DOI:
10.1039/C6DT00625F
(Paper)
Dalton Trans., 2016,
45, 12409-12422
Unique mononuclear MnII complexes of an end-off compartmental Schiff base ligand: experimental and theoretical studies on their bio-relevant catalytic promiscuity†
Received
16th February 2016
, Accepted 1st July 2016
First published on 4th July 2016
Abstract
Three new mononuclear manganese(II) complexes, namely [Mn(HL)2]·2ClO4 (1), [Mn(HL)(N(CN)2)(H2O)2]·ClO4 (2) and [Mn(HL)(SCN)2] (3) [LH = 4-tert-butyl-2,6-bis-[(2-pyridin-2-yl-ethylimino)-methyl]-phenol], have been synthesized and structurally characterized. An “end-off” compartmental ligand (LH) possesses two symmetrical compartments with N2O binding sites but accommodates only one manganese atom instead of two due to the protonation of the imine nitrogen of one compartment. Although all three complexes are mononuclear, complex 1 is unique as it has a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 metal to ligand stoichiometry. The catalytic promiscuity of complexes 1–3 in terms of two different bio-relevant catalytic activities namely catecholase and phenoxazinone synthase has been thoroughly investigated. EPR and cyclic voltametric studies reveal that radical formation rather than metal centered redox participation is responsible for their catecholase-like and phenoxazinone synthase-like catalytic activity. A computational approach suggests that imine bond bound radical generation rather than phenoxo radical formation is most likely responsible for the oxidizing properties of the complexes.
:2 metal to ligand stoichiometry. The catalytic promiscuity of complexes 1–3 in terms of two different bio-relevant catalytic activities namely catecholase and phenoxazinone synthase has been thoroughly investigated. EPR and cyclic voltametric studies reveal that radical formation rather than metal centered redox participation is responsible for their catecholase-like and phenoxazinone synthase-like catalytic activity. A computational approach suggests that imine bond bound radical generation rather than phenoxo radical formation is most likely responsible for the oxidizing properties of the complexes.
Introduction
Schiff-base complexes have been comprehensively studied in coordination chemistry mainly due to their ease of synthesis, excellent structural diversities, and enormous potential towards various applications including bio-activity.1–5 Among the Schiff base ligands, phenol based compartmental ligands are of interest to the bio-inorganic chemists to synthesize small coordination compounds as structural and/or functional models of different metallobiosites like catechol oxidase, phenoxazinone synthase, urease, catalase etc.6–20 Mn-containing redox enzymes are omnipresent in nature and perform a number of vital functions.21 The most important examples are manganese superoxide dismutase,22a manganese catalases,22b peroxidase,22c and manganese-dependent dioxygenases.22d Photosynthetic water oxidation by photosystem-II is controlled by a tetra-manganese cluster.22e,f The modeling approach has been widely used to reveal the structure and function of manganese redox enzymes.23 In recent time, dioxygen activation catalyzed by Mn(II) Schiff-base complexes in terms of oxidation of catechol or o-amino phenol are being investigated by several groups including us.24 However, the exact mechanistic pathways involved in these oxidation processes are not well explored experimentally as well as theoretically. In order to have a better understanding of these mechanistic pathways we have undertaken this project. Complexation has been carried out by employing a phenol based bi-compartmental Schiff base ligand, 4-tert-butyl-2,6-bis-[(2-pyridin-2-yl-ethylimino)-methyl]-phenol (LH) and manganese(II) ions. Interestingly, we have got three mononuclear MnII Schiff base complexes of bi-compartmental ligand LH (Scheme 1) instead of dinuclear complexes with different metal:ligand stoichiometries. Dioxygen activation in terms of catechol and o-aminophenol oxidation using 3,5-di-tert-butyl catechol (3,5-DTBC) and o-aminophenol (OAPH), respectively, as model substrates catalyzed by our Mn(II) complexes have been thoroughly investigated. A new mechanistic pathway involved in this oxidation has been unveiled by combined experimental and theoretical approaches as is depicted in this communication.
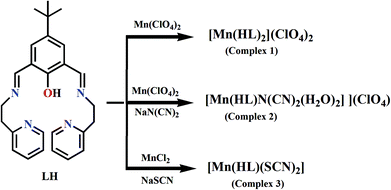 |
| | Scheme 1 Synthetic route of complexes 1–3. | |
Experimental sections
Methods and materials
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a Perkin-Elmer 240C elemental analyzer. Infrared spectra (4000–500 cm−1) were recorded at 27 °C using a Shimadzu FTIR-8400S with KBr pellets. Magnetic susceptibilities were measured at 27 °C using an EG and G PAR 155 vibrating sample magnetometer with Hg[Co(SCN)4] as reference; diamagnetic corrections were made using Pascal's constants. Electronic spectra (800–200 nm) were obtained at 27 °C using a Shimadzu UV-2450 with methanol as the solvent and reference. The cyclic voltammetric measurements were carried out in dry acetonitrile solutions with 0.2 M TEAP as the supporting electrolyte (scan rate = 100 mV s−1) employing a CH1106A potentiostat. A three electrode system was used in which the counter and working electrodes were platinum foils and the reference electrode was a Ag/AgCl electrode. EPR experiments were performed at liquid nitrogen temperature (77 K) in methanol, using a JEOL JES-FA200 spectrometer at the X band (9.13 GHz).
All chemicals were obtained from Sigma-Aldrich and used as received.
Syntheses of the complexes
Synthesis of [Mn(HL)2](ClO4)2 (1).
Complex 1 was synthesized by dropwise addition of an aqueous solution (20 mL) of Mn(ClO4)2·6H2O (0.662 g, 2 mmol) to a hot methanolic solution (50 mL) of LH (0.415 g, 1 mmol). The solution turned into light brown color from light yellow. The solution was refluxed for another 2 h. The resulting solution was put in the dark for crystallization. Light brown single crystals suitable for X-ray data collection were obtained from the filtrate after a week. Yield 72%. Anal. calcd for C52H59Cl2MnN8O10: C, 57.67; H, 5.45; N, 10.35. Found: C, 57.62; H, 5.40; N, 10.28. IR (KBr): ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) 1653 cm−1; ν(skeletal vibration) 1596 cm−1; ν(ClO4−) 1086 cm−1.
N) 1653 cm−1; ν(skeletal vibration) 1596 cm−1; ν(ClO4−) 1086 cm−1.
Synthesis of [Mn(HL)(N(CN)2)(H2O)2]·ClO4 (2).
Complex 2 was prepared following the same procedure used for complex 1, with an addition of 10 mL aqueous solution of NaN(CN)2 (0.178 g, 2 mmol). The resulting solution was stirred for 1 h and the final solution was allowed for slow evaporation. X-ray suitable single crystals of complex 2 were obtained after one week. Yield 79%. C28H34ClMnN7O7: C, 50.00; H, 5.06; N, 14.06. Found: C, 49.96; H, 5.01; N, 14.02. IR (KBr): ν(CN) 1648 cm−1; ν(skeletal vibration) 1533 cm−1; ν(ClO4−) 1059 cm−1; ν(N(CN)2−) 2169, 2219, 2280 cm−1.
Synthesis of [Mn(HL)(SCN)2] (3).
Complex 3 was synthesized by dropwise addition of an aqueous solution (20 mL) of MnCl2 (0.125 g, 1 mmol) to a hot methanolic solution (50 mL) of LH (0.415 g, 1 mmol). The solution was allowed to reflux for 1 h in water bath. Then the solution was cooled and allowed to stir in an open atmosphere for an additional 1 h after adding an aqueous solution of NaSCN (0.162 g, 2 mmol). A pale brown colored product appeared from the filtrate after a few days. Single crystals suitable for X-ray diffraction were obtained after the dissolution of the product in acetonitrile. Yield 83%. Anal. calcd for C28H30MnN6OS2: C, 57.42; H, 5.16; N, 14.35. Found: C, 57.38; H, 5.28; N, 14.43 IR (KBr): ν(CN) 1641 cm−1; ν(skeletal vibration) 1532 cm−1; ν(SCN−) 2063 cm−1.
Caution! Transition metal perchlorate complexes are potentially explosive and should be handled in small amounts and with necessary precautions.
Crystal structure determination
Prism crystals of 1, 2 and 3 were mounted on a Bruker automatic diffractometer equipped with a CCD detector, and used for data collection. X-ray intensity data were collected under graphite monochromated MoKα radiation (λ = 0.71073 Å) at temperature 291 K, with an ω scan mode. Due to the extremely weak scattering of the X-ray radiation on the crystals of compounds 1 and 3 the reflections inside the Ewald sphere were collected up to θ = 23.04 and 22.67° (at higher θ angles all reflections were unobservable, and, in the case of compound 1, at lower angles the reflections were weak but well-shaped with relatively large values of I/σ). It must be noted that such a limitation of the angle did not affect the lucidity of the structural determinations. Details concerning crystal data and refinement are given in Table 1. Examination of reflections on two reference frames monitored after each 20 frames measured showed no loss of the intensity for all measurements. Lorentz, polarization, and numerical absorption multi-scan corrections were applied. The structures were solved by a partial structure expansion procedure. All the non-hydrogen atoms were refined anisotropically using the full-matrix least-squares technique on F2. All the hydrogen atoms were found from difference Fourier synthesis after four cycles of anisotropic refinement, and refined as “riding” on the adjacent atom with the individual isotropic displacement factor equal to 1.2 times the value of the equivalent displacement factor of the parent non-methyl carbon, oxygen and nitrogen atoms and 1.5 times for the parent methyl carbon atoms, except for the hydrogen atoms of compound 2 bonded to the oxygen and nitrogen atoms. The crystal of compound 2 scatters X-ray radiation enough to allow reliable refinement of the isotropic displacement parameters and positions of the hydrogen atoms bonded to the oxygen and nitrogen atoms. The tert-butyl group of compound 3 is disordered over two positions with 0.66:0.34 participation of domains. The displacement ellipsoids of minor domain atoms exhibit some prolation and oblation effects, probably as a result of further disorder. The introduction of a third domain leads to unstable refinement, which suggests the presence of some dynamic disorder. The “riding” hydrogen atom positions were idealised after each cycle of refinement. The SHELXS, SHELXL and SHELXTL25 programs were used for all the calculations. Atomic scattering factors were incorporated in the computer programs. Selected inter-atomic bond distances and angles are listed in Table S1 (ESI†), and the geometrical parameters of intermolecular interactions are listed in Tables S2 and S3 (ESI†). The perchlorate anion in compound 2 is disordered over two positions (the second domain is rotated along the local three-fold axis passing through the Cl2 and O7 atoms at about 60°) with a 57:43 domain ratio. In all compounds some terminal atoms show symptoms of dynamic disorder, visible in the prolating displacement ellipsoids of these atoms.
