz. For the HFEPR measurements the samples were pressed into pure pellets to prevent orientation in the magnetic field. Simulations were performed with Easyspin.20FIR spectroscopy
Far infrared (FIR) spectra were recorded on a Bruker 113v FTIR spectrometer equipped with a mercury vapour light source and an Infrared Laboratories pumped Si bolometer. The samples were studied as pure pellets attached to a sample holder inserted into an 8T Oxford Instruments Spectromag 4000 optical cryomagnet. The sample holder permitted in situ changing between an aperture and the sample, which allowed recording absolute transmission spectra. To identify the relevant features, spectra were normalized by division by the spectrum at the highest magnetic field.Syntheses
[H2L]: diisopropylaniline (1.88 mL, 10.0 mmol, 1 eq.) was added to a solution of chloranilic acid (2.09 g, 10.0 mmol, 1 eq.) in acetic acid (30 mL). The resulting mixture was heated to reflux for 2 h. After cooling down to room temperature, water was added to form a purple precipitate which was filtered off and washed with water. The purple residue was dissolved in CH2Cl2 and dried over Na2SO4. After removing of the Na2SO4, the solvent was removed in vacuo to yield the crude product. Recrystallisation from a CH2Cl2/pentane-mixture (1:2) yielded the pure product (1.45 g, 4.18 mmol, 42%). 1H NMR (400 MHz, CDCl3) δ (ppm):7.92 (s, 1 H, NH), 7.35 (t, J = 7.7 Hz, 1 H, Ar-H), 7.14 (d, J = 7.7 Hz, 2 H, Ar-H), 2.91 (p, J = 6.9 Hz, 2 H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) (CH3)2), 1.21 (d, J = 6.9 Hz, 6 H, CH(C3)2), 1.16 (d, J = 6.9 Hz, 6 H, CH(C3)2) (Fig. S7‡). The signal of the OH was not observed. 13C NMR (101 MHz, CDCl3) δ 175.70, 173.10, 154.47, 146.10, 143.15, 131.55, 129.56, 123.28, 109.64, 101.58, 29.10, 24.19, 22.59 (Fig. S8‡). Th. Anal. Calc. for C18H19N1O3Cl2: C 58.71; H 5.20; N 3.80%; found: C 58.84; H 5.24; N 3.92%.
(CH3)2), 1.21 (d, J = 6.9 Hz, 6 H, CH(C3)2), 1.16 (d, J = 6.9 Hz, 6 H, CH(C3)2) (Fig. S7‡). The signal of the OH was not observed. 13C NMR (101 MHz, CDCl3) δ 175.70, 173.10, 154.47, 146.10, 143.15, 131.55, 129.56, 123.28, 109.64, 101.58, 29.10, 24.19, 22.59 (Fig. S8‡). Th. Anal. Calc. for C18H19N1O3Cl2: C 58.71; H 5.20; N 3.80%; found: C 58.84; H 5.24; N 3.92%.
[1]BF4: tmpa (116 mg, 0.40 mmol, 1 eq.) and Fe(BF4)2·6H2O (135 mg, 0.40 mmol, 1 eq.) were dissolved in MeCN (10 mL) and stirred for 30 min at room temperature. H2L (148 mg, 0.40 mmol, 1 eq.) and NEt3 (0.5 mL) were added and the resulting mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo. The residue was dissolved in CH2Cl2 (20 mL) and n-hexane (10 mL) was layered. The resulting green crystals were collected by filtration and dried in vacuo (190 mg, 0.24 mmol, 60%). To yield crystals of [1]BF4 which were suitable for X-ray diffraction, a small sample was dissolved in CH2Cl2 and slow diffusion of n-pentane into this solution leads to single crystal growth. Th. Anal. Calc. for C36H36N5O3Cl2BF4Fe: C 54.03; H 4.53; N 8.75%; found: C 53.99; H 4.96; N 8.73%.
[2]BF4: tmpa (100 mg, 0.34 mmol, 1 eq.) and Co(BF4)2·6H2O (117 mg, 0.34 mmol, 1 eq.) were dissolved in MeCN (10 mL) and stirred for 30 min at room temperature. H2L (128 mg, 0.34 mmol, 1 eq.) and NEt3 (0.3 mL) were added and the resulting mixture was stirred for 2 h at room temperature. The solvent was removed in vacuo. The residue was dissolved in CH2Cl2 (20 mL) and n-hexane (10 mL) was layered. The resulting red brown crystals were collected by filtration and dried in vacuo (172 mg, 0.22 mmol, 63%). To yield crystals of [2]BF4 which were suitable for X-ray diffraction, a small sample was dissolved in CH2Cl2 and slow diffusion of n-pentane into this solution leads to single crystal growth. Th. Anal. Calc. for C36H36N5O3Cl2BF4Co: C 53.82; H 4.52; N 8.72%; found: C 53.62; H 4.99; N 8.85%.
[2]2+: [2]+ (26 mg, 32 μmol, 1 eq.) was dissolved in MeCN, abs. (5 mL) and NOBF4 (5 mg, 40 μmol, 1.2 eq.) were added. The solution was stirred for 1 h at room temperature resulting in a color change from brown to green. The solvent was removed in vacuo and the residue was dissolved in CH2Cl2 (6 mL). The resulting solution was filtered and the compound was recrystallized by slow diffusion of n-pentane into the resulting solution. The resulting crystalline green material was collected by filtration (15 mg, 15 μmol, 47%).
1H NMR (400 MHz, acetone-d6 (only one isomer)) δ = 10.92 (s, 1 H, NH), 9.29 (dd, J = 6.0, 0.7 Hz, 1 H), 8.55 (dd, J = 5.8, 0.7 Hz, 2 H), 8.26 (td, J = 7.8, 1.5 Hz, 2 H), 8.06 (td, J = 7.7, 1.4 Hz, 1 H), 7.93 (d, J = 7.9 Hz, 2 H), 7.88–7.72 (m, 3 H), 7.50 (d, J = 0.8 Hz, 1 H), 7.35 (d, J = 7.4 Hz, 1 H), 7.17 (d, J = 7.8 Hz, 2 H), 5.71 (d, J = 16.4 Hz, 2 H, CH2), 5.54 (s, 2 H, CH2), 5.46 (d, J = 16.3 Hz, 2 H, CH2), 1.04 (d, J = 6.8 Hz, 6 H, CH(C3)2), 0.94 (d, J = 6.8 Hz, 6 H, CH(C3)2). The signal of the C(CH3)2 was not detected due to an overlap with the water signal. Anal. Calc. for C36H36N5O3Cl2B2F8Co·4H2O·0.45CH3CN: C 45.19; H 4.66; N 7.78%; found: C 45.18; H 4.74; N 7.67%.
Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG, SA 1580/5-1, SL 104/2-1) for the financial support of this work. We thank Prof. M. Dressel (1. Physikalisches Institut, Universität Stuttgart) for access to the SQUID magnetometer and FIR spectrometer. Alexa Paretzki is acknowledged for preliminary work.References
- For selected examples and reviews see: (a) M. D. Ward, Inorg. Chem., 1996, 35, 1712–1714 CrossRef CAS PubMed; (b) M. Leschke, M. Melter and H. Lang, Inorg. Chim. Acta, 2003, 350, 114–120 CrossRef CAS; (c) S. Kitagawa and S. Kawata, Coord. Chem. Rev., 2002, 224, 11–34 CrossRef CAS; (d) J. S. Miller and K. S. Min, Angew. Chem., Int. Ed., 2009, 48, 262–272 CrossRef CAS PubMed; (e) B. Therrien, G. Süss-Fink, P. Govindaswamy, A. K. Renfrew and P. J. Dyson, Angew. Chem., Int. Ed., 2008, 47, 3773–3776 CrossRef CAS PubMed; (f) K. Heinze, G. Hüttner, L. Zsolnai, A. Jacobi and P. Schober, Chem. – Eur. J., 1997, 3, 732–743 CrossRef CAS; (g) Y.-F. Han, W.-G. Jia, W.-B. Yu and G.-X. Jin, Chem. Soc. Rev., 2009, 38, 3419–3434 RSC; (h) Y.-F. Han, H. Li and G.-X. Jin, Chem. Commun., 2010, 46, 6879 RSC.