Table 1 Crystal data and structure refinement details for studied complexes 1–3
| Compound |
(1) |
(2) |
(3) |
| Empirical formula |
C52H59Cl2MnN8O10 |
C28H34ClMnN7O7 |
C28H30MnN6OS2 |
| Formula weight |
1081.91 |
671.01 |
585.64 |
| Crystal system, space group |
Orthorhombic, Fdd2 |
Triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
Triclinic, P |
| Unit cell dimensions [Å, °] |
a = 19.971(2) |
a = 9.8005(5) |
a = 10.4398(10) |
|
b = 40.861(6) |
b = 12.8410(6) |
b = 10.9965(10) |
|
c = 13.081(2) |
c = 14.5139(7) |
c = 13.7776(18) |
|
α = 90.00 |
α = 65.006(1) |
α = 80.040(17) |
|
β = 90.00 |
β = 80.937(1) |
β = 79.204(12) |
|
γ = 90.00 |
γ = 70.876(1) |
γ = 72.490(9) |
| Volume [Å3] |
10675(3) |
1563.74(13) |
1470.2(3) |
|
Z, calculated density [Mg m−3] |
8, 1.348 |
2, 1.425 |
2, 1.323 |
| Absorption coefficient [mm−1] |
0.412 |
0.564 |
0.622 |
|
F(000) |
4536 |
698 |
610 |
| Crystal size [mm] |
0.290 × 0.221 × 0.086 |
0.254 × 0.116 × 0.069 |
0.205 × 0.154 × 0.084 |
|
θ range for data collection [°] |
1.93 to 22.67 |
1.83 to 26.45 |
2.32 to 28.41 |
| Index ranges |
−21 ≤ h ≤ 21, −44 ≤ k ≤ 44, −14 ≤ 1 ≤ 14 |
−12 ≤ h ≤ 12, −15 ≤ k ≤ 16, −18 ≤ 1 ≤ 17 |
−13 ≤ h ≤ 13, −14 ≤ k ≤ 14, −18 ≤ 1 ≤ 18 |
| Reflections collected/unique |
14575/3482 [R(int) = 0.0355] |
19093/6222 [R(int) = 0.0287] |
19656/7266 [R(int) = 0.0684] |
| Min. and max. transmission |
0.879 and 0.972 |
0.914 and 0.970 |
0.903 and 0.984 |
| Data/restraints/parameters |
3482/1/333 |
6222/0/448 |
7266/0/380 |
| Goodness-of-fit on F2 |
1.030 |
1.050 |
1.016 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0372, wR2 = 0.0918 |
R1 = 0.0378, wR2 = 0.1053 |
R1 = 0.0672, wR2 = 0.1655 |
|
R indices (all data) |
R1 = 0.0467, wR2 = 0.0989 |
R1 = 0.0501, wR2 = 0.1151 |
R1 = 0.1067, wR2 = 0.1946 |
| Largest diff. peak and hole [e Å−3] |
0.169 and −0.300 |
0.244 and −0.316 |
0.929 and −0.549 |
Catecholase like activity of complexes 1–3
In order to confirm the ability of the manganese(II) complexes to oxidize 3,5-DTBC, 1 × 10−4 mol dm−3 methanolic solutions of 1–3 were treated with 1 × 10−2 mol dm−3 (100 equiv.) of 3,5-DTBC under aerobic conditions. The reaction was followed by UV–vis spectroscopy. The kinetics of the 3,5-DTBC oxidation was measured by monitoring the increase of the concentration of the product 3,5-DTBQ. The kinetics for the oxidation of the substrate 3,5-DTBC were determined by the initial rate method at 25 °C. The concentration of the substrate 3,5-DTBC was always kept 10 times higher than that of the complex and the increase of the respective quinone concentration was determined at an ∼410 nm wavelength for each complex. Solutions of substrates of concentrations ranging from 0.001 to 0.05 mol dm−3 were prepared from a concentrated stock solution in methanol. 2 mL of the substrate solution were taken in a 1 cm spectrophotometer quartz cell and followed by the addition of 0.04 mL of 0.005 mol dm−3 complex solution. The dependence of the initial rate on the concentration of the substrate was monitored spectrophotometrically at a particular wavelength. Moreover the initial rate method showed a first-order dependence on the complex concentration and exhibited saturation kinetics at higher substrate concentrations. Since all the complexes showed saturation kinetics, a treatment based on the Michaelis–Menten model seemed to be appropriate. The binding constant (KM), maximum velocity (Vmax), and rate constant for the dissociation of substrates (i.e., turnover number, kcat) were calculated for the complexes by using the Lineweaver–Burk graph of 1/V vs. 1/[S], using the equation 1/V = {KM/Vmax}{1/[S]} + 1/Vmax.
Detection of hydrogen peroxide in the catalytic reactions
The formation of H2O2 during the catechol oxidation reaction was identified following the advancement of the characteristic band for I3− spectrophotometrically (λmax = 353 nm; ε = 26000 M−1 cm−1), upon reaction with I−.26,27 The oxidation reactions of 3,5-DTBC in the presence of different catalysts were carried out as in the kinetic experiments ([Complex] = 2.5 × 10−5 M; [3,5-DTBC] = 50 × 10−5 M). After completion of the reaction the same volume of water was added and the quinone formed was extracted several times with dichloromethane. The aqueous layer was acidified with H2SO4 until the pH is equal to 2 to stop further oxidation, and 1 mL of a 10% solution of KI and a few drops of 3% solution of ammonium molybdate were added. In the presence of hydrogen peroxide, iodine was produced that formed tri-iodide ions with an excess of iodide ions. The reaction rate is slow but increases with increasing concentrations of the acid, and the addition of an ammonium molybdate solution makes the reaction almost instantaneous. The formation of I3− could be examined spectrophotometrically following the development of the I3− band at λ = 353 nm (ε = 26000 M−1 cm−1).4,28
Computational details
All DFT calculations were performed with the Amsterdam Density Functional (ADF) suite of programs.29,30 MOs were expanded in an uncontracted set of Slater type orbitals31 (STOs) of triple-ζ quality containing diffuse functions and two sets of polarization functions (TZ2P), or a mixture of triple-ζ quality on the metal (TZP) and double-ζ quality (DZP) on all other atoms, in both cases with one set of polarization functions, which we name here as TDZP.31 Core electrons (1s for 2nd period and 1s2s2p for 3rd and 4th period) were not treated explicitly during the geometry optimizations (frozen core approximation), as it was shown to have a negligible effect on the obtained geometries.32 An auxiliary set of s, p, d, f, and g STOs was used to fit the molecular density and to represent the Coulomb and exchange potentials accurately for each SCF cycle.
Geometries were optimized with the QUILD program33 which uses superior optimization routines based on the adapted delocalized coordinates34 until the maximum gradient component was less than 10−4 a.u. This computational setup was shown to work well for transition-metal complexes.35 The starting point for geometry optimizations was the structures obtained from the X-ray diffraction analysis. Energies and gradients were calculated using the BP86 functional36,37 and TDZP basis set with the inclusion of Grimme's dispersion (D3) correction.38 The COSMO39–41 dielectric continuum model was used for implicit treatment of the environment (with methanol as a solvent).42,43 Scalar relativistic corrections have been included self-consistently by using the zeroth-order regular approximation (ZORA).44–46 Subsequent single point calculations that utilize all electron TZ2P basis sets have been performed on all optimized geometries, with S12g47 level of theory.
Catalytic oxidation of o-aminophenol
Phenoxazinone synthase like activity of the complexes was examined by the reaction of 1.0 × 10−4 M solutions of the complexes with 0.01 M solution of o-aminophenol (OAPH) in dioxygen-saturated methanol at 300 K. The reaction was followed spectrophotometrically by monitoring the development of the absorbance at the wavelength of 433 nm which is a characteristic band of the phenoxazinone chromophore.48,49 To determine the dependence of the rate of the reaction on the substrate concentration and to evaluate various kinetic parameters, 1.0 × 10−4 M solution of the complexes were mixed with the substrate under the pseudo-first-order conditions. Moreover, to check the rate dependency on catalyst concentrations a similar set of experiments were performed at a fixed concentration of the substrate with varying amounts of catalyst. The rate of a reaction was evaluated from the initial rate method, and the average initial rate over three independent measurements was recorded.
Results and discussion
Preparation and characterization
A Schiff-base (LH) was synthesized via a 1:2 condensation of 2,6-diformyl-4-tertiary-butylphenol and 2-(2-aminoethyl)pyridine in methanol.7 Complex 1 has been prepared by adding manganese(II) perchlorate hydrate to the methanolic solution of LH. Complex 2 was obtained by adding an excess of sodium dicyanamide to a solution of 1. In the synthesis of complex 3, manganese chloride was used in place of a metal perchlorate salt, followed by the addition of excess sodium thiocyanate. FTIR spectra of complexes 1–3 show bands in the range of 1641–1653 and 1532–1538 cm−1 which can be assigned to the CN stretching and skeletal vibration, respectively (Fig. S1–S3, ESI†). The FTIR bands in the range of 1086–1059 cm−1 for complexes 1 and 2 are due to the presence of the ClO4− group.50 Complex 2 shows that three IR bands centered at 2159, 2219 and 2280 cm−1 correspond to the CN present at the N(CN)2− moiety. One sharp IR band at 2063 cm−1 is due to the CN stretching of the SCN− moiety present in complex 3. The effective magnetic moment for complexes 1–3 in the range of 5.85–5.90 B.M. at 300 K is highly consistent with the spin only value (5.92 B.M.) for high spin MnII. The oxidation state of the metal complexes has further been examined by the EPR study in methanol. All the complexes 1–3 exhibit characteristic six line EPR spectra as is expected for octahedral MnII species (Fig. S4 and S5, ESI†). ESI-MS spectra of all the complexes are presented in Fig. S6–S8 (ESI†). In the ESI-MS spectrum, three peaks for complex 1 can be assigned to [H(LH)]+, [Mn(L)]+ and [Mn(LH)ClO4]+, respectively. Complex 2 exhibits peaks at m/z = 415.25, 468.17, 507.29 and 568.12 which may be assigned to [H(LH)]+, [Mn(LH)]+, [Mn(L)K]+ and [Mn(LH){N(CN)2}]+, respectively. Similarly for complex 3 peaks at 415.33, 501.34 and 527.32 may be assigned to [(LH)H]+, [Mn(LH)CH3OH]+ and [Mn(LH)SCN]+, respectively.
Description of the crystal structures of complexes 1–3
The Cambridge Structural Database51 search showed that the bis-Schiff bases derived from 2,6-diformylphenol derivatives and 2-(2-pyridyl)ethylamine usually form dinuclear coordination compounds with metal ions. The mononuclear compounds have been obtained only for MnII and FeIII ions when the inorganic ions where used as a co-ligand.52–55 The incorporation of a bridging acetate ligand leads to formation of dinuclear manganese(II) coordination compounds.52,54
The crystal structures of complexes 1–3 are presented in Fig. 1–3, respectively. The crystal structure of 1 consists of a [Mn(HL)2]2+ mononuclear cationic species and perchlorate counter-anions, joined by C–H⋯O hydrogen bonds (Table S3 and Fig. S9, ESI†). It should be noted that a metal to ligand stoichiometry of 1:2 has never been observed in the metal complexes of flexible 2,6-bis[N-(2-pyridylethyl)iminomethyl]phenol ligands.51 The central manganese atom lies on a two-fold axis, thus the coordination entity occupies two asymmetric units and consequently each manganese ion is coordinated by two imino-nitrogen atoms, two pyridine nitrogen atoms and two phenolate oxygen atoms from two ligand molecules. The Mn1–O1 bonds are the strongest ones.56–59 The bidentate Schiff-base moiety of this compound is not planar, the imine nitrogen atom N1 deviates from the plane defined by O1–C8–C7–C11 atoms by 0.301(6) Å. The MnN4O2 octahedron is rhombically distorted, with the cis-N/O–Mn–N/O angles ranging from 81.08(15) to 105.40(17)°. As in 1 and 2, the second imine nitrogen atom from the Schiff base ligand is not coordinated to the metal centre and it forms an intra-molecular hydrogen bond with the phenolate group. The dihedral angle between the phenolate group plane and the side-arm pyridine ring plane is 37.24(22)°, whereas the dihedral angle between the phenolate group plane and coordinated pyridine ring plane is 55.89(13)°.
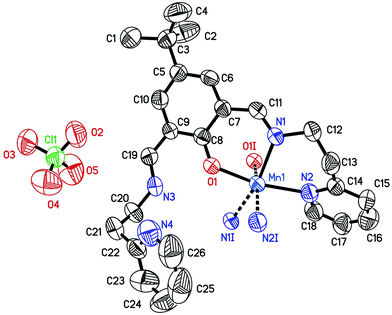 |
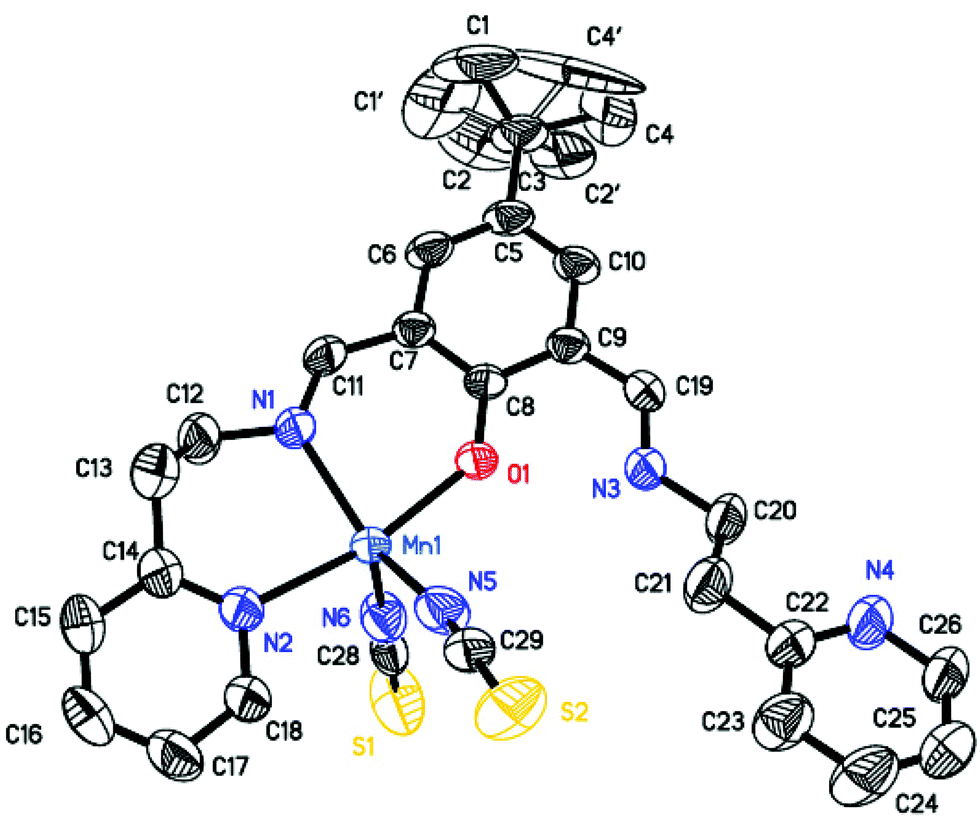
| | Fig. 1 The asymmetric unit of compound 1. The hydrogen atoms are omitted for clarity. The ellipsoids are drawn at the 50% probability level. The symmetry generated atoms N1I, N2I, O1I symmetry code: −x + 3/2, −y + 1/2, z. Selected bond lengths (Å) and angles (°): Mn1–O1 2.111(4), Mn1–N1 2.257(4), Mn1–N2 2.226(5), N1–C11 1.273(7), N3–C19 1.283(7), C11–N1–C12 116.1(4), C19–N3–C20 124.5(5). | |
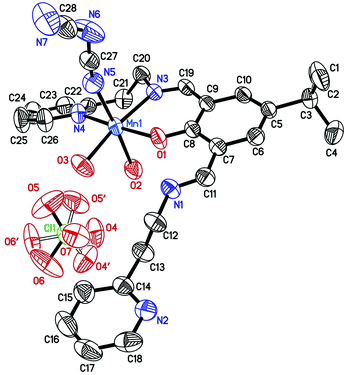 |
| | Fig. 2 Molecular structure of compound 2. The disordered domain of the perchlorate ion is indicated by the hollow lines. The hydrogen atoms are omitted for clarity. The ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Mn1–O1 2.0784(15), Mn1–O3 2.1784(17), Mn1–N5 2.223(2), Mn1–O2 2.2247(18), Mn1–N3 2.2306(16), Mn1–N4 2.2408(19), N1–C11 1.284(3), N3–C19 1.274(3), C11–N1–C12 126.3(2), C19–N3–C20 117.66(17). | |
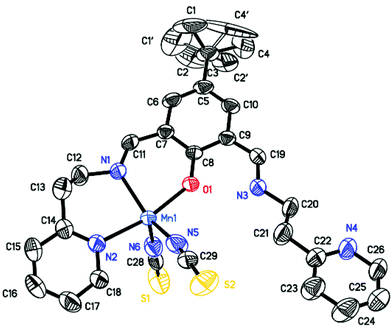 |
| | Fig. 3 Molecular structure of compound 3. The disordered domain of the tert-butyl group is indicated by hollow lines. The hydrogen atoms are omitted for clarity. The ellipsoids are drawn at the 50% probability level. Selected bond lengths (Å) and angles (°): Mn1–O1 2.073(2), Mn1–N1 2.185(3), Mn1–N2 2.249(3), Mn1–N5 2.111(3), Mn1–N6 2.091(3), N1–C11 1.271(4), N3–C19 1.280(4), C11–N1–C12 115.7(3), C19–N3–C20 125.6(3). | |
The asymmetric unit of 2 contains one [Mn(HL)(N3C2)(H2O)2]+ complex cation and a disordered perchlorate ion as the counter ion. The organic ligand coordinates to the manganese(II) ion in the formally neutral zwitterionic form. The coordination sphere geometry of the central atom can be described as a slightly distorted octahedron. The calculated bond valences56–59 indicated that the Mn1–O1 bond (of the phenolate group) is the strongest one whereas the Mn1–O2 (of the water molecule) bond is the weakest one. The methanol oxygen atom in 1 is also weakly bonded to the central ion. As in 1, the coordinated pyridine ring is twisted out of the bis-iminomethylphenolate plane by 31.29(8)°. The dicyanamide ions and water molecules are linked by the O–H⋯N hydrogen bonds (Table S3 and Fig. S10, ESI†) forming a dimer via the R22(10) motif. Further intermolecular O–H⋯N interactions between the pyridine ring and water molecules expand the complex dimer to the one dimensional chain extending along the crystallographic [01−1] axis. The perchlorate anions are the terminal units of the supramolecular chain, bonded to complex cations via O–H⋯O hydrogen bonds.
The asymmetric unit of 3 contains one neutral [Mn(HL)(SCN)2] unit. The manganese(II) ion displays a distorted tetragonal–pyramidal coordination geometry formed by the phenolate oxygen atom, the imine and the pyridine nitrogen atoms of 2,6-bis[N-(2-pyridylethyl)iminomethyl]-4-tert-butylphenolate ligand, and two nitrogen atoms of thiocyanate anions. One of the side-arm uncoordinatated imine nitrogen atom is protonated, which allows the formation of an intramolecular N–H⋯O hydrogen bond with the phenolate oxygen atom. The Mn–O and Mn–N bond lengths vary from 2.073(2) to 2.249(3) Å, and they are similar to those found for other mononuclear MnII coordination compounds.52–54 The intermolecular assembly of 3 is dominated by π⋯π stacking interactions of 6-membered rings of adjacent molecules (Table S3 and Fig. S11, ESI†). The coordinated pyridine ring is twisted out of the bis-iminomethylphenolate plane by 18.27(20)°.
The molecular structure of the two synthesized 1:1 complexes, 2 and 3, is similar to that of the previously reported mononuclear manganese(II) coordination compounds of 2,6-bis[N-(2-pyridylethyl)iminomethyl]-4-methylphenol possessing the azide,52 the nitrate53 and the thiocyanate52–54 ions, respectively. Complex 1 is the first example of a compound crystallizing with a 1:2 metal to Schiff base ligand stoichiometry.
Electrochemical studies
Cyclic voltammograms for the complexes are presented in Fig. S12–S14 (ESI†). All the complexes show a strong, irreversible reductive wave at −1.25 to −1.29 V, which is assigned to the reduction of the imino moiety of the Schiff base ligand. At more negative potential, complexes 2 and 3 show another quasi-reversible reductive couple at around −1.65, with a peak to peak separation of 110–160 mV, which is attributed to the addition of electrons to the pyridine π*-orbital of the Schiff base ligand. For complex 1, containing two Schiff base ligands, two pyridine centered reductions are observed at −1.7 and −1.8 V, but the electrode processes are nearly irreversible in nature. On the positive side, for complexes 2 and 3 two irreversible ligand centered oxidations are observed at 0.92 and 1.4 V for complex 2 and at 1.1 V and 1.75 V for complex 3. In the case of complex 1, two quasi-reversible oxidations are observed at 0.45 V (ΔEp = 100 mV) and 0.75 V (ΔEp = 100 mV), followed by an irreversible oxidation at 1.5 V. The couple at 0.45 V is tentatively assigned to the Mn(III)/Mn(II) couple, whereas the other two oxidative responses are probably ligand oxidations. The presence of two phenolato oxygens in the coordination sphere of Mn(II) in complex 1 probably makes it easier to oxidize to Mn(III) compared to the other two complexes where the Mn(III)/Mn(II) couple could not be detected.
Catecholase like activity
We have examined the catalytic activity of the complexes by evaluating the oxidation of 3,5-di-tert-butylcatechol (3,5-DTBC) via O2 activation to o-quinone in air-saturated methanol solution at 25 °C. All the three mononuclear MnII complexes show significant catalytic oxidation of the substrates as monitored by means of UV–vis spectroscopy. The kinetics for the oxidation of the substrates 3,5-DTBC has been determined by monitoring the increased concentration of the products, 3,5-DTBQ, following the procedure reported in the Experimental section. The spectral scans for complex 1, 2 and 3 are presented in Fig. 4, S15 and S16,† respectively. Enzyme kinetics plots data for complexes 1–3 are reported in Fig. S17–S22 (ESI†). Analysis of the experimental data shows that the Michaelis binding constant (KM) values are significantly different from each other and they vary in a wide range from 1.7 × 10−2 to 1.08 × 10−3, whereas the Vmax values for all the complexes are very similar and vary in a narrow range from 2.883 × 10−5 to 1.68 × 10−5. Kinetic parameters for complexes 1–3 are shown in Table 2. Table 3 represents the kcat values of some previously reported transition metal complexes.4,16,24,53,60 Upon comparison of Tables 2 and 3, it may be stated that our synthesized complexes belong to the highly efficient catalyst group, where the order of their activity is 1 > 2 > 3. We can also conclude that the transition metal complexes undergoing ligand centred catechol oxidation have a higher kcat value than that of the metal complexes having metal centered catechol oxidation properties. On the basis of the ESI-MS study, complexes 1, 2 and 3 should exist in solution as [Mn(LH)ClO4]+, [Mn(LH){N(CN)2}]+, and [Mn(LH)SCN]+, respectively. Due to their non-coordinating nature perchlorate ions generally act as counter anions and consequently the overall charge of complex 1 is +2. On the other hand due to the coordinating ability of N(CN)2− and SCN− they should be present in the inner coordination sphere imparting +1 overall charge for both complexes 2 and 3, respectively. It is quite evident that higher the effective charge on the metal ion higher will be the catalyst–substrate interactions followed by higher efficiency, as is observed in the case of complex 1. Structural characterization reveals that Mn–N bond distances with N(CN)2 and (NCS) are 2.223 and 2.165 Å for complexes 2 and 3, respectively. A longer Mn–N bond distance for complex 2 causes easy dissociation to accommodate the incoming substrate 3,5-DTBC to a greater extent, thereby showing higher activity over complex 3. In order to get insight into the nature of possible complex–substrate intermediates, the ESI-MS positive spectra of a 1:50 mixture of complex 1 (most active) and 3,5-DTBC in methanol are recorded after 5 minutes of mixing, and the spectra is depicted in Fig. S23 (ESI†). The peak at m/z = 690.3265 can be assigned to the 1:1 complex–substrate aggregate, [Mn(L)(3,5-DTBC)]+. The rest of the peaks at m/z = 468.1732, 415.2570 and 243.1356 can be considered as [Mn(L)]+, [H(LH)]+ and [3,5-DTBQ-Na]+, respectively.