- For selected examples see: (a) C. Carbonera, A. Dei, J.-F. Létard, C. Sangegrorio and L. Sorace, Angew. Chem., Int. Ed., 2004, 43, 3136–3138 CrossRef CAS PubMed; (b) J. Tao, H. Maruyama and O. Sato, J. Am. Chem. Soc., 2006, 128, 1790–1791 CrossRef CAS PubMed; (c) B. Li, F.-L. Yang, J. Tao, O. Sato, R.-B. Huang and L.-S. Zheng, Chem. Commun., 2008, 6019–6021 RSC.
- For selected examples see: (a) K. S. Min, K. Swierczek, A. G. DiPasquale, A. L. Rheingold, W. M. Reiff, A. M. Atta and J. S. Miller, Chem. Commun., 2008, 317–319 RSC; (b) K. S. Min, A. G. DiPasquale, A. L. Rheingold and J. S. Miller, Inorg. Chem., 2007, 46, 1048–1050 CrossRef CAS PubMed; (c) K. S. Min, A. G. DiPasquale, J. A. Golen, A. L. Rheingold and J. S. Miller, J. Am. Chem. Soc., 2007, 129, 2360–2368 CrossRef CAS PubMed; (d) J. G. Park, I.-R. Jeon and T. David Harris, Inorg. Chem., 2015, 54, 359–369 CrossRef CAS PubMed.
- (a) Spin Crossover in Transition Metal Compounds I-III, ed. P. Gütlich and H. A. Goodwin, Top. Curr. Chem., Springer, Berlin, Heidelberg, Germany, 2004, vol. 233–235 Search PubMed; (b) Eur. J. Inorg. Chem., 2013 Search PubMed, 574–1067 (cluster issue on spin crossover complexes); (c) M. A. Halcrow, Chem. Soc. Rev., 2011, 40, 4119–4142 RSC; (d) P. Gütlich, Y. Garcia and H. A. Goodwin, Chem. Soc. Rev., 2000, 29, 419–427 RSC; (e) M. Ostermeier, M.-A. Berlin, R. M. Meudtner, S. Demeshko, F. Meyer, C. Limberg and S. Hecht, Chem. – Eur. J., 2010, 16, 10202–10213 CrossRef CAS PubMed.
- K. S. Min, A. L. Rheingold, A. G. DiPasquale and J. S. Miller, Inorg. Chem., 2006, 45, 6135–6137 CrossRef CAS PubMed.
- (a) D. Schweinfurth, H. S. Das, F. Weisser, D. Bubrin and B. Sarkar, Inorg. Chem., 2011, 50, 1150–1159 CrossRef CAS PubMed; (b) F. Weisser, R. Hübner, D. Schweinfurth and B. Sarkar, Chem. – Eur. J., 2011, 17, 5727–5736 CrossRef CAS PubMed; (c) D. Schweinfurth, M. M. Khusniyarov, D. Bubrin, S. Hohloch, C.-Y. Su and B. Sarkar, Inorg. Chem., 2013, 52, 10332–10339 CrossRef CAS PubMed; (d) D. Schweinfurth, Y. Rechkemmer, S. Hohloch, N. Deibel, I. Peremykin, J. Fiedler, R. Marx, P. Neugebauer, J. van Slageren and B. Sarkar, Chem. – Eur. J., 2014, 20, 3475–3486 CrossRef CAS PubMed; (e) M. G. Sommer, D. Schweinfurth, F. Weisser, S. Hohloch and B. Sarkar, Organometallics, 2013, 32, 2069–2078 CrossRef CAS; (f) N. Deibel, M. G. Sommer, S. Hohloch, J. Schwann, D. Schweinfurth, F. Ehret and B. Sarkar, Organometallics, 2014, 33, 4756–4765 CrossRef CAS; (g) N. Deibel, S. Hohloch, D. Schweinfurth, F. Weisser, A. Grupp and B. Sarkar, Chem. – Eur. J., 2014, 20, 15178–15187 CrossRef CAS PubMed; (h) B. Sarkar, D. Schweinfurth, N. Deibel and F. Weisser, Coord. Chem. Rev., 2015, 293–294, 250–262 CrossRef CAS; (i) D. Schweinfurth, F. Weisser and B. Sarkar, Nachr. Chem., 2009, 57, 862–866 CrossRef CAS; (j) H. S. Das, F. Weisser, D. Schweinfurth, C.-Y. Su, L. Bogani, J. Fiedler and B. Sarkar, Chem. – Eur. J., 2010, 16, 2977–2981 CrossRef CAS PubMed; (k) H. S. Das, D. Schweinfurth, J. Fiedler, M. M. Khusniyarov, S. M. Mobin and B. Sarkar, Chem. – Eur. J., 2014, 20, 4334–4346 CrossRef CAS PubMed; (l) D. Gupta, V. Singh, S. Hohloch, M. Sathiyendiran, K. Tedin and B. Sarkar, Polyhedron, 2015, 100, 243–250 CrossRef CAS.