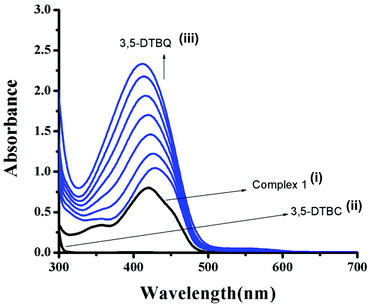 |
| | Fig. 4 UV–vis spectra of (i) complex 1, (ii) 3,5-DTBC, and (iii) changes in UV–vis spectra of complex 1 upon addition of 3,5-DTBC observed after each 5 min interval. | |
Table 2 Kinetic parameters of catecholase like activity for complex 1–3
| Catalyst |
V
max (M s−1) |
K
M (M) |
k
cat (h−1) |
|
1
|
2.883 × 10−5 |
1.7 × 10−2 |
1038 |
|
2
|
2.420 × 10−5 |
1.08 × 10−3 |
871.2 |
|
3
|
1.680 × 10−5 |
4.105 × 10−3 |
604.8 |
Table 3
k
cat values of some previously reported transition metal complexes
| Catalysta |
Solvent |
k
cat (h−1) |
Ref.year |
|
(1) HL1 = 2-[(2-piperazin-1-ylethylimino)methyl]phenol; (2) HL1 = 2-[1-(3-methylaminopropylamino)ethyl]phenol; (3) HL7 = 6-(bispyrazol-1-ylmethylamino)hexan-1-ol; (4) H3L = 2,6-bis[{{(2-hydroxybenzyl)(N′,N′-(dimethylamino)ethyl)}amino}methyl]-4-methylphenol; (5) diep = 2,8-dimethyl-5,11-bis(pyridin-2-ethyl)-1,4,5,6,7,10,11,12-octahydroimidazo[4,5-h]imidazo[4,5-c][1,6]-diazecine; (6) 6′Me2indH = 1,3-bis(6′-methyl-2′-pyridylimino)isoindoline; (7–9) HL = 2,6-bis{2-(N-ethyl)pyridineiminomethyl}-4-methylphenolato; (10, 11) HL1 = 2-formyl-4-chloro-6-N-ethylmorpholine-iminomethyl-phenol; (12) HL2 = 2-formyl-4-methyl-6-N-ethylpyrrolidine-iminomethyl-phenol; (13) LMe,Me = 1,3-bis[(N,N,N′-trimethylaminoethyl)aminomethyl]benzene; (14) L1 = 2,7-bis(2-hydroxyphenyl)-2,6-diazaocta-2,6-diene; (15, 16) L2 = 1,7-bis(2-hydroxyphenyl)-2,6-diazahepta-1,6-diene); (17) L2 = N,N-1-methylethylenebis(3-formyl-5-methylsalicylaldimine); (18) L3 = N,N-1,1-dimethylethylenebis(3-formyl-5-methylsalicylaldimine); (19) L4 = N,N-cyclohexenebis(3-formyl-5-methylsalicylaldimine); (20) L1 = 1,3-bis(4,6-di-tert-butyl-2-iminophenol)benzene; (21) L2 = 2-anilino-4,6-di-tert-butylphenol; (22) L3 = 2-(3,5-di-t-butyl-anilino)-4,6-di-tert-butylphenol; (23) L4 = 2-(3,5-ditrifluromethane-anilino)-4,6-di-tert-butylphenol; (24) L5 = 2-(3,5-dimethyl-anilino)-4,6-di-tert-butylphenol; (25) L6 = 2-(3,5-dichloro-anilino)-4,6-di-tert-butylphenol; (26) L7 = 2-(3,5-dimethoxy-anilino)-4,6-di-tert-butylphenol.
|
| (1) [NiIIL1(H2O)3]I2·H2O |
Methanol |
92.7 |
4
2013
|
| (2) [NiII2(L1)2(NCS)2] |
Acetonitrile |
64.1 |
60a
2012
|
| (3) [CuII(L7)Cl]Cl |
Methanol |
11.16 |
60b
2011
|
| (4) [CuII2(H2L)(μ-OH)](ClO4)2 |
Methanol |
28.74 |
60c
2008
|
| (5) CuII2diep |
Water–methanol |
63 |
60d
2012
|
| (6) [MnII(6′Me2indH)(H2O)2(CH3CN)](ClO4)2 |
DMF |
48.9 |
60e
2008
|
| (7) [MnII(HL)(H2O)3](NO3)2·(H2O)] |
Methanol |
2160 |
53
2011
|
| (8) [MnII(HL)(SCN)2(H2O)]·0.5H2O] |
Methanol |
1440 |
53
2011
|
| (9) [MnII(HL)(N(CN)2)(H2O)2](NO3)·H2O] |
Methanol |
720 |
53
2011
|
| (10) [MnII(L1)(SCN)2(H2O)] |
DMSO |
607.08 |
24c
2016
|
| (11) [MnII2(L1)(OAc)2](BPh4)] |
DMSO |
853.38 |
24c
2016
|
| (12) [MnII2(L2)(OAc)2(dca)]n |
DMSO |
255.19 |
24c
2016
|
| (13) [CuII2(LMe,Me–O)(OH)(OClO3)][ClO4]·MeCN |
Methanol |
564 |
16
2012
|
| (14) [MnIIIL1(OOCH)(OH2)] |
Acetonitrile |
936.64 |
60f
2012
|
| (15) [MnIIIL2(OH2)2][Mn2(L22(NO2)3] |
Acetonitrile |
365.34 |
60f
2012
|
| (16) [MnIII2L2(NO2)2] |
Acetonitrile |
1432.74 |
60f
2012
|
| (17) [MnIIIL2Cl·4H2O] |
Methanol |
247 |
60g
2009
|
| (18) [MnIIIL3Cl·4H2O] |
Methanol |
360 |
60g
2009
|
| (19) [MnIIIL4Cl·4H2O] |
Methanol |
720 |
60g
2009
|
| (20) [MnIV2(L1A)2(L1˙˙) |
DCM |
12.60 |
60h
2004
|
| (21) [MnIV(L2A)(L2˙)2] |
DCM |
21.30 |
60h
2004
|
| (22) [MnIV(L3A)(L3˙)2] |
DCM |
55.20 |
60h
2004
|
| (23) [MnIV(L4A)(L4˙)2] |
DCM |
14.80 |
60h
2004
|
| (24) [MnIV(L5A)(L5˙)2] |
DCM |
34.14 |
60h
2004
|
| (25) [MnIV(L6A)(L6˙)2] |
DCM |
16.32 |
60h
2004
|
| (26) [MnIV(L7A)(L7˙)2] |
DCM |
15.60 |
60h
2004
|
To find out the exact pathway of 3,5-DTBC oxidation, we performed the EPR experiment of only complex as well as a mixture of a complex and 3,5-DTBC. The only complex has six line spectra, which implies the Mn2+ systems. But the EPR signal of a mixture of 3,5-DTBC and complex 1 (as representative) has one extra peak at 337 mT (g = 1.999) along with six lines (Fig. 5). The EPR signal at g = 1.999 is undoubtedly due to the generation of an organic radical. We have already reported that the generation of an imine bond radical (g ≈ 2) is responsible for catechol oxidation.4,7,61 It is now essential to know whether dioxygen reduces to water or H2O2 during the oxidation process. The oxidation of I− to I2 followed by the generation of I3−, as is evident from the UV–vis spectral study of the solution (Fig. S24, ESI†) obtained after a suitable workup of the mixture of catechol, complex, and KI (see the Experimental section), clearly hints that dioxygen is reduced to H2O2, as reported by other investigators also.60a An experiment to detect the formation of H2O2 was also carried out separately in the absence of oxygen as well as in the absence of 3,5-DTBC. In both cases we did not find any indication for the formation of H2O2, suggesting that both the substrate and oxygen are required together for the generation of H2O2. Quantitative estimation of H2O2 performed at different time intervals was carried out as reported earlier.15 The results suggest that the amount of H2O2 is nearly twice that of the amount of 3,5-DTBQ (Fig. S25, ESI†). This observation also strengthens our proposed mechanism for the catecholase like oxidation (vide infra).
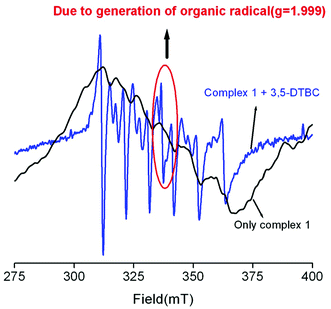 |
| | Fig. 5 EPR spectra of complex 1 and the mixture of complex 1 and 3,5-DTBC in methanol. | |
In addition to the generation of imine bond radicals, there are also some reports of phenoxo radical species which are accountable for catechol oxidation.62–66 Now to rationalize the exact pathway for our complexes we performed DFT calculation (vide infra).
A blank experiment, carried out with only Mn2+ salt and 3,5-DTBC in the absence of the ligand and another blank experiment with only ligand and 3,5-DTBC in the absence of the Mn2+ salt, showed no band around ∼400 nm in the UV–vis spectra, suggesting the effectiveness of our complexes.
Computational study
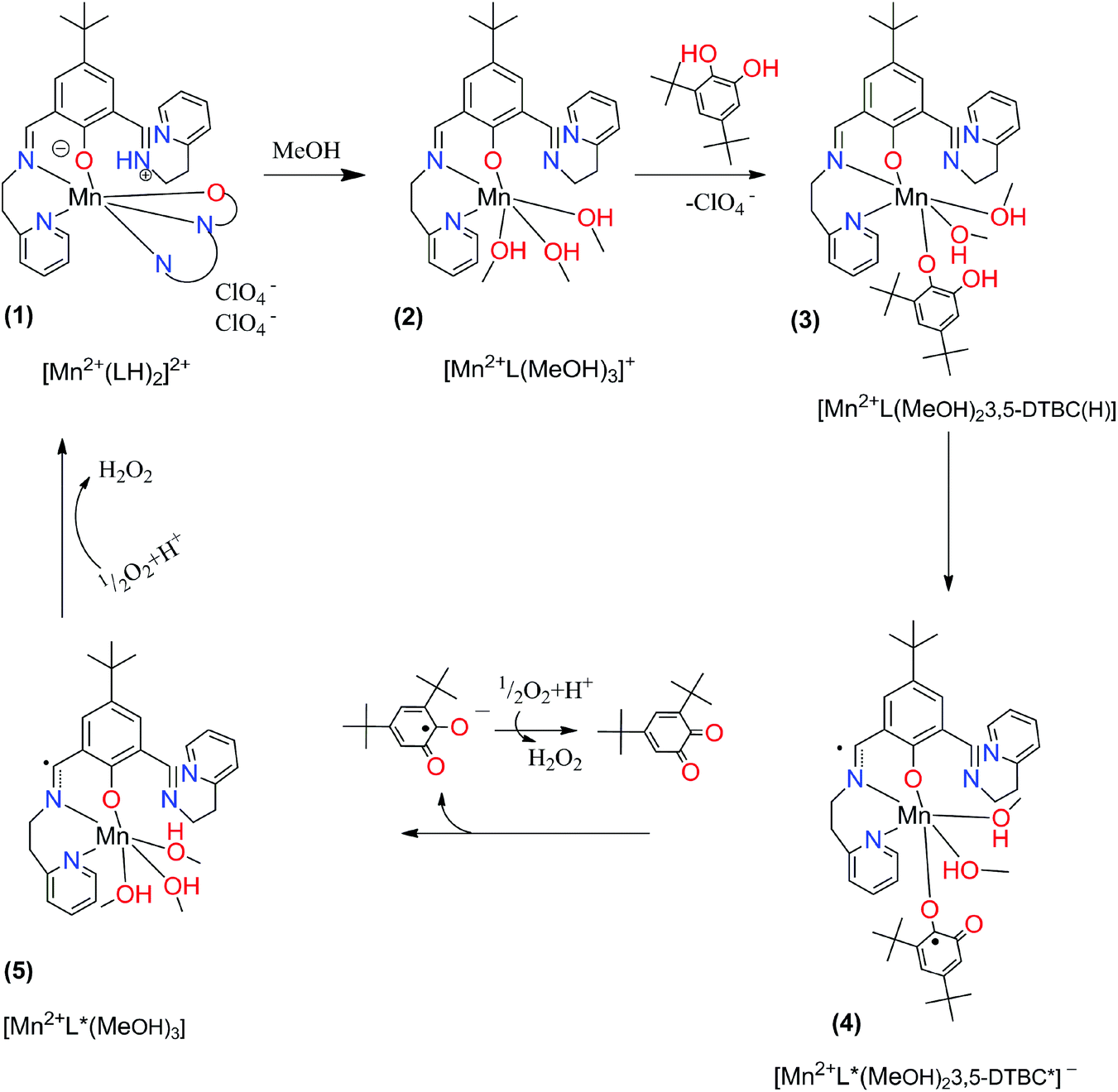
In order to investigate the assumed reaction mechanism we employed density functional theory (DFT) calculations. A characteristic feature of open-shell transition-metal ions in general is that several electronic configurations are accessible that may give rise to a number of different spin states, where the preferred one is determined by various factors. Since Mn2+ can be either in high-spin (HS), intermediate (IS), or low-spin (LS) state, in order to determine the ground state we performed calculations at the S12g/TZ2P//BP86-D3/TDZP levels of theory,31,36–38,46 including scalar relativistic29,67 and solvation effects,39,41,42 which has been proven to be accurate for the spin-state energetics.35,68,69 The proposed mechanistic cycle is given in Fig. 6.
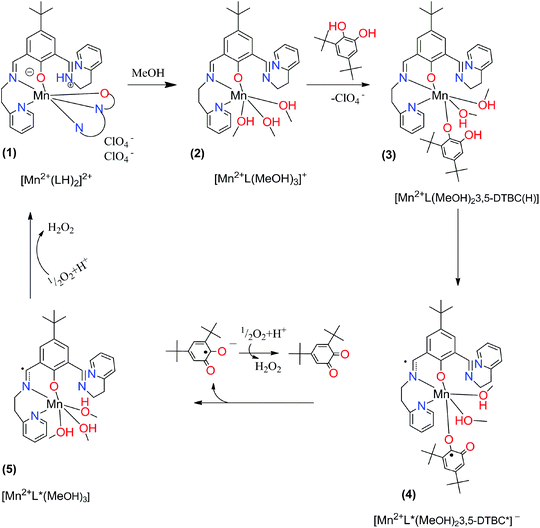 |
| | Fig. 6 Proposed mechanistic cycle for catecholase-like activity of complex (1) with 3,5-di-tert-butylcatechol. | |
The radicals in structures (4) and (5) can in principle be localized at the imine bond or at the phenoxy oxygen, see Fig. 7.
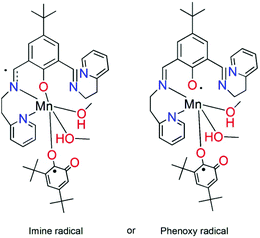 |
| | Fig. 7 Plausible structures of radical intermediate (4). | |
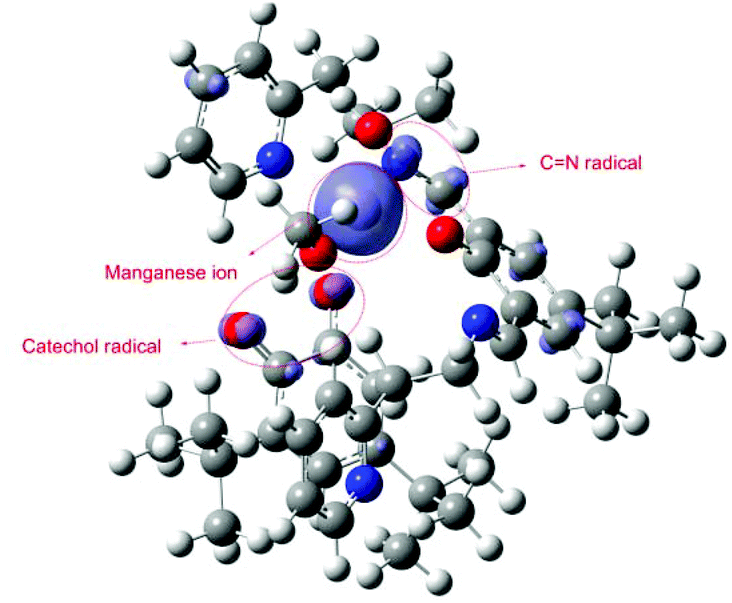
Our DFT calculations revealed that the active intermediate should be the high spin Mn(II) complex with the radical localized at the imine, Fig. 8. The spin density from the metal is slightly delocalized over the ligating atoms that can stabilize it with conjugation (pyridine and imine nitrogens and catechol oxygen). Besides at the metal, an excess of spin density can be observed at the imine and catechol carbons (which is further delocalized in tiny portions over conjugated positions). Hence according to our calculations the formation of an imine radical (4) is preferred.
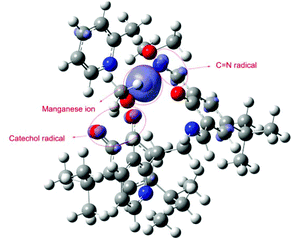 |
| | Fig. 8 Spin density of intermediate (4). | |
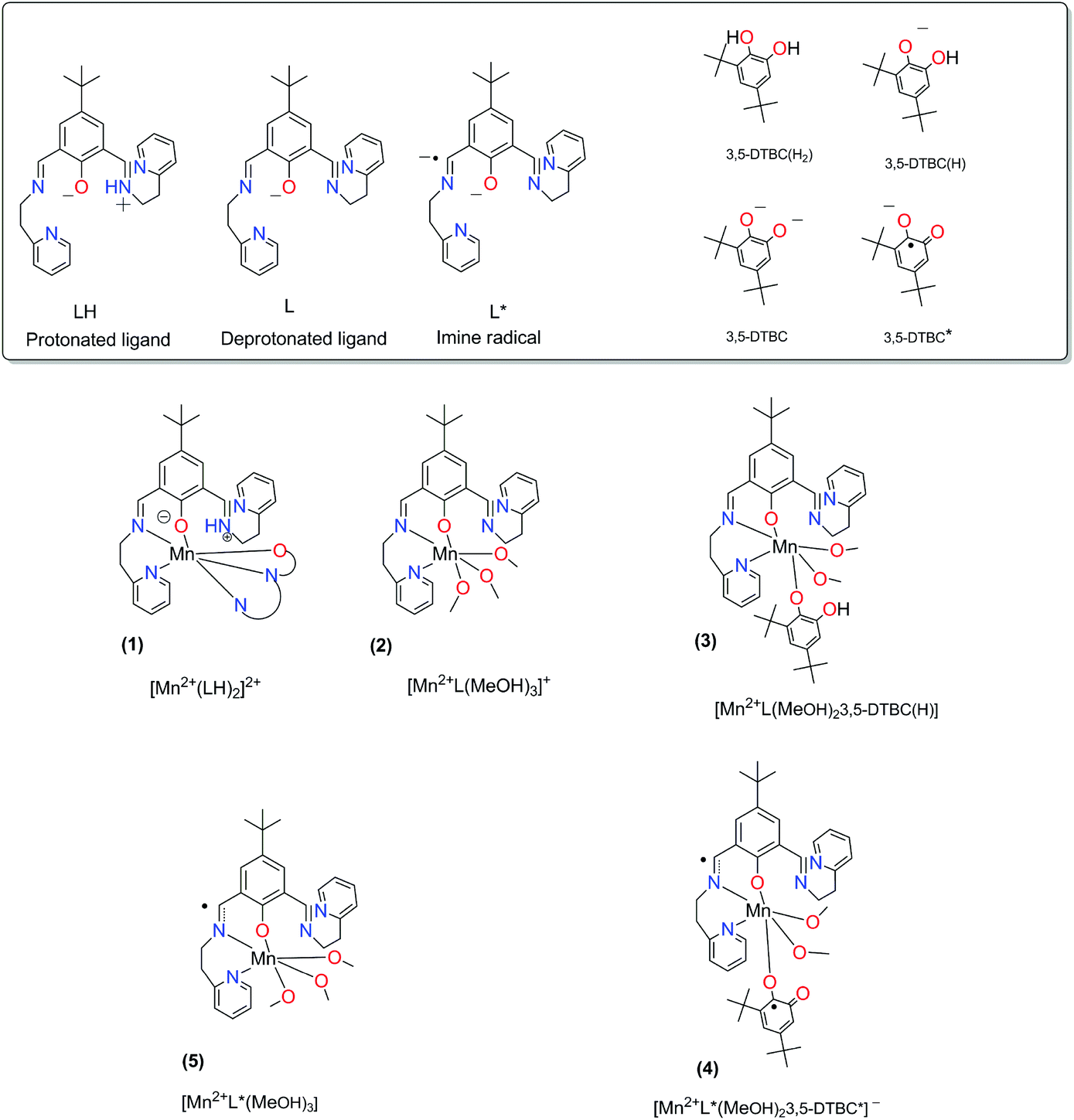
The results for the spin state calculations are given in Table 4. The abbreviations for the complexes and ligands are explained in Fig. 9.
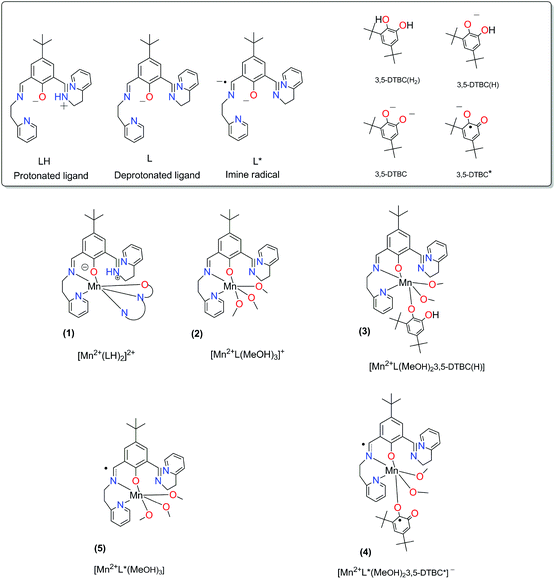 |
| | Fig. 9 The abbreviations for the complexes and ligands that are used in Table 4. | |
Table 4 Spin state energiesa (S12g/TZ2P//BP86-D3/TDZP) for complex species (1–5) from the catalytic cycle described in Fig. 6
| Spin state |
[Mn2+(LH)2]2+ (1) |
[Mn2+L(MeOH)3]+ (2) |
[Mn2+L(MeOH)23,5-DTBC(H)] (3) |
[Mn2+L*(MeOH)23,5-DTBC*]− (4) |
[Mn2+L*(MeOH)3] (5) |
|
Scalar relativistic corrections and solvation effects included in both geometry optimization and single-point energy calculations.