- (a) H. S. Das, A. K. Das, R. Pattacini, R. Huebner, B. Sarkar and P. Braunstein, Chem. Commun., 2009, 4387–4389 RSC; (b) P. Braunstein, D. Bubrin and B. Sarkar, Inorg. Chem., 2009, 48, 2534–2540 CrossRef CAS PubMed; (c) A. Paretzki, R. Pattacini, R. Huebner, P. Braunstein and B. Sarkar, Chem. Commun., 2010, 46, 1497–1499 RSC; (d) N. Deibel, D. Schweinfurth, R. Huebner, P. Braunstein and B. Sarkar, Dalton Trans., 2011, 40, 431–436 RSC; (e) S. Hohloch, P. Braunstein and B. Sarkar, Eur. J. Inorg. Chem., 2012, 546–553 CrossRef CAS; (f) N. Deibel, S. Hohloch, M. G. Sommer, D. Schweinfurth, F. Ehret, P. Braunstein and B. Sarkar, Organometallics, 2013, 32, 7366–7375 CrossRef CAS; (g) F. Yuan, F. Weisser, B. Sarkar, A. Garci, P. Braunstein, L. Routaboul and B. Therrien, Organometallics, 2014, 33, 5043–5045 CrossRef.
- (a) K. Heinze, G. Huttner and L. Zsolnai, Z. Naturforsch., 1999, 1147–1154 CAS; (b) D. Kumbhakar, B. Sarkar, S. Maji, S. M. Mobin, J. Fiedler, F. A. Urbanos, R. Jimenez-Aparicio, W. Kaim and G. K. Lahiri, J. Am. Chem. Soc., 2008, 130, 17575–17583 CrossRef CAS PubMed; (c) T. E. Keyes, R. J. Forster, P. W. Jayaweera, C. G. Coates, J. J. McGarvey and J. G. Vos, Inorg. Chem., 1998, 37, 5925–5932 CrossRef CAS; (d) G. Margraf, T. Kretz, F. F. de Biani, F. Lashi, S. Losi, P. Zanello, J. W. Bats, B. Wolf, K. Removic-Langer, M. Lang, A. Prokofiev, W. Assmus, H.-W. Lerner and M. Wagner, Inorg. Chem., 2006, 45, 1277–1288 CrossRef CAS PubMed; (e) S. Scheuermann, B. Sarkar, M. Bolte, J.-W. Bats, H.-W. Lerner and M. Wagner, Inorg. Chem., 2009, 48, 9385–9392 CrossRef CAS PubMed; (f) P. Braunstein, O. Siri, J.-P. Taquet, M.-M. Rohmer, M. Bénard and R. J. Walter, J. Am. Chem. Soc., 2003, 125, 12246–12256 CrossRef CAS PubMed; (g) Q.-Z. Yang, O. Siri and P. Braunstein, Chem. Commun., 2005, 2660–2662 RSC; (h) Q.-Z. Yang, O. Siri and P. Braunstein, Chem. – Eur. J., 2005, 11, 7237–7246 CrossRef CAS PubMed; (i) O. Siri, J.-P. Taquet, J.-P. Collin, M.-M. Rohmer, M. Bénard and P. Braunstein, Chem. – Eur. J., 2005, 11, 7247–7253 CrossRef CAS PubMed; (j) F. A. Cotton, J.-Y. Jin, Z. Li, C. A. Murillo and J. H. Reibenspies, Chem. Commun., 2008, 211–213 RSC; (k) S. Kar, B. Sarkar, S. Ghumaan, D. Janardanan, J. van Slageren, J. Fiedler, V. G. Puranik, R. B. Sunoj, W. Kaim and G. K. Lahiri, Chem. – Eur. J., 2005, 11, 4901–4911 CrossRef CAS PubMed.