This structure does not correspond to intermediate 4, there is no radical on the catechol.
Structure with no excess of spin density on any atom, i.e. a pure closed-shell singlet state.
Methanol dissociated away.
|
| LS |
9.28 |
25.25 |
28.23 |
0.34b |
18.53c |
| IS |
10.53 |
8.47 |
14.13 |
−12.01b,d |
−2.81d |
| HS |
0 |
0 |
0 |
0 |
0 |
As can be seen, for structures (1–3), Mn2+ is in the HS state, as expected. For structures (4–5), only the HS state has the required electronic structure (with an excess of spin density on imine and catechol) in order to be considered as a catalytically active intermediate. Lower spin states cannot be in the appropriate electronic structure (and it is unlikely that they participate in the catalytic cycle). Additionally, in the case of intermediate spin states, the MeOH dissociated (bond distances are about 2.5 angstroms and even more).
Phenoxazinone synthase activity
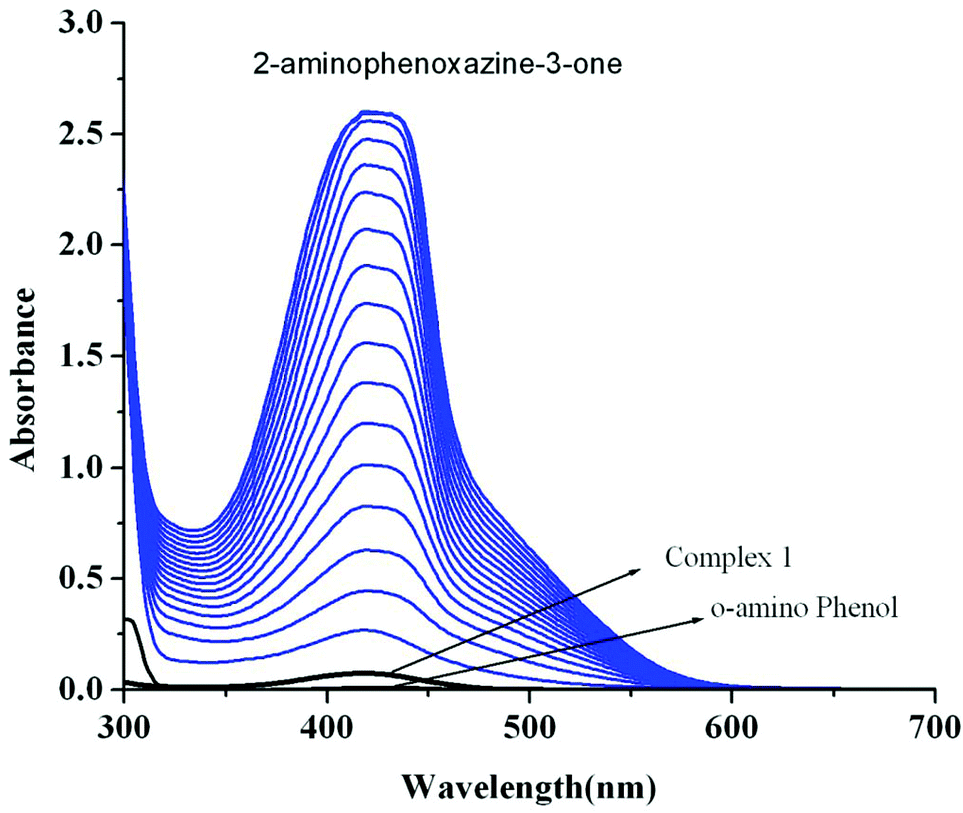
Catalytic oxidation of 2-aminophenol (OAPH) was examined spectrophotometrically in dioxygen-saturated methanol solution at 30 °C. In order to confirm the ability of the manganese(II) complexes to oxidize OAPH, 1.0 × 10−4 M methanolic solutions of the three complexes were treated with a 0.01 M solution of OAPH under aerobic conditions. The course of the reaction was followed by UV–Vis spectroscopy, and a representative time-resolved spectral profile for a period of 2 h after the addition of OAPH is shown in Fig. 10 for 1. The spectral scans for complexes 2 and 3 are shown in Fig. S26 and S27,† respectively (ESI†). The spectral scan reveals the progressive increase of the peak intensity at ca. 424 nm, a characteristic of phenoxazinone chromophores, suggesting the catalytic oxidation of OAPH to 2-aminophenoxazine-3-one under aerobic conditions.70 A blank experiment without a catalyst under identical conditions does not show significant increase in the band intensity at 424 nm. These spectral behaviors unambiguously demonstrate that all the complexes are active catalysts for the aerial oxidation of OAPH to the corresponding phenoxazinone chromophore.
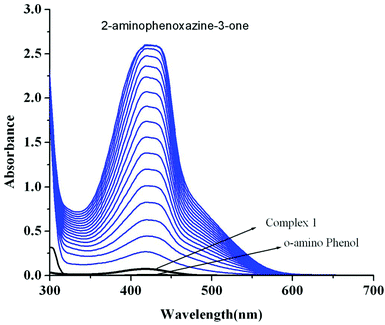 |
| | Fig. 10 UV–Vis spectral scans showing an increase in the phenoxazinone chromophore band at 420 nm after the addition of o-aminophenol (0.001 M) to a solution of complex 1 (1 × 10−4 M) in methanol at 25 °C. The spectra were recorded for a period of 2 h. | |
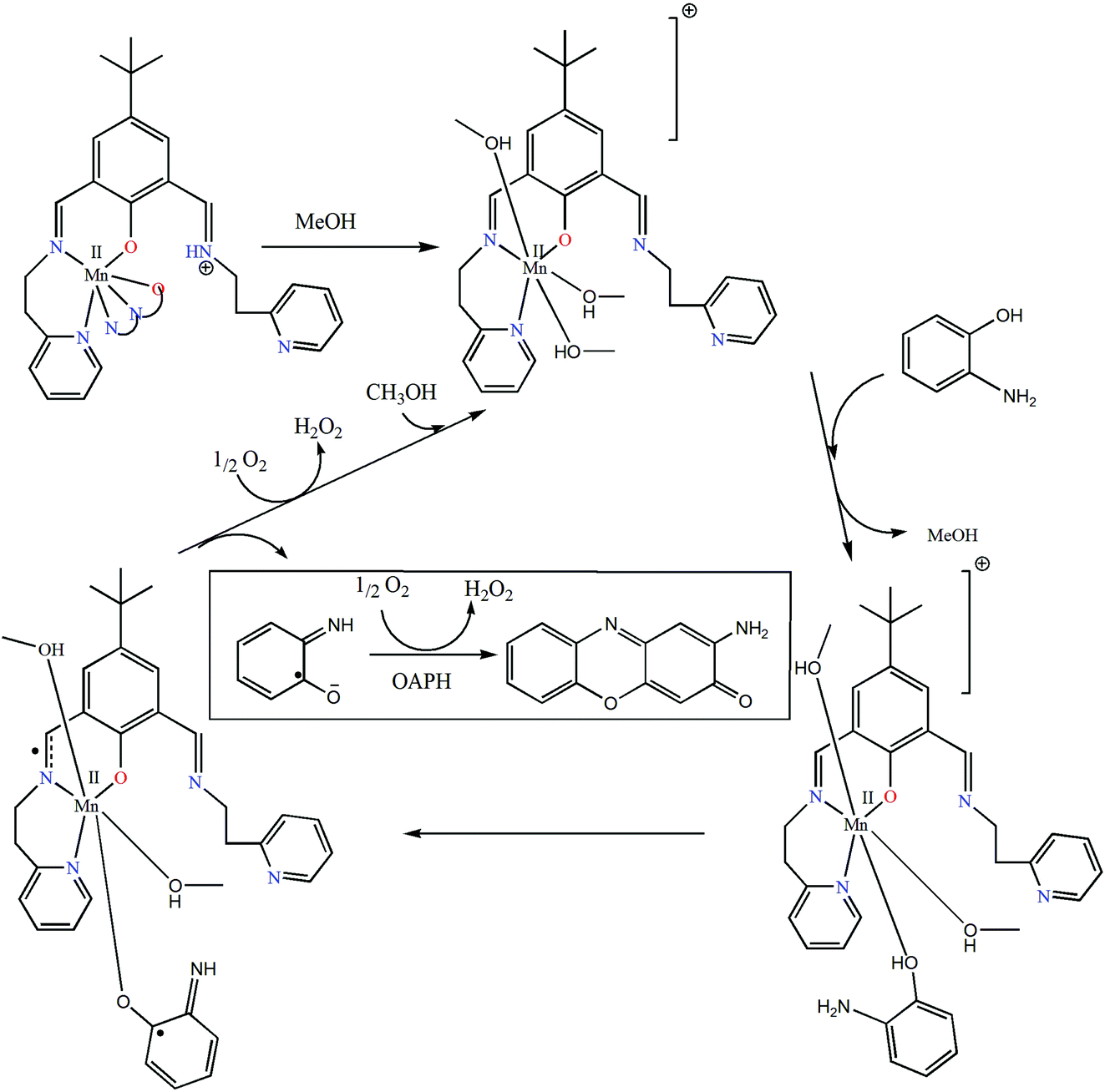
Reaction kinetics was performed to understand the extent of catalytic efficiency. For this purpose, 1.0 × 10−4 M solution of the complexes was treated with at least 10-fold excess of the substrate to follow the pseudo-first-order conditions. Kinetic studies were performed at a constant temperature of 25 °C, monitored with a thermostat under aerobic conditions. For a particular complex–substrate mixture, a time scan at the maximum band (424 nm) of 2-aminophenoxazine-3-one was carried out for a period of 20 min, and the initial rate was determined by linear regression from the slope of the absorbance versus time, and each experiment was performed thrice and the average values were noted. ESI-MS positive spectra of a 1:50 mixture of the complex 1 (most active) and o-aminophenol in methanol was recorded after 5 minutes of mixing, and the spectra are depicted in Fig. S28 (ESI†). The peak at m/z = 577.68 (for 1) can be assigned to [MnL-(OAPH)]+, which is consistent with the rate saturation kinetics as discussed earlier. Other major peaks at m/z = 468.42, 415.45, 235.16 and 213.18 are assigned to [Mn(L)]+, [H(LH)]+, [Na(2-aminophenoxazine-3-one)]+ and [(2-aminophenoxazine-3-one)H]+, respectively. The initial rate of the reactions versus concentrations of the substrate plot shows rate saturation kinetics as depicted in Fig. S29–S34 (ESI†). This observation indicates that the reaction proceeds through the formation of an intermediate complex–substrate adduct in a pre-equilibrium stage and that the irreversible substrate oxidation is the rate determining step of the catalytic cycle. This type of saturation rate dependency on the concentration of the substrate can be treated with the Michaelis–Menten model, which on linearization gives a double reciprocal Lineweaver–Burk plot to analyze the values of the parameters, Vmax, KM, and kcat. Analyses of the experimental data yielded the Michaelis binding constant (KM) values of 3.39 × 10−3, 3.55 × 10−3 and 3.34 × 10−3 M for 1–3, respectively, while the Vmax values of 3.85 × 10−6, 1.78 × 10−6 and 3.97 × 10−7 M s−1 for 1–3, respectively. The turnover number (kcat) value is obtained by dividing the Vmax by the concentration of the complex used, and is calculated to be 138.62, 64.07 and 14.2 h−1 for 1–3, respectively (Table 5). Table 6 represents the kcat values for the oxidation of OAPH by previously reported catalyst transition metal complexes.11,49,60e,70,71Table 6 clearly suggests that our complexes belong to a highly efficient catalyst group. In cyclic voltammetric studies we did not get any indication of oxidation from MnII to MnIII for both complexes 2 and 3. Therefore, it may be stated that both complexes are not oxidized during the oxidation of OAPH as proposed previously.70a However, both our complexes are able to catalyze the oxidation of OAPH. So there may be some new pathway that has been followed during catalytic oxidation of OAPH. In order to unveil that pathway we have performed an EPR experiment (Fig. 11). From the EPR experiment we have observed that the EPR signal of only complex 2 in methanol shows a six line spectrum as expected for the MnII system. However, the 1:50 mixture of complex 2 and OAPH exhibits a broad signal at g ≈ 1.98. Comparing these two EPR spectra we can conclude that the six line spectrum of MnII obtained from complex 2 is masked by a broad peak of the organic radical generated from complex 2 as reported earlier.72 We have also proposed a probable mechanistic pathway of OAPH oxidation by complex 1 (Fig. 12). Generation of H2O2 during the OAPH oxidation has been confirmed following the same procedure as mentioned previously in the Experimental section. Quantitative estimation of H2O2 at different time intervals implies that nearly 2 mol of H2O2 were shown to be produced per mole of OAPH oxidation (Fig. S35, ESI†). The result also supports our proposed mechanism for OAPH oxidation.
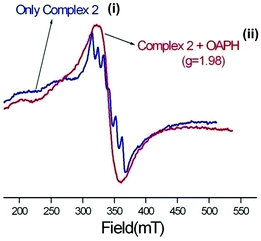 |
| | Fig. 11 EPR spectra of (i) complex 2 and (ii) 1:50 mixture of complex 2 and OAPH in methanol. | |
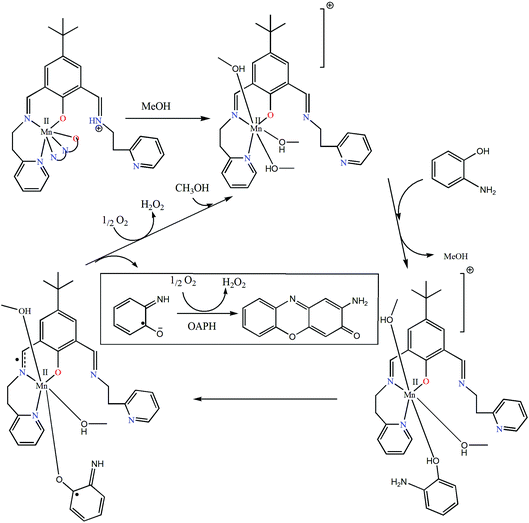 |
| | Fig. 12 Plausible mechanistic pathway showing the formation of 2-aminophenoxazine-3-one in which complex 1 is chosen as the model complex. | |
Table 5 Kinetic parameters (Vmax, KM, kcat) of complexes 1–3 for the oxidation of OAPH
| Catalyst |
V
max (M s−1) |
K
M (M) |
k
cat (h−1) |
|
1
|
3.85 × 10−6 |
3.39 × 10−3 |
138.62 |
|
2
|
1.78 × 10−6 |
3.55 × 10−3 |
64.07 |
|
3
|
3.97 × 10−7 |
3.34 × 10−3 |
14.2 |
Table 6
k
cat values for the phenoxazinone synthase like activity of different metal complexes previously reported
| Catalysta |
Solvent |
k
cat (h−1) |
Ref.year |
|
(1) HL5 = 1,3-bis(5′-methyl-2′-thiazolylimino)isoindoline; (2) L2 = (2-pyridylmethyl)(2-pyridylethyl)amine; (3) amp = 2-aminomethylpyridine; imp = 2-iminomethylpyridine anion; (4) L1 = N,N′-bis(pyridin-2-ylmethylene)-2,2-dimethylpropane-1,3-diamine; (5) L1 = pyridin-2-ylmethylene-[2-(2-pyridin-2-yl-tetrahydro-pyrimidin-1-yl)-ethyl]-amine; (6) L2 = (methoxy-pyridin-2-yl-methyl)-[2-(2-pyridin-2-yl-imidazolidin-1-yl)-ethyl]-amine; (7) L3 = {2-[2-(6-methyl-pyridin-2-yl)-imidazolidin-1-yl]-ethyl}-(6-methyl-pyridin-2-ylmethylene)-amine; (8) L4 = hexahydro-1-(2-(tetrahydro-2-(pyridin-2-yl)pyrimidin-(2H)-yl)ethyl)-2-(pyridin-2-yl)pyrimidine; (9) L1 = N,N′-bis(2-methylbenzimidazolyl)pyridinediamide; (10) 6′Me2indH = 1,3-bis(6′-methyl-2′-pyridylimino)isoindoline; (11) Hhebmz = 2-(α-hydroxyethyl)benzimidazole; (12) L1 = pyridin-2-ylmethylene-[3-(2-pyridin-2-yl-tetrahydro-pyrimidin-1-yl)-propyl]-amine; (13) L2 = N-(3-amino-propyl)-N′-(1-pyridin-2-yl-ethylidene)-propane-1,3-diamine.
|
| (1) [FeIIICl2(L5)] |
DMF |
137 |
71a
2014
|
| (2) [Co(L2)(N3)3] |
Methanol |
33.26 |
71b
2014
|
| (3) [Co2(amp)2(μ-imp)2Cl2]Cl2·2H2O |
Methanol |
13.75 |
11
2013
|
| (4) [Co(L1)Cl(H2O)]Cl·H2O |
Methanol |
13.68 |
49
2014
|
| (5) [Mn(L1)Cl2] |
Methanol |
11.90 |
71c
2014
|
| (6) [Mn(L2)Cl2] |
Methanol |
9.66 |
71c
2014
|
| (7) [Mn(L3)Cl2] |
Methanol |
8.20 |
71c
2014
|
| (8) [Mn(L4)Cl2]·H2O |
Methanol |
26.32 |
71c
2014
|
| (9) [Fe(L1)Cl3] |
Methanol |
56 |
71d
2012
|
| (10) [Mn(6′Me2indH)(H2O)2(CH3CN)](ClO4)2 |
DMF |
2.93 |
60e
2008
|
| (11) [Cu(hebmz)2] |
DMF |
29.7 |
71e
2005
|
| (12) [Mn(L1)Cl2]·2MeOH |
Methanol |
23.54 |
70a
2013
|
| (13) [Mn(L2)Cl2] |
Methanol |
27.32 |
70a
2013
|
Conclusion
Three mononuclear Mn(II) complexes of an “end-off” compartment ligand have been synthesized where the metal:ligand stoichiometry is unusually observed to be 1:2 in complex 1. All three complexes have been comprehensively structurally characterized by X-ray single crystal structural analyses. All three complexes have been utilized as a potential dioxygen activator to catalyze the aerobic oxidation of 3,5-DTBC and o-aminophenol as model substrates and thereby to exhibit catecholase-like and phenoxazinone synthase-like activities. All three complexes are highly active with different degrees i.e.1 > 2 > 3 and the efficiency order has been rationalized in terms of effective positive charge on the complexes and metal–ligand bond lability. EPR and CV studies suggest that oxidation of catechol and o-aminophenol catalyzed by our Mn(II) complexes are due to ligand bound radical generation rather than metal centered redox participation. Detailed DFT calculation on catechol oxidation is in favor of an imine bound radical instead of phenoxo radical formation as the basis of oxidation.
References
- E. Tsuchida and K. Oyaizu, Coord. Chem. Rev., 2003, 237, 213–228 CrossRef CAS.
- L. Canali and D. C. Sherrington, Chem. Soc. Rev., 1999, 28, 85–93 RSC.
- J. Tisato, F. Refosco and F. Bandoli, Coord. Chem. Rev., 1994, 135, 325–397 CrossRef.
- J. Adhikary, P. Chakraborty, S. Das, T. Chattopadhyay, A. Bauzá, S. K. Chattopadhyay, B. Ghosh, F. A. Mautner, A. Frontera and D. Das, Inorg. Chem., 2013, 52, 13442 CrossRef CAS PubMed.
- J. Adhikary, P. Kundu, S. Dasgupta, S. Mukherjee, S. Chattopadhyay, G. Aullón and D. Das, Polyhedron, 2015, 101, 93 CrossRef CAS.
- S. Anbu, M. Kandaswamy and B. Varghese, Dalton Trans., 2010, 39, 3823 RSC.
- T. Ghosh, J. Adhikary, P. Chakraborty, P. K. Sukul, M. S. Jana, T. K. Mondal, E. Zangrando and D. Das, Dalton Trans., 2014, 43, 841 RSC.
- V. K. Bhardwaj and A. Singh, Inorg. Chem., 2014, 53, 10731 CrossRef CAS PubMed.
- P. Chakraborty, J. Adhikary, B. Ghosh, R. Sanyal, S. K. Chattopadhyay, A. Bauzá, A. Frontera, E. Zangrando and D. Das, Inorg. Chem., 2014, 53, 8257 CrossRef CAS PubMed.
- A. Banerjee, A. Guha, J. Adhikary, A. Khan, K. Manna, S. Dey, E. Zangrando and D. Das, Polyhedron, 2013, 60, 102 CrossRef CAS.
- A. Panja and P. Guionneau, Dalton Trans., 2013, 42, 5068 RSC.
- N. A. Rey, A. Neves, A. J. Bortoluzzi, C. T. Pich and H. Terenzi, Inorg. Chem., 2007, 46, 348 CrossRef CAS PubMed.
- R. Marion, M. Zaarour, N. A. Qachachi, N. M. Saleh, F. Justaud, D. Floner, O. Lavastre and F. Geneste, J. Biol. Inorg. Chem., 2011, 105, 1391–1397 CAS.
- A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023–4031 CrossRef CAS PubMed.
-
(a) A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111–10121 CrossRef CAS PubMed;
(b) K. Selmeczi, M. Réglier, M. Giorgi and G. Speier, Coord. Chem. Rev., 2003, 245, 191–201 CrossRef CAS.
- S. Mandal, J. Mukherjee, F. Lloret and R. N. Mukherjee, Inorg. Chem., 2012, 51, 13148–13161 CrossRef CAS PubMed.
- M. R. Mendoza-Quijano, G. Ferrer-Sueta, M. Flores-Álamo, N. Aliaga-Alcalde, V. Gómez-Vidales, V. M. Ugalde-Saldívara and L. Gasque, Dalton Trans., 2012, 41, 4985–4997 RSC.
- M. Suzuki, H. Furutachi and H. Okawa, Coord. Chem. Rev., 2000, 105, 200–202 Search PubMed.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717 CrossRef CAS.
-
(a) I. A. Koval, P. Gamez, C. Belle, K. Selmeczi and J. Reedijk, Chem. Soc. Rev., 2006, 35, 814 RSC.
-
A. Sigel and H. Sigel, Manganese and Its Role in Biological Processes, in Metal Ions in Biological Systems, Marcel Dekker, New York, 2000, vol. 37 Search PubMed.
-
(a)
J. H. Whittaker, ref. 21; Chapter 18, pp. 587–611;
(b)
D. W. Yoder, J. Hwang and J. E. Penner-Hahn, ref. 21; Chapter 16, pp. 527–557;
(c)
M. H. Gold, H. L. Youngs and M. D. Sollewijn Gelpke, ref. 21; Chapter 17, pp. 559–586;
(d)
L. Que Jr. and M. F. Reynolds, ref. 21; Chapter 15, pp. 505–525;
(e)
C. W. Hoganson and G. T. Babcock, ref. 21; Chapter 19, pp. 613–656;
(f) K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS PubMed.
-
V. L. Pecoraro and W.-Y. Hsieh, ref. 21; Chapter 14, pp. 429–504.
-
(a) V. L. Pecoraro, M. J. Baldwin and A. Gelasco, Chem. Rev., 1994, 94, 807–826 CrossRef CAS;
(b) A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903–938 CrossRef CAS PubMed;
(c) P. Chakraborty, I. Majumder, K. S. Banu, B. Ghosh, H. Kara, E. Zangrando and D. Das, Dalton Trans., 2016, 45, 742–752 RSC.
-
(a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed;
(b) G. M. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8 CrossRef PubMed.
-
(a) E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553–562 CrossRef CAS PubMed;
(b) E. Monzani, G. Battaini, A. Perotti, L. Casella, M. Gullotti, L. Santagostini, G. Nardin, L. Randaccio, S. Geremia, P. Zanello and G. Opromolla, Inorg. Chem., 1999, 38, 5359–5369 CrossRef CAS.
- J. Ackermann, F. Meyer, E. Kaifer and H. Pritzkow, Chem. – Eur. J., 2002, 8, 247–258 CrossRef CAS.
- E. Monzani, L. Quinti, A. Perotti, L. Casella, M. Gullotti, L. Randaccio, S. Geremia, G. Nardin, P. Faleschini and G. Tabbi, Inorg. Chem., 1998, 37, 553–562 CrossRef CAS PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, Chemistry with ADF, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
-
E. J. Baerends, J. Autschbach, A. Bérces, J. A. Berger, F. M. Bickelhaupt, C. Bo, P. L. de Boeij, P. M. Boerrigter, L. Cavallo, D. P. Chong, L. Deng, R. M. Dickson, D. E. Ellis, M. van Faassen, L. Fan, T. H. Fischer, C. Fonseca Guerra, S. J. A. van Gisbergen, J. A. Groeneveld, O. V. Gritsenko, M. Grüning, F. E. Harris, P. van den Hoek, C. R. Jacob, H. Jacobsen, L. Jensen, E. S. Kadantsev, G. van Kessel, R. Klooster, F. Kootstra, E. van Lenthe, D. A. McCormack, A. Michalak, J. Eugebauer, V. P. Nicu, V. P. Osinga, S. Patchkovskii, P. H. T. Philipsen, D. Post, C. C. Pye, W. Ravenek, P. Romaniello, P. Ros, P. R. T. Schipper, G. Schreckenbach, J. G. Snijders, M. Solà, M. Swart, D. Swerhone, G. teVelde, P. Vernooijs, L. Versluis, L. Visscher, O. Visser, F. Wang, T. A. Wesolowski, E. M. van Wezenbeek, G. Wiesenekker, S. K. Wolff, T. K. Woo, A. L. Yakovlev and T. Ziegler, 2013.01, SCM, Amsterdam, 2013 Search PubMed.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- M. Swart and J. G. Snijders, Theor. Chem. Acc., 2003, 110, 34–41 CrossRef CAS , Erratum: M. Swart and J. G. Snijders, Theor. Chem. Acc., 2003, 111, 56 CrossRef.
- M. Swart and F. M. Bickelhaupt, J. Comput. Chem., 2008, 29, 724–734 CrossRef CAS PubMed.
- M. Swart and F. M. Bickelhaupt, Int. J. Quantum Chem., 2006, 106, 2536–2544 CrossRef CAS.
- M. Swart, Chem. Commun., 2013, 49, 6650–6652 RSC.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter, 1986, 33, 8822–8824 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- A. Klamt and G. Schuurmann, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805 RSC.
- A. Klamt and V. Jonas, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS.
- A. Klamt, J. Chem. Phys., 1995, 99, 2224–2235 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 CrossRef CAS.
- M. Swart, E. Rösler and F. M. Bickelhaupt, Eur. J. Inorg. Chem., 2007, 23, 3646–3654 CrossRef.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- M. Swart, Chem. Phys. Lett., 2013, 580, 166–171 CrossRef CAS.
- C. Mukherjee, T. Weyhermuller, E. Bothe, E. Rentschler and P. Chaudhuri, Inorg. Chem., 2007, 46, 9895 CrossRef CAS PubMed.
- A. Panja, Polyhedron, 2014, 80, 81 CrossRef CAS.
-
K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 3rd edn, 1978 Search PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef.
- M. Mikuriya, T. Fujii, T. Tokii and A. Kawamori, Bull. Chem. Soc. Jpn., 1993, 66, 1675 CrossRef CAS.
- A. Guha, K. S. Banu, A. Banerjee, T. Ghosh, S. Bhattacharya, E. Zangrando and D. Das, J. Mol. Catal. A: Chem., 2011, 338, 51 CAS.
- M. Mikuriya, T. Fujii, S. Kamisawa, Y. Kawasaki, T. Tokii and H. Oshio, Chem. Lett., 1990, 1181 CrossRef CAS.
- M. Mikuriya, K. Kushida, H. Nakayama, W. Mori and M. Kishita, Inorg. Chim. Acta, 1989, 165, 35 CrossRef CAS.
- W. H. Zachariasen, J. Less-Common Met., 1978, 62, 1 CrossRef CAS.
- I. D. Brown, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 53, 381 CrossRef.
- I. D. Brown, Acta Crystallogr., Sect. B: Struct. Sci., 1992, 48, 553 CrossRef.
- M. O'Keeffe and N. E. Brese, Acta Crystallogr., Sect. B: Struct. Sci., 1997, 47, 192 Search PubMed.
-
(a) A. Biswas, L. K. Das, M. G. B. Drew, G. Aromí, P. Gamez and A. Ghosh, Inorg. Chem., 2012, 51, 7993–8001 CrossRef CAS PubMed;
(b) R. Marion, M. Zaarour, N. A. Qachachi, N. M. Saleh, F. Justaud, D. Floner, O. Lavastre and F. Geneste, J. Biol. Inorg. Chem., 2011, 105, 1391–1397 CAS;
(c) A. Banerjee, S. Sarkar, D. Chopra, E. Colacio and K. K. Rajak, Inorg. Chem., 2008, 47, 4023–4031 CrossRef CAS PubMed;
(d) M. R. Mendoza-Quijano, G. Ferrer-Sueta, M. Flores-Álamo, N. Aliaga-Alcalde, V. Gómez-Vidales, V. M. Ugalde-Saldívara and L. Gasque, Dalton Trans., 2012, 41, 4985–4997 RSC;
(e) J. Kaizer, G. Baráth, R. Csonka, G. Speier, L. Korecz, A. Rockenbauer and L. Párkányi, J. Inorg. Biochem., 2008, 102, 773–780 CrossRef CAS PubMed;
(f) P. Seth, M. G. B. Drew and A. Ghosh, J. Mol. Catal. A: Chem., 2012, 365, 154 CrossRef CAS;
(g) K. S. Banu, T. Chattopadhyay, A. Banerjee, M. Mukherjee, S. Bhattacharya, G. K. Patra, E. Zangrando and D. Das, Dalton Trans., 2009, 8755 RSC;
(h) S. Mukherjee, T. Weyhermüller, E. Bothe, K. Wieghardt and P. Chaudhuri, Dalton Trans., 2004, 3842 RSC.
- A. Guha, T. Chattopadhyay, N. D. Paul, M. Mukherjee, S. Goswami, T. K. Mondal, E. Zangrando and D. Das, Inorg. Chem., 2012, 51, 8750–8759 CrossRef CAS PubMed.
- K. Ghosh, P. Kumar, N. Tyagi and U. P. Singh, Inorg. Chem., 2010, 49, 7614–7616 CrossRef CAS PubMed.
- A. Mukherjee, F. Lloret and R. Mukherjee, Inorg. Chem., 2008, 47, 4471–4480 CrossRef CAS PubMed.
- P. Chaudhuri, M. Hess, J. Muller, K. Hildenbrand, E. Bill, T. Weyhermuller and K. Wieghardt, J. Am. Chem. Soc., 1999, 121, 9599–9610 CrossRef CAS.
- G. M. Zats, H. Arora, R. Lavi, D. Yufit and L. Benisvy, Dalton Trans., 2012, 41, 47–49 RSC.
- R. Sanyal, S. K. Dash, S. Das, S. Chattopadhyay, S. Roy and D. Das, J. Biol. Inorg. Chem., 2014, 19, 1099–1111 CrossRef CAS PubMed.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef CAS.
- M. Swart, J. Chem. Theor. Comput., 2008, 4, 2057–2066 CrossRef CAS PubMed.
- M. Gruden, S. Stepanovic and M. Swart, J. Serb. Chem. Soc., 2015, 80, 1399–1410 CrossRef.
-
(a) A. Panja, RSC Adv., 2013, 3, 4954–4963 RSC;
(b) J. Kaizer, G. Baráth, R. Csonka, G. Speier, L. Korecz, A. Rockenbauer and L. Párkányi, J. Inorg. Biochem., 2008, 102, 773–780 CrossRef CAS PubMed;
(c) G. Blay, I. Fernández, J. R. Pedro, R. Ruiz-Garcia, T. Temporal-Sánchez, E. Pardo, F. Lloret and M. C. Munoz, J. Mol. Catal. A: Chem., 2006, 250, 20–26 CrossRef CAS;
(d) I. C. Szigyártó, T. M. Simándi, L. I. Simándi, L. Korecz and N. Nagy, J. Mol. Catal. A: Chem., 2006, 251, 270–276 CrossRef.
-
(a) M. Szávuly, R. R. Csonka, G. Speier, R. Barabás, M. Giorgi and J. Kaizer, J. Mol. Catal. A: Chem., 2014, 392, 120–126 CrossRef;
(b) A. Panja, M. Shyamal, A. Saha and T. K. Mandal, Dalton Trans., 2014, 43, 5443–5452 RSC;
(c) A. Panja, Polyhedron, 2014, 79, 258–268 CrossRef CAS;
(d) R. Bakshi, R. Kumar and P. Mathur, Catal. Commun., 2012, 17, 140–145 CrossRef CAS;
(e) M. R. Maurya, S. Sikarwar, T. Joseph and S. B. Halligudi, J. Mol. Catal. A: Chem., 2005, 236, 132–138 CrossRef CAS.
-
(a) K. Mitra, S. Biswas, S. K. Chattopadhyay and B. Adhikary, Transition Met. Chem., 2005, 30, 185–190 CrossRef;
(b) S. Naskar, S. Biswas, D. Mishra, B. Adhikary, L. R. Falvello, T. Soler, C. H. Schwalbe and S. K. Chattopadhyay, Inorg. Chim. Acta, 2004, 357, 4257–4264 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR spectra, crystallographic data, kinetic plots, electronic spectra of the formation of I3−, cyclic voltammograms, ESI-MS spectra and DFT data. CCDC 1025942–1025944 (1–3). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6dt00625f |
| ‡ Present address: Department of Chemical Sciences, Ariel University, Ariel 40700, Israel. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence fg and
Debasis
Das
*a
fg and
Debasis
Das
*a