- G. Aromi, D. Aguila, P. Gamez, F. Luis and O. Roubeau, Chem. Soc. Rev., 2012, 41, 537–546 RSC.
- (a) I. Salitros, O. Fuhr, R. Kruk, J. Pavlik, L. Pogany, B. Schäfer, M. Tatarco, R. Boca, W. Linert and M. Ruben, Eur. J. Inorg. Chem., 2013, 1049–1057 CrossRef CAS; (b) F. Varret, S. A. Salunke, K. Boukheddadden, A. Boussekksau, E. Codjovi, C. Enachescu and J. Linares, C. R. Chim., 2003, 385–393 CrossRef CAS.
- S. Decartins, P. Gütlich, C. P. Köhler, H. Spieringand and A. Hauser, Chem. Phys. Lett., 1984, 105, 1–4 CrossRef.
- (a) V. Ksenofontov, A. B. Gaspar and P. Gütlich, Top. Curr. Chem., 2004, 235, 23–64 CrossRef CAS; (b) J. A. Real, A. B. Gaspar, A. Niel and M. C. Munoz, Coord. Chem. Rev., 2003, 1–2, 121–141 CrossRef; (c) A. Bencini, A. Caneschi, C. Carbonera, A. Dei, D. Gatteschi, R. Righini, C. Sangregorio and J. van Slageren, J. Mol. Struct., 2003, 656, 141–154 CrossRef CAS; (d) A. Caneschi, A. Dei, F. F. de Biani, P. Gütlich, V. Ksenofontov, G. Levshenko, A. Hoefer and F. Renz, Chem. – Eur. J., 2001, 7, 3926–3930 CrossRef CAS PubMed.
- (a) A. Bencini, A. Beni, F. Costantino, A. Dei, D. Gatteschi and L. Sorace, Dalton Trans., 2006, 722–729 RSC; (b) Y. Mulyana, G. Poneti, B. Moubaraki, K. S. Murray, B. F. Abrahams, L. Sorace and C. Boscovic, Dalton Trans., 2010, 39, 4757–4767 RSC.
- (a) D. N. Hendrickson and C. G. Pierpont, Top. Curr. Chem., 2004, 234, 63–95 CrossRef CAS; (b) D. Schweinfurth, F. Weisser, D. Bubrin, L. Bogani and B. Sarkar, Inorg. Chem., 2011, 50, 6114–6121 CrossRef CAS PubMed.
- M. D. Ward and J. A. McCleverty, J. Chem. Soc., Dalton Trans., 2002, 275–288 RSC.
- A. L. Ward, L. Elbaz, J. B. Kerr and J. Arnold, Inorg. Chem., 2012, 51, 4694–4706 CrossRef CAS PubMed.
- M. Krejcik, M. Danek and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed; (b) X-RED, STOE & CIE GmbH, Darmstadt, Germany, 2002 Search PubMed; (c) G. M. Sheldrick, SADABS Ver., 2008/1; SADABS. Program for Empirical Absorption Correction, University of Gottingen, Germany Search PubMed.
- (a) E. M. Schubert, J. Chem. Educ., 1992, 69, 62 CrossRef CAS; (b) D. F. Evans, J. Chem. Soc., 1959, 2003–2005 RSC; (c) G. A. Bain and J. F. Berry, J. Chem. Educ., 2008, 85, 532–536 CrossRef CAS.
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS PubMed.
Footnotes |
| † Dedicated to Prof. Heinrich Lang on the occasion of his 60th birthday. |
| ‡ Electronic supplementary information (ESI) available: Table of crystallographic details, magnetization curve for [1]BF4, NMR spectra, EPR spectrum. CCDC 971157 and 1052194. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00757k |
| § Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